

Abstract
Osteoprotegerin (OPG) is a secreted protein that inhibits osteoclast formation. In this study the physiological role of OPG is investigated by generating OPG-deficient mice. Adolescent and adult OPG−/− mice exhibit a decrease in total bone density characterized by severe trabecular and cortical bone porosity, marked thinning of the parietal bones of the skull, and a high incidence of fractures. These findings demonstrate that OPG is a critical regulator of postnatal bone mass. Unexpectedly, OPG-deficient mice also exhibit medial calcification of the aorta and renal arteries, suggesting that regulation of OPG, its signaling pathway, or its ligand(s) may play a role in the long observed association between osteoporosis and vascular calcification.
Keywords: Osteoprotegerin, osteoporosis, knockout mice, arterial calcification
Development and maintenance of the mammalian skeleton involves the regulation and interaction of its component cell types (Erlebacher et al. 1995; Marks 1997). Major contributors to skeletal architecture include chondrocytes, which form cartilage; osteoblasts, which synthesize and deposit bone matrix; and osteoclasts, which resorb bone. Chondrocytes are derived from mesenchymal cells and function to generate an initial cartilage template required for endochondral bone formation. Osteoblasts, derived from mesenchymal osteoprogenitor cells, are located on the surface of bone where they synthesize, transport, and arrange the matrix proteins. Osteoclasts are derived from granulocyte–monocyte precursors present in the hematopoietic marrow (Mundy 1993a; Manolagas and Jilka 1995; Roodman 1996). After establishing a tight adherence to the bone surface, osteoclasts form resorption zones that are acidified by a specialized structure known as the ruffled border. The ruffled border functions as a secretory conduit in which secreted protons and acid proteases decalcify and then digest bone matrix. During the process of osteoclast-mediated resorption, it is thought that protein factors are elaborated that act as signaling molecules to initiate bone renewal by osteoblasts. Osteoblasts, in turn, can influence osteoclast function through the expression of soluble or membrane-bound regulators (Takahashi et al 1988). The coupling between osteoblast and osteoclast functions is critical for skeletal modeling, remodeling, and repair (Mundy 1993b; Mundy et al. 1995).
Postmenopausal osteoporosis, the most common bone disease in the developed world, has been causally linked to estrogen loss (for review, see Pacifici 1996). Postmenopausal bone loss can be attributed to loss of regulatory control exerted by estrogen on the production of cytokines and other factors that regulate osteoclast development. The resultant shift in the balance of osteoclast and osteoblast activity favors a net loss of bone mass, ultimately leading to osteoporosis.
Osteoporosis in human populations has been associated with a higher incidence of arterial calcification, a component of many atherosclerotic lesions (Banks et al. 1994; Parhami and Demer 1997; Parhami et al. 1997). Common factors may underlie the pathogenesis of these two diseases. Some arterial calcium mineral deposits appear identical to fully formed lamellar bone, including trabeculae, lacunae, and islands of marrow (Bunting 1906; Haust and Geer 1970). Furthermore, calcified arteries have been shown to express several bone matrix proteins, including collagen type I, matrix GLA protein, osteocalcin, osteonectin, and bone morphogenic protein type 2 (Bostrom et al. 1993, 1995; Giachelli et al. 1993; O’Brien et al. 1995). These findings have led to speculation that arterial calcification is an organized, regulated process with cellular and molecular mechanisms similar to bone formation (Demer 1995; Parhami et al. 1996).
Osteoprotegerin (OPG), a recently identified member of the tumor necrosis factor receptor gene superfamily, is a secreted factor that inhibits osteoclast development both in vitro and in vivo (Simonet et al. 1997). Transgenic mice overexpressing OPG in the liver have high levels of OPG protein in their systemic circulation and exhibit a marked increase in bone density (osteopetrosis). OPG in normal mouse embryos has been localized within cartilage rudiments of developing bones, as well as in the small intestine and the muscular wall of several major arteries. We show here that OPG is a physiological regulator of normal postnatal bone mass by demonstrating that targeted deletion of OPG in mice results in severe, early-onset osteoporosis. Interestingly, loss of OPG also results in calcification of the aorta and renal arteries, sites of endogenous OPG expression in normal animals. These findings demonstrate that OPG plays an important role in regulating physiological bone formation in the postnatal animal and suggest an additional role for OPG in regulating pathological calcification of arteries.
Results
We have shown previously that systemic OPG administration or overexpression of OPG in mice results in a marked increase of trabecular bone within the long bones and vertebrae, with no detectable effects on the bones of the skull. Severely affected animals lack a clearly defined cortex because of the presence of nonremodeled trabecular bone. Although OPG levels have not been measurable in normal mice, OPG mRNA is detectable predominantly in liver and lung in adult normal mice, suggesting that low circulating levels of this protein may be important in aspects of skeletal metabolism. Therefore, to discern the physiological role and importance of OPG in normal skeletal development and other processes, the endogenous OPG gene was deleted. Data presented here show for the first time that endogenous OPG is not required for embryonic bone formation yet is essential for maintenance of postnatal trabecular and cortical bone mass throughout the skeleton, including the skull. Unexpectedly, loss of OPG results in marked calcification of the aorta and renal arteries.
