

Abstract
Proteins with short nonpolar carboxyl termini are unstable in Escherichia coli. This proteolytic pathway is used to dispose of polypeptides synthesized from truncated mRNA molecules. Such proteins are tagged with an 11-amino-acid nonpolar destabilizing tail via a mechanism involving the 10Sa (SsrA) stable RNA and then degraded. We show here that the ATP-dependent zinc protease HflB (FtsH) is involved in the degradation of four unstable derivatives of the amino-terminal domain of the λcI repressor: three with nonpolar pentapeptide tails (cI104, cI105, cI108) and one with the SsrA tag (cI–SsrA). cI105 and cI-SsrA are also degraded by the ClpP-dependent proteases. Loss of ClpP can be compensated for by overproducing HflB. In an in vitro system, cI108 and cI–SsrA are degraded by HflB in an energy-dependent reaction, indicating that HflB itself recognizes the carboxyl terminus. These results establish a tail-specific pathway for removing abnormal cytoplasmic proteins via the HflB and Clp proteases.
Keywords: 10Sa RNA, AAA ATPase family, intracellular proteolysis, tail-specific proteolysis, Clp protease, proteasome 26S
Living cells have elaborate mechanisms such as check points and coupling devices to evaluate their physiological state and environment to maintain a harmonious cell cycle. In addition, cell growth requires efficient systems to clean up erroneous metabolites and macromolecules that would otherwise accumulate in the cytoplasm and ultimately arrest growth. Abnormal proteins are a frequent and cumbersome type of cytoplasmic junk, and ingenious proteolytic systems have evolved to recognize and remove them. Abnormal proteins can arise in various ways: from denaturing treatments, from improper folding of newly synthesized polypeptides (often the case for foreign proteins), and from premature termination of transcription or translation (Gottesman 1996; Miller 1996).
Cytoplasmic proteolysis serves several purposes (Gottesman 1996; Herman and D’Ari 1998). It permits rapid regulatory responses to specific signals, it creates protein turnover in times of starvation, and it removes abnormal proteins. The latter housekeeping role for cytoplasmic proteolysis is a major cell need, and inability to perform it quickly perturbs growth.
Regulatory proteolysis is generally quite specific, each unstable regulator being the preferential substrate of a particular protease, whereas in housekeeping proteolysis, abnormal proteins are often substrates of several proteases (Maurizi et al. 1985). Such lack of specificity is perhaps not surprising, since virtually any protein can become “abnormal” by denaturation, in which case recognition presumably exploits general criteria, shared by most proteases. However, the specific features of proteins that target them for degradation remain largely unknown.
In a systematic search for protease recognition signals in Escherichia coli, Parsell et al. (1990) showed that a stable protein can be destabilized by addition of nonpolar carboxy-terminal pentapeptide tags. A natural tagging mechanism of this sort was described recently in E. coli, by which a nonpolar amino acid tail is added to peptides translated from truncated mRNA molecules (Keiler et al. 1996), making nonpolar tails an important signal in housekeeping proteolysis. Carboxy-terminal nonpolar tags do not cause protein unfolding (Parsell et al. 1990), so they are presumably recognized directly by proteolytic systems. The periplasmic protease Tsp (or Prc) specifically recognizes these tagged polypeptides both in vitro and, for periplasmic proteins, in vivo (Keiler and Sauer 1996), but of course it does not degrade tagged cytoplasmic proteins in vivo (Silber and Sauer 1994). The proteases responsible for their degradation, which have not been identified previously, are the subject of this report.
We examined the ability of the HflB- and ClpP-dependent proteases to degrade cI repressor variants with nonpolar carboxy-terminal tails. HflB is the only essential protease in E. coli. It is a membrane-anchored protein with its active site in the cytoplasm (Tomoyasu et al. 1993) and is responsible for degradation of the the heat shock sigma factor σ32 and of the regulatory proteins cII and cIII of phage λ (Herman et al. 1995; Tomoyasu et al. 1995; Herman et al. 1997; Shotland et al. 1997); few other substrates are known (Kihara et al. 1995). ClpP is the proteolytic subunit of two different cytoplasmic proteases, ClpXP and ClpAP, each of which has distinct recognition signals. For example, ClpXP degrades the σ factor involved in the stationary phase, whereas ClpAP degrades the chromosomal addiction module MazE (Aizenman et al. 1996; Schweder et al. 1996).
In the present work, we study four carboxy-terminally tagged proteins. The amino-terminal domain of phage λ cI repressor, normally a stable cytoplasmic protein, is rapidly degraded when tagged with either a nonpolar pentapeptide tail or the SsrA natural 11-amino-acid carboxy-terminal tag (Parsell et al. 1990; Keiler et al. 1996). We show that these tagged proteins are substrates of HflB- and, in some cases, also of ClpP-dependent proteases, strongly suggesting that HflB- and ClpP-dependent proteases can select substrates by their carboxy-terminal tails.
Results
Tail-specific cytoplasmic degradation in the hflB1(Ts) mutant
We first examined the role of HflB in the degradation of three unstable cI variants carrying nonpolar pentapeptide tails—cI104, cI105, and cI108 (Parsell et al. 1990)—and the cI variant carrying the natural nonpolar tail, cI–SsrA; the stable variant cI102 served as control (see Table 1). To determine whether HflB is responsible for the cytoplasmic degradation of these unstable cI variants, we carried out pulse chase experiments at 42°C in a thermosensitive hflB1(Ts) mutant carrying a plasmid coding for one or another cI variant. In wild-type cells at 42°C, the half-life of all four unstable variants was ∼2 min, whereas the stable control, cI102, had a half-life of 200 min (Fig. 1). In the hflB1(Ts) mutant at 42°C (Fig. 1), two variants, cI104 and cI108, were strongly stabilized (30-min half-lives), one variant, cI105, was partially stabilized (7-min half-life), and cI–SsrA was not stabilized at all (2-min half-life). These data show that HflB is involved in the degradation of carboxy-terminal-tagged cI104, cI108, and, to a lesser extent, cI105.
