

Abstract
Expression of HPV16 early region genes in basal keratinocytes of transgenic mice elicits a multistage pathway to squamous carcinoma. We report that infiltration by mast cells and activation of the matrix metalloproteinase MMP-9/gelatinase B coincides with the angiogenic switch in premalignant lesions. Mast cells infiltrate hyperplasias, dysplasias, and invasive fronts of carcinomas, but not the core of solid tumors, where they degranulate in close apposition to capillaries and epithelial basement membranes, releasing mast-cell-specific serine proteases MCP-4 (chymase) and MCP-6 (tryptase). MCP-6 is shown to be a mitogen for dermal fibroblasts that proliferate in the reactive stroma, whereas MCP-4 can activate progelatinase B and induce hyperplastic skin to become angiogenic in an in vitro bioassay. Notably, premalignant angiogenesis is abated in a mast-cell-deficient (KITW/KITWWv) HPV16 transgenic mouse. The data indicate that neoplastic progression in this model involves exploitation of an inflammatory response to tissue abnormality. Thus, regulation of angiogenesis during squamous carcinogenesis is biphasic: In hyperplasias, dysplasias, and invading cancer fronts, inflammatory mast cells are conscripted to reorganize stromal architecture and hyperactivate angiogenesis; within the cancer core, upregulation of angiogenesis factors in tumor cells apparently renders them self-sufficient at sustaining neovascularization.
Keywords: angiogenesis, cancer, gelatinase B, HPV16, inflammation, mast cells
Tumorigenesis is invariably a multistep process (Fearon and Vogelstein 1990; Folkman and Hanahan 1991; Christofori and Hanahan 1994), that involves not only transformed cells but also an assemblage of normal support cells, including stromal fibroblasts, and endothelial cells (Dvorak 1986; Hanahan 1998; Kinzler and Vogelstein 1998). Tumor growth demonstrably depends on angiogenesis, whereby concomitant increases in the tumor vasculature supply nutrients and oxygen to the expanding neoplastic mass (Folkman 1990).
The skin is composed of an avascular epidermal compartment and a subjacent vascularized dermis. Whereas the skin vasculature is typically quiescent (Detmar 1996), it maintains a capability to initiate transitory neovascular responses to diverse epithelial stimuli, e.g., pathogen assault and wounding. The skin microenvironment likely facilitates angiogenic responses; among its salient characteristics is the propensity for rapid inflammatory responses to abnormal tissue conditions, resulting in increased blood supply, increased vascular permeability, and extravasation of diverse cytokine-producing leukocytes, e.g., macrophages, lymphocytes, and mast cells (for review, see Roitt et al. 1989), seeking to resolve the abnormality.
Mast cells (MCs) play an important role in acute inflammation owing to their release of stored and newly synthesized inflammatory mediators following activation (Yong 1997). MC density within skin connective tissue correlates with blood vessel density (Eady et al. 1979), and activation and degranulation of MCs is associated with connective tissue degradation around sites of neovascularization (Dabbous et al. 1986). As neovascularization of otherwise quiescent tissues, such as skin, involves the action of angiogenic growth factors and the localized proteolytic modification of the extracellular matrix (ECM), MCs are implicated as accessory cells for angiogenesis (Meininger 1995; Yong 1997).
We have investigated mechanisms regulating angiogenesis in a mouse model of epithelial carcinogenesis wherein the early region genes of human papillomavirus type 16 (HPV16) are expressed under the control of the keratin 14 (K14) promoter/enhancer (Arbeit et al. 1994). In this skin cancer model, mice are born phenotypically normal; by 1 month of age mice develop epidermal hyperplasia with 100% penetrance, which advances focally into angiogenic dysplasias between 3 and 6 months. By one year, 50% of the mice develop invasive squamous cell carcinomas (SCC) (Coussens et al. 1996). Characteristically, cancers appear on the ears and upper trunk. We have determined previously that neoplastic progression in this model is accompanied by up-regulation of genes encoding several pro-angiogenic growth factors (Arbeit et al. 1996), implicating these molecules in tumor angiogenesis in the skin. We now present evidence that inflammatory mast cells are involved in activating premalignant neovascularization in the skin, and propose functional roles for MC-specific proteases in angiogenesis and stromal remodeling in this transgenic mouse model of epithelial carcinogenesis.
Results
Premalignant dysplasias are characterized by alterations in vascular architecture
In the skin of K14–HPV16 transgenic mice (Smith-McCune et al. 1997), as in human cervical carcinogenesis (Smith-McCune and Weidner 1994; Smith-McCune et al. 1997), neoplastic progression involves an early increase in capillary density first evident subjacent to hyperplastic epithelium. We now report that dramatic changes in dermal capillary location, density, and architecture accompany both premalignant and malignant progression. Normal mouse skin contained infrequent capillaries located deep within the dermis as visualized by immunolocalization of platelet endothelial cell (EC) adhesion molecule (PECAM-1/CD31), an endothelial cell-specific marker (DeLisser et al. 1994) (Fig. 1A). Hyperplastic lesions showed a modest increase in the density and dilation of capillaries that remain distal to the neoplastic epidermis (Fig. 1B). Dysplastic lesions contained dilated and enlarged capillaries that are increased in number, and localized proximal to the epithelial basement membrane (Fig. 1C). This vascular pattern is indicative of an angiogenic switch from vascular quiescence to modest neovascularization in early low-grade lesions (hyperplasias), followed by a striking upregulation of angiogenesis in high-grade lesions (dysplasias). The core of SCCs, both well differentiated (WDSC; Fig. 1D) and moderately to poorly differentiated (M-PDSC; Fig. 1E), as well as the invasive fronts of the tumors (Fig. 1F), were likewise well vascularized, containing a chaotic array of small and dilated capillary webs.
Figure 1.

Altered morphology and architecture of capillaries during premalignant neovascularization. Immunohistochemical staining of 5-μm paraffin-embedded tissue sections for CD31 expressed on capillary endothelial cells (red staining) counterstained with methyl green in (A) normal nontransgenic (-lm) ear skin, (B) hyperplastic (hyp) ear skin, (C) dysplastic (dys) ear skin, (D) ear WDSC, (E) truncal M-PDSC center, and (F) the invasive front (Inv front) of a truncal M-PDSC with arrows indicating direction of tumor expansion. (Dashed line) Epidermal–dermal interface or location of skin basement membrane zone; (e) epidermis. Bar, 44.6 μm (A–F).
Stromal infiltration of mast cells accompanies activation of angiogenesis and progression to dysplasia
Based on clues revealed by whole-mount visualization of the vasculature, we asked whether MCs were associated with the distinctive stages of carcinogenesis by utilizing chloroacetate esterase (CAE) histochemistry on paraffin-embedded sections of staged neoplastic skin (Fig. 2). CAE detects the presence of chymotrypsin-like serine esterases (chymases) present within connective tissue MCs (Leder 1979; Caughey et al. 1988). Normal skin contained few MCs distributed within the dermis distal to the epidermal basement membrane (Fig. 2A). MC numbers increased at the hyperplastic stage, and were associated predominately with small blood vessels within dermal stroma (Fig. 2B). With the switch to intense angiogenesis in dysplasias, MC density increased markedly in the dermal connective tissue in both early (3-month-old) and late (6-month-old) angiogenic lesions (Fig. 2C,D, respectively). In contrast to the angiogenic dysplasias, tumor stroma within M-PDSCs was essentially devoid of MCs (Fig. 2E). However, the stroma around invading fronts of the tumors contained abundant MCs, most frequently abutting capillaries (Fig. 2F). MCs were never observed within the epithelial compartment at any stage of neoplastic progression.
Figure 2.
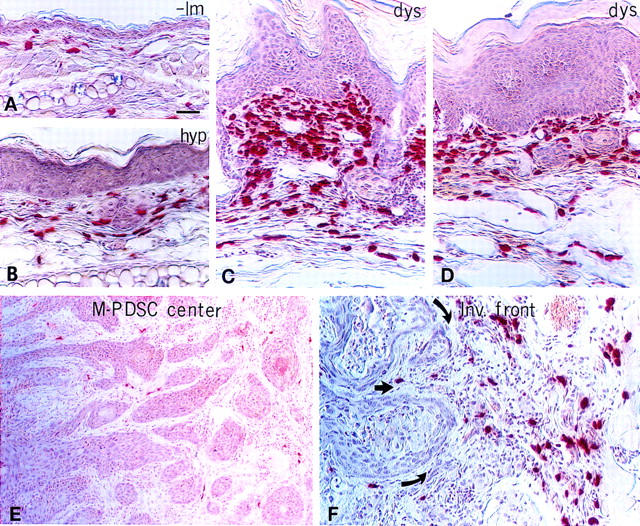

Mast cell infiltration during activation of angiogenesis. Chloroacetate esterase histochemistry (red staining) on 5-μm paraffin-embedded tissue sections counterstained with hematoxylin (blue) identifies location of infiltrating MCs during neoplastic progression in (A) nontransgenic litter mate (-lm) ear skin, (B) hyperplastic (hyp) ear skin, (C and D) dysplastic (dys) ear skin, (E) the center of a chest M-PDSC, (F) and at the invading front of a truncal M-PDSC. Arrows indicate direction of tumor expansion. (G) Whole-mount microscopy of lectin-perfused transgenic mouse with dysplastic truncal lesions. Darkly stained mast cells (arrows) appear to adhere directly to basement membranes surrounding the dermal capillaries (c) in angiogenic dysplasias. (H) Metachromatic granules in MCs stained with toluidine blue. Degranulating mast cells (arrows) present within a dysplastic lesion are juxtaposed tightly to skin basement membrane (bm) and dermal capillaries (c, arrowhead). (d) Dermis, (e) epidermis. Bar, 44.6 μm (A–D, F); 89 μm (E); 19.7 μm (G); 12.9 μm (H).
Exploiting the avidin and toluidine blue binding capabilities of tissue MCs to view their location at high resolution (Befus et al. 1982; Tharp et al. 1985), we saw an intimate association between infiltrating MCs in angiogenic dysplasias with both subendothelial and epithelial basement membranes (Fig. 2, G and H, respectively).
We assessed the possibility that intense MC infiltration of dysplastic lesions was an antibody-mediated response to chronic bacterial infection of the skin, by treating one cohort of transgenic mice systemically with antibiotics [sulfamethoxole (60 mg/tablet), trimethoprim (10 mg/tablet), 1 tablet/2 mice per week], and by breeding a second cohort into the immunodeficient Rag1/null background (Mombaerts et al. 1992). These treatments did not alter influx of MCs into the stroma beneath dysplastic epidermis (data not shown). Thus, the presence of MCs in angiogenic dysplasias does not appear to be a response to activation of the humoral immune system.
