

Abstract
Verotoxin II (VTII: or Shiga-like toxin 2) is a key factor for Escherichia coli O157:H7–induced multiple tissue failure and contains a pentameric sequence (NWGRI) similar to the Bcl-2 homolog domain, BH1. In the current study, we demonstrate that VTII, but not VTI, interacts with Bcl-2 through each BH1 domain pentameric sequence (NWGRI) and that the VTII/Bcl-2 complex is necessary for cell-death induction in target cells. VTII translocates to mitochondria and induces cell death only when target cells are expressing Bcl-2. In addition, interruption of VTII-Bcl-2 complex formation by a pentameric BH1 synthetic peptide suppresses VTII-induced cell death. In the present article, we propose that Bcl-2 mediates VTII-induced target cell death by the interaction with each pentameric sequence of BH1 domain.
Keywords: Bcl-2, Escherichia coli O157:H7–derived verotoxin II, cell death, multiple tissue failure
Infection with Escherichia coli O157:H7 is occasionally lethal in patients because of extensive cell death in the intestines and kidneys (Sandvig et al. 1997; Paton and Paton 1998). Verotoxin I and II (VTI and VTII) are key factors in E. coli O157:H7–initiated tissue failure (Sandvig et al. 1997; Paton and Paton 1998;). In recent years, some possible role for verotoxins in cell-death induction has been suggested. Verotoxins suppress protein synthesis and mitogen (Brigotti et al. 1997; Van Settenet al. 1997), and Gb3/CD77 glycolipid antigen has been identified as the receptor for verotoxin, especially for 5-verotoxin subunit B, and the stimulation of Gb3/CD77-induced apoptotic cell death (Tyrrellet al. 1992; Mangeneyet al. 1993; Arabet al. 1998). However, details of molecular mechanism have not been elucidated. It also has been reported that cell-death induction by verotoxins is initiated independently of their ability to suppress protein synthesis, suggesting two distinct mechanisms (Van Setten et al. 1997).
Cell death is essential for cell homeostasis and for cell growth and has been well documented during embryonic and postembryonic development (Wyllie et al. 1980; Nagata 1997). There are two distinct processes leading to cell death, known as apoptotic and necrotic cell death (Wyllie et al. 1980). Apoptotic cell death is accompanied by condensation and/or fragmentation of nuclei, apoptotic body formation, and chromosomal DNA fragmentation into 180-bp oligomers (Wyllie et al. 1980). Many studies have demonstrated the important role of apoptotic cell death in different disease states and physiologic cell death (Nagata and Golstein 1995), and many factors involved with the death signaling have been identified.
Bcl-2 proto-oncoprotein was identified originally through study of the t(14; 18) translocation present in human B-cell follicular lymphomas (Tsujimoto et al. 1984). Bcl-2 localizes on the membrane surface of organelles, and its expression can be encountered on the nuclear membrane, endoplasmic reticulum and mitochondrial membrane (Akao et al. 1994). Bcl-2 is unique in that it inhibits apoptosis rather than promoting cell proliferation (Vaux et al. 1988; Tsujimoto 1989). Recently, multiple genes have been identified within the Bcl-2 family; some of these genes, such as Bcl-xs, Bax, and Bak (Oltvai et al. 1993; Chittenden et al. 1996), drive the death mechanism and others, such as Bcl-2 and Bcl-xL (Vaux et al. 1988; Tsujimoto 1989; Boiseet al. 1993), act against apoptotic cell death. Bcl-2 contains four unique domains, BH1–4 (Yin et al. 1994). The Bcl–2-BH1 domain is important for the interaction with other Bcl-2 family members for cell death suppression (Seto et al. 1988; Yin et al. 1994). The mitochondrial channel VDAC recently was identified as the target molecule of Bcl-2 (Shimizu et al. 1999).
Caspase is the nomenclature that refers to the interleukin-1? converting enzyme (ICE)/CED-3 cysteine proteinase family (Alnemri et al. 1996). During death induction, sequential activation of the caspase 1 and caspase 3 subfamilies has been reported (Enari et al. 1996), and this phenomenon is known as the “ICE cascade.” At present, 14 genes have been identified within the caspase family, and the caspase 3 subfamily, including caspase 3 and caspase 8, in particular acts as the dominant regulator in the death signaling. Therefore, the regulation of caspase 3 subfamily activation is an especially important focus for cell-death research.
In the current study, we investigated the molecular machinery of cell-death induction by VTs. We report that VTII subunit A, but not VTI subunit A, contains a pentameric sequence (NWGRI) from the BH1 domain and interacts with mitochondrial Bcl-2 to induce target cell death resulting from caspase 3 activation.
Results
Heterodimerization of Bcl-2 and VTII with BH1 domain
Bcl-2 contains five functional domains, BH1–4 and a transmembrane domain (Fig. 1A). The BH1 domain is located between residues 136 and 155, and the sequence from 143 to 147 (NWGRI and Fig. 1A) is essential for BH1 domain function (Seto et al. 1988). As shown in Figure 1A, VTII also contains NWGRI sequence residues 223–227. The corresponding sequence in VTI is NWGRL at residues 234–238. Both isoleucine (I) and leucine (L) belong to the same amino acid group. Calculated protein identities and similarities were also indicated in Figure 1A (VTI vs. VTII: 53%, 17%; VTI vs. Bcl-2: 14%, 9%; VTII vs. Bcl-2: 14%, 11%).