Table 1.
Carboxy-terminal amino acid sequence of the cI variants
| cI variant | Carboxyl terminus |
| cI102 | –Arg–Ser–Glu–Tyr–Glu |
| cI104 | –Ile–His–Trp–Val–Thr |
| cI105 | –Trp–Val–Ala–Ala–Ala |
| cI108 | –Ser–Leu–Leu–Trp–Ser |
| cI–SsrA | –Ala–Ala–Asn–Asp–Glu–Asn–Tyr–Ala–Leu–Ala–Ala |
(Boldface type) Nonpolar amino acids; (underline) uncharged polar amino acids; (others) acidic and basic amino acids.
Figure 1.

Stability of cI variants in the hflB1(Ts) mutant. The strains used were JM105 (WT) or its hflB1(Ts) derivative carrying one of the following plasmids: pDP102 (cI102), p104 (cI104), p105 (cI105), p108 (cI108), or pλN-SPT (cI–SsrA). (+) Wild-type, cI102; (▵) wild type, cI108; (▴) hflB1, cI108; (○) wild type, cI104; (•) hflB1, cI104; (⋄) wild type, cI105; (♦) hflB1, cI05; (□) wild type, cI–SsrA; (▪) hflB1, cI–SsrA. Cultures growing exponentially at 30°C in M63 supplemented with glycerol and ampicillin were shifted to 42°C, induced, pulse labeled, and chased, and the amount of labeled cI variant was measured periodically, as described in Materials and Methods.
Tail-specific cytoplasmic degradation after HflB depletion
Previous experiments have indicated that the HflB1 mutant protein still has degradative activity toward some of its specific substrates at 42°C (Herman et al. 1997), which could explain the continued instability of cI105 and cI–SsrA in the mutant. Therefore, we examined the stability of these cI variants in a different conditional system in which the HflB protein is depleted. The HflB depletion strain carries a plasmid-borne copy of hflB under control of the tightly regulated arabinose promoter and a deletion of the chromosomal hflB gene. Cells growing in glycerol plus arabinose at 37°C were transferred to glucose medium lacking arabinose to shut off hflB expression. After 6 hr in glucose, the half-lives of induced cI108, cI105, and cI–SsrA were 45 min, 18 min, and 16 min, respectively, compared to 6 min in glycerol plus arabinose medium (Fig. 2). These data again show that HflB is involved in the degradation of cI108 and cI105 and show further that it is also involved in cI–SsrA degradation. However, the fact that cI105 and cI–SsrA are only partially stabilized suggests that these proteins are recognized by another protease as well.
Figure 2.

Stability of cI variants in the ΔhflB3::kan mutant. The strains used were JM105 (WT) or its ΔhflB derivative, carrying two plasmids, pBHB1 (paraBAD–hflB) and p108 (cI108), p105 (cI105), or pλN-SPT (cI–SsrA). (○) Wild-type, cI108; (•) ΔhflB, cI108; (▵) wild type, cI105; (▴) ΔhflB, cI105; (□) wild type, cI–ssrA; (▪) Δ hflb, cI–SsrA. Cultures were grown to exponential phase at 37°C in M63 supplemented with glycerol, arabinose, ampicillin, and chloramphenicol. HflB protein was depleted by 6 hr incubation in glucose medium lacking arabinose, after which the cultures were pulse labeled and chased and the amount of labeled cI variant was measured periodically, as described in Materials and Methods.
Role of ClpP in tail-specific cytoplasmic degradation
We tested the effect of a clpP mutation (which eliminates the ClpXP and ClpAP proteases) in combination with the hflB1(Ts) mutation on cI105 and cI–SsrA degradation. Both cI variants were strikingly stabilized in the hflB1(Ts) clpP::Tn9 double mutant at 42°C, with half-lives of 31 and 120 min for cI105 and cI–SsrA, respectively, compared to ∼2 min in wild type (Fig. 3). Strains mutant for each protease separately exhibited less stabilization than that observed in the double mutant. In the single mutants, the cI105 protein (Fig. 3A) was slightly stabilized in clpP::Tn9 (3 min) and in hflB1(Ts) (6 min), and the cI–SsrA protein (Fig. 3B) was partially stabilized in clpP::Tn9 (9 min) but not at all in hflB1(Ts) (2 min).
Figure 3.