Angiogenic dysplasias are highly enriched in mast cell-specific serine proteases
We next quantified the activities of MC-specific tryptic and chymotryptic serine proteases (tryptases and chymases, respectively) in extracts of tissue from each neoplastic stage with selective solution assays (Fig. 3A). Although chymase-like activity was detectable in normal negative littermate control tissue [n = 2; 62.5 ± 17.5 nmoles of pNA/min per mg protein (s.e.m.)] and in hyperplastic tissue (n = 4; 39.8 ± 13.5 nmoles of pNA/min per mg protein), there was a significant increase in activity in angiogenic dysplasias (n = 6; 204 ± 3.6 nmoles of pNA/min/mg protein; P < 0.003). In addition, angiogenic dysplasias contained higher levels of chymase-like activity than SCCs (n = 3; 15.3 ± 8.2 nmoles of pNA/min/mg protein). Chymase-like activity in extracts was completely inhibited by 10 μm soybean trypsin inhibitor (SBTI), but not by 100 μg/ml aprotinin, as is characteristic of MC chymases (Caughey 1995).
Figure 3.





Chymase and tryptase in neoplastic skin. (A) Chymotryptic and tryptic activity in neoplastic skin. Values represent mean chymase and tryptase activity in ear skin biopsies from negative littermate (N), hyperplastic (H), dysplastic (D), and tumor (T) solubilized by extraction at high ionic strength, normalized to the amount of protein in extracts ±s.e.m.); 5-μl each of tissue sample were incubated with 1 mm chymotryptic or tryptic substrate. The change in absorbance at 410 nm at 37°C is shown where units indicate μmoles of substrate cleaved/min per mg protein. (B) RT–PCR analysis for all mMCPs in dysplastic ear mRNA. Sizes shown indicate base pairs. All primer pairs were tested for specific priming of control RNA. (C) SDS-PAGE of mMCP-4 purification from K14–HPV16 mouse ears. Samples were reduced with dithiothreitol and visualized with Coomassie brilliant blue. (Lane 1), 2 m NaCl extract of K14-HPV mouse ears (5 μg of protein); (lane 2) 2 m NaCl extract SBTI-agarose unbound fraction (1 μg of protein); (lane 3), 5 μg of pure mMCP-4, SBTI-agarose acid eluate. (D) Anti-rat MCP-1 IgG reacts specifically with mMCP-4. (Lane 1) Crude high-salt extract of ears (2 μl); (lane 2), 2.5 ng of purified mMCP-4 from SBTI-agarose acid eluate. (E) SDS-PAGE of mMCP-6 purification from K14–HPV16 mouse ears. Samples were reduced with dithiothreitol and visualized with Coomassie brilliant blue. (Lane 1) Unbound fraction from p-aminobenzamidine–agarose chromatography, 2 μl; (lane 2), 2 m NaCl extract, unbound fraction, 2 μl; (lane 3) MMCP-6 peak (240 ng) from heparin–HPLC NaCl gradient elution. Molecular masses are shown in kD.
Similarly, tryptase-like activity was detectable in both normal (n = 2; 13 ± 13 nmoles of pNA/min per mg protein) and hyperplastic (n = 4; 15.7 ± 10.6 nmoles of pNA/min/mg protein) skin; again, angiogenic dysplasias possessed significantly elevated activity (n = 6; 131 ± 49.3 nmoles of pNA/min per mg protein; P < 0.04), compared to the former and to frank carcinomas (n = 3; 30 ± 12.6 nmoles of pNA/min/mg protein). The tryptase-like activity in extracts was inhibited by 1.5 mm benzamidine, a general inhibitor of tryptic serine proteases, but not by 10% mouse plasma (data not shown). Resistance to serine protease inhibitors present in plasma is an idiosyncratic feature of MC tryptases.
MCs in different anatomic locations and in different pathologic conditions selectively express distinct chymase transcripts [mouse mast cell protease (mMCP) 1, -2, -4 and/or -5] and tryptase transcripts (mMCP-6 and/or -7) (Reynolds et al. 1990; Stevens et al. 1994). We detected mRNA representing only mMCP-4, a β-chymase, and mMCP-6, a tryptase, by RT–PCR analysis of RNA isolated from dysplastic ear skin (Fig. 3B). We confirmed the presence of their encoded gene products by immunoblot analysis of neoplastic skin lysates; an ∼28 kD mMCP-4 was detected with affinity-purified anti-rat MCP-1 IgG (rat MCP-1 is the ortholog of mMCP-4; Caughey 1995) and an ∼34 kD mMCP-6 was detected with affinity-purified anti-mMCP-6 IgG (Ghildyal et al. 1996) (data not shown).
The chymase purified from angiogenic dysplasias represented 99% of the chymotryptic activity loaded onto an SBTI-agarose column. It had a specific activity of 5.5-μmole of succinyl-l-Ala-Ala-Pro-Phe-4-nitroanilide hydrolyzed/min per mg, migrated as a predominant band at ∼28 kD by SDS-PAGE (Fig. 3C), and reacted with anti-rat MCP-1 IgG in immunoblotting (Fig. 3D). The amino-terminal sequence of the purified chymase was IIGGVESRP, which is identical to the published amino acid sequence of mMCP-4 (Huang et al. 1991). Purified mMCP-4 was inhibited completely by SBTI in a 1:100 enzyme-to-inhibitor molar ratio, but inhibited only weakly by aprotinin in a 500-fold molar excess (data not shown).
mMCP-6 was purified from dysplastic K14–HPV16 transgenic mouse ears by high-salt buffer extraction followed by p-amino benzamidine and heparin affinity chromatography. The purified tryptase migrated as a single protein band at ∼34 kD (Fig. 3E), and immunoblotted with anti-mMCP-6 antibody (data not shown). The amino-terminal sequence was IVGGHEASES, identical to mMCP-6 (Reynolds et al. 1991) and distinct from the other mouse tryptase, mMCP-7. The specific activity of purified mMCP-6 was 56 μmole of tosyl-Gly-Pro-Lys-4-nitroanilide hydrolyzed/min per mg. This activity represented 99% of the tryptic activity present in the high-salt buffer loaded onto the benzamidine-agarose column (data not shown). MMCP-6 was inhibited by bis (5-amidino-2-benzimidazoyl) methane (BABIM) at a 1:500 molar ratio, a potent inhibitor of mast cell tryptases (Caughey et al. 1993), but was resistant to inhibition by aprotinin, at a 1:75 enzyme-to-inhibitor molar ratio.
Mouse MCP-6 is a mitogen for dermal fibroblasts
We observed that the cellularity of dermal fibroblasts and the deposition of collagen into the ECM, as demonstrated by Masson’s trichrome staining (data not shown), increased incrementally during early neoplastic progression. This is notable, as stromal fibroblasts in skin, much like ECs, are typically quiescent (Gregoire and Lieubeau 1995). In wound healing and neoplastic situations, proliferating fibroblasts are the source of newly synthesized type I procollagen and are responsible for desmoplastic phenotypes (Vuorio and de Crombrugghe 1990; Gregoire and Lieubeau 1995). Tryptase is a mitogen (Hartmann et al. 1992) and potent stimulator of type α1(I) procollagen mRNA synthesis (Cairns and Wells 1997; Gruber et al. 1997) in human fibroblasts in vitro. These activities of tryptase may be mediated through interaction with protease activated receptor-2 (PAR-2) present on fibroblast plasma membranes (Molino et al. 1997; Schechter et al. 1998).
Accordingly, we tested the ability of mMCP-6 tryptase and mMCP-4 chymase to stimulate DNA synthesis of quiescent serum-deprived subconfluent primary murine dermal fibroblasts (PMDFs) and quiescent human umbilical vein endothelial cells (HUVECs), in the absence of exogenous growth factors. MMCP-6 stimulated fibroblast DNA synthesis selectively in a concentrationdependent manner yet did not affect proliferation of HUVECs. (Fig. 4A,B). In contrast, mMCP-4 did not stimulate DNA synthesis in either cell population at any concentration tested (Fig. 4A,B). These results demonstrate that mMCP-6 tryptase has cell-type-specific mitogenic potency.
Figure 4.

Effects of tryptase on skin fibroblasts. Effect of MCPs on HUVEC proliferation (A) and on PMDF proliferation (B). Agonists were added to subconfluent, serum-deprived cultures (VEGF, 1 and 10 ng/ml; bFGF, 1 and 10 ng/ml; mMCP-6, 1 and 10 nm; mMCP-4, 2 and 20 nm; heparin at 74 and 740 ng/ml) for 16 hr. [3H]Thymidine incorporation was then determined. Results shown indicate mean values (±s.e.m.; n = 3). (C–E) Dark-field photographs of ear-tissue sections from (C) negative litter mate (-lm) normal skin, (D) hyperplastic (hyp) skin, and (E) dysplastic (dys) skin hybridized with a (α1)I procollagen antisense mRNA probe. Arrows point to procollagen expressing cells within the stroma. (e) Epidermis; (d) dermis, (c) central ear cartilage. Bar, 44.6 μm (C–E).
In vivo, we observed that normal skin exhibited patchy expression of type α1(I) procollagen mRNA in stroma adjacent to capillaries (Fig. 4C), whereas mRNA expression was up-regulated incrementally in hyperplastic lesions adjacent to capillaries and subjacent to epithelium (Fig. 4D). In angiogenic dysplasias heavily infiltrated with MCs, type α1(I) procollagen mRNA was expressed widely in stroma (Fig. 4E), consistent with high levels of tryptase, increased cellularity of dermal fibroblasts, and increased deposition of collagen.
MMCP-4 induces ECM remodeling and angiogenesis by direct and indirect mechanisms
The cell proliferation assay pointed to a role for tryptase in activating quiescent fibroblasts to become a proliferative component of the reactive neoplastic stroma; that same assay revealed no mitogenic activity of chymase on fibroblasts or endothelial cells. Therefore, we asked whether chymase contributed by altering tissue microenvironment in some other way. Interestingly, canine and human MC α-chymases are known to activate the latent extracellular protease progelatinase B (proMMP-9) (Fang et al. 1996 1997). Gelatinase B is a member of the matrix metalloproteinase (MMP) family and is involved in both ECM remodeling (Coussens and Werb 1996) and regulation of angiogenesis (Vu et al. 1998).
We hypothesized, therefore, that mMCP-4 contributed to activation of angiogenesis by inducing ECM remodeling through the activation of progelatinase B. To address this, we first assessed the relative activity of gelatinase B in tissue lysates representing distinct neoplastic stages utilizing gelatin-substrate zymography (Fig 5A). Progelatinase B was not detectable in normal skin, but was up-regulated incrementally in hyperplastic, dysplastic, and tumor tissue; however, active gelatinase B appeared only in the dysplastic and tumor lysates. In addition, this assay revealed the activity profile of another MMP family member, namely pro- and active gelatinase A (MMP-2), whose activity changed only modestly in the different neoplastic stages. The appearance of activated gelatinase B in dysplastic lesions correlated with dramatic MC infiltration and elevated levels of mMCP-4 chymase activity.
Figure 5.