The strategy for targeted deletion of the mouse OPG gene is outlined in Figure 1. OPG-deficient mice (OPG−/−) are viable and appear histologically normal at birth (data not shown). Analysis of 265 mice generated from OPG+/− hemizygous matings showed that the percentage of +/+, +/−, and −/− mice was 29%, 53%, and 18%, respectively. The frequency of homozygous knockout mice was close to the expected Mendelian ratio of 25% during the first week of life. This number decreased to ∼15% in litters genotyped at 3–4 weeks of age, indicating some mortality in young OPG−/− mice. Interestingly, ∼10% of postnatal OPG−/− mice are runted during adolescent and adult life. The decrease in OPG−/− mouse viability observed in adolescence is associated with an increase in the incidence of vertebral or endochondral bone fractures (see below) in the first 2 weeks of life. Despite the presence of brittle bones during early adolescence, OPG−/− mice that survived past weaning age were fertile. Female OPG−/− mice carry litters to term and give birth to OPG−/− offspring that are histologically normal, indicating that endogenous and/or maternal OPG are not required for normal embryonic development. Both male and female OPG−/− mice have survived to 5–6 months of age. Otherwise, the mice appear normal and no hematological or biochemical abnormalities other than elevated alkaline phosphatase levels are observed. In contrast to the splenomegaly observed in OPG transgenic mice (Simonet et al. 1997), the spleens of OPG knockout mice appear normal.
Figure 1.

Gene targeting at the OPG locus. (A) Schematic representation of the genomic structure of the murine OPG locus. Restriction sites are indicated as follows: (RI) EcoRI; (X) XmnI; (P) PstI. Exons are indicated as black boxes and numbered by Roman numerals. Introns are shown as thin black lines. (B) The targeting vector used to disrupt the OPG gene. The PGK–neo casette was placed in reverse orientation within exon 2, replacing the portion of exon 2 encoding the first 93 amino acids of the mature OPG protein. This strategy effectively eliminates the coding region for the first two cysteine-rich domains of OPG that are required for its activity and places a translation stop codon in-frame, preventing translation of any downstream OPG sequence. (C) The structure of a targeted OPG allele following recombination of the targeting vector at the OPG locus. (D) Southern blot analysis of PstI-digested genomic DNA from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) OPG knockout mice. The wild-type allele is a 2.3-kb PstI fragment, and the targeted allele is a 3.0-kb PstI fragment. The small open box in A and C represents the DNA probe used to screen for recombinant ES cell clones. (E) Northern blot analysis of RNA from offspring derived from heterozygous matings. Ten micrograms of total RNA collected from liver was probed for OPG and β-actin expression. The absence of OPG expression in OPG−/− mice confirmed that OPG is a null allele.
A decrease in bone mineral density is first evident by radiography at 1 month of age in OPG−/− mice. Changes are more pronounced by 2 months of age in both males and females (Fig. 2), and become more severe as the mice age. Gross morphological abnormalities are evident as early as 1 month after birth. Some OPG−/− mice exhibit multiple fractures (Fig. 2). Older mice have marked kyphosis of the spine, sometimes seen with fractures. Another common deformity is flattening of the distal articular region of the femur, indicating partial collapse or compression of the epiphysis. Heterozygous knockout mice exhibit normal radiological appearance.
Figure 2.

Radiographic analysis of the bones of OPG−/− and wild-type mice. (A,B) Radiographs of 2-month-old female wild-type and OPG−/− mice, respectively. (C,D) Radiographs of the leg, hemipelvis and vertebrae of 2-month-old female wild-type and OPG−/− mice respectively. OPG−/− mice were x-rayed adjacent to wild-type and heterozygous mice using the same x-ray film, to allow for direct comparison of bone density. The strongest phenotype is evident in the vertebrae and long bones. The cortical bone in the femur and pelvis are thinned, and the femoral growth plate is not visible. OPG+/− mice are not different from OPG+/+ mice at this time point. (E) Gross anomalies of the skeleton are seen as early as 1 month after birth in the form of multiple fractures (arrows). (F) Radiograph of 6-month-old female wild-type mouse. (G) Radiograph of 5-month-old OPG−/− female mouse. Note severe deformity of the vertebral column due to the collapse of several vertebral bodies (arrow). This was confirmed by subsequent lateral radiographs of the spine.
Histologically, the lumbar vertebrae and metaphyseal regions of the humerus, femurs, and tibias are profoundly osteoporotic in the OPG−/− mice, with almost complete absence of trabeculae by 1–2 months of age (Fig. 3). Loss of cancellous bone and the onset of vertebral osteoporosis are evident within a week after birth and become more severe through adolescent life. Cortical shafts of the long bones showed increased porosity beginning at ∼1 month of age. The porosity is characterized by an increased number of vessels within the cortex. Numerous osteoclasts and osteoblasts are present on the bone surfaces adjacent to these vessels. Polarization microscopy revealed that cortical bone is composed chiefly of woven bone (Fig. 4C,D) and that osteocyte density is higher than normal (Fig. 4J). These histologic features are consistent with a high turnover state, and because woven bone is not as strong as lamellar bone, an increase in fracture incidence would be expected. The parietal bones of the skull also show marked thinning and increased porosity (data not shown). The proximal epiphysis of the femur and humerus of OPG−/− mice exhibit resorption of subchondral bone and collapse of the joint surface, indicating mechanical damage at the end of the bone (Fig. 3H). Interestingly, OPG mRNA is detectable by in situ hybridization in adult articular chondrocytes (data not shown), suggesting that OPG has an effect on the structural integrity of the joint surfaces by maintenance of subchondral bone mass.
Figure 3.