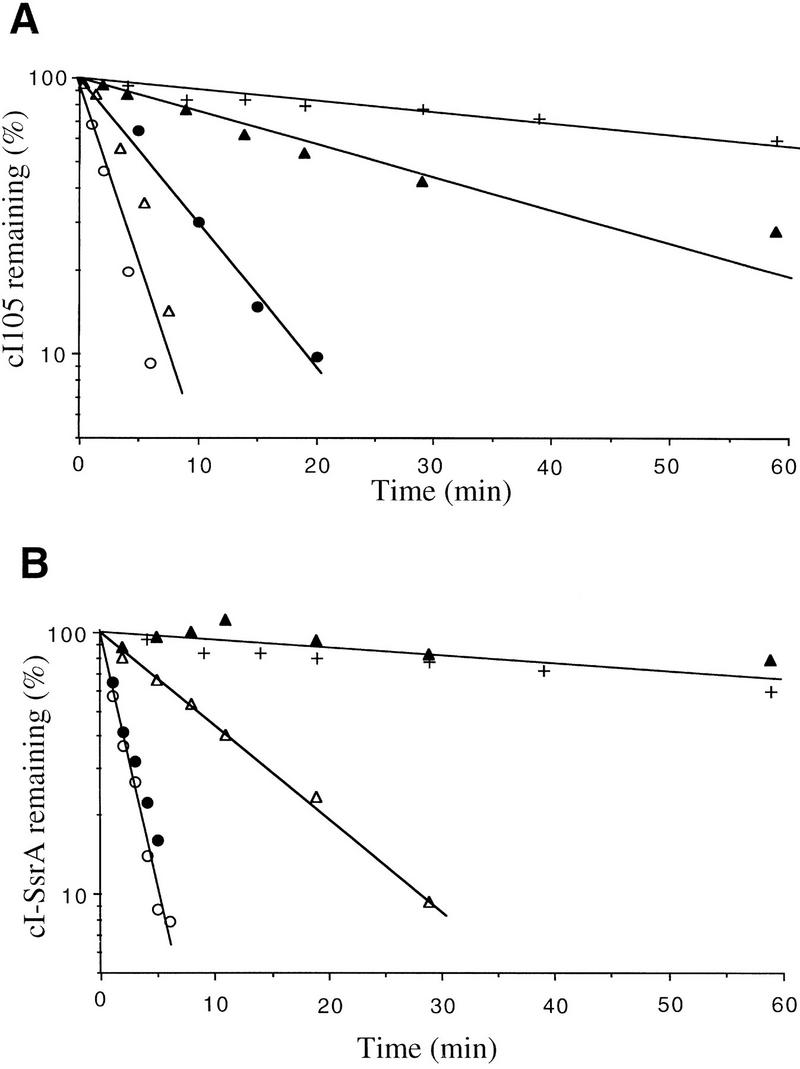
Degradation of cI105 and cI–SsrA by both HflB and Clp. The strains used were JM105 (WT) (○), its hflB1 derivative (•), its clpP::Tn9 derivative (▵), and its hflB1 clpP::Tn9 derivative (▴). (A) These strains carry plasmid p105 (cI105); (B) they carry plasmid pλN-SPT (cI–SsrA); a wild-type control (+) carrying plasmid pDP102 (cI102), coding for a stable cI variant, is shown in A and B. Cultures were grown, pulse labeled, chased, and analyzed as in the experiments shown in Fig. 1.
These data show clearly that the degradation of cI105 and cI–SsrA involves both ClpP and HflB. Stabilization of cI–SsrA in a clpP mutant has also been observed by Gottesman et al. (1998), who further showed that a related molecule (with a small insert between the cI and SsrA moieties) can be degraded in vitro by both ClpXP and ClpAP. It is interesting to note that cI105 is not totally stabilized in the hflB1(Ts) clpP::Tn9 double mutant, suggesting that yet a third protease may be involved in its degradation.
Overexpression of HflB compensates for the proteolysis defect of the clpP mutant
As shown above, cI–SsrA is recognized by the ClpP-dependent proteases and hflB protease. To investigate their respective roles, we measured the half-life of cI–SsrA in the clpP::Tn9 mutant at 37°C so that we could compare it to that in HflB-depleted cells. In clpP::Tn9 cells there was a 5-fold increase in stability (30 vs. 6 min in wild type), compared to the 2.5-fold increase in HflB-depleted cells (16 vs. 6 min in wild type). We have previously shown that the HflB concentration is a limiting factor in the degradation of its substrates σ32 and λcIII (Herman et al. 1995, 1997). We therefore asked whether overproduction of HflB could overcome the proteolytic defect of the clpP::Tn9 mutant. We introduced into the clpP::Tn9 strain a plasmid in which the expression of hflB is under control of plac. In the resulting strain, the addition of IPTG causes overexpression of both HflB and cI–SsrA. Under these conditions, the half-life of cI–SsrA was reduced to 7 min, compared to 28 min in the clpP::Tn9 control strain in which HflB was not overproduced (Fig. 4). The half-lives reported here are longer than those shown in Fig. 3 because of the lower temperature (see below).
Figure 4.
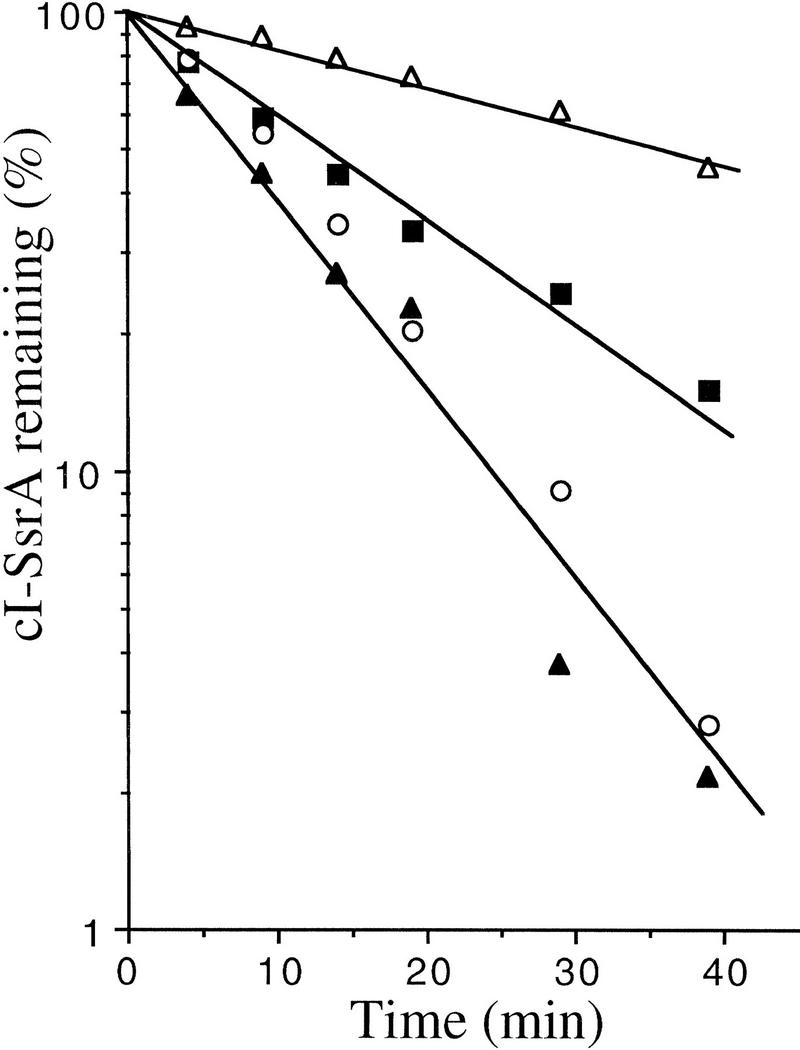
Excess HflB compensates for loss of ClpP in the degradation of cI–SsrA. The strains used were JM105 (○), its ΔhflB3::kan derivative (▪), and its clpP::Tn9 derivative carrying either the HflB-overproducing plasmid pULB6234 (▴) or the control plasmid pRK7813 (▵). All strains carried plasmid pλN-SPT, coding for cI–SsrA. Cultures were grown to exponential phase at 37°C in M63 supplemented with glycerol, ampicillin, and chloramphenicol. The cultures were pulse labeled and chased and the amount of labeled cI variant was measured periodically, as described in Materials and Methods.