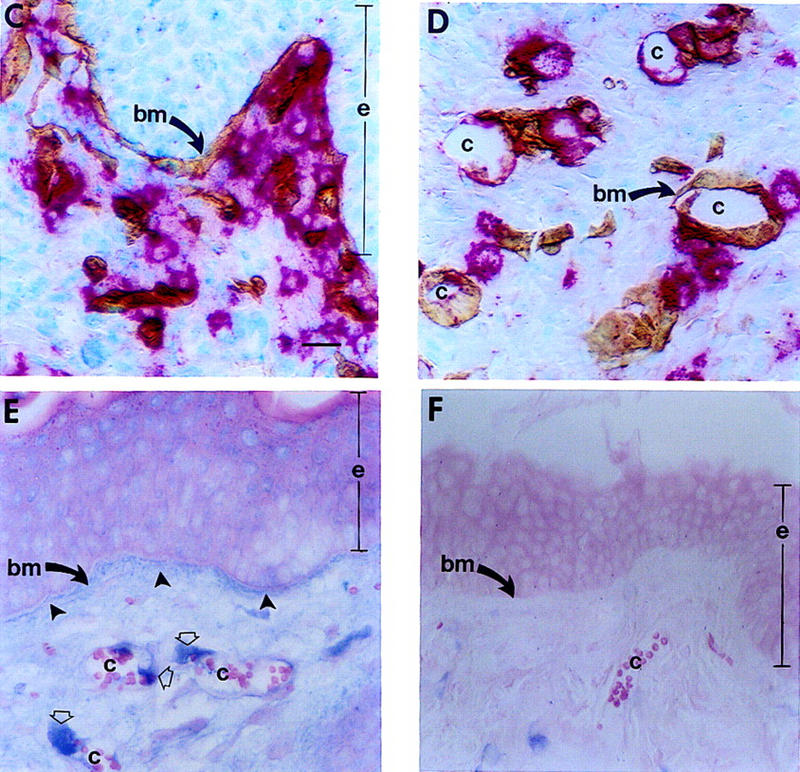
Effect of mMCP-4 on progelatinase B present in hyperplastic tissue and localization of mMCP-4 and gelatinase B in vivo. (A) Gelatinolytic activity in tissue lysates (2 μl) from normal (N), hyperplastic (H), dysplastic (D) and carcinoma (T) biopsies. Incubation of gelatin zymograms with 1,10 phenanthroline (an inhibitor of MMPs), but not PMSF (an inhibitor of serine proteases) following electrophoresis abolished the 68, 72, 80, and 90-kD bands completely (data not shown). (B) In vitro reconstitution of gelatinase B activity. Two microliters of hyperplastic (H) lysate alone or 2 μl of hyperplastic lysate incubated for 30 min at 37°C with 40 ng of purified mMCP-4. Molecular masses are shown in kD. (C–D) Immunolocalization of mMCP-4 (red staining) and laminin (brown staining) at basement membranes (bm, arrows) adjacent to epithelium (e) and capillaries (c) in dysplastic skin. (E) Immunolocalization of gelatinase B (blue staining) in dysplastic skin localizes to basement membranes (bm) adjacent to epithelium (e, closed arrows) and around capillaries (c, open arrows) in dermis. (F) Control for nonspecific binding using a control rabbit IgG; background staining was negligible. Bar, 44.6 μm (C–F).
To test if mMCP-4 could activate progelatinase B, similar to canine or human α-chymases, lysates from hyperplastic skin (containing predominantly the pro-form of gelatinase B) were incubated with purified mMCP-4 prior to substrate zymography. Incubation of the lysates with mMCP-4 led to a dramatic increase in the amount of activated gelatinase B, whereas neither the pro nor active forms of gelatinase A were changed (Fig. 5B). Addition of ecotin (an inhibitor of mMCP-4 but not urokinase) abolished mMCP-4-induced activation of gelatinase B completely, whereas 1 mm amiloride (an inhibitor of urokinase) had no effect (data not shown). Furthermore, immunolocalization of mMCP-4 (red staining; Fig. 5C,D) and gelatinase B (blue staining; Fig. 5E) in dysplastic tissue sections revealed colocalization of both enzymes proximal to epithelial and subendothelial basement membranes as demonstrated by immunolocalization of laminin-β1 (brown staining, Fig. 5C, D). Thus, mMCP-4 colocalizes with and induces activation of progelatinase B, but not progelatinase A, in early neoplastic tissue.
We next asked if mMCP-4 chymase, mMCP-6 tryptase, or gelatinase B could convert an otherwise nonangiogenic tissue to an angiogenic one utilizing an in vitro angiogenesis bioassay (Fig. 6). Hyperplastic ear skin from 1-month-old transgenic mice was incubated in medium alone or medium containing mMCP-4, mMCP-6, or gelatinase B. Subsequently, the hyperplastic skin was embedded into a collagen gel alone, or a collagen gel containing randomly dispersed bovine capillary endothelial cells (BCEs). Hyperplastic skin pieces, both control and protease-treated, were placed into collagen gels lacking BCEs. They showed negligible cell emigration from biopsies (data not shown). BCEs remained randomly distributed around untreated hyperplastic skin (Fig. 6A). Treatment of skin with mMCP-6 also yielded no response (Fig. 6B), even though human tryptase is a known EC morphogen (Blair et al. 1997). In contrast, mMCP-4-treated skin induced a dramatic response of the BCEs, with radial alignment, proliferation, migration, and tube formation toward the dermal surface of the skin pieces with no evidence of migration or tube formation in the direction of the epidermis (Fig. 6C,D). Gelatinase B-treated hyperplastic skin showed a similar response to that observed with mMCP-4 (data not shown). Neither mMCP-4 nor gelatinase B added directly to BCEs in the collagen gel induced a response (data not shown), indicating that these activities were caused directly by release of sequestered activity from the skin pieces. Thus, both mMCP-4 and gelatinase B induce ECM remodeling of hyperplastic tissue directly leading to release of sequestered angiogenic activity.
Figure 6.

Effect of mMCP-4 on sequestered angiogenic activity in premalignant tissue. Hyperplastic ear skin (S) from a 1-month old K14–HPV16 mouse was incubated for 48 hr in the presence or absence of enzymes at 37°C followed by implantation into a collagen gel with EC coculture. Cultures were maintained for 3 weeks, with photographs of tube ingrowths taken after 12 days of coculture. (A) untreated ear skin implanted into EC-collagen gel; (B) mMCP-6-treated (10 nm) ear skin implanted into EC-collagen gel; (C and D) mMCP-4-treated (20 nm) ear skin implanted into EC-collagen gel showing an angiogenic response with extensive EC tube formation. Lower magnification (D) shows the polarized direction of tubular structures towards the dermal (d) surface, as opposed to the epidermal (e) surface of the skin. Small arrows (A and B) indicate the more-or-less random growth of ECs, whereas thick arrows (C and D) indicate radially aligned tubules growing toward the skin piece. (h) Hair. Bar: 58 μm (A–C); 116 μm (D).
MC deficiency abates premalignant progression
Our data suggest that MC-derived factors, including chymase and tryptase, are involved in the early events of neoplastic progression, e.g., fibroblast activation, ECM remodeling, and activation of angiogenesis. To test this hypothesis, we sought to take advantage of mice that were genetically MC deficient. The receptor tyrosine kinase c-Kit, is encoded by the white spotting (W) locus on mouse chromosome 5 (Chabot et al. 1988). Complete loss of c-Kit function is lethal; however, mice carrying KITW mutations (W37, Wv, W41, and W) are viable to varying degrees, but manifest pleiotropic developmental defects, including sterility, coat color abnormalities, severe macrocytic anemia, and mast-cell deficiency (Bernstein et al. 1990; Nocka et al. 1990). The tissues of adult WBB6F1 KITW/KITWv mice contain <1% the number of MCs compared to their normal (+/+) littermates (Kitamura et al. 1978). Thus, we attempted to generate K14–HPV16 KITW/KITWv mice to assess the importance of MCs as extrinsic potentiators of early neoplastic progression.
Because WBB6F1 KITW/KITWv mice are sterile (Nocka et al. 1990), and because neoplastic progression in K14–HPV16 is only permissive in FVB/n (Coussens et al. 1996), we first crossed both KITW/+ (WB/ReJ) and KITWv/+ (C57BL/6J) alleles into FVB/n for four generations. At the fourth generation, we intercrossed each W allele with K14–HPV16; genotypes of the expected Mendelian ratios were obtained. To obtain K14–HPV16 KITW/KITWv genotypes, we crossed HPV16 KITW/+ with KITWv/+, and HPV16 KITWv/+ with KITW/+, generating 104 litters and 712 pups. The expected simultaneous transmission frequency of all three desired loci is 12.5%. Thus, of the 712 live pups, 179 should have been KITW/KITWv and of those, 89 would be predicted to be K14–HPV16 KITW/KITWv. Instead, one K14–HPV16 KITW/KITWv mouse and no KITW/KITWv mice were born. When we analyzed litter size of the KITW/+ with KITWv/+ crosses, we realized that on average, litters contained 25% fewer live births [6.85 ± 2.94 P < 0.0001.] as compared to K14–HPV16 with FVB/n crosses [8.79 ± 3.31 (±s.d.), n = 80 litters, 703 mice), leading us to conclude that in the FVB/n background, KITW/KITWv produces a near-lethal phenotype. Hence, much like with many targeted gene knockouts, genetic background has a profound effect on viability.
We analyzed the neoplastic phenotype in the K14–HPV16 KITW/KITWv mouse at 2 months of age (shortly before it died) in comparison to MC-proficient littermates (KIT+/KIT+ and K14–HPV16 KIT+/KIT+; Fig. 7). At this point in neoplastic progression, the hyperplastic phenotype in K14–HPV16 mice is 100% penetrant (n = 380), and is characterized by a thickened epidermis with underlying dermal MC infiltration (CAE, red staining; Fig. 7B), hyperproliferation of keratinocytes (Ki-67, brown staining; Fig. 7E), and altered vascular architecture including increased vascular density and dilation (CD-31, brown staining; Fig. 7H). In the K14–HPV16 KITW/KITWv mouse, MCs were absent from the dermis (Fig. 7C). Keratinocyte proliferation, as measured by immunoreactivity of Ki-67, although enhanced [Fig. 7F, brown staining; 35% ± 2.8 (s.e.m.), n = 10, 20× fields] compared to skin of negative littermates [Fig. 7D, brown staining; 12% ± 1.2 (s.e.m.), n = 10, 20× fields/mouse, 3 mice], was markedly decreased compared to K14–HPV16 KIT+/KIT+ littermates (Fig. 7E, brown staining; 48% ± 1.6% (s.e.m.), n = 10, 20× fields/mouse, 8 mice]. Likewise, dermal vasculature in the K14–HPV16 KITW/KITWv skin (Fig. 7I) was more similar to quiescent vasculature typical of normal skin (Fig. 7G) than to that observed in angiogenic skin (Fig. 7H), with infrequent, nondilated vessels found distal to the epidermal basement membrane. Thus, by three different criteria (epidermal thickness, keratinocyte proliferation, and vascularization) the absence of MCs functionally compromises early neoplastic progression.
Figure 7.