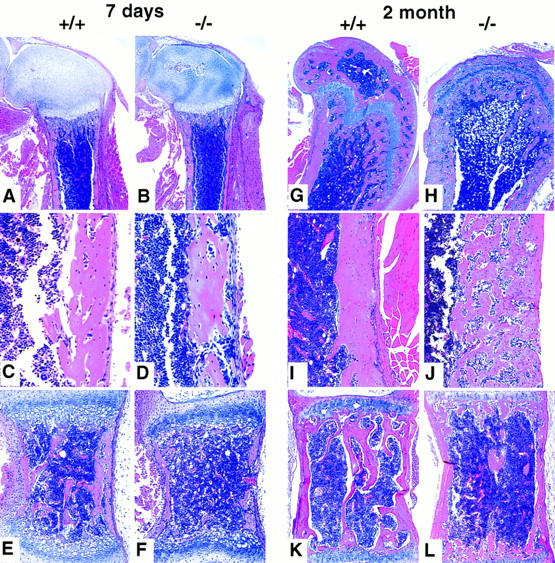
Histological evaluation of bone morphology in 7-day and 2-month-old female OPG−/− vs. OPG+/+ mice. (A) OPG+/+ femur; normal femur morphology. (B) OPG−/− femur; note minimal loss of the primary and secondary spongiosa. (C) OPG+/+ femur, cortical area; normal morphology. (D) OPG−/− femur, cortical area, note somewhat increased bone resorption and remodeling. (E) OPG+/+ vertebral body; normal morphology. (F) OPG−/− vertebral body; note osteoporosis characterized by loss of trabecular bone and thinning of the cortical bone. Magnification, 4× in A and B; 30× in C and D; 20× in E and F; H & E stained. (G) OPG+/+ femur; normal femur morphology. (H) OPG−/− femur; note marked loss of the primary and secondary spongiosa, and collapse of the normally rounded articular surface. (I) OPG+/+ femur, cortical area; normal morphology. (J) OPG−/− femur, cortical area; note severe cortical bone porosity with bone resorption and active remodeling. (K) OPG+/+ vertebral body; normal morphology. (L) OPG−/− vertebral body; note severe osteoporosis characterized by loss of trabecular bone and thinning of the cortical bone. Magnification, 4× in G and H; 15× in I and J; 10× in K and L; H & E stained.
Figure 4.
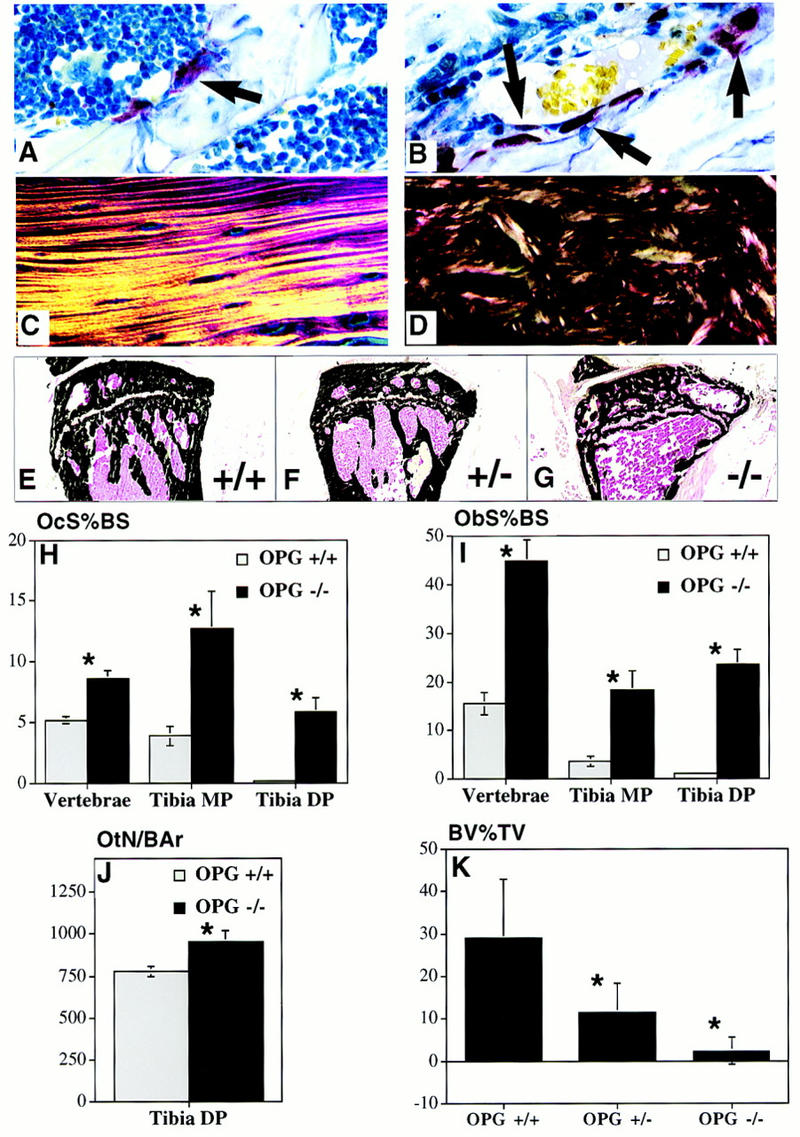
Bone remodeling rate, collagen structure, and trabecular density changes in OPG−/− mice. (A,B) TRAP-stained sections (60×) of the metaphyseal region of the proximal humerus showing increased numbers of osteoclasts (arrows) in the OPG−/− (B) compared to OPG+/+ mice (A). (C,D) Polarized light microscopy of cortical bone of the humeral diaphysis showing lamellar collagen deposition in bone of the OPG+/+ (C) compared to the woven pattern in OPG−/− mice (D). (E–G) Von Kossa-stained frozen sections of the proximal tibial metaphysis of 6-month-old OPG+/+, OPG+/−, and OPG−/− mice. Note normal morphology in the OPG+/+ mice (E), lower density in the metaphysis of the OPG+/− mice (F), and even further reduced density in the OPG−/− mice (G). Osteoclast surface as a percent of bone surface (OcS%BS) (H) and osteoblast surface as a percent of bone surface (ObS%BS) (I) are increased in the vertebrae, tibial metaphysis (Tibia MP) and the tibial diaphysis (Tibia DP) of the OPG−/− (solid bars) vs. the OPG+/+ (shaded bars) mice. (*) Different from OPG+/+, P < 0.005. (J) Osteocyte number per mm2 bone area (OtN/BAr) in the tibial diaphysis is increased in OPG−/− vs. the OPG+/+ mice, P < 0.05. (K) Quantitative representation of the trabecular bone density in the metaphyseal region of the tibia expressed as a percent of the total tissue area (BV%TV) in 6-month-old OPG+/+, OPG+/−, and OPG−/− mice. The trabecular bone density was markedly reduced in the proximal tibial metaphysis in the homozygous knockout mice, n = 6, vs. heterozygous mice, n = 9, and wild type mice, n = 7. (*) Significantly lower than wild type, P < 0.001.