Figure 1.

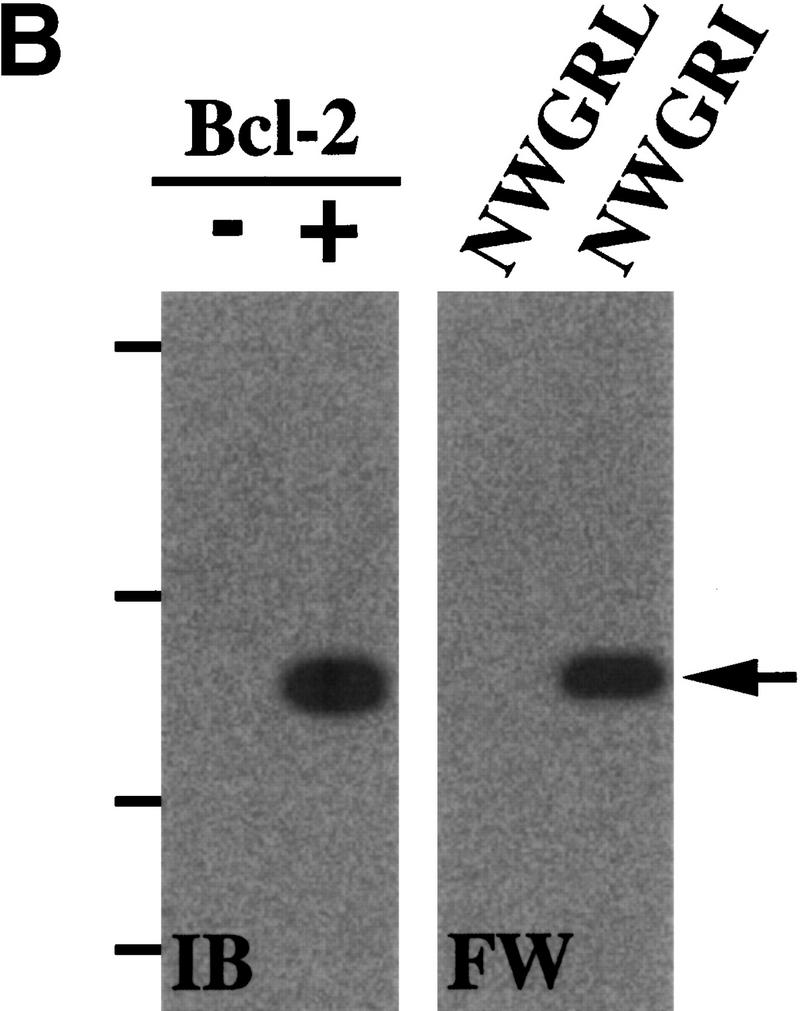
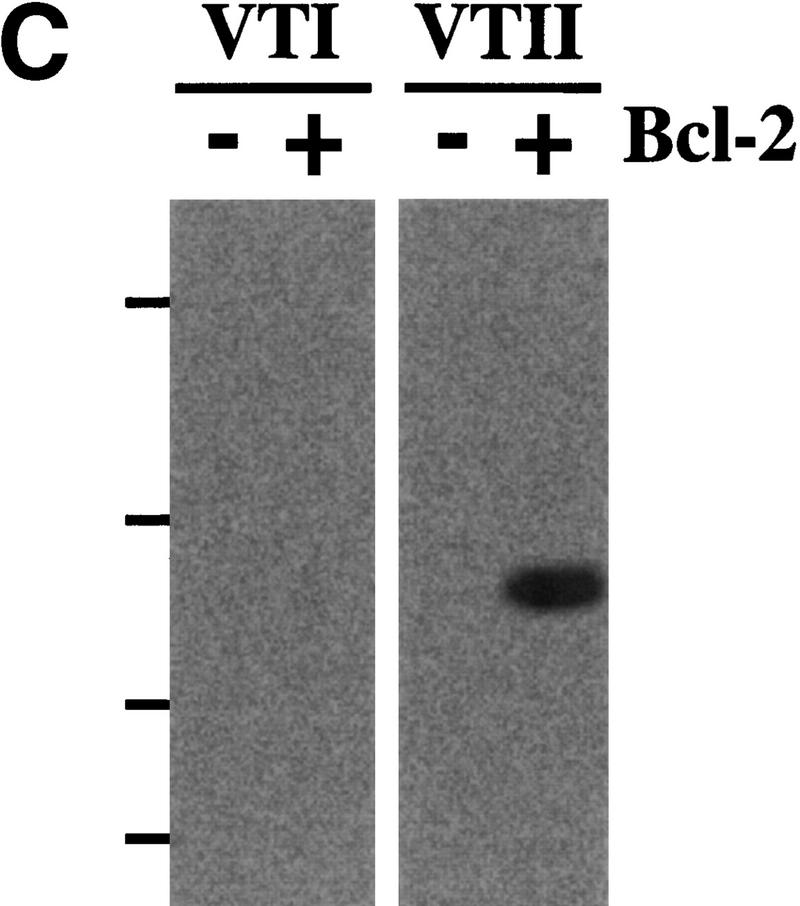
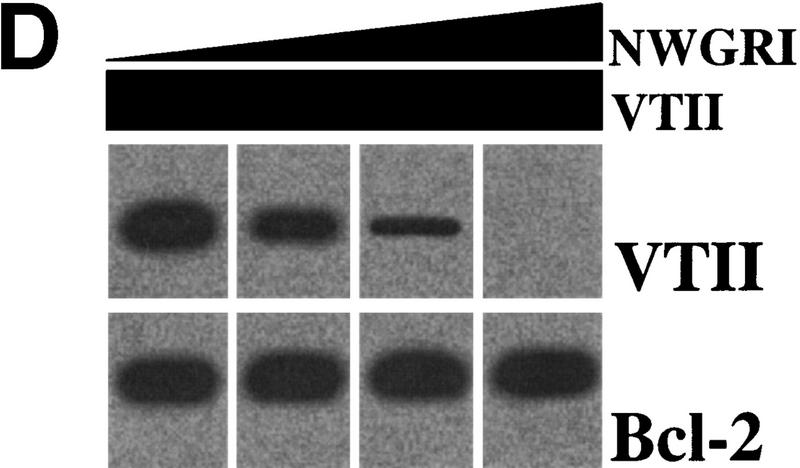
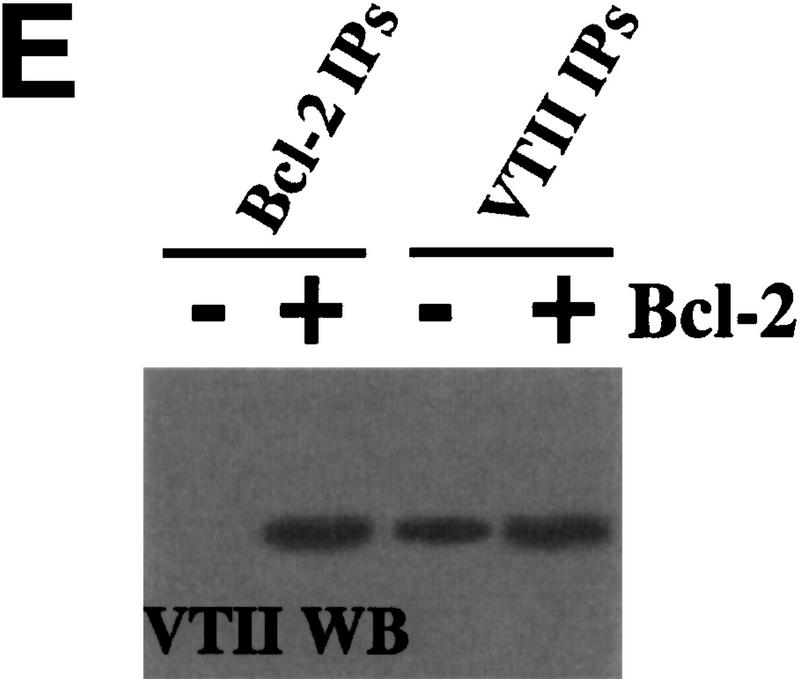
VTII-Bcl-2 complex formation. (A) VTII, but not VTI, contains the pentameric sequence from the Bcl-2 BH1 domain. Shown are the schematic model, sequence alignment, and protein identities and similarities of human Bcl-2, VTI and VTII. The dotted line indicates the region of splicing variants. (B,C). VTII, but not VTI, interacts with Bcl-2. Expression of Bcl-2 in V (-) and B10 (+) cells was detected with immunoblotting (IB). B10 (B) and/or V (B,C) cell extracts were separated by SDS-PAGE, and far-western blotting (FW) was performed with biotinylated peptides (NWGRL and NWGRI in B) or VTs (VTI and VTII in C). Interacted protein was detected with avidin-HRP and ECL. Bars on the left show positions of protein markers 46, 30, 20, and 14 kDa, from top to bottom. (D) VTII interacts with Bcl-2 through the NWGRI sequence. Far-western blotting analysis was performed with biotinylated VTII and B10 cell extracts in the presence of NWGRI (0, 1, 5, and 10 nm). (E) Coimmunoprecipitation analysis. V (-) or B10 (+) cells were treated with VTII, and then immunoprecipitates with antibodies for Bcl-2 (Bcl-2 IPs) or VTII (VTII IPs) were separated on SDS-PAGE, and immunoblotting analysis with VTII antibody was performed (VTII WB).
To examine whether VTI (NWGRL) or VTII (NWGRI) interact with Bcl-2, we prepared a human hepatoma HepG2 cell line that overexpresses human Bcl-2. As shown in Figure 1B, Bcl-2–expressing HepG2 cells (B10) expressed Bcl-2, but the pseudovector transfected clone (V) did not. Using these cell lines, far-western blotting showed that NWGRI (VTII), but not NWGRL (VTI), interacts with Bcl-2 (Fig. 1B, C). We demonstrated that NWGRI interrupted an interaction between VTII and Bcl-2 in a dose-dependent manner (Fig. 1D). When B10 cells were treated with VTII, coimmunoprecipitation analysis showed that VTII interacted with Bcl-2 (Fig. 1E).