These data show that excess HflB is able to replace the ClpP-dependent proteolytic pathway for cI–SsrA degradation. If the role of HflB is direct, the results suggest that HflB can fully recognize the SsrA tag and that the concentration of SsrA-specific proteases determines the half-life of SsrA-tagged proteins.
In vitro degradation of cI108 and cI–SsrA
To determine whether the action of HflB on unstable variants is direct, we purified HflB-His6-Myc from the membrane fractions of an overproducing strain (Kihara et al. 1996). The purification steps included a nickel column followed by a monoQ column. His6–cI108, His6–cI–SsrA, and His6–cI102 were purified in a single step, using a nickel affinity column; the preparations were free of ATP-dependent proteases since addition of ATP to the purified cI derivatives did not stimulate their degradation.
With these preparations, we investigated the ability of HflB to degrade tail-tagged λ cI repressor molecules in vitro. As shown in Figure 5A, purified HflB degraded cI108 in the presence of ATP or CTP, under protease-limiting conditions (the molar ratio of HflB to cI108 was 1:45). A major product of degradation, about half the protein (5 kD), was visible, suggesting that HflB acts as an endoprotease. In contrast, the stable variant cI102 was not degraded under the same conditions, in agreement with the in vivo results and suggesting that HflB is specific for cI molecules with a nonpolar tail, such as cI108. No degradation was observed when ATP was replaced with GTP or TTP. The degradation is HflB-dependent since addition of a zinc chelator inhibited the reaction whereas a serine protease inhibitor did not. The nonhydrolyzable ATP analog ATP-γ-S did not allow cI108 degradation, suggesting that this proteolysis requires ATP hydrolysis and thus is energy dependent. Finally, we compared the kinetics of degradation of cI108 and cI–SsrA; with a 45-fold molar excess of substrate at 37°C, the in vitro half-life of cI108 was 10 min, whereas that of cI–SsrA was ∼15 min (Fig. 5B).
Figure 5.
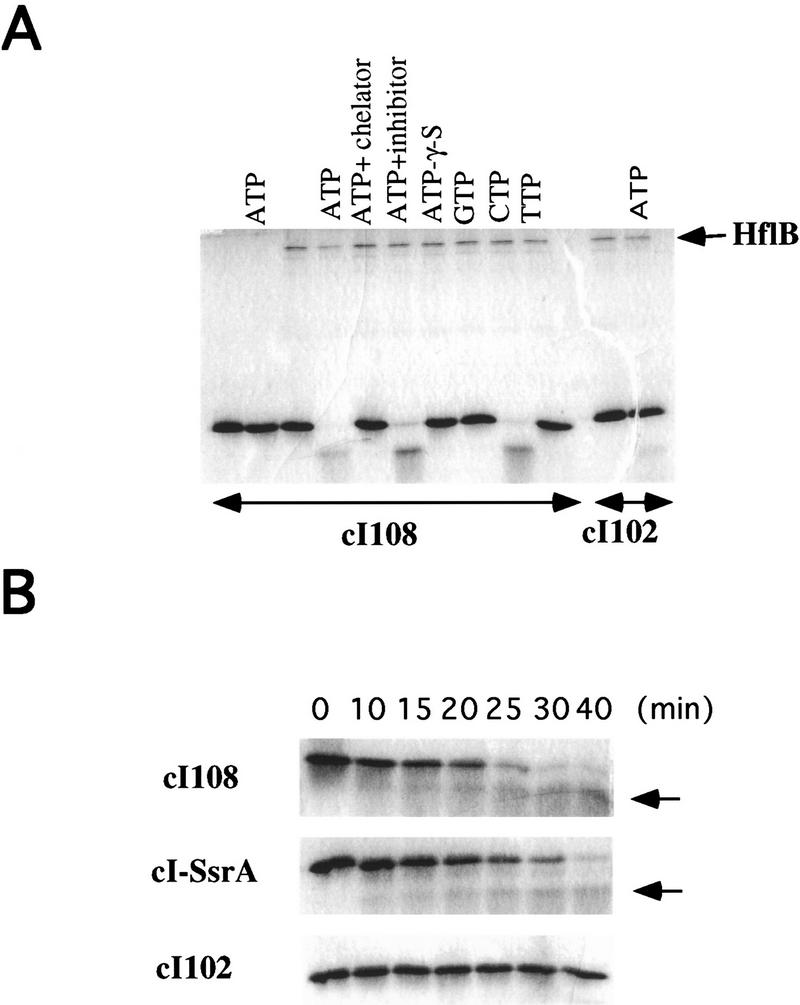
Degradation of cI–SsrA and cI108 by HflB in vitro. His-tagged HflB, cI–SsrA, cI108, and cI102 proteins were purified as described in Materials and Methods. (A) The indicated reactants were mixed in P buffer for 30 min at 37°C, and the reaction mixture was electrophoresed. Lanes 1–10 contained the unstable variant cI108; lanes 11 and 12 contained the stable variant cI102; HflB was present everywhere except in the controls shown in lanes 1 and 2. The other reactants in the different lanes were (lane 1) none; (lane 2) ATP; (lane 3) none; (lane 4) ATP; (lane 5) ATP + 1-10 phenanthrolin; (lane 6) ATP + pefabloc; (lane 7) ATP–γ-S; (lane 8) GTP; (lane 9) CTP; (lane 10) TTP; (lane 11) none; and (lane 12) ATP. (B) Kinetic analysis of reaction mixtures containing HflB, ATP and cI–SsrA, cI108, or cI102. Arrow indicate a major breakdown peptide appearing in the first two mixtures. Concentrations of the various reactants are indicated in Materials and Methods.