Quantification of bone density in 2- and 6-month-old OPG-deficient mice and their heterozygous and wild-type littermates was performed by peripheral quantitative computed tomography (pQCT) measurements (Beamer et al. 1996) of one tibia and by image analysis of Von Kossa-stained sections of the other tibia (Fig. 4E–G). At 2 months of age, trabecular bone density in the metaphysis and cortical bone density in the diaphysis are significantly reduced in OPG−/− mice relative to OPG−/+ and OPG+/+ mice when assessed by pQCT. Trabecular bone density in the OPG−/− mice at 2 month of age is 273.11 ± 44 mg/cm3 versus 448.38 ± 65 mg/cm3 in OPG+/− and 496.55 ± 58 mg/cm3 in OPG+/+ mice, P < 0.005. Cortical bone density in the OPG−/− mice is 549.58 ± 41 mg/cm3 vs. 669.07 ± 43 mg/cm3 in OPG+/− and 676.21 ± 78 mg/cm3 in OPG+/+ mice, P < 0.005. Interestingly, heterozygous (OPG+/−) mice exhibit a significant loss of total bone density relative to wild-type littermates by 6 months of age (487.6 ± 27 vs. 521.2 ± 29 mg/cm3, P < 0.05), indicating that loss of a single OPG allele results in significant loss of bone mass over a period of several months. Histomorphometric analysis shows that the trabecular area in the proximal tibia is markedly reduced in OPG-deficient animals, to <5% compared to ∼30% in controls (Fig. 4K). Again, heterozygotes have an intermediate level of trabecular area compared to OPG−/− and OPG+/+ mice, thereby confirming the changes seen using the pQCT method.
Osteoclast and osteoblast numbers were determined in decalcified hematoxylin and eosin (H & E) stained tibia and vertebrae (Fig. 4H,I). Osteoclast surface as a percent of total bone surface is increased in vertebrae and both the trabecular and cortical regions of the tibiae (Fig. 4H). The highest osteoclast surface is observed in the tibial metaphysis of OPG−/− mice in which the number of osteoclasts per millimeter of tibial metaphyseal trabecular bone perimeter in 2-month-old OPG−/− mice is increased about threefold (4.90 ± 2.8 vs. 1.4 ± 0.6, P < 0.01). Osteoblast-lined bone perimeter as a percentage of total perimeter is also increased in the vertebrae and both the trabecular and cortical regions of the tibiae (Fig. 4I). Proportionally, the largest effect is seen in the tibial diaphysis; however, on the basis of absolute values the vertebrae have the highest percentage of bone surfaced by osteoblasts. Consistent with this increase of osteoblast activity seen histologically, serum alkaline phosphatase levels (which can be a marker of osteoblast activity) are elevated in both male and female OPG−/− mice from 2 weeks onward, likely reflecting the coupling between osteoclast-mediated resorption and osteoblast-mediated bone synthesis. At 2 months of age, OPG−/− mice have significantly elevated serum alkaline phosphatase values of 477.2 ± 200.9 IU/liter (n = 5, P < 0.0001) compared to 106.1 ± 39.6 IU/liter (n = 7) and 112.9 ± 15.7 IU/liter (n = 8) for the wild-type and heterozygous mice, respectively.
We demonstrated previously that OPG inhibits the terminal stages of osteoclast development in vitro and in vivo but does not effect the development of osteoclast precursors (Simonet et al. 1997). The levels of osteoclast precursors contained in OPG−/− or wild-type bone marrow and spleen cells were found to be similar when measured by an in vitro osteoclast-forming assay (Lacey et al. 1995; data not shown). This finding suggests that the osteoporosis in OPG−/− mice is most likely due to dysregulated osteoclast recruitment and activation in vivo rather than alterations in the number of osteoclast precursors.
Expression of OPG in mice has been localized within embryonic cartilage rudiments, focally adjacent to lymphatics in the small intestinal mucosa, and in large arteries (Fig. 5A,B) (Simonet et al. 1997). The gastrointestinal tract is normal in OPG−/− mice. The large arteries, including the aorta, however, exhibit some calcification as early as 2 weeks after birth and marked calcification by 2 months of age (Fig. 5), when it affected two-thirds of the OPG−/− animals examined (both males and females). The aortic calcification is mainly in the media and is associated with mild to moderate intimal and medial proliferation and partial aortic dissection in 2- to 6-month-old OPG−/− mice (Fig. 5E). Calcified lesions are also observed in renal arteries of OPG−/− mice but not in smaller arteries, veins, or capillaries. No atherosclerotic plaques are seen. Endogenous OPG expression is localized within the smooth muscle layer (media) of the aortic and renal arteries, suggesting a role for OPG in maintaining normal structure in larger arteries. Loss of OPG somehow contributes to arterial calcification.