Cell-death induction and caspase 3 activation by VTII, but not by VTI
We showed that VTII, but not VTI, specifically interacts with Bcl-2 through the NWGRI sequence. To investigate the physiologic role of this complex, cell-death induction and caspase 3 activation were analyzed. When B10 and V cells were treated with VTI or VTII, a decrease in cell viability was observed only in VTII-treated B10 cells (Fig. 2A). Death induction accompanied by nuclear condensation and fragmentation was also observed only in VTII-treated B10 cells (Fig. 2A), which is consistent with apoptotic cell death.
Figure 2.


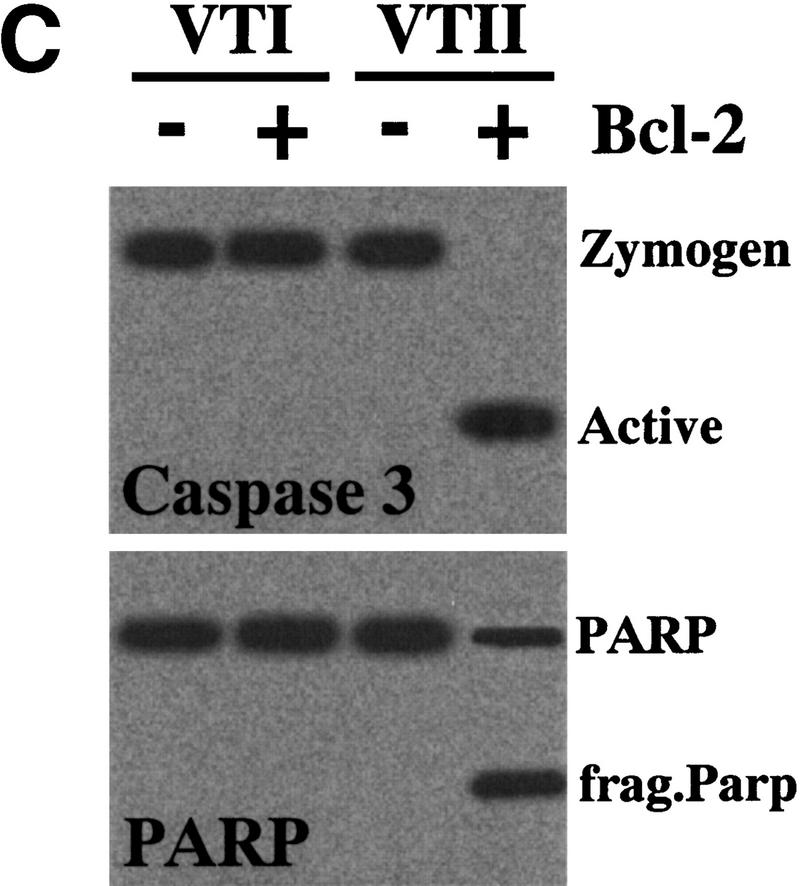
Cell-death induction by the VTII-Bcl-2 complex. (A) VTII-induced cell death in B10 cells. B10 (closed symbols) or V (open symbols) cells were treated with increasing concentrations of VTI (circles) or VTII (squares) for 24 hours. Cell viability was measured with an MTT assay (left). B10 (closed bars) or V (open bars) cells were treated with 10 ng/ml VTI or VTII for 24 hours, and the percentage of death induction was measured with Hoechst 33342/PI staining. (B) Caspase 3 activation measured with tetrapeptide substrate. B10 cells were treated with 10 ng/ml VTI (open cycle) or VTII (closed cycle), and the enzymatic activity of caspase 3 was measured as Ac-DEVD-MCA cleavage. (C) Caspase 3 activation detected with caspase 3 processing and PARP cleavage. B10 (+) or V (-) cells were treated with 10 ng/ml VTI or VTII for 24 hours, and proteins from treated cells were used for immunoblotting with antibody for caspase 3 or PARP.
We also found that VTII, but not VTI, induced Ac-DEVD-MCA cleavage in a time-dependent manner (Fig. 2B). Only VTII-treated B10 cells showed caspase 3 activation (molecular shift of the 32-kDa zymogen to 17-kDa active form) and poly(ADP-ribose) polymerase (PARP) cleavage (Fig. 2C), which is consistent with VTII-induced cell death mediated by caspase 3 activation in the B10 cells.
Mitochondrial translocation of VTII
We investigated the subcellular translocation of VTI and VTII in both V and B10 cells. When the cells were treated with VTI or VTII, there was predominantly nuclear staining (Figs. 3A,B). However, B10 cells treated with VTII also showed cytosol staining. Subcellular fractionation of VTI- or VTII-treated B10 cells showed VTI predominantly in the nuclear fraction (Fig. 3C). In contrast, VTII was detected predominantly in mitochondria (Fig. 3C).
Figure 3.