These results confirm that the unstable cI variants with nonpolar tails are substrates of the protease HflB, suggesting that the nonpolar tail is directly recognized by HflB.
Temperature dependence of HflB degradation
In the course of the above work, we noticed that the half-lives of the different cI variants varied with the temperature. To characterize this phenomemon quantitatively, we measured the half-life of cI108 in vivo as a function of temperature. As shown in Figure 6, the half-life of cI108 was ∼20 min at 30°C and 34°C, 6 min at 37°C, 3 min at 40°C and, as already shown, 2 min at 42°C.
Figure 6.

Temperature dependence of cI108 degradation. Strain JM105 carrying plasmid p108, coding for cI108, was grown to exponential phase at the indicated temperatures [ (▵) 30°C; (▴) 34°C; (○) 37°C; (•) 40°C] in M63 supplemented with glycerol and ampicillin, then pulse labeled and chased at that temperature and analyzed as in the experiments shown in Fig. 1.
These data demonstrate that proteolysis of cI108 increases with temperature. The effect is larger than the known heat-shock induction of the protease HflB (Herman et al. 1995); this could reflect temperature-dependent changes in substrate conformation or in HflB activity.
Discussion
Removing abnormal proteins from the cytoplasm is a vital housekeeping function (Miller 1996). In bacteria, mRNA is highly unstable; thus, truncated messenger molecules may be a significant source of such abnormal proteins. In E. coli, peptides arising from such mRNA molecules have recently been shown to be tagged by a process in which the stable 10Sa RNA encoded by ssrA is used to add an 11-amino-acid carboxy-terminal sequence with a nonpolar tail and release them from the ribosome; these SsrA-tagged truncated peptides are targeted for degradation (Keiler et al. 1996), and conservation of the nonpolar tail suggests that this system is common to all bacteria (Williams and Bartel 1996). In a systematic screen for protease recognition signals, it was found that proteins could be destabilized by the addition of similar nonpolar pentapeptide tails without changing their thermal stability (Parsell et al. 1990). The periplasmic protease Tsp (or Prc) recognizes proteins with nonpolar tails in vitro and in vivo as well for periplasmic proteins (Silber et al. 1992; Keiler and Sauer 1996). We show here that cytoplasmic proteins destabilized by such carboxy-terminal tags become substrates of the ATP-dependent protease HflB, an essential, membrane-anchored zinc protease. In some cases the ClpP-dependent proteases are also involved in their degradation. Using purified HflB in vitro, the half-life of proteins tagged with nonpolar tails, in the presence of excess substrate was similar to the in vivo half-life, strongly suggesting that the carboxy-terminal nonpolar sequence is recognized by the protease itself.
All cytoplasmic proteases seem to be polyvalent, with both regulatory and housekeeping functions. Proteolysis involved in regulation is often specific, for example, the heat shock specific sigma factor σ32 is degraded primarily by the protease HflB (Herman et al. 1995), whereas the stationary phase sigma factor σS is degraded primarily by ClpXP (Schweder et al. 1996). Housekeeping proteolysis, on the contrary, does not usually exhibit such sharp specificity, and abnormal proteins are often substrates of several proteases. For example, puromycin-truncated proteins are degraded in E. coli by both Lon and HslUV (Maurizi et al. 1985; Straus et al. 1988; Missiakas et al. 1996). The tagged carboxy-terminal cI variants provide an experimental approach to study how proteases can have both specific regulatory functions and nonspecific housekeeping functions. Two of the cI variants studied here are degraded exclusively by HflB. Two others exhibit little specificity; they are substrates of HflB, Tsp, and ClpP-dependent proteases (see Fig. 7). Since these variants differ only in their carboxyl terminus, the information in this small tail would seem to be sufficient to confer specificity to a single or several proteases. It is interesting to note that universal tags (cI105, cI–SsrA) are composed of only nonpolar amino acids (usually buried inside the protein), whereas specific tags are composed of polar and nonpolar amino acids (see Table 1). Using artificial tail-tagged cI variants, it should be possible to define the molecular mechanism by which proteases recognize both regulatory and housekeeping substrates.
Figure 7.

Proteolysis of carboxy-terminal-tagged proteins in E. coli. The degradation of carboxy-terminal-tagged polypeptides is schematized. The three cytoplasmic proteases, all ATP-dependent, are connected to their substrates by arrows. Thicker arrows indicate higher affinity; broken arrows indicate hypothetical reactions.
Among the proteases able to degrade cI–SsrA in the cytoplasmic compartment, the ClpXP and ClpAP proteases are collectively more important than HflB, as we observed greater stabilization by loss of ClpP (5-fold) than by loss of hflB (2.5-fold). The importance of ClpP-dependent proteases in cI–SsrA degradation has also been established by Gottesman et al. (1998). Since HflB is in the membrane, its paramount role may be to degrade abnormal membrane proteins, SsrA-tagged or not, that have their carboxyl termini in the cytoplasm (Fig. 7). This idea is consistent with the fact that HflB plays a further role in membrane quality control by degrading uncomplexed membrane proteins (Kihara et al. 1995). The SsrA tag is thus a signal for protease recognition in all cell compartments—the periplasm (Tsp), the membrane (HflB), and the cytoplasm (ClpXP, ClpAP, and HflB), permitting the rapid elimination of tagged proteins, wherever they may be, as one would expect for a housekeeping function.