Mast cell deficiency abates early neoplastic progression. Presence of mast cells (CAE, red staining; A–C), proliferation of keratinocytes (Ki-67, brown staining; D–F), and capillary architecture and densities (CD-31, brown staining; panels G–I) were compared between wild-type littermates (-LM; n = 3; A, D, and G), K14–HPV16 KIT+/KIT+ (n = 8; B, E, and H), and K14–HPV16 KITW/KITWWv (n = 1; C, F, and I) mice. Ear tissue biopsies (5 × 2-mm) were removed from 2-month old littermates, embedded in OCT freezing medium, and sectioned (10 μm). Dashed line indicates epidermal–dermal interface. Arrows point to dilated capillaries subjacent to hyperplastic skin. (f) Hair follicle. Bar, 44.6 μm (A–I).
Discussion
Inflammation is a protective reaction elicited by the host in response to infection, injury, or tissue damage. Under normal physiologic circumstances, this response serves to destroy pathogens, and initiate repair. Inflammatory reactions are also observed commonly in human neoplasia (Wernert 1997; Ohtani 1998). Thus, several parallels exist between the generation of tumor stroma and healing of wounds, supporting the concept that tumor stroma is, in part, wound healing gone awry (Dvorak 1986). In this report, we demonstrate that an inflammatory reaction, characterized by stromal infiltration of mast cells, occurs coincident with activation of angiogenesis in a mouse model of squamous epithelial carcinogenesis and demonstrate a role for MCs in jump-starting the angiogenic switch. We show that MCs within angiogenic dysplasias are juxtaposed tightly to epithelial and subendothelial basement membranes, both of which are sites of active ECM remodeling (Coussens et al. 1996). The data support a model in which MCs contribute to premalignant progression in part by releasing two MC specific serine proteases, mMCP-6 (a tryptase), mMCP-4 (a chymase), as well as progelatinase B. MC deficiency in K14–HPV16 animals results in a severe attenuation of early neoplasia, strengthening the notion that MCs are important functionally. Thus, during the hyperplastic and dysplastic stages of squamous carcinogenesis, an ostensibly antitumor inflammatory response is hijacked, instead facilitating the angiogenic switch and the conversion of a normal dermis into an aberrant stromal support, setting the stage for subsequent progression to malignancy.
Inflammation, mast cells, and angiogenesis: parameters of early neoplastic progression
Angiogenesis in the adult organism is limited normally to conditions of tissue repair/remodeling such as exist during menstruation, mammary gland involution, wound healing, inflammation, and neoplasia (Salamonsen and Wooley 1996; Hanahan and Folkman 1996). Angiogenesis involves the activation, proliferation, and migration of endothelial cells in concert with localized proteolytic modification of the ECM (Ingber and Folkman 1989). Proteolysis of the ECM facilitates endothelial cell migration and proliferation, releasing stored angiogenic signaling molecules from sequestered latent reserves (Meininger 1995; Brown et al. 1997; Friedl et al. 1997; Poltarek et al. 1997), and loosening the stromal milieu to facilitate migration (Coussens and Werb 1996). In cutaneous organs, activation of angiogenesis and infiltration of stroma by inflammatory cells occurs within seconds following tissue injury. The implication is that skin contains regulatory machinery that can both maintain vascular quiescence, as well as initiate rapid neovascular responses. How might this be accomplished?
MCs release diverse factors known to enhance angiogenic phenotypes, including heparin, heparinase, histamine, metallo- and serine proteinases, and various polypeptide growth factors, including bFGF [basic (fibroblast growth factor)] and VEGF/VPF (vascular endothelial growth factor/vascular permeability factor) (Bashkin et al. 1990; Vlodavsky et al. 1992; Meininger 1995; Reed et al. 1995; Grutzkau et al. 1998). Thus, MCs provide direct mitogens for fibroblasts, endothelial, and epithelial cells, as well as diverse enzymatic activities. Activation of angiogenesis in K14–HPV16 transgenic mice was coincident with marked infiltration of dermal stroma by MCs. MCs accumulated around capillaries, as well as subjacent to epithelial basement membranes, where degranulation and release of MC products was evident. Strikingly, MCs were essentially absent within solid tumor stroma, but were prominent at tumor-host interfaces. MC exclusion from tumors reveals a distinction between the core tumor microenvironment and the microenvironments associated with premalignant progression and with cancer cell invasion into normal tissue.
A clear goal for the future is to identify the signals that recruit the MC inflammation. As the HPV-expressing skin becomes hyperkeratotic quite early, signals resulting from the loss of skin barrier function are candidates. One such effector could be activation of humoral immunity in response to consequential infections. However, when K14–HPV16 transgenic mice were treated with antibiotics or bred into Rag1/null background, they had similar spatial/temporal infiltration of dysplasias by MCs, arguing against infection as the stimulus. Instead, the signal may emanate from the incipient epidermal neoplasia itself, or perhaps arise in response to other consequential abnormalities of the affected skin.
Two neutral MC-specific serine proteolytic activities were identified in angiogenic dysplasias, mMCP-4 and mMCP-6. Mouse MCP-4, a β-chymase, is the major chymotryptic enzyme present in angiogenic dysplasias. The limited diffusability of the mMCP-4 enzyme, as demonstrated by immunolocalization, suggests that in vivo substrates are likewise located proximal to basement membranes. Indeed, our data show that mMCP-4 activates progelatinase B, and in addition, can switch otherwise nonangiogenic tissue to the angiogenic phenotype as revealed by endothelial coculture assays for angiogenesis. We propose that mMCP-4 directly, and indirectly through its ability to activate progelatinase B, releases sequestered angiogenic factors such as VEGF/VPF and a/bFGF from neoplastic skin, thereby effecting induction of angiogenesis. Support for the angiogenic factor sequestration/release mechanism comes from the work of Whitlock et al. (1996), who demonstrated that stromelysin-1, collagenase-3, and heparinase each degrade the ECM component perlecan resulting in release of bound bFGF. Direct evidence that gelatinase B is important for generation of angiogenic signals comes from studies of bone growth plates in gelatinase B-knockout mice, which show delayed angiogenesis (Vu et al. 1998). The identity of the angiogenic growth factor(s) released by gelatinase B and/or mMCP-4 from skin ECM remains to be determined.
Our data on MCP-6 suggest that this serine protease does not affect EC growth directly, but instead activates dermal fibroblasts, thus contributing to induction of α1(I) procollagen synthesis, supporting a role for tryptase in stroma generation and matrix remodeling associated with neovascularization.
Analysis of the hyperplastic stage in MC-deficient mice documents the importance of an early MC-inflammatory response. Notably, the vascular density and architecture was more characteristic of a quiescent vasculature than that observed typically underlying hyperplastic epithelium in age-matched MC-proficient HPV mice. We speculate that the impaired vascular response is caused by the absence of angiogenic growth factors (e.g., bFGF and VEGF/VPF) and MC-proteases delivered normally by MCs as they focally degranulate around vascular beds and epidermal basement membranes.
Generality of mast cells and neoplasia
The presence of MCs at the borders of tumors was first reported by Westphal in 1891 (Westphal 1891). Subsequently, numerous investigators have confirmed the presence of MCs at the periphery of both experimentally induced rodent tumors (Norrby and Wooley 1993; Meininger 1995), and diverse human neoplasms (Aaltomaa et al. 1993; Shea and Prieto 1994; Duncan et al. 1998; J.M. McKerrow, V. Bhargava, E. Hansell, T. Kuwahara, M. Matley, L. Coussens, and R. Warren, in prep.); thus, MC recruitment to sites of neoplasia would appear to be HPV16-independent. Interestingly, three of the most aggressive human cancers, malignant melanoma, breast carcinoma, and colorectal adenocarcinoma, are associated commonly with a dramatic host response comprised of various inflammatory cells, especially MCs at the tumor periphery (Norrby and Wooley 1993).
Are MCs merely a smoking gun? Our analysis of neoplastic progression in a MC-deficient background, albeit one mouse, suggests that MCs are much more than passive bystanders. More likely, MCs and their products are subverted in the neoplastic tissue and functionally assist in jump-starting angiogenesis, thereby acting as key extrinsic modulators of neoplasia. Two other studies likewise support a causal role for MCs in tumorigenesis: MC-deficient KITW/KITWv mice were demonstrated to have decreased tumor-associated angiogenesis (Starkey et al. 1988); and, rats treated with a compound (FPL 55618) that inhibits MC degranulation, resulted in a 70% reduction in growth of mammary adenocarcinomas (Dabbous et al. 1991).
Conclusion
A primary distinction between angiogenesis associated with wound healing and that of neoplasia lies in the ability to turn off pro-angiogenic signals in the former. Following wounding, sequestered angiogenic activators can be released by degranulation of inflammatory cells and made available rapidly, thus abrogating the necessity for transcriptional induction of angiogenic growth factor genes by the wounded tissue. Turning this switch effectively ‘off’ could be as simple as shifting the balance between pro- and anti-inflammatory cytokines regulating infiltration and/or stability of inflammatory cells. With such programs readily available, it is not surprising that neoplastic tissues may take advantage of this machinery to initiate tissue remodeling and angiogenesis, thus providing nutrients and generating a tumor stroma for a rapidly expanding neoplastic mass. Once a tumor forms, transcription of angiogenic factors increases (Arbeit et al. 1996; Hanahan and Folkman 1996; Hanahan et al. 1996; Bergers et al. 1998). Thus, overtly malignant tissues that constitutively express high levels of angiogenic factors and ECM-degrading proteinases may no longer require the assistance of inflammatory cell-derived factors. Moreover, the presence within tumors of inflammatory cells whose mission may have been to resist (and not assist) neoplasia might also have deleterious effects, such that their exclusion could be beneficial for expansive tumor growth.
The data presented herein suggest that, in squamous carcinogenesis, angiogenic regulation is biphasic (Fig. 8). In the premalignant early phase of hyperplasia and dysplasia, infiltrating MCs degranulate and activate dermal fibroblasts. And, both directly and via activation of progelatinase B, the MCs turn on and progressively intensify angiogenesis by releasing sequestered angiogenic activators. As neoplastic progression proceeds, angiogenic growth factor gene expression is up-regulated in the cancer cells, marking progression to the second cancer phase, wherein the tumor cells control their angiogenic phenotype directly instead of depending on the manipulation of inflammatory cells to indirectly effect neovascularization. Interestingly, invasion from solid tumors apparently again benefits from MC assistance: MCs accumulate at the leading edges of cancers in which tumor cells invade into normal tissue stroma, likely again to help convert it into a neoplastic stroma with concomitant angiogenesis.
Figure 8.