Mitochondrial localization of VTII in the presence of Bcl-2. (A,B) VT subcellular localization observed by microscopy. B10 (A) or V (B) cells were treated with 10 ng/ml VTI (3) or VTII (4) for 6 hours. VTI and VTII were detected with each antibody and phycoerythrin-conjugated second antibody. Nuclei (PI: 1) and mitochondria (MitoTracker: 2) were also stained. (C) VT subcellular localization observed by immunoblotting. B10 cells were treated with 10 ng/ml VTI (left) or VTII (right), and fractionated proteins were collected by using previously described methods (Fleisher and Kervina 1974; Suzuki et al. 1999). VTI and VTII in each subfraction were detected by immunoblotting with antibodies for VTI and VTII. As a positive control, purified VTI and VTII, which were used for the treatments, were also analyzed.
Mitochondrial Bcl-2 is necessary for VTII-induced cell death
To further investigate the physiologic role of the Bcl-2-VTII complex, we examined the effect of the Bcl-2-VTII complex in HepG2 cells lacking mitochondrial DNA (HepG2Δm). Cell death, which was accompanied by nuclear condensation and fragmentation, was observed only in the presence of both Bcl-2 and VTII (Fig. 4A). In addition, cell-death induction was observed when B10-derived mitochondria and VTII were microinjected into cells (Fig. 4A). Mitochondria alone or VTII-mixed V-cell mitochondria could not induce cell death.
Figure 4.


Cell death induced by the VTII-Bcl-2 complex and its suppression by the NWGRI pentameric peptide. (A) Cell-death induction by VTII and mitochondrial Bcl-2. Mitochondria extracted from B10 (B10-mit) or V (V-mit) were treated with or without 10 ng/ml VTII and microinjected into HepG2Δm cells. Death induction was measured as the ratio of apoptotic nuclei to total nuclei (about 5000). As the control, B10 (Bcl-2+/VTII+) and V (Bcl-2-/VTII+) cells were also treated with VTII. (B) Suppression of VTII-induced cell death by the NWGRI peptide. B10 cells pretreated with NWGRI (0, 10, 50, and 100 μm) were treated with 10 ng/ml VTII for 24 hours, and death induction was measured as the ratio of apoptotic nuclei in total nuclei (about 5000). As the controls, B10 cells were treated with VTII or NWGRI (100 μm) alone.
Our current results showed that the NWGRI peptide of the BH1 domain interrupted Bcl-2-VTII interactions. Therefore, we investigated an effect of NWGRI in Bcl-2-VTII complex-induced cell death in HepG2Δm cells. When B10 cells were pretreated with NWGRI, followed by VTII incubation, cell-death induction was suppressed in a dose-dependent manner (Fig. 4B).
Discussion
Systemic bacterial infection is a significant source of morbidity and mortality, because it triggers various secondary effects such as disseminated intravascular coagulation (DIC) and multiple organ failure (Sandvig et al. 1997; Paton and Paton 1998). A recent outbreak of E. coli O157:H7 in Japan caused many children and old people to die (Guest 1996; Nathan 1997). Verotoxins were identified as the bacterial toxins of E. coli O157:H7, and they induce extensive cell death in the intestine and kidneys, although the molecular basis is not known.
The kidney is a particular target organ of E. coli O157:H7, especially for VTs. The Bcl-2 null mouse presents with polycystic kidney (Veiset al. 1993; Kamadaet al. 1995), suggesting an important role for Bcl-2 in renal physiology. Verotoxin-induced kidney injury is accompanied by nephritis rather than by polycystic kidney as a result of extensive cell death; however, these two distinct types of kidney failure are sufficient to link VTs and Bcl-2 in cell-death induction. Bcl-2 contains multiple domains, BH1–4, and a transmembrane domain (Williams and Smith 1993), and the BH domains are essential for interaction with other Bcl-2 family members. For example, Bid was identified as BH3, the only Bcl-2 family member death agonist (Wang et al. 1996), and Bax and Bak interacts with BH1 and BH2 (Oltvai et al. 1993; Chittenden et al. 1995, 1996; Wang et al. 1996). VTI subunit A versus VTII subunit A showed high homology (53% shared identities), but VTI subunit A versus Bcl-2 (14% shared identities) and VTII subunit A versus Bcl-2 (14% shared identities) was lower. Interestingly, however, the first structure of VT subunit A showed the presence of a pentameter sequence very similar to that found in the Bcl-2 BH1 domain. BH1 and BH2 domains in Bcl-2 are required for the cell-death suppression (Yin et al. 1994), and the NWGRI sequence in the BH1 domain is essential (Seto et al. 1988). Although the sequences in VTI (NWGRL) and VTII (NWGRI) are remarkably similar to the Bcl-2 BH1 domain sequence (NWGRI), we demonstrated that VTII, but not VTI, interacts with Bcl-2 via NWGRI in vitro and in vivo. Further, coimmunoprecipitation analysis directly demonstrated VTII subunit A-Bcl-2 complex formation. It has been reported that the toxicity of VTII, is higher than that of VTI with a 50% lethal dose (LD50) of VTII being approximately 400 times lower than that of VTI in a murine model (Tesh et al. 1993), and the difference in toxicity can be traced to differences in amino-acid sequence between the VTs (Gannon and Gyles 1990). In recent years, the receptor protein (Gb3/CD77) for VT subunit B has been identified (Tyrrell et al. 1992; Mangeney et al. 1993; Arab et al. 1998). Verotoxins induce apoptotic cell death through protein synthesis suppression; however, a distinct mechanism has also been recently suggested, because VT-induced cell death is independently initiated by protein synthesis suppression (Van Setten et al. 1997). Therefore, our current results suggest that the VTII-Bcl-2 interaction is one of essential death-induction systems triggered by E. coli O157:H7 and that the previously suggested distinct system is VTII-Bcl-2 complex formation.