HflB, ClpXP, and ClpAP all carry out tail-specific cytoplasmic proteolysis, and all can degrade SsrA-tagged substrates in vitro (this work; Gottesman et al. 1998). These proteases recognize nonpolar tails, which presumably provide an anchoring site to which they bind. It has recently been shown that ClpX interacts with the SsrA tail through a PDZ-like domain (Levchenko et al. 1997). PDZ domains, defined in eukaryotes, are modules of ∼90 residues that directly bind the carboxy-terminal ends of targeted proteins (Saras and Heldin 1996). Sequence analysis of HflB reveals homology with the PDZ-like domain of Clp in its well conserved “AAA” module (data not shown). The latter, a 220–250-amino-acid sequence, including an ATPase motif, defines a large family of >200 proteins (e.g., regulatory subunits of the 26S proteasome) (Confalonieri and Duguet 1995; Beyer 1997).
HflB, ClpXP, and ClpAP are tail-specific proteases. HslU, which belongs to the Clp family, also possesses a putative tail-specific binding domain (Levchenko et al. 1997). The Lon protease, too, seems to bind to tails (Higashitani et al. 1997; Sen and Rothfield 1998). Tail-specific recognition thus seems to be quite general among E. coli cytoplasmic proteases. The resemblance of HflB to a proteasome regulatory subunit suggests that tail-specific recognition may occur in higher organisms as well, and indeed it has been reported in several eukaryotic systems. For example, the S4 proteasome subunit in human cells binds to the carboxy-terminal part of the papillomavirus oncoprotein E7, apparently as part of a mechanism by which E7 delivers the retinoblastoma tumor suppressor protein to the proteasome (Berezutskaya and Bagchi 1997). Similarly, mammalian ornithine decarboxylase, normally degraded by the 26S proteasome (Murakami et al. 1992), was stabilized when the 37 carboxy-terminal residues were removed (Ghoda et al. 1989), and the degradation of mouse thymidine kinase also depends on its carboxy-terminal residues (Sutterluety et al. 1996). Tail-specific recognition may be a major entry into proteolytic pathways in all organisms.
Materials and methods
Bacterial strains
The strains used in this work are all E. coli K12 derivatives. There are isogenic strains derived from JM105, which carries a lacIq allele causing Lac repressor overproduction (Sambrook et al. 1989). HflB–His6–myc was purified from strain AK1181 (Kihara et al. 1996).
P1 vir-mediated transduction was carried out as described by Miller (1992). Plasmid preparations, transformation, cloning, and ligation were carried out as described by Sambrook et al. (1989). The ΔhflB3::kan (ΔftsH3::kan) allele (Akiyama and Shirai 1994) was introduced by transduction to kanamycin resistance in the presence of 70 μm l-arabinose into strains carrying plasmid pBHB1. Other protease mutations were introduced by Pl transduction, selecting for an associated antibiotic resistance marker: hflB1(Ts) zgj-203::Tn10, tetracycline resistance, from strain SL93 (Herman et al. 1993), and clpP::Tn9, chloramphenicol resistance, from strain SL246 (Herman et al. 1995).
Plasmids
Plasmids pDP102, p104, p105, p108 (Parsell et al. 1990), and pλN-SPT (Keiler et al. 1996) carry the different cI variant genes under control of the IPTG-inducible tac promoter of an ampicillin-resistant pBR322 derivative.
Plasmid pHis–cI108 codes for the cI108 protein fused to a histidine tag at its amino terminus; it was constructed by amplifying plasmid pcI108 with primers CINH2 (5′-GGCGGATCCATGAGCACAAAAAAGAAACC-3′) and CICOOHHIII (5′-GCCAAGCTTGCAATTTAACTGTGATAAACT-3′). The resulting product was cut with BamHI and HindIII and ligated to the pQE8 (Qiagen) BamHI–HindIII backbone fragment. The expected structure of the construct was verified by DNA sequencing.
Plasmids pHis–cI102 and pHis–cI–SsrA overexpress the histine-tagged cI102 and cI–SsrA, respectively. To construct these plasmids, we replaced the NsiI–NdeI fragment of pHis–cI108 with the short NsiI–NdeI fragment of pDP102 or pλN-SPT. The proteolytic susceptibility of each His-tagged variant was tested by pulse chase in a wild-type strain and shown to be identical to that of the untagged variant.
Plasmid pULB6234 (Herman et al. 1993) carries the intact hflB gene cloned downstream of the lac promoter in a low copy number, tetracycline-resistant plasmid vector, pRK7813 (Jones and Gutterson 1987).
Plasmid pBHB1 (Herman et al. 1997) carries the intact hflB gene cloned downstream of the ara promoter in a low copy number, chloramphenicol-resistant pACYC184 derivative, pBAD33 (Guzman et al. 1995).
Plasmid pSTD113 (Kihara et al. 1996) carries the hflB–his6–myc gene fusion under control of the IPTG-inducible lac promoter of an ampicillin-resistant pBR322 derivative.
Culture conditions
Cells were grown in M63 supplemented with 0.4% glycerol and 1 μg/ml thiamine or in LB broth (Miller 1992). For HflB depletion, cells were grown in M63 glucose (0.4%) plus 19 amino acids (lacking methionine) at 100 μg/ml each. When necessary, antibiotics were added at the following concentrations: 50 μg/ml ampicillin, 15 μg/ml chloramphenicol; 40 μg/ml kanamycin; and 10 μg/ml tetracycline.