Figure 5.
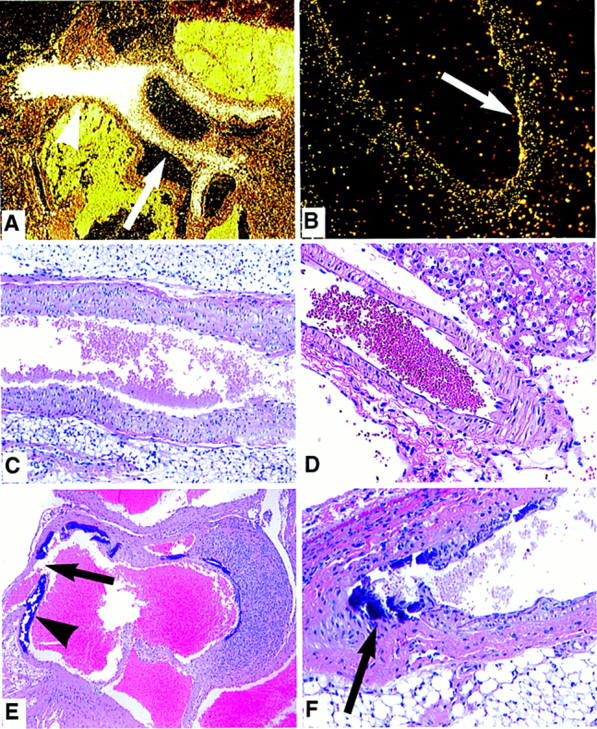
Arterial pathology in OPG−/− mice. (A) In situ hybridization with radioactively labeled antisense OPG probe shows high expression of OPG in the incipient part of the aorta of a E18.5 mouse embryo (arrow). Very high signal is detected in a tangential section of the aorta through the smooth muscle wall (arrowhead). (B) OPG expression is also detected by in situ hybridization in the smooth muscle wall of the renal artery of the adult rat. (C) Note normal morphology of a OPG+/+ mouse abdominal aorta; H & E stained, magnification, 4×. (D) Normal morphology of the renal artery of an adult OPG+/+ mouse; H&E stained, 10×. (E) Extensive calcification (arrowhead) in the medial and subintimal region of the ascending aorta of a 2-month-old OPG−/− mouse. Acute dissection of the aortic wall is marked by an arrow; on the opposite wall of the aorta, granulation tissue is present at the site of a former dissection; H & E stained, 2×. (F) Medial/subintimal calcification (arrow) in the renal artery of a 2-month-old OPG−/− mouse with moderate intimal proliferation; H & E stained, 20×.
Discussion
Osteoprotegerin inhibits osteoclast formation when administered in vivo to mice or when added to an in vitro osteoclast-forming assay (Simonet et al. 1997). Ablation of OPG by targeted gene deletion in mice leads to early-onset osteoporosis, demonstrating for the first time a critical requirement for endogenous OPG in the maintenance of normal postnatal bone mass and skeletal architecture. Transgenic mice overexpressing OPG had increased bone density predominately in the trabecular region of long bones and vertebrae due to lack of remodeling of the primary spongiosa. This suggested a specific role for OPG in regulating trabecular bone osteoclast activity (Simonet et al. 1997). The phenotype of OPG-deficient mice reveals an additional role for OPG in regulating remodeling within the cortical shafts of long bones and within bones of the skull. OPG−/− mice exhibit significant trabecular bone loss during early adolescence, with the subsequent formation of porous cortical bones by weaning age. Many OPG−/− mice exhibit gross skeletal abnormalities from shortly after birth up to 5–6 months of age. Interestingly, OPG+/− mice also exhibit significant loss of trabecular bone by 6 months, suggesting that modest decreases in OPG expression lead to long-term declines in bone mineral density. It is tempting to speculate that OPG, its signaling pathway, or ligand(s) may be sensitive to estrogen regulation. Imbalances in this pathway may occur during postmenopausal periods associated with bone loss, potentially implicating a role for OPG in the pathogenesis of osteoporosis. The osteoporotic phenotype of OPG-deficient mice supports a potential utility for OPG in treating osteoporosis and other diseases of bone loss.
Compared to other rodent osteoporosis models reported in the literature, OPG−/− mice exhibit rapid postnatal bone loss and more severe bone porosity. Ablation of OPG from the mouse genome is the first targeted mutation to exhibit osteoporosis of this severity. Other engineered osteoporosis models involve overexpression of specific cytokines and growth factors. Ectopic overexpression of IL4 or TGFβ in the bone have been reported to result in osteopenia, both of which are likely mediated by primary effects on osteoblast activity (Lewis et al. 1993; Erlebacher and Derynck 1996). Overexpression of granulocyte colony-stimulating factor (GCSF) in transgenic mice leads to an osteoporosis-like phenotype attributable to an increase in osteoclast activity (Takahashi et al. 1996). Numerous gene mutations [macrophage CSF (MCSF), c-src and c-fos] have been associated with impaired osteoclast function, resulting in osteopetrotic phenotypes in mice (Yoshida et al. 1990; Soriano et al. 1991; Wang et al. 1992). These mutations are associated with defects in osteoclast development or activation.