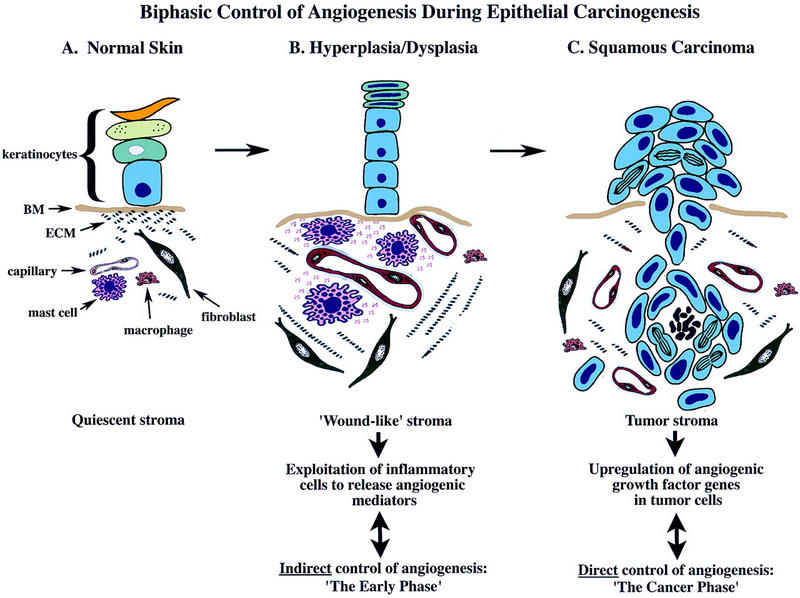
A model for biphasic control of angiogenesis during squamous carcinogenesis. Skin is compartmentalized into an avascular epidermis, composed of keratinocytes and dendritic cells, and a vascularized dermis composed of fibroblasts, various hemopoietic cell types, and endothelial and smooth muscle cells forming blood vessels, embedded in a quiescent stromal milieu. During premalignant progression, stroma adjacent to neoplastic epithelium resembles that observed in chronic wounds characterized by proliferating fibroblasts, increased synthesis of α1(I) procollagen, increased vessel density, vascular permeability, increased expression and activity of ECM-degrading proteinases, increased presence of diverse leukocytes, and degranulation of MCs. MCs are exploited by neoplastic epithelia in early lesions and act to jump-start angiogenesis by their release of several bioactive molecules, e.g., bFGF, VEGF, heparin, histamine, chymase, and tryptase. Tryptase increases vascular permeability, and is a potent mitogen and activator of fibroblasts and inducer of α1(I) procollagen synthesis. Chymase, although not a direct mitogen, induces activation of angiogenesis by releasing sequestered angiogenic activity from stromal reservoirs, through gelatinase B-dependent and independent mechanisms. Gelatinase B made by reactive stromal cells, although likely involved in ECM-remodeling, releases ECM-sequestered angiogenic activity also stimulating EC chemotaxis, proliferation, and tube formation. In contrast, maintenance of neovascularization within tumor stroma is MC-independent. Sustaining angiogenesis within the cancer phase is likely accomplished directly via dramatic up-regulation of multiple heparin-binding growth factor genes in fully malignant epithelial cells.
Our study demonstrates the significance of the MC as a key accessory during the premalignant stages of squamous carcinogenesis. We show a link between MC inflammation and the angiogenic switch by a mechanism that includes the release of pro-angiogenic proteases. Thus, MC chymase can activate progelatinase B, itself a component of MC granules, as well as release angiogenic activity sequestered in the skin. MC tryptase induces normally quiescent fibroblasts to participate in formation of neoplastic stroma. The involvement of MCs and their proteases has implications for the pathogenic mechanism and potential therapeutic intervention in cutaneous malignancy, in particular for anti-angiogenic strategies aimed at arresting neoplastic progression prior to the emergence of overtly malignant tumors.
Materials and methods
Transgenic mice and tissue preparation
The K14–HPV16 transgenic mice (Arbeit et al. 1994), the characterization of neoplastic stages based on keratin intermediate filament expression, and preparation of tissue sections for histologic examination (hematoxylin and eosin) have been reported previously (Coussens et al. 1996). To identify metachromatic granules of MCs, 1 μm thick sections of EPON 812-embedded skin biopsies were stained for 5 min in 1% toluidine blue (in water), rinsed in deionized water, dried, and mounted under cover slips in Cytoseal 60 (Stephens Scientific).
Immunohistochemistry
Tissue pieces from transgenic animals were either quick-frozen in OCT Freezing Medium (Sakura) and stored at −80°C or immersion-fixed in 3.75% paraformaldehyde and phosphate-buffered saline (PBS) followed by dehydration through graded alcohols and xylene, and embedded in paraffin. OCT sections (10-μm thick) were cut using a Hacker cryostat set at −20°C. Prior to using, cut frozen sections were dried at room temperature, followed by fixation in ice-cold acetone for 10 min. Alternatively, 5-μm-thick paraffin sections were cut using a Leica 2135 microtome. Sections were deparaffinized and subjected to immunohistochemical staining as described previously (Coussens et al. 1996). Dilution of primary antibodies was 1:200 for rabbit anti-rat MCP-1 (recognizing mMCP-4; Moredun Research Institute), 1:100 for rabbit anti-mouse gelatinase B (Behrendtsen et al. 1992), 1:100 for rat anti-mouse laminin-β1 (Upstate Biotechnology, Inc.), 1:100 for rat anti-mouse CD31 (Pharmingen), and 1:50 for rabbit anti-mouse Ki-67 (Dianova) in blocking solution containing PBS (pH 7.4), 2.5% goat serum and 1% bovine serum albumin (BSA). Incubation with primary antibody was 4 hr at room temperature. Following incubation with a biotinylated secondary antibody (goat anti-rabbit IgG or goat anti-rat IgG, 1:200, Pierce Chemical) for 30 min at ambient temperature, antigens were revealed with either 3,3′-diaminobenzidine (DAB; Sigma,) or alkaline phosphatase substrate (Vector) containing Levamisole (Vector). Sections were counterstained in 1% methyl green (1 min), dehydrated in graded alcohols (70%, 95%, 100% ethanol), mounted in Cytoseal 60 (Stephens Scientific) and visualized with Nomarski optics. All immunolocalization experiments were repeated three times on multiple tissue sections and included negative controls for determination of background staining, which was negligible.
Perfusion fixation and lectin binding
For visualization of the vascular bed in situ, lectin perfusion and visualization of the bound lectin was done essentially as described previously (Tharp et al. 1985; Thurston et al. 1996). Briefly, animals were anesthetized with 50 mg/kg pentobarbital sodium by intraperitoneal injection and then perfused via the ascending aorta with fixative [1% paraformaldehyde (PFA), 0.5% glutaraldehyde, in PBS at pH 7.4], for 5 min at a pressure of 130 mmHg. Fixation was followed by perfusion of (1) 50 ml PBS (1 min); (2) 25 ml PBS, 1% BSA (80 sec); (3) 50 ml 5-μg/ml biotinylated Lycopersicon esculentum lectin (Vector) in PBS, 1% BSA (80 sec); (4) 25 ml PBS, 1% BSA (80 sec). Tissues were then removed and immersion-fixed overnight in PBS, 4% PFA. This was followed by permeabilization in PBS, 0.3% Triton X-100 overnight at room temperature on a rotating platform. To localize lectin binding, tissues were incubated on a rotating platform for 48 hr with avidin-peroxidase complex (Vector) diluted 1:200, washed for 2 hr with 50 mm Tris at pH 7.4, 1% Triton X-100, and exposed for 10 min to 0.5 mg/ml DAB (Sigma), 50 mm Tris at pH 7.4, 0.01% H2O2 at room temperature. After staining, tissues were washed in H2O and dehydrated through graded alcohols (50%, 70%, 95%, and 100% ethanol). Tissues were then flattened between two glass slides in 100% ethanol overnight, cleared in toluene, and mounted in Cytoseal 60 (Stephens Scientific). Whole mounts of tissues were visualized by Nomarski optics.
Enzyme histochemistry
MCs were visualized in either 10-μm OCT-embedded acetone-fixed sections, or, 5-μm paraffin-embedded tissue sections deparaffinized in xylene, hydrated through graded alcohols (100%, 95%, 70%, 50% ethanol), and equilibrated in H2O. Chloroacetate esterase histochemistry was performed as described previously to reveal presence of the chymotrypsin-like serine esterase activity of MCs (Leder 1979; Caughey et al. 1988). Slides were then washed extensively in water, counterstained in Gills Hematoxylin #3 (3 sec), dehydrated in 100% alcohol, and mounted under coverslips in 100% glycerol. Data shown are representative of results obtained following examination of tissues removed from a minimum of 10 different K14–HPV16 transgenic animals each representing a distinctive stage of neoplastic progression.
RNA isolation and RT–PCR analysis
Total cellular RNA was isolated from control tissues (auxiliary lymph node and intestine) or pieces representing distinct histologic stages removed from K14–HPV16 transgenic mice using Trizol reagent (GIBCO–BRL) according to the manufacturer’s specifications and was quantified by ultraviolet absorbance at 260 nm. Reverse transcription of total RNA (1–5 μg) was performed with oligo(dT) primer and Superscript II RT (GIBCO–BRL) according to the manufacturer’s specifications. Equal volumes of cDNA were then amplified in a thermocycler (Perkin-Elmer/Cetus) in a final volume of 50 μl containing 50 mm KCl, 10 mm Tris at pH 8.3, 4 mm MgCl2, 200 μm each of all four dNTPs, 0.4 μm 5′ and 3′ oligonucleotide primers, and 0.6 unit of Taq polymerase (Perkin-Elmer Corp.). Samples were first denatured at 95°C for 5 min and amplified for 30 cycles (1 cycle: denature at 95°C 1 min, annealing at 55°C for 30 sec, and extension at 72°C for 1 min) with a final extension of 5 min at 72°C. Samples were then removed and electrophoresed on 5% polyacrylamide gels.
Oligonucleotide primers
Primers used in PCR reactions were mMCP-1 (Wastling et al. 1997) sense 5′-CAGCTGGGGACAGAATGGGG-3′, antisense 5′-GAGCTCTCTGGTACTCTTTG-3′ (100 bp); mMCP-2 (Xia et al. 1996) sense 5′-ACTGGCAAAATGCAGGCC-3′, antisense 5′-CATCATCACAGACATGTG-3′ (910 bp); mMCP-4 (Jippo et al. 1994) sense 5′-GGAGACTCTGGAGGACCTCT-3′, antisense 5′-ACAGGGAACAGTCCATCATC-3′ (336 bp); mMCP-5 (McNeil et al. 1991) sense 5′-ACTCTGGAGCTTTTGCCAG-3′, antisense 5′-CAGTCGACAATCTGGGTCT-3′ (200 bp); mMCP-6 (Hunt et al. 1996) sense 5′-GCACATCAAAAGCCCACAGC-3′ antisense 5′-TAGACAGGGGAGACAGAGGAC -3′ (700 bp); mMCP-7 (Hunt et al. 1996) sense 5′-GCACTACTCCTCACTGTG-3′, antisense 5′-CGCATTTTATTGAGGCATAGCAGA-3′ (1003 bp).
Protein extraction
Ear and chest skin from K14–HPV transgenic and nontransgenic wild-type FVB/n mice of various ages and stages of neoplastic progression were harvested and frozen at −80°C. While still frozen, samples were minced and ground under liquid N2 to a fine powder, followed by weighing and resuspension in 10 mm bis-Tris-HCl at pH 6.1 (low-salt buffer) at a concentration of 10 μl/mg of tissue. After vortexing for 3 min the suspension was centrifuged for 5 min at 16,000g. The supernatant was saved; the pellet was resuspended in the above buffer containing 2 m NaCl (high-salt buffer) at 10 μl/mg of tissue, then vortexed and centrifuged as above. Recovered supernatants were saved for assay.