Using two human heptoblastoma HepG2 cell lines, Bcl-2-positive B10 and Bcl-2-negative V, MTT assay showed that VTII treatment reduced MTT value (cell viability) only in the presence of Bcl-2, suggesting that VTII induced cell-growth suppression and/or cell-death induction in the presence of Bcl-2. Because Hoechst 33342/propidium iodide (PI) staining analysis showed up-regulation of cell-death induction, which was accompanied by nuclear condensation and/or fragmentation, by VTII treatment, we suspect that the reduction of the MTT value is caused as a result of apoptotic cell death induction and that VTII induces apoptotic cell-death in the presence of Bcl-2. We also demonstrated that VTII activated caspase 3 only in the presence of Bcl-2. Caspase 3 is well documented as an essential death factor (Suzuki et al. 1998, 1999), and it activates caspase 3 proteolyses poly(ADP-ribose), polymerase (PARP; Lazebnicet al. 1994), ICAD (Enariet al. 1998) and Acinus (Sahara et al. 1999). These results suggest that VTII induces cell death as a result of caspase 3 activation and that the death signaling is triggered by the presence of Bcl-2.
When V or B10 cells were treated with VTs, their subcellular localization showed a different pattern; VTI was detected only in the nuclear fraction both of V and B10 cells, but VTII in mitochondrial fraction was predominantly observed only in the presence of Bcl-2 (in B10 cells). Bcl-2 expression is encountered in the nuclear outer membrane, endoplasmic reticulum, and mitochondrial membrane (Akao et al. 1994). The mitochondrial channel VDAC has been reported as a target molecule of Bcl-2 (Shimizu et al. 1999). During cell-death induction, cytochrome c released from mitochondria activates caspase 3 with Apaf-1 (Zou et al. 1997), and Bcl-2 suppresses cytochrome c release (Yang et al. 1997). In the current study, we have demonstrated that VTII-treated mitochondria derived from Bcl-2-positive cells induced cell death signaling, suggesting that the mitochondrial Bcl-2-VTII complex acts as a death inducer. The molecular basis of VTII-induced cell death via Bcl-2 is uncertain, but it may be due to the blocking of mitochondrial Bcl-2, because the absence of Bcl-2 results in cell-death induction in the kidney (Veis et al. 1993; Kamada et al. 1995).
Specific cell-death induction by VTI (Shiga-like toxin-1) in breast cancer, lymphoma, and myeloma, but not in CD34(+) hematopoetic stem cells, has been reported recently (LaCasse et al. 1999). This cell-death induction was due to the specific expression of a VTI-receptor, Gb3/CD77 (LaCasse et al. 1999). It has been documented that cell-death induction by the stimulation of Gb3/CD77 was initiated as a result of protein synthesis suppression (Tyrrell et al. 1992; Mangeney et al. 1993; Arab et al. 1998). In recent years, however, another cell-death induction machinery by VTs has been reported, that is, VT-induced cell death was initiated independently by protein synthesis suppression (Van Setten et al. 1997). In the current study, we demonstrated that VTII, but not VTI, contained the pentameric sequence from the Bcl-2 BH1 domain (NWGRI) and specifically induced cell death in Bcl-2-expressing cells through the formation of the VTII-Bcl-2 complex. On the basis of our current results, we propose that VTII-Bcl-2 complex formation is the other cell-death induction system suggested by Van Setten et al.
Cell and procedures
Cells
Human hepatoma HepG2 with human Bcl-2 (transfected clone: B10) and its control clone (pseudovector transfectant: V) were supplied by Dr. Yoshihide Tsujimoto and were maintained in RPMI 1640 medium (GIBCO BRL, Gaithersburg, MD, USA) supplemented with 10% heat-inactivated fetal calf serum (GIBCO BRL) in a humidified atmosphere of 5% CO2 and 95% air.
Assay of cell viability
Apoptotic cell death is accompanied by condensation and fragmentation of chromosomal DNA (Wyllie et al. 1980). In the present study, nuclei of treated cells were stained with Hoechst 33342/PI using procedures described previously (Hasegawa et al. 1996; Suzuki et al. 1998, 1999). Nuclei without condensed and/or fragmented nuclei were recognized as normal. Cell viability was indicated as the percentage of normal nuclei per total nuclei (about 5000 nuclei). Hoechst 33342 and PI were purchased from Molecular Probes, Inc. (Eugene, OR). After stimulation with anti-human Fas antibody (CH-11 clone, MBL, Nagoya, Japan; Yonehara et al. 1989), cells were collected and stained with Hoechst 33342/PI, and then observed by fluorescence microscopy.
Cell viability was also measured with an MTT assay, which was performed as previously described (Suzuki et al. 1998, 1999).