Measurement of cI variant stability
All strains carried a lacIq allele and a plasmid with the cI variant gene under control of the tac (IPTG-inducible) promoter. The protocol was essentially that described by Parsell et al. (1990). Cells were grown to exponential phase at 30°C in glycerol minimal medium supplemented with antibiotics, then shifted to 42°C (t = 36 min), induced with 1 mm IPTG (t = 16 min), pulse-labeled (t = 1 min) with [35S]methionine + [35S]cysteine (38.3 TBq/mmole, 0.22 MBq/ml, ICN Biomedicals), and chased (t = 0) with 100 μg/ml each cold methionine plus cysteine. For pulse-chase experiments done at 37°C, cells were also pregrown at 37°C. Samples were electrophoresed on 15% SDS–polyacrylamide Tris-tricine gels. Radioactivity in the cI variant band was revealed with a Molecular Dynamics PhosphorImager and quantified with the ImageQuant 1.33 program (Molecular Dynamics). Half-lives were determined as the slope of the least squares best fit through the points plotted on a semilog graph. All experiments were repeated two to four times; there were no significant variations in the results.
Protein purification
Purification of HflB–his6–myc was essentially as described by Kihara et al. (1996), with the following modifications. Cells of AK1272 carrying pSTD113 were grown at 37°C to midexponential phase in 1 l of TB with ampicillin (50 mg/liter). IPTG (final concentration 1 mm) was added 3 hr before cells were harvested by centrifugation. The DEAE–Sepharose fast flow column was replace by a monoQ fast flow column.
The His6–cI102, His6–cI108, and His6–cI–SsrA proteins were purified by denaturing nickel NTA chromatography according to the Qiagen protocol.
In vitro degradation of cI variants by HflB–his6–myc
Purified cI variant preparations (30 μm) were incubated with HflB (0.67 μm) in buffer P (50 mm Tris-HCl at pH 7.6, 5 mm MgCl2, 25 μm zinc acetate, 1 mm DTT, 0.1% NP-40). The mixture was incubated at 37°C and stopped with 2 × SDS sample buffer (Sambrook et al. 1989). ATP, CTP, GTP, TTP, and ATP-γ-S were added at a final concentration of 5 mm. When indicated, pefabloc or 1-10 phenanthrolin was added at a final concentration of 10 mm. Samples were electrophoresed on 15% sodium SDS–polyacrylamide Tris-glycine gels and revealed by Coomassie stain. Gels were digitalized with an Apple scanner.
Acknowledgments
We are indebted to Patrick Waller, Akio Kihara, and Yoshinori Akiyama for sending plasmids and strains and to Susan Gottesman for communicating results prior to publication. We thank Danièle Joseleau-Petit, Sarah Ades, Lynn Connolly, members of Graham Walker’s laboratory, Robert Sauer, and Carol Gross for helpful discussions and warm support. C.H. is Chargé de Recherche of the Fonds National de la Recherche Scientifique (Belgium) and received a Belgian American Educational Foundation award for his stay at MIT. This work was supported in part by grant 6696 from the Association pour la Recherche sur le Cancer to R.D., grant “Contrôle des interactions protéine-protéine dans les réponses cellulaires et les cycles viraux” from the CNRS to P.B., grants GM28988 and CA21695 from the U.S. Public Health Service to G.C.W., and grant 36278 from the National Institutes of Health Service Grants to Carol A. Gross.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lherman@itsa.ucsf.edu; FAX (415) 476-4204.
References
- Aizenman E, Engelberg-Kulka H, Glaser G. An Escherichia coli chromosomal “addiction module” regulated by 3′,5′-bipyrophosphate: A model for programmed bacterial cell death. Proc Natl Acad Sci. 1996;93:6059–6063. doi: 10.1073/pnas.93.12.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama Y, Shirai Y. Involvement of FtsH in protein assembly into and through the membrane. II. Dominant mutations affecting FtsH functions. J Biol Chem. 1994;269:5225–5229. [PubMed] [Google Scholar]
- Berezutskaya E, Bagchi S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26 S proteasome. J Biol Chem. 1997;272:30135–30140. doi: 10.1074/jbc.272.48.30135. [DOI] [PubMed] [Google Scholar]
- Beyer A. Sequence analysis of the AAA protein family. Prot Sci. 1997;6:2043–2058. doi: 10.1002/pro.5560061001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Confalonieri R, Duguet M. A 200 amino acid-ATPase module in search for a basic function. BioEssays. 1995;17:639–650. doi: 10.1002/bies.950170710. [DOI] [PubMed] [Google Scholar]
- Ghoda L, van Daalen Wetters T, Macrae M, Ascherman D, Coffino P. Prevention of rapid intracellular degradation of ODC by a carboxyl-terminal truncation. Science. 1989;243:1493–1495. doi: 10.1126/science.2928784. [DOI] [PubMed] [Google Scholar]
- Gottesman S. Proteases and their targets in Escherichia coli. Annu Rev Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- Gottesman, S., E. Roche, Y.-N. Zhou, and R.T. Sauer. 1998. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA tagging system. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Guzman L-M, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation and high-level expression by vectors containing the arabinose pBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman C, D’Ari R. Proteolysis and chaperones: The destruction/reconstruction dilemma. Curr Opin Micriobiol. 1998;1:204–209. doi: 10.1016/s1369-5274(98)80012-x. [DOI] [PubMed] [Google Scholar]