Some systemic and local factors that regulate development and interplay between osteoclasts and osteoblasts have been identified (Mundy 1993a,b; Erlebacher and Derynck 1996; Manolagas and Jilka 1995; Hughes et al. 1996; Pacifici 1996). IL1, IL3, IL4, IL6, and IL11 and several CSFs, including MCSF, have been implicated in the control of osteoclast development (Jilka et al. 1992; Lewis et al. 1993; Girasole et al. 1994). Systemic hormones, such as parathyroid hormone and Vitamin D3, and local factors like tumor necrosis factor, promote the development of osteoclasts through their ability to stimulate production of cytokines like IL6 and IL11 by osteoblasts or their precursors (Kitazawa et al. 1994; De La Mata et al. 1995; Uy et al. 1995). OPG is a secreted regulator of osteoclast-mediated bone resorption, and the results of the current gene knockout study clearly implicate OPG as a physiological regulator of osteoclast-mediated bone resorption during postnatal bone growth and remodeling of the skeleton.
OPG−/− mice exhibit an unexpected increase in vascular calcification in the aorta and renal arteries. Most of the OPG−/− mice died or were moribund by 6 months of age due to complications associated with gross skeletal changes. The association of vascular calcification with osteoporosis in patient populations is a recognized phenomenon (Banks et al. 1994; Parhami and Demer 1997; Parhami et al. 1997). The mineral within calcified plaques in the vasculature has been determined to be hydroxyapatite, the same mineral found in bone (Schmid et al. 1980). Additionally, calcified arteries have been shown to express several bone matrix proteins, including collagen type I, matrix GLA protein, osteocalcin, osteonectin, and bone morphogenetic protein type 2, raising the possibility that dysregulation of vessel wall protein and/or growth factor synthesis by smooth muscle or other cells leads to vessel wall calcification.
Interestingly, the arteries exhibiting calcification in OPG−/− mice are sites of endogenous OPG expression (Simonet et al. 1997), suggesting that OPG may have a role in protecting these arteries from pathological calcification. The abundant levels of OPG mRNA detected in embryonic cartilage and arteries suggest that OPG may regulate calcification of these structures during development. However, embryonic bone formation was unaffected in the OPG−/− mice, which were normal at birth. The data reveal that endogenous or maternal OPG is not required for endochondral ossification, or that other factors may compensate for the loss of OPG during embryonic bone formation. Postnatal loss of bone was associated with very active bone remodeling, suggesting that OPG exerts its effects by regulating osteoclast development and recruitment. Bone synthesis by osteoblasts appears to be elevated in OPG−/− mice as evident by sites of active bone remodelling and elevated serum alkaline phosphatase levels. This apparent paradox for OPG loss, that is, loss of mineral from the skeleton with inappropriate mineral deposition in certain vessels, is difficult to reconcile. Regarding the vascular lesions, one must conclude that the loss of OPG ultimately leads to a disturbance in vessel wall protein synthesis in a way that favors the deposition of calcium. Obviously, further studies will be required to determine the mechanism of this effect.
Genetic studies looking at engineered and inbred strains of mice indicate that aortic calcification is likely regulated by two or more genetic components (Qiao et al. 1994). Matrix GLA protein (MGP), an extracellular matrix mineral binding protein, has been implicated as one possible genetic determinant of arterial calcification. MGP-deficient mice exhibit spontaneous calcification of arteries and cartilage (Luo et al. 1997). Unlike the OPG−/− mice, in which arterial calcification is restricted to the aorta and large renal artery, calcification in the MGP mutant mice occurs in elastic and muscular arteries as well. Blood vessel rupture is significantly more severe in the MGP-deficient mice, leading to death within 2 months after birth. However, bone loss is markedly more severe in OPG−/− mice. These findings suggest that OPG has a primary role as a regulator of postnatal bone mass and may also be a contributing factor in degenerative arterial disease.
Materials and methods
Generation of OPG knockout mice
Murine OPG genomic clones were obtained from a 129 SVJ mouse genomic library in Lambda Fix II Vector (Stratagene, La Jolla, CA) by screening with a radiolabeled DNA fragment corresponding to nucleotides 90–1296 of the murine OPG cDNA (GenBank accession no. U94331). A targeting vector was constructed that contained a 1.1-kb short arm of homology and a 5.5-kb long arm of homology flanking a PGK–neo cassette. The PGK–neo cassette replaced a 279-bp region of exon 2 that coded for the first 93 amino acids of the mature OPG protein. A PGK–tk cassette was adjacent to the long arm. The targeting vector was electroporated into RW4 embryonic stem cells (Genome Systems, St. Louis, MO), which were selected in the presence of Gancyclovir and G418, or G418 only. Southern blot analysis using genomic DNA prepared from the 35 isolated cell colonies, identified three recombinant ES cell clones with a single targeted allele. Targeted cells were injected into fertilized blastocysts from C57BL/6 female mice. Chimeric male offspring were crossed with C57BL/6 or Swiss black females. Germ-line transmission of the mutated OPG locus was obtained with chimeras generated from all three targeted ES cell clones. Following heterozygous matings, homozygotes (OPG−/−) were identified and distinguished from heterozygotes (OPG+/−) and wild-type (OPG+/+) mice by Southern blot analysis of genomic DNA cut with PstI and probed with an EcoRI–XmnI OPG-specific probe that was used to confirm homologous recombination.
Histology and bone density measurements
Groups of OPG−/−, OPG+/−, and OPG+/+ mice were necropsied at embryonic day 18, and postnatal day 7, 14, 30, 60, and 180. Radiography was performed prior to the gross dissection on a Faxitron X-ray system (model 43855A, Faxitron X-ray Corp., Buffalo Grove, IL). Blood from the mice was analyzed for clinical chemistries and full hematology. Total body and major organs were weighed and all tissues fixed in formalin.