Far-western blotting analysis
Proteins extracted from cells were separated on 5–20% Sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose filter using a semidry blotting system. Proteins were renatured with phosphate-buffered saline (PBS) containing 3% NP-40, and filters were blocked with skim milk and incubated with 1 μg/ml of biotinylated VTs or synthesized peptides. Biotinylation of proteins or peptides was performed with the Biotinylation kit (Pierce, Rockfor, IL). Detection of bound biotin VTs or peptides were performed with avidin–horse radish peroxidase (HRR) and enhanced chemiluminesence (ECL).
Immunoblotting analysis
Sample proteins separated by SDS-PAGE were transferred onto nitrocellulose membranes with a semidry blotting system. The membranes were blocked with PBS containing 5% (w/v) skim milk at room temperature for 1 hour, washed with a mixture of PBS and 0.05% Tween 20 (Tween-PBS; Sigma, St. Louis, MO), and then incubated overnight at room temperature with each antibody diluted with PBS. After washing with Tween-PBS, the membranes were incubated with a 1000-fold diluted biotinylated second antibody, washed with Tween-PBS, and then incubated with avidin-HRP (Vector Laboratories, Burlingame, CA) at room temperature for 1 hour. The membranes were washed with Tween-PBS and then developed with the ECL system.
Preparation of fractionated proteins
Cells were collected and washed with ice-cold PBS and then suspended in buffer I (2 mm EDTA and 10 mm Tris-HCl at pH 7.5). After incubation on ice for 10 min, an equal volume of buffer II (0.5 m sucrose, 0.1 m KCl, 10 mm MgCl2, 2 mm CaCl2, 2 mm EDTA, 10 mm Tris-HCl at pH 7.5) was added. The nuclear-rich fraction was pelleted by centrifugation (600g/10 min). The supernatant was removed to a separate tube and again centrifuged (100,000g/90 min). The supernatant was collected as a cytosol-rich fraction, and the pellet was dissolved in buffer III (8 mm CHAPS, 150 mm NaCl, 0.1 m sucrose, 2 mm EDTA, 10 mm Tris-HCl at pH 7.5) and incubated at 4°C for 2 hr. The membrane-rich fraction was then collected by centrifugation (100,000g/60 min). Mitochondria were collected by using a previously described method (Fleisher and Kervina 1974), and its proteins were extracted with 1% NP40-containing PBS. Each fractionation was separated by 5–20% SDS-PAGE and VTs were detected by immunoblotting analysis and ECL detection system (Amersham, Buckinghamshire, UK).
Microinjection analysis
Microinjection analysis was performed for the investigation of mitochondrial Bcl-2 effects in cells (about 10,000 cells). Collected mitochondria were suspended in PBS, and microinjection was performed with a Transjector 5246 (Eppendorf, Hamburg, Germany). Microinjection was performed at 100 hPa for 3. In the present study, no gross abnormalities were observed for 2 weeks after the cells were microinjected with PBS.
Preparation of the mitochondrial DNA–lacking cells
Preparation of mitochondrial DNA–lacking cells (HepG2Δm) was done by using previously described methods (Suzuki et al. 1999). Cells were cultured in RPMI 1640 medium containing ethidium bromide (0.4 μg/ml) for about 2 months. Loss of mitochondrial DNA was determined by Southern blotting analysis, cell-cycle arrest in conditioned medium, and cell-growth recovery in uridine-containing medium.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL leb00373@nifty.ne.jp; FAX +81-3-5696-8336.
References
- Akao Y, Otsuki Y, Kataoka S, Ito Y, Tsujimoto Y. Multiple subcellular localization of bcl-2: Detection in nuclear outer membrane, endoplasmic reticulum, and mitochondrial membranes. Cancer Res. 1994;54:2468–2471. [PubMed] [Google Scholar]
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornbery NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Arab S, Murakami M, Dirks P, Boyd B, Hubbard SL, Lingwood CA, Rutka JT. Verotoxins inhibit the growth of and induce apoptosis in human astrocytoma cells. J Neurooncol. 1998;40:137–150. doi: 10.1023/a:1006010019064. [DOI] [PubMed] [Google Scholar]
- Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Brigotti M, Carnicelli D, Alvergna P, Mazzaracchio R, Sperti S, Montanaro L. The RNA-N-plycosidase activity of Shiga-like toxin I: Kinetic parameters of the native and activated toxin. Toxicon. 1997;35:1431–1437. doi: 10.1016/s0041-0101(96)00225-5. [DOI] [PubMed] [Google Scholar]
- Chittenden T, Flemington C, Houghton AB, Ebb RG, Gallo GJ, Elangovan B, Chinnadurai G, Lutz RJ. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO J. 1995;14:5589–5596. doi: 10.1002/j.1460-2075.1995.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittenden T, Harrington EA, O‘Connor R, Flemington C, Lutz RJ, Evan GI, Guild BC. Induction of apoptosis by the Bcl-2 homologue Bak. Nature. 1996;374:733–736. doi: 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- Fleisher S, Kervina M. Subcellular fractionation of rat liver. Methods Enzymol. 1974;31:6–41. doi: 10.1016/0076-6879(74)31005-1. [DOI] [PubMed] [Google Scholar]