Protease solution assays
Chymase-like activity was measured by addition of 5-μl aliquots of samples to assay buffer containing 1 mm succinyl-l-Ala-Ala-Pro-Phe-4-nitroanilide (Sigma), 1.8 m NaCl, and 9% dimethylsulfoxide in 0.45 m Tris-HCl (pH 8.0). Tryptase-like activity was measured by the addition of 5-μl aliquots of samples to assay buffer containing 0.1 mmole tosyl-Gly-l-Pro-Lys-4-nitroanilide (Sigma) in 50 mm Tris-HCl at pH 7.6, 120 mm NaCl, 30 μg/ml bovine lung heparin. Change in absorbance at 410 nm was monitored spectrophotometrically at 37°C. Amounts of substrate cleaved were estimated using an extinction coefficient for free 4-nitroaniline of 8800/m per cm.
Immunoblotting
High-salt extracts of protein samples were electrophoresed in the presence of dithiothreitol on 12.5% SDS–polyacrylamide gels, then electroblotted to polyvinylidene difluoride (PVDF) membranes, which were blocked for 30 min at room temperature in a solution of 0.3% Tween-20 and 0.5 m NaCl in 50 mm Tris-HCl (pH 7.2), followed by incubation in the same buffer for 1 hr with 1:1000 dilutions of rabbit antisera or purified rabbit antibodies. Blots were washed three times for 5 min with the Tris/NaCl/ Tween-20 buffer indicated above. Bound primary antibody was detected with goat anti-rabbit IgG-alkaline phosphatase antibody and Fast Red TR/Naphthol AS-MX Phosphate (Sigma). Richard Stevens (Harvard University, Cambridge, MA) provided the rabbit antiserum to mMCP-6.
Mouse MCP-4 purification
MC chymase in the angiogenic dysplasias was purified using modifications to the methods for human skin chymase purification described by Schechter et al. (1986). Ears (840 mg total) of 6-month-old K14–HPV16 transgenic mice were extracted sequentially in low- and high-salt buffer as above. High-salt extracts were applied to a 1-ml column of SBTI-agarose (Sigma) equilibrated with high-salt buffer. The column was washed first with high-salt buffer, then with H2O, until absorbance of the eluate at 280 nm reached baseline, after which 1 mm HCl was applied to the column. Eluted fractions were neutralized immediately with one-fifth volume of 0.5 m Tris-HCl (pH 8.0). Protein in eluted fractions was separated by SDS-PAGE and visualized by Coomassie blue staining. Protein concentration in fractions was determined using protein assay reagent (Bio-Rad) with BSA as standard. The purified chymase protein concentration was determined by using the theoretical molar extinction coefficient 33,760/m per cm at 280 nm, computed from the deduced amino acid sequence by the method of Creighton (1984).
Mouse MCP-6 purification
Thirty-five milliliters of high-salt extracts from 5 to 6 month-old K14–HPV16 transgenic mouse ears were applied to a 4-ml p-aminobenzamidine-agarose column (Sigma) equilibrated in 10 mm bis-Tris-HCl (pH 6.1) with 2 m NaCl (high-salt buffer). No tryptase-like activity was detected in the unbound fraction. The column was eluted with 150 mm benzamidine in high-salt buffer. The eluate was then diluted threefold with 10 mm bis-Tris-HCl (pH 6.1) and applied to a 7.5 × 75-mm heparin 5PW HPLC column (Toso-Haas) equilibrated with 0.7 m NaCl, 10 mm bis-Tris-HCl (pH 6.1). Active mMCP-6 eluted with the application of a linear gradient of 0.7 m NaCl to 2 m NaCl over 15 ml at 1.4 m NaCl. Amino acid sequencing, SDS-PAGE analysis, and immunoblotting with anti-mMCP-6 polyclonal antisera confirmed the identity and purity of the eluted activity. The purified mMCP-6 protein concentration was determined by using the theoretical molar extinction coefficient 68,860/m per cm at 280 nm, computed from the deduced amino acid sequence by the method of Creighton (1984).
Protein sequencing
Amino-terminal sequence analysis was performed by the Biomolecular Resource Center, University of California, San Francisco. Purified mMCP-4 and mMCP-6 (2.5-μg of proteins) were applied individually to a PVDF membrane disk and subjected to Edman degradation in a 470A gas-phase sequencer with an online 120A PTH analyzer (Applied Biosystems).
Substrate zymography
Tissue samples representing distinct histological stages of neoplastic progression, as verified by histological analysis of paraffin-embedded sections from adjacent skin, were weighed and then homogenized (1:4 weight to volume) in lysis buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1% NP-40, 0.5% deoxycholate, and 0.1% SDS. Soluble and insoluble extracts were separated by centrifugation and subsequently stored at −20°C. Equivalent amounts of soluble extract were analyzed by gelatin zymography (Herron et al. 1986a,b) on 10% SDS–polyacrylamide gels copolymerized with substrate (1 mg/ml of gelatin) in sample buffer (10% SDS, 0.25 m Tris-HCl, and 0.1% bromophenol blue at pH 6.8). After electrophoresis, gels were washed twice for 15 min in 2.5% Triton X-100, incubated for 16 hr at 37°C in 50 mm Tris-HCl, 10 mm CaCl2 (pH 7.6), and then stained in 0.5% Coomassie blue and destained in 50% methanol. Negative staining indicates the location of active protease bands. Exposure of proenzymes within tissue extracts to SDS during gel separation procedures leads to activation without proteolytic cleavage (Talhouk et al. 1991). For inhibition of proteolytic activities, substrate gels were incubated in substrate buffer with either 4 mm 1,10-phenanthroline (Sigma), 5 mm PMSF (Sigma), or 10 μg/ml recombinant ecotin (McGrath et al. 1991; provided by Charles Craik, University of California, San Francisco) as described (Adler et al. 1990). Data shown are representative of results obtained following examination of 91 tissue pieces representing various stages of neoplastic progression in K14–HPV16 transgenic mice.
Cell culture
PMDFs were isolated from FVB/n mice by the method of Bossy-Wetzel et al. (1992) and maintained in culture at subconfluence in DMEM containing 4.5 grams/liter glucose, and supplemented with 10% fetal bovine serum (GIBCO-BRL), 50 U/ml penicillin, 50 μg/ml streptomycin, at 37°C in 5% CO2. HUVECs (Clonetics) were maintained in EBM complete medium (Clonetics) supplemented with 5% fetal calf serum. BCEs were a gift from the laboratory of Judah Folkman (Harvard University, Boston, MA), and were maintained in DMEM containing 10% heat-inactivated calf serum.
DNA synthesis assay
Stimulation of DNA synthesis in subconfluent cultures of PMDFs or HUVECs was determined by radiolabeled thymidine incorporation. Cells were plated at 2500 cells/cm2 in multiwell plates containing growth medium supplemented with fetal calf serum [PMDFs, 10%; HUVECs, 5% (GIBCO-BRL)]; prior to plating HUVECs, culture dishes were precoated with 1.5% gelatin in PBS. After 24 hr, cells were washed three times with PBS and covered with serum-free DMEM (GIBCO-BRL) containing 0.5% bovine serum albumin (GIBCO-BRL). After an additional 24 hr, cells were incubated for 16 hr with agonists [murine bFGF, 1 and 10 ng/ml, (GIBCO-BRL); human VEGF at 1 and 10 ng/ml (Sigma); purified mMCP-4, 2 and 20 nm in 50 mm Tris-HCl at pH 8.0; purified mMCP-6, 1 and 10 nm in 10 mm bis-Tris at pH 6.1, 0.7 m NaCl, 225 μg/ml heparin (Sigma); heparin at 74 and 740 ng/ml ], followed by the addition of [methyl-3H]thymidine (2 mCi/ml, NEN) for an additional 6 hr. Trichloroacetic acid-precipitable material was solubilized in 0.3 n NaOH, and liquid scintillation counting assessed the incorporated radioactivity. Each assay condition was in triplicate; results shown are representative from two independent experiments.
In situ hybridization
In situ hybridization analysis on 5-μm paraffin-embedded normal and transgenic skin was carried out as described previously (Coussens et al. 1996). The riboprobe used to detect α1(I) collagen (pmCol1–129) mRNA was provided by Paul Bornstein (University of Washington, Seattle, WA).
Endothelial tube formation assay
BCEs were trypsinized and resuspended in DMEM with 10% calf serum. Cells (4 × 105/ml) were mixed 1:1 with a chilled collagen solution (Vitrogen 100; Collagen Corp.) in MEM medium containing 0.1% NaHCO3, 2 mm glutamine, and 10 mm HEPES. Skin biopsies were removed from 1-month-old transgenic mice, washed in PBS, and cut into <1 mm2 pieces and then incubated for 24 hr at 37°C in 5% CO2 balanced air incubator in DMEM containing 0.5% BSA and either 10 mm mMCP-6, 20 mm mMCP-4, 1 ng/μl gelatinase B (Calbiochem), or medium/BSA alone. After 24 hr, medium was removed and fresh medium containing appropriate agonist was replaced and allowed to incubate for an additional 24 hr. Aliquots (250 μl) of the collagen-cell mixture were pipetted into 48-well tissue culture plates, and skin pieces were added to each well before the solution was allowed to gel at 37°C. Culture plates were incubated in 10% CO2 balanced air atmosphere and observed for endothelial cell growth, migration, and tube formation over a period of 2 weeks. In addition, 10 mm mMCP-6, 20 mm mMCP-4, and 1 ng/μl gelatinase B (Calbiochem) were added individually to collagen–EC mixtures, and monitored for tube formation over a 1-week period.
KITW/KITWv genotyping
WB/ReJ KITW/+ and C57BL/6J-KITWv/+ mice (Jackson Laboratories) were each intercrossed into the FVB/n strain four generations. Genotypes were determined from tail DNA by a PCR-based assay (Laig-Webster et al. 1998). Briefly, to trace the KITW mutation, oligonucleotide primers specific for intron 9 (5′-CCAAGAGAAAGCTTTGTTCCCTGAATGTGC-3′) and intron 10 (5′-AGAACAATTCAATGCTCAT-3′) from the mouse c-KIT gene (Hayashi et al. 1991; Gokkel et al. 1992) were generated. To trace the KITWv mutation, oligonucleotide primers specific for exon 12 (5′-CATTGGGAGCTGGTGCCTTCGGGAAGGT-3′) and exon 13 (5′-AGACTACCTCCCACCA-3′) from the mouse c-KIT gene (Gokkel et al. 1992) were generated. Utilizing these primers, mutant alleles, but not wild-type alleles, were amplified preferentially in 50-μl reactions containing 3 μl of genomic tail DNA (100 ng), 2.5 units of AmpliTaq Gold (Perkin-Elmer), 10 pmoles of each oligonucleotide primer, 200 μm dNTPs and 1.5 mm MgCl2. KITW and KITWv mutations were detected by PCR amplification (KITW: 1 cycle at 94°C for 5 min, 20 cycles at 94°C for 30 sec and 72°C for 1 min, 40 cycles at 94°C for 30 sec, 58°C for 1 min and 72°C for 30 sec; KITWv: 1 cycle at 94°C for 5 min, 10 cycles at 94°C for 30 sec and 72°C for 1 min, 50 cycles at 94°C for 30 sec, 51°C for 1 min, and 72°C for 30 sec. PCR products were visualized with ultraviolet light on 2% agarose gels stained with ethidium bromide. KITW/KITWvgenotypes were confirmed by DNA sequence analysis.