- Herman C, Ogura T, Tomoyasu T, Hiraga S, Akiyama Y, Ito K, Thomas R, D’Ari R, Bouloc P. Cell growth and λ phage development controlled by the same essential Escherichia coli gene, ftsH/hflB. Proc Natl Acad Sci. 1993;90:10861–10865. doi: 10.1073/pnas.90.22.10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman C, Thévenet D, DAri R, Bouloc P. Degradation of σ32, the heat shock regulator in Escherichia coli, is governed by HflB. Proc Natl Acad Sci. 1995;92:3516–3520. doi: 10.1073/pnas.92.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman C, Thévenet D, DAri R, Bouloc P. The HflB protease of Escherichia coli degrades its inhibitor λcIII. J Bacteriol. 1997;179:358–363. doi: 10.1128/jb.179.2.358-363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashitani A, Ishii Y, Kato Y, Koriuchi K. Functional dissection of a cell-division inhibitor, SulA, of Escherichia coli and its negative regulation by Lon. Mol Gen Genet. 1997;254:351–357. doi: 10.1007/s004380050426. [DOI] [PubMed] [Google Scholar]
- Jones JDG, Gutterson N. An efficient mobilisable cosmid vector pRK7813, and its use in rapid method for marker exchange in Pseudomonas fluorescens strain HV37a. Gene. 1987;61:299–306. doi: 10.1016/0378-1119(87)90193-4. [DOI] [PubMed] [Google Scholar]
- Keiler KC, Sauer RT. Sequence determinants of C-terminal substrate recognition by the Tsp protease. J Biol Chem. 1996;271:2589–2593. doi: 10.1074/jbc.271.5.2589. [DOI] [PubMed] [Google Scholar]
- Keiler KC, Waller PRH, Sauer RT. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- Kihara A, Akiyama Y, Ito K. FtsH is required for proteolytic elimination of uncomplexed forms of SecY, an essential protein translocase subunit. Proc Natl Acad Sci. 1995;92:4532–4536. doi: 10.1073/pnas.92.10.4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— A protease complex in the Escherichia coli plasma membrane: HflKC (HflA) forms a complex with FtsH (HflB), regulating its proteolytic activity against SecY. EMBO J. 1996;15:6122–6131. [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Smith CK, Walsh NP, Sauer RT, Baker TA. PDZ-like domains mediate binding specificity in the Clp/Hsp100 family of chaperones and protease regulatory subunits. Cell. 1997;91:939–947. doi: 10.1016/s0092-8674(00)80485-7. [DOI] [PubMed] [Google Scholar]
- Maurizi MR, Trisler P, Gottesman S. Insertional mutagenesis of the lon gene in Escherichia coli: lon is dispensable. J Bacteriol. 1985;164:1124–1135. doi: 10.1128/jb.164.3.1124-1135.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CG. Protein degradation and proteolytic modification. In: Neidhardt FC, Curtiss III R, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. Escherichia coli and Salmonella. Cellular and molecular biology. Washington, D.C.: ASM Press; 1996. pp. 938–954. [Google Scholar]
- Miller JH. A short course in bacterial genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Missiakas D, Schwager F, Betton JM, Georgopoulos C, Raina S. Identification and characterization of HsIV HsIU (ClpQ ClpY) proteins involved in overall proteolysis of misfolded proteins in Escherichia coli. EMBO J. 1996;15:6899–6909. [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Matsufuji S, Kameji T, Hayashi S, Igarashi K, Tamura T, Tanaka K, Ichihara A. Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature. 1992;360:597–599. doi: 10.1038/360597a0. [DOI] [PubMed] [Google Scholar]
- Parsell DA, Silber KR, Sauer RT. Carboxy-terminal determinants of intracellular protein degradation. Genes & Dev. 1990;4:277–286. doi: 10.1101/gad.4.2.277. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Saras J, Heldin C-H. PDZ domains bind carboxy-terminal sequences of target proteins. Trends Biochem Sci. 1996;21:455–458. doi: 10.1016/s0968-0004(96)30044-3. [DOI] [PubMed] [Google Scholar]
- Schweder T, Lee K-H, Lomovskaya O, Matin A. Regulation of Escherichia coli starvation sigma factor (σS) by ClpXP protease. J Bacteriol. 1996;178:470–476. doi: 10.1128/jb.178.2.470-476.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen M, Rothfield LI. Stability of the Escherichia coli division inhibitor protein MinC requires determinants in the carboxy-terminal region of the protein. J Bacteriol. 1998;180:175–177. doi: 10.1128/jb.180.1.175-177.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shotland Y, Koby S, Teff D, Mansur N, Oren DA, Tatematsu K, Tomoyasu T, Kessel M, Bukau B, Ogura T, Oppenheim AB. Proteolysis of the phage lambda CII regulatory protein by FtsH (HflB) of Escherichia coli. Mol Microbiol. 1997;24:1303–1310. doi: 10.1046/j.1365-2958.1997.4231796.x. [DOI] [PubMed] [Google Scholar]
- Silber KR, Keiler KC, Sauer RT. Tsp: A tail-specific protease that selectively degrades proteins with nonpolar C termini. Proc Natl Acad Sci. 1992;89:295–299. doi: 10.1073/pnas.89.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber KR, Sauer RT. Deletion of the prc (tsp) gene provides evidence for additional tail-specific proteolytic activity in Escherichia coli K-12. Mol & Gen Genet. 1994;242:237–240. doi: 10.1007/BF00391018. [DOI] [PubMed] [Google Scholar]
- Straus DB, Walter WA, Gross CA. Escherichia coli heat shock gene mutants are defective in proteolysis. Genes & Dev. 1988;2:1851–1858. doi: 10.1101/gad.2.12b.1851. [DOI] [PubMed] [Google Scholar]
- Sutterluety H, Bartl S, Karlseder J, Wintersberger E, Seiser C. Carboxy-terminal residues of mouse thymidine kinase are essential for rapid degradation in quiescent cells. J Mol Biol. 1996;259:383–392. doi: 10.1006/jmbi.1996.0327. [DOI] [PubMed] [Google Scholar]
- Tomoyasu T, Yamanaka K, Murata K, Suzaki T, Bouloc P, Kato A, Niki H, Hiraga S, Ogura T. Topology and subcellular localization of FtsH protein in Escherichia coli. J Bacteriol. 1993;175:1352–1357. doi: 10.1128/jb.175.5.1352-1357.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoyasu T, Gamer J, Bukau B, Kanemory M, Mori H, Rutman AJ, Oppenheim AB, Yura T, Yamanaka K, Niki H, Hiraga S, Ogura T. A membrane-bound, ATP-dependent protease FtsH degrades the heat-shock transcription factor σ32 in Escherichia coli. EMBO J. 1995;14:2551–2560. doi: 10.1002/j.1460-2075.1995.tb07253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KP, Bartel DP. Phylogenetic analysis of tmRNA secondary structure. RNA. 1996;2:1306–1310. [PMC free article] [PubMed] [Google Scholar]