Bone tissue was decalcified using a formic acid solution and embedded in paraffin. Enzyme histochemistry was performed to determine the expression of tartrate-resistant acid phosphatase activity (TRAP) that specifically stains osteoclasts red (Simonet et al. 1997). In situ hybridization using OPG riboprobes was performed as described previously (Simonet et al. 1997). Measurements and counts were done in a 1-mm2 area distal to the growth plate in the tibia and vertebrae. The field of measurement started adjacent to the growth plate in the center of the marrow cavity, not including the cartilage or cortical bone. Osteoclast number and osteoblast surface were determined. Osteocyte number was determined in three measurement fields per sample at 20× magnification in the cortex of the tibial diaphysis and related to bone area. All measurements were made by tracing the section image onto a digitizing platen with the aid of a camera lucida attachment on the microscope and Osteomeasure (Osteometrics, Inc.) bone analysis software.
Mineral density was determined from bones fixed in 70% ETOH at the proximal tibial metaphysis and tibial cortical shaft of OPG+/+, OPG+/−, and OPG−/− mice by pQCT (XCT-960M, Norland Medical Systems, Ft. Atkinson, WI). Two 0.5-mm cross sections of bone, 1.5 and 2.0 mm from the proximal end of the tibia, were analyzed (XMICE 5.2, Stratec, Germany) to determine total and trabecular bone mineral density in the metaphysis. A single 0.5-mm cross section of bone, 4.0 mm from the proximal end of the tibia, was analyzed to determine the cortical bone density in the tibial diaphysis. In the metaphyseal measurement, total bone density and trabecular bone density (defined as the innermost 20% of the bone cross section) were determined, and an average value for both cross sections is reported. A soft tissue separation threshold of 1500 was used to define the boundary of the metaphyseal bone, and a separation threshold of 2000 was used to define cortical bone.
Matrix mineralization was determined in frozen sections of nondecalcified bone stained by the Von Kossa method. The mineralized matrix is black, whereas other tissues stain red. Bone density was determined in a region 1.0 × 1.5 mm midway between the cortices adjacent to the edge of the growth plate in the proximal tibia by automated image analysis (Metamorph, Universal Imaging Systems, West Chester, PA).
Acknowledgments
We thank Margaret DeRose for assistance in mapping OPG genomic clones, Marlese Pisegna for assistance in genotyping, Laura Martin for injection of blastocysts, Kathy Christensen for colony maintenance, Dave Hill for pathology support, and Hosung Min for critical review of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL ssimonet@amgen.com; FAX (805) 447-1982.
References
- Banks LM, Macsweeney JE, Stevenson JC. Effect of degenerative spinal and aortic calcification on bone density measurements in postmenopausal women: Links between osteoporosis and cardiovascular disease? Eur J Clin Invest. 1994;24:813–817. doi: 10.1111/j.1365-2362.1994.tb02024.x. [DOI] [PubMed] [Google Scholar]
- Beamer WG, Donahue LR, Rosen CJ, Baylink DJ. Genetic variability in adult bone density among inbred strains of mice. Bone. 1996;18:397–403. doi: 10.1016/8756-3282(96)00047-6. [DOI] [PubMed] [Google Scholar]
- Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91:1800–1809. doi: 10.1172/JCI116391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom K, Watson KE, Stanford WP, Demer LL. Atheroclerotic calcification: Relation to developmental osteogenesis. Am J Cardiol. 1995;75:88B–91B. doi: 10.1016/0002-9149(95)80020-s. [DOI] [PubMed] [Google Scholar]
- Bunting CH. The formation of true bone with cellular (red) marrow in a sclerotic aorta. J Exp Med. 1906;8:365–376. doi: 10.1084/jem.8.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Mata J, Uy HL, Guise TA, Story B, Boyce BF, Mundy GR, Roodman GD. Interleukin-6 enhances hypercalcemia and bone resorption mediated by parathyroid hormone-related protein in vivo. J Clin Invest. 1995;95:2846–2852. doi: 10.1172/JCI117990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demer LL. A skeleton in the atherosclerosis closet. Circulation. 1995;92:2029–2032. doi: 10.1161/01.cir.92.8.2029. [DOI] [PubMed] [Google Scholar]
- Erlebacher A, Derynck R. Increased expression of TGF-β2 in osteoblasts results in an osteoporosis-like phenotype. J Cell Biol. 1996;132:195–210. doi: 10.1083/jcb.132.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlebacher A, Filvaroff EH, Gitelman SE, Derynck R. Toward a molecular understanding of skeletal development. Cell. 1995;80:371–378. doi: 10.1016/0092-8674(95)90487-5. [DOI] [PubMed] [Google Scholar]
- Giachelli CM, Bae N, Almeida M, Denhardt DT, Alpers CE, Schwartz SM. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J Clin Invest. 1993;92:1686–1696. doi: 10.1172/JCI116755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girasole G, Passeri G, Jilka RL, Manolagas SC. Interleukin-11: A new cytokine critical for osteoclast development. J Clin Invest. 1994;93:1516–1524. doi: 10.1172/JCI117130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haust MD, Geer JC. Mechanism of calcification in spontaneous aortic arteriosclerotic lesions of the rabbit: an electron microscopic study. Am J Pathol. 1970;60:329–346. [PMC free article] [PubMed] [Google Scholar]
- Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-β. Nature Medicine. 1996;2:1132–1136. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- Jilka RL, Hangoc G, Girasole G, Passeri G, Williams DC, Abrams JS, Boyce B, Broxmeyer H, Manolagas SC. Increased osteoclast development after estrogen loss: mediation by IL-6. Science. 1992;257:88–91. doi: 10.1126/science.1621100. [DOI] [PubMed] [Google Scholar]
- Kitazawa R, Kimble RB, Vannice JL, Kung VT, Pacifici R. Interleukin-1 receptor antagonist and tumor necrosis factor binding protein decrease osteoclast formation and bone resorption in ovariectomized mice. J Clin Invest. 1994;94:2397–2406. doi: 10.1172/JCI117606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey DL, Erdmann JM, Teitelbaum SL, Tan HL, Ohara J, Shioi A. Interleukin 4, interferon-γ, and prostaglandin E impact the osteoclastic cell-forming potential of murine bone marrow macrophages. Endocrinology. 1995;136:2367–2376. doi: 10.1210/endo.136.6.7750457. [DOI] [PubMed] [Google Scholar]
- Lewis DB, Liggitt HD, Effmann EL, Motley ST, Teitelbaum SL, Jepsen KJ, Goldstein SA, Bonadio J, Carpenter J, Perlmutter RM. Osteoporosis induced in mice by overproduction of interleukin 4. Proc Natl Acad Sci. 1993;90:11618–11622. doi: 10.1073/pnas.90.24.11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G, Ducy P, Mckee MD, Pinero G J, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- Manolagas SC, Jilka RL. Bone marrow, cytokines, and bone remodeling; Emerging insights into the pathophysiology of osteoporosis. New Engl J Med. 1995;332:305–311. doi: 10.1056/NEJM199502023320506. [DOI] [PubMed] [Google Scholar]
- Marks SC. The structural basis for bone cell biology. Acta Med Dent Helv. 1997;2:141–157. [Google Scholar]
- Mundy GR. Cytokines and growth factors in the regulation of bone remodeling. J Bone Min Res. 1993a;8:S505–S510. doi: 10.1002/jbmr.5650081315. [DOI] [PubMed] [Google Scholar]
- ————— Role of cytokines in bone resorption. J Cell Biochem. 1993b;53:296–300. doi: 10.1002/jcb.240530405. [DOI] [PubMed] [Google Scholar]
- Mundy GR, Boyce B, Hughes D, Wright K, Bonewald L, Dallas S, Harris S, Ghosh-Choudhury N, Chen D, Dunstan C, Izbicka E, Yoneda T. The effects of cytokines and growth factors on osteoblastic cells. Bone. 1995;17:71S–75S. doi: 10.1016/8756-3282(95)00182-d. [DOI] [PubMed] [Google Scholar]
- O’Brien KD, Kuusisto J, Reichenbach DD, Ferguson M, Giachelli C, Alpers CE, Otto CM. Osteopontin is expressed in human aortic valvular lesions. Circulation. 1995;92:2163–2168. doi: 10.1161/01.cir.92.8.2163. [DOI] [PubMed] [Google Scholar]
- Pacifici R. Estrogen, cytokines, and pathogenesis of postmenopausal osteoporosis. J Bone Min Res. 1996;11:1043–1051. doi: 10.1002/jbmr.5650110802. [DOI] [PubMed] [Google Scholar]
- Parhami F, Bostrom K, Watson K, Demer LL. Role of molecular regulation in vascular calcification. J Atheroscler Thromb. 1996;3:90–94. doi: 10.5551/jat1994.3.90. [DOI] [PubMed] [Google Scholar]
- Parhami F, Demer LL. Arterial calcification in face of osteoporosis in ageing: Can we blame oxidized lipids? Curr Opin Lipidol. 1997;8:312–314. doi: 10.1097/00041433-199710000-00010. [DOI] [PubMed] [Google Scholar]
- Parhami F, Morrow AD, Balucan J, Leitinger N, Watson AD, Tintut Y, Berliner JA, Demer LL. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation: A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol. 1997;17:680–687. doi: 10.1161/01.atv.17.4.680. [DOI] [PubMed] [Google Scholar]
- Qiao J, Xie P, Fishbein MC, Kreuzer J, Drake TA, Demer LL, Lusis AJ. Pathology of atheromatous lesions in inbred and genetically engineered mice: Genetic determination of arterial calcification. Arterioscler Thromb. 1994;14:1480–1497. doi: 10.1161/01.atv.14.9.1480. [DOI] [PubMed] [Google Scholar]
- Roodman GD. Advances in bone biology: The osteoclast. Endocr Rev. 1996;17:308–332. doi: 10.1210/edrv-17-4-308. [DOI] [PubMed] [Google Scholar]
- Schmid K, McSharry WO, Pameijer CH, Binette JP. Chemical and physicochemical studies on the mineral deposits of the human atherosclerotic aorta. Atherosclerosis. 1980;37:199–210. doi: 10.1016/0021-9150(80)90005-2. [DOI] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang M-S, Luethy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted Disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley JM, Martin TJ, Suda T. Osteoblastic cells are involved in osteoclast formation. Endocrinology. 1988;123:2600–2602. doi: 10.1210/endo-123-5-2600. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Wada T, Mori M, Kokai Y, Ishii S. Overexpression of the granulocyte colony stimulating factor gene leads to osteoporosis in mice. Lab Invest. 1996;74:827–834. [PubMed] [Google Scholar]
- Uy HL, Guise TA, De La Mata J, Taylor SD, Story BM, Dallas MR, Boyce BF, Mundy GR, Roodman GD. Effects of parathyroid hormone (PTH)-related protein and PTH on osteoclasts and osteoclast precursors in vivo. Endocrinology. 1995;136:3207–3212. doi: 10.1210/endo.136.8.7628353. [DOI] [PubMed] [Google Scholar]
- Wang Z-Q, Ovitt C, Grigoriadis AE, Moehle-Steinlein U, Ruether U, Wagner EF. Bone and haematopoitic defects in mice lacking c-fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okumura H, Sudo T, Shultz LD, Nishikawa S-I. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]