Acknowledgments
We thank Donald McDonald, Gavin Thurston, and Tom Murphy for assistance with lectin perfusions and whole mount microscopy, Richard Stevens for rabbit anti-mMCP-6 IgG, Paul Bornstein for type I collagen cDNA, Charles Craik and Sushma Selvarajan for recombinant ecotin, John Blount for genotype analysis, and Emily Bergsland, Jeffrey Hager, and Dylan Daniel for valuable discussion. This study was supported by grants from the National Institutes of Health (HL24136, G.H.C; CA72006, Z.W.), by grants from the National Cancer Institure (D.H.), the American Social Health Association/Pfizer Fellowship in Sexually Transmitted Diseases (L.M.C.), and a Howard Hughes Medical Institute Transgenic Animal Research Grant (G.H.C.)
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL: coussens@cc.ucsf.edu; FAX (415) 502-6779.
References
- Aaltomaa S, Lipponen P, Papinaho S, Kosma V-M. Mast cells in breast cancer. Anticancer Res. 1993;13:785–788. [PubMed] [Google Scholar]
- Adler RR, Brenner CA, Werb Z. Expression of extracellular matrix-degrading metalloproteinases and metalloproteinase inhibitors is developmentally regulated during endoderm differentiation of embryonal carcinoma cells. Development. 1990;110:211–220. doi: 10.1242/dev.110.1.211. [DOI] [PubMed] [Google Scholar]
- Arbeit J, Munger K, Howley P, Hanahan D. Progressive squamous epithelial neoplasia in K14-human papillomavirus type 16 transgenic mice. J Virol. 1994;68:358–368. doi: 10.1128/jvi.68.7.4358-4368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeit JM, Olson D, Hanahan D. Upregulation of fibroblast growth factors and their receptors during multi-stage epidermal carcinogenesis in K14–HPV16 transgenic mice. Oncogene. 1996;13:1847–1857. [PubMed] [Google Scholar]
- Bashkin P, Razin E, Eldor A, Vlodavsky I. Degranulating mast cells secrete an endoglycosidase which degrades heparin sulfate in subendothelial extracellular matrix. Blood. 1990;75:2204–32212. [PubMed] [Google Scholar]
- Befus AD, Pearce FL, Gauldie J, Horsewood P, Bienenstock J. Mucosal mast cells: Isolation and functional characteristics of rat intestinal mast cells. J Immunol. 1982;128:2475–2486. [PubMed] [Google Scholar]
- Behrendtsen O, Alexander CM, Werb Z. Metalloproteinases mediate extracellular matrix degradation by cells from mouse blastocyst outgrowths. Development. 1992;114:447–456. doi: 10.1242/dev.114.2.447. [DOI] [PubMed] [Google Scholar]
- Bergers G, Hanahan D, Coussens LM. Angiogenesis and apoptosis are cellular parameters of neoplastic progression in transgenic mouse models of tumorigenesis. Int J Dev Biol. 1998;42:995–1002. [PubMed] [Google Scholar]
- Bernstein A, Chabot B, Durbreuil P, Reith A, Mocka K, Majumder S, Rat P, Bessmer P. The mouse W/c-KIT locus. Ciba Found Symp. 1990;148:148–166. [PubMed] [Google Scholar]
- Blair RJ, Meng H, Marchese MJ, Ren S, Schwartz LB, Tonnesen MG, Gruber BL. Human mast cells stimulate vascular tube formation. J Clin Invest. 1997;99:2691–2700. doi: 10.1172/JCI119458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Bravo R, Hanahan D. Transcription factors JunB and c-Jun are selectively up-regulated and functionally implicated in fibrosarcoma development. Genes & Dev. 1992;6:2340–2351. doi: 10.1101/gad.6.12a.2340. [DOI] [PubMed] [Google Scholar]
- Brown L, Detmar M, Claffey K, Nagy J, Feng D, Dvorak A, Dvorak H. Vascular permeability factor/vascular endothelial growth factor. A multifunctional angiogenic cytokine. In: Goldberg I, Rosen E, editors. Regulation of angiogenesis. Basel, Switzerland: Birkhauser Verlag; 1997. pp. 233–269. [DOI] [PubMed] [Google Scholar]
- Cairns JA, Wells AF. Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J Clin Invest. 1997;99:1313–1321. doi: 10.1172/JCI119290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey GH. Mast cell chymases and tryptases: Phylogeny, family relations, and biogenesis. In: Caughey GH, editor. Mast cell proteases in immunology and biology. New York, NY: Marcel Dekker; 1995. pp. 305–329. [Google Scholar]
- Caughey GH, Viro NF, Calonico LD, McDonald DM, Lazarus SC, Gold WM. Chymase and tryptase in dog mastocytoma cells: Asynchronous expression as revealed by enzyme cytochemical staining. J Histochem Cytochem. 1988;36:1053–1060. doi: 10.1177/36.8.3134486. [DOI] [PubMed] [Google Scholar]
- Caughey GH, Raymond WW, Bacci E, Lombardy RJ, Tidwell RR. Bis(5-amidino-2-benzimidazolyl)methane and related amidines are potent, reversible inhibitors of mast cell tryptases. J Pharmacol Exp Ther. 1993;264:676–682. [PubMed] [Google Scholar]
- Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:88–89. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- Christofori G, Hanahan D. Molecular dissection of multi-stage tumorigenesis in transgenic mice. In: Merlino Glenn., editor. Seminars in cancer biology. London, UK: Academic Press; 1994. pp. 3–12. [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Matrix metalloproteinases and the development of cancer. Chem Biol. 1996;3:895–904. doi: 10.1016/s1074-5521(96)90178-7. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Hanahan D, Arbeit JM. Genetic predisposition and parameters of malignant progression in K14–HPV16 transgenic mice. Am J Pathol. 1996;149:1899–1917. [PMC free article] [PubMed] [Google Scholar]
- Creighton TF. Proteins, structures and molecular properties. New York, NY: W.H. Freeman and Company; 1984. Chemical properties of polypeptides; p. 14. [Google Scholar]
- Dabbous MK, Walker R, Haney L, Carter LM, Nicolson GL, Wooley DE. Mast cells and matrix degradation at sites of tumor invasion in rat mammary adenocarcinomas. Br J Cancer. 1986;54:459–465. doi: 10.1038/bjc.1986.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabbous MK, Laney L, Nicolson GL, Eckley D, Wooley DE. Mast cell modulation of tumor cell proliferation in rat mammary adenocarcinoma 13762NF. Br J Cancer. 1991;63:873–878. doi: 10.1038/bjc.1991.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLisser HM, Newman PJ, Albelda SM. Molecular and functional aspects of PECAM-1/CD-31. Immunol Today. 1994;15:490–495. doi: 10.1016/0167-5699(94)90195-3. [DOI] [PubMed] [Google Scholar]
- Detmar M. Molecular regulation of angiogenesis in the skin. J Invest Derm. 1996;149:1899–1917. doi: 10.1111/1523-1747.ep12340457. [DOI] [PubMed] [Google Scholar]
- Duncan LM, Richards LA, Mihm MC. Increased mast cell density in invasive melanoma. J Cutan Pathol. 1998;25:11–15. doi: 10.1111/j.1600-0560.1998.tb01683.x. [DOI] [PubMed] [Google Scholar]
- Dvorak HF. Tumors: Wounds that do not heal. New Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- Eady RAJ, Cowen T, Marshall TF, Plummer V, Greaves MW. Mast cell population density, blood vessel density and histamine content in normal skin. Br J Dermatol. 1979;100:635–640. doi: 10.1111/j.1365-2133.1979.tb08065.x. [DOI] [PubMed] [Google Scholar]
- Fang K, Raymond WW, Lazarus SC, Caughey GH. Dog mastocytoma cells secrete a 92-kD gelatinase activated extracellularly by mast cell chymase. J Clin Invest. 1996;97:1589–1596. doi: 10.1172/JCI118583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang K, Raymond WW, Blount J, Caughey GH. Dog mast cell α-chymase activates progelatinase B by cleaving the Phe88-Gln89 and Phe91-Glu92 bonds of the catalytic domain. J Biol Chem. 1997;272:25628–25635. doi: 10.1074/jbc.272.41.25628. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- Folkman J, Hanahan D. Expression of the angiogenic phenotype during development of murine and human cancer. In: Brugge J, Curran J, Harlow E, McCormick F, editors. Origins of human cancer: A comprehensive review. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1991. pp. 803–814. [Google Scholar]
- Friedl A, Chang Z, Tierney A, Rapraeger AC. Differential binding of fibroblast growth factor-2 and -7 to basement membrane heparin sulfate. Am J Pathol. 1997;150:1443–1455. [PMC free article] [PubMed] [Google Scholar]
- Ghildyal N, Friend DS, Stevens RL, Austen KF, Huang C, Penrose JF, Sali, Gurush MF. Fate of two mast cell tryptases in V3 mastocytosis and normal BALB/c mice undergoing passive systemic anaphylaxis: Prolonged retention of exocytosed mMCP-6 in connective tissue, and rapid accumulation of enzymatically active mMCP-7 in the blood. J Exp Med. 1996;184:1061–1073. doi: 10.1084/jem.184.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokkel E, Grossman Z, Ramot B, Yarden Y, Rechavi G, Givol D. Structural organization of the murine c-kit proto-oncogene. Oncogene. 1992;7:1423–1429. [PubMed] [Google Scholar]
- Gregoire M, Lieubeau B. The role of fibroblasts in tumor behavior. Cancer Metast Rev. 1995;14:339–350. doi: 10.1007/BF00690602. [DOI] [PubMed] [Google Scholar]
- Gruber BL, Kew RR, Jelaska A, Marchese MJ, Garlick J, Ren S, Schwartz LB, Korn JH. Human mast cells activate fibroblasts. J Immunol. 1997;158:2310–2317. [PubMed] [Google Scholar]
- Grutzkau GA, Kruger-Krasagakes S, Baumeister H, Schwartz C, Kogel H, Welker P, Lippert U, Henz BM, Moller A. Synthesis, storage, and release of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) by human mast cells: Implications for the biological significance of VEGF206. Mol Biol Cell. 1998;9:875–884. doi: 10.1091/mbc.9.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. A flanking attack on cancer. Nat Med. 1998;4:1–2. doi: 10.1038/nm0198-013. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Christofori G, Naik P, Arbeit J. Transgenic mouse models of tumor angiogenesis: the angiogenic switch, its molecular controls, and prospects for preclinical therapeutic models. Eur J Cancer. 1996;32:2386–2393. doi: 10.1016/s0959-8049(96)00401-7. [DOI] [PubMed] [Google Scholar]