- Gannon VP, Gyles CL. Characteristics of the Shiga-like toxin produceed by Escherichia coli associated with porcine edema disease. Vet Microbiol. 1990;24:89–100. doi: 10.1016/0378-1135(90)90054-y. [DOI] [PubMed] [Google Scholar]
- Guest R. Four die in food poisoning outbreak in Japan. Br Med J. 1996;313:187. doi: 10.1136/bmj.313.7051.187. [DOI] [PubMed] [Google Scholar]
- Hasegawa J, Kamada S, Kamiike W, Shimizu S, Imazu T, Matsuda H, Tsujimoto Y. Involvement of CPP32/Yama(-like) proteases in Fas-mediated apoptosis. Cancer Res. 1996;56:1713–1718. [PubMed] [Google Scholar]
- Kamada S, Shimono A, Shinto Y, Tsujimura T, Takahashi T, Noda T, Kitamura Y, Kondoh H, Tsujimoto Y. Bcl-2 deficiency in mice leads to pleiotropic abnormalities: Accelerated lymphoid cell death in thymus and spleen, polycystic kidney, hair hypopigmentation, and distorted small intestine. Cancer Res. 1995;55:354–359. [PubMed] [Google Scholar]
- LaCasse EC, Bray MR, Patterson B, Lim WM, Perampalam S, Radvanyl LG, Keating A, Stewart AK, Buckstein R, Sandhu JS, et al. Shiga-like toxin-1 receptor on human breast cancer, lymphoma, and myeloma and absence from CD34(+) hematopoetic stem cells: Implications for ex vivo tumor purging and autologous stem cell transplantation. Blood. 1999;94:2901–2910. [PubMed] [Google Scholar]
- Lazebnic YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly (ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Mangeney M, Lingwood CA, Taga S, Caillou B, Tursz T, Wiels J. Apoptosis induced in Burkitt’s lymphoma cells via Gb3/CD77, a plycolipid antigen. Cancer Res. 1993;53:5314–5319. [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- Nathan R. American seeds suspected in Japanese food poisoning epidemic. Nat Med. 1997;3:705–706. doi: 10.1038/nm0797-705b. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Paton JC, Paton AW. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin Microbiol Rev. 1998;11:450–479. doi: 10.1128/cmr.11.3.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara S, Aoto M, Eguchi Y, Imamoto N, Yoneda Y, Tsujimoto Y. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature. 1999;401:168–173. doi: 10.1038/43678. [DOI] [PubMed] [Google Scholar]
- Sandvig K, Garred O, Van Deurs B. Intracellular transport and processing of protein toxins produced by enteric bacteria. Adv Exp Med Biol. 1997;412:225–232. doi: 10.1007/978-1-4899-1828-4_34. [DOI] [PubMed] [Google Scholar]
- Seto M, Jaeger U, Hockett RD, Graninger W, Bennett S, Goldman P, Korsmeyer SJ. Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. EMBO J. 1988;7:123–131. doi: 10.1002/j.1460-2075.1988.tb02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Tsutomi Y, Akahane K, Araki T, Miura M. Resistance to Fas-mediated apoptosis: Activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene. 1998;17:931–940. doi: 10.1038/sj.onc.1202021. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Tsutomi Y, Yamamoto N, Shibutani T, Akahane K. Mitochondrial regulation of cell death:Mitochondria are essential for procaspase 3/p21 complex formation to resist Fas-mediated cell death. Mol Cell Biol. 1999;19:3842–3847. doi: 10.1128/mcb.19.5.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesh VL, Burris JA, Owens JW, Gordon VW, Wadolkowski EA, O‘Brien AD, Samuel JE. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect Immunol. 1993;61:3392–3402. doi: 10.1128/iai.61.8.3392-3402.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y. Stress-resistance conferred by high level of bcl-2 alpha protein in human B lymphoblastoid cell. Oncogene. 1989;4:1331–1336. [PubMed] [Google Scholar]
- Tsujimoto Y, Yunis J, Nowell PC, Croce CM. Cloning of the chromosomal breakpoint of neoplastic B cells with the t(14; 18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- Tyrrell GJ, Ramotar K, Toye B, Boyd B, Lingwood CA, Brunton JL. Alteration of the carbohydrate binding specificity of verotoxins from Galα 1-4Gal to GalNAcβ 1-3Galα 1-4Gal and vice versa by site-directed mutagenesis of the binding subunit. Proc Natl Acad Sci. 1992;89:524–528. doi: 10.1073/pnas.89.2.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Setten PA, van Hinsbergh VW, Van den Heuvel LP, van der Velden TJ, van den Kar NC, Krebbers RJ, Karmali MA, Monnens LA. Verotoxin inhibits mitogenesis and protein synthesis in purified human glomerular mesangial cells without affecting cell viability: Evidence for two distinct mechanisms. J Am Soc Nephrol. 1997;8:1877–1888. doi: 10.1681/ASN.V8121877. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haematopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Veis JD, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: A novel BH3 domain-only death agonist. Genes & Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- Williams GT, Smith CA. Molecular regulation of apoptosis: Genetic controls on cell death. Cell. 1993;74:777–779. doi: 10.1016/0092-8674(93)90457-2. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death: The significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- Yonehara S, Ishii A, Yonehara M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J Exp Med. 1989;169:1747–1756. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]