- Hartmann T, Ruoss SJ, Raymond WW, Seuwen K, Caughey GH. Human tryptase as a potent, cell-specific mitogen: role of signaling pathways in synergistic responses. Am J Physiol. 1992;262:528–534. doi: 10.1152/ajplung.1992.262.5.L528. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Kunisada T, Ogawa M, Yamaguchi K, Nishikawa S. Exon skipping by mutation of an authentic splice site of c-kit gene in W/W mouse. Nucleic Acids Res. 1991;19:1267–1271. doi: 10.1093/nar/19.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron GS, Werb Z, Dwyer K, Banda MJ. Secretion of metalloproteinases by stimulated capillary endothelial cells. I. Production of procollagenases and prostromelysin exceeds expression of proteolytic activity. J Biol Chem. 1986a;261:2810–2813. [PubMed] [Google Scholar]
- Herron GS, Banda MJ, Clark EJ, Gavrilovic J, Werb Z. Secretion of metalloproteinases by stimulated capillary endothelial cells. II. Expression of collagenase and stromelysin activities is regulated by endogenous inhibitors. J Biol Chem. 1986b;261:2814–2818. [PubMed] [Google Scholar]
- Huang R, Blom T, Hellman L. Cloning and structural analysis of MMCP-1, MMCP-4 and MMCP-5, three mouse mast cell-specific serine proteases. Eur J Immunol. 1991;21:1611–1621. doi: 10.1002/eji.1830210706. [DOI] [PubMed] [Google Scholar]
- Hunt JE, Stevens R, Austen K, Zhang J, Xia Z, Ghildyal N. Natural distribution of the mouse mast cell protease 7 gene in the C57BL/6 mouse. J Biol Chem. 1996;271:2851–2855. doi: 10.1074/jbc.271.5.2851. [DOI] [PubMed] [Google Scholar]
- Ingber DE, Folkman J. How does extracellular matrix control capillary morphogenesis? Cell. 1989;58:803–805. doi: 10.1016/0092-8674(89)90928-8. [DOI] [PubMed] [Google Scholar]
- Jippo T, Ushio H, Hirota S, Mizuno H, Yamatodani A, Nomura S, Matsuda H, Kitamura Y. Poor response of cultured mast cells derived from mi/mi mutant mice to nerve growth factor. Blood. 1994;84:2977–2983. [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280:1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood. 1978;52:447–452. [PubMed] [Google Scholar]
- Laig-Webster M, Coussens LM, Hanahan D, Caughey G. A general method for genotyping mast cell-deficient KITW/KITW-v mice by allele-specific polymerase chain reaction. FASEB J. 1998;12:A895. [Google Scholar]
- Leder LD. The chloroacetate esterase reaction. A useful means of histological diagnosis of hematological disorders from paraffin sections of skin. Am J Dermatopath. 1979;1:39–42. [PubMed] [Google Scholar]
- McGrath ME, Hines WM, Sakanari JA, Fletterick RJ, Craik CS. The sequence and reactive site of ecotin. A general inhibitor of pancreatic serine proteases from Escherichia coli. J Biol Chem. 1991;266:6620–6625. [PubMed] [Google Scholar]
- McNeil H, Austen K, Someville L, Gurish M, Stevens R. Molecular cloning of the mouse mast cell protease-5 gene. J Biol Chem. 1991;266:20316–20322. [PubMed] [Google Scholar]
- Meininger C. Mast cells and tumor-associated angiogenesis. In: Marone G, editor. Human basophils and mast cells: Clinical aspects. Basel, Switzerland: Karger; 1995. pp. 238–256. [Google Scholar]
- Molino M, Barnatham ES, Numerof R, Clark J, Dreyer M, Cumashi A, Hoxie JA, Schechter N, Woolkalis M, Brass LF. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1 deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Nocka K, Tan JC, Chiu E, Chu TY, Ray P, Traktman P, Bessmer P. Molecular basis of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41, and W. EMBO J. 1990;9:1805–1813. doi: 10.1002/j.1460-2075.1990.tb08305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrby K, Wooley DE. Role of mast cells in mitogenesis and angiogenesis in normal tissue and tumor tissue. Adv Biosci. 1993;89:71–115. [Google Scholar]
- Ohtani H. Stromal reaction in cancer tissue: Pathophysiologic significance of the expression of matrix-degrading enzymes in relation to matrix turnover and immune/inflammatory reactions. Pathol Int. 1998;48:1–9. doi: 10.1111/j.1440-1827.1998.tb03820.x. [DOI] [PubMed] [Google Scholar]
- Poltarek Z, Cohen T, Sivan R, Kandelis Y, Spira G, Vlodovsky I, Keshet E, Neufeld G. VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J Biol Chem. 1997;272:7151–7158. doi: 10.1074/jbc.272.11.7151. [DOI] [PubMed] [Google Scholar]
- Reed JA, Albino AP, McNutt NS. Human cutaneous mast cells express basic fibroblast growth factor. Lab Invest. 1995;72:215–222. [PubMed] [Google Scholar]
- Reynolds DS, Stevens RL, Lane WS, Carr MH, Austen KF, Serafin WE. Different mouse mast cell populations express various combinations of at least six distinct mast cell serine proteases. Proc Natl Acad Sci. 1990;87:3230–3234. doi: 10.1073/pnas.87.8.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds DS, Gurley DS, Austen KF, Serafin WE. Cloning of the cDNA and gene of mouse mast cell protease-6. Transcription by progenitor mast cells and mast cells of the connective tissue subclass. J Biol Chem. 1991;266:3847–3853. [PubMed] [Google Scholar]
- Roitt IM, Brostoff J, Male DK, editors. Immunology. London, UK: Gower; 1989. Cells involved in the immune response; pp. 11–28. [Google Scholar]
- Salamonsen L, Wooley DE. Matrix metalloproteinases in normal menstruation. Hum Reprod. 1996;11:124–133. doi: 10.1093/humrep/11.suppl_2.124. [DOI] [PubMed] [Google Scholar]
- Schechter NM, Choi JK, Slavin DA, Deresienski DT, Sayama S, Dong G, Lavker RM, Proud D, Lazarus GS. Identification of a chymotrypsin-like proteinase in human mast cells. J Immunol. 1986;137:962–970. [PubMed] [Google Scholar]
- Schechter NM, Brass LF, Lavker RM, Jensen PJ. Reaction of mast cell protease tryptase and chymase with protease activated receptors (PARs) on keratinocytes and fibroblasts. J Cell Physiol. 1998;176:365–373. doi: 10.1002/(SICI)1097-4652(199808)176:2<365::AID-JCP15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Shea CR, Prieto VG. Mast cells in angiolipomas and hemangiomas of human skin: Are they important for angiogenesis? J Cutan Pathol. 1994;21:247–251. doi: 10.1111/j.1600-0560.1994.tb00268.x. [DOI] [PubMed] [Google Scholar]
- Smith-McCune K, Weidner N. Demonstration and characterization of the angiogenic properties of cervical dysplasia. Cancer Res. 1994;54:800–804. [PubMed] [Google Scholar]
- Smith-McCune K, Zhu Y-H, Hanahan D, Arbeit J. Cross-species comparison of angiogenesis during the premalignant stages of squamous carcinogenesis in the human cervix and K14–HPV16 transgenic mice. Cancer Res. 1997;57:1294–1300. [PubMed] [Google Scholar]
- Starkey JR, Crowle PK, Taubenberger S. Mast-cell deficient W/Wv mice exhibit a decreased rate of tumor angiogenesis. Int J Cancer. 1988;42:48–52. doi: 10.1002/ijc.2910420110. [DOI] [PubMed] [Google Scholar]
- Stevens RL, Friend DS, McNeil HP, Schiller V, Ghildyal N, Austen KF. Strain-specific and tissue-specific expression of mouse mast cell secretory granule proteases. Proc Natl Acad Sci. 1994;91:128–132. doi: 10.1073/pnas.91.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhouk RS, Chin JR, Unemori EN, Werb Z, Bissell MJ. Proteinases of the mammary gland: Developmental regulation in vivo and vectorial secretion in culture. Development. 1991;112:439–449. doi: 10.1242/dev.112.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharp MD, Seelig LL, Tigelaar RE, Bergstresser PR. Conjugated avidin binds to mast cell granules. J Histochem Cytochem. 1985;33:27–32. doi: 10.1177/33.1.2578142. [DOI] [PubMed] [Google Scholar]
- Thurston G, Baluk P, Hirata A, McDonald D. Permeability-related changes revealed at endothelial cell borders in inflamed venules by lectin binding. Am J Physiol. 1996;271:2547–2562. doi: 10.1152/ajpheart.1996.271.6.H2547. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Eldor A, Haimovitz-Friedman A, Matzner Y, Ishai-Michaeli R, Lider O, Naparstek T, Cohen IR, Fuks Z. Expression of heparanase by platelets and circulating cells of the immune system: Possible involvement in diapedesis and extravasation. Inv Metast. 1992;12:112–127. [PubMed] [Google Scholar]
- Vu TV, Shipley JM, Bergers G, Berger J, Helms JA, Hanahan D, Shapiro SD, Senior R, Werb Z. Matrix metalloproteinase gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorio E, de Crombrugghe B. The family of collagen genes. Annu Rev Biochem. 1990;59:837–872. doi: 10.1146/annurev.bi.59.070190.004201. [DOI] [PubMed] [Google Scholar]
- Wastling J, Scudamore C, Thornton E, Newlands G, Miller H. Constitutive expression of mouse mast cell protease-1 in normal BALB/c mice and its up-regulation during intestinal nematode infection. Immunology. 1997;90:308–313. doi: 10.1046/j.1365-2567.1997.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernert N. The multiple roles of tumor stroma. Virchows Arch. 1997;430:433–443. doi: 10.1007/s004280050053. [DOI] [PubMed] [Google Scholar]
- Westphal E. Uber mastzellen. In: Ehrlich P, editor. Farbenanalytische Utersuchuugen zur Histologie und Klinik des Blutes: gesammelte Mitt(h)eilungen. Vol. 1. Berlin, Germany: Hirschwald Press; 1891. p. 17. [Google Scholar]
- Whitlock JM, Murdoch AD, Iozzo RV, Underwood PA. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J Biol Chem. 1996;271:10079–10086. doi: 10.1074/jbc.271.17.10079. [DOI] [PubMed] [Google Scholar]
- Xia Z, Ghildyal N, Austen KF, Stevens RL. Post-transcriptional regulation of chymase expression in mast cells. J Biol Chem. 1996;271:8747–8753. doi: 10.1074/jbc.271.15.8747. [DOI] [PubMed] [Google Scholar]
- Yong LC. The mast cell: Origin, morphology, distribution, and function. Exp Toxicol Pathol. 1997;49:409–424. doi: 10.1016/S0940-2993(97)80129-7. [DOI] [PubMed] [Google Scholar]