

Abstract
During embryonic development, a large number of cells die naturally to shape the new organism. Members of the caspase family of proteases are essential intracellular death effectors. Herein, we generated caspase-2-deficient mice to evaluate the requirement for this enzyme in various paradigms of apoptosis. Excess numbers of germ cells were endowed in ovaries of mutant mice and the oocytes were found to be resistant to cell death following exposure to chemotherapeutic drugs. Apoptosis mediated by granzyme B and perforin was defective in caspase-2-deficient B lymphoblasts. In contrast, cell death of motor neurons during development was accelerated in caspase-2-deficient mice. In addition, caspase-2-deficient sympathetic neurons underwent apoptosis more effectively than wild-type neurons when deprived of NGF. Thus, caspase-2 acts both as a positive and negative cell death effector, depending upon cell lineage and stage of development.
Keywords: Caspase-2, Ich1, Nedd2, apoptosis, caspases, cell death
The execution of programmed cell death in the nematode, Caenorhabditis elegans, requires the cysteine protease CED-3 (Yuan and Horvitz 1990; Yuan et al. 1993). Homologs of CED-3, termed caspases, have been implicated in the execution of apoptosis in vertebrates (Kumar 1995a; Martin and Green 1995). To date, 12 structurally related mammalian caspases have been identified and subgrouped into three families based on homology and substrate specificity (Nicholson and Thornberry 1997). Caspases are synthesized as proenzymes that are cleaved at key aspartic acid residues by other members of the family to generate active proteases. Once activated, caspases cleave cellular proteins, thus leading to the demise of the cell. The most compelling evidence that activity of caspases is critical in the control of programmed cell death comes from the observation that specific viral and peptide inhibitors of caspases are effective at preventing apoptosis induced by various stimuli. However, the role played by individual caspases in programmed cell death during development, homeostasis, and injury remains unclear. To date, mice deficient in caspase-1 (interleukin-1-converting enzyme) (Kuida et al. 1995; Li et al. 1995) and caspase-3 (CPP32) (Kuida et al. 1996) have been characterized. caspase-1 mutant mice develop normally but caspase-1−/− thymocytes show a slight delay in Fas-induced apoptosis. caspase-3 mutant mice are affected more severely with decreased apoptosis in the brain resulting in postnatal mortality. caspase-2, described previously as Nedd2/Ich1, was initially identified as a gene highly expressed in embryonic brain and down-regulated in adult brain (Kumar et al. 1992). Four lines of evidence point to caspase-2 as a major player in the programmed cell death pathway. It is highly expressed during embryonic development when extensive cell death occurs (Kumar et al. 1994); it is activated early in the process of apoptosis (Harvey et al. 1997; Li et al. 1997); it induces cell death when overexpressed in mammalian cells (Kumar et al. 1994; Wang et al. 1994); and decreasing caspase-2 levels using antisense technology delays cell death induced by trophic factor deprivation in hematopoetic and neuronal cell lines (Kumar 1995b; Troy et al. 1997). Interestingly, alternative splicing generates two caspase-2 messages that encode caspase-2L, which induces cell death, and caspase-2S, a truncated protein that can antagonize cell death (Wang et al. 1994; Kumar et al. 1997). Caspase-2S is generated by the addition of a 61-bp exon following the enzyme active site, resulting in a stop codon downstream. Caspase-2L is the dominant form expressed in all tissues examined. On the other hand, caspase-2S is predominantly expressed in brain, skeletal muscle, and heart (Wang et al. 1994; Kumar et al. 1997). We evaluated the requirement of caspase-2 in the induction of apoptosis in various tissues through gene targeting in mice.
Results
Generation of caspase-2-deficient mice
To generate caspase-2-deficient mice, we inactivated the caspase-2 gene by homologous recombination in embryonic stem (ES) cells with a targeting vector that replaced a 1.65-kb fragment with a neomycin resistance gene (Fig. 1A). The deleted fragment comprises the exon that encodes the QACRG active site of the enzyme and part of the next exon that encodes the caspase-2S sequence, thereby inactivating both the long and the short forms of caspase-2. ES cell clones harboring homologous recombination at the caspase-2 locus were identified by Southern blot analysis. Two independent clones (72 and 511) were injected into C57BL6 blastocysts to generate chimeric mice. Chimeric males with >80% agouti coat color were crossed with C57BL6 females to generate the colony. caspase-2 mutant mice reached adulthood with no gross abnormality.
Figure 1.




Targeted disruption of the caspase-2 gene. (A) The structure of the 3′ end of the mouse caspase-2 gene is depicted. The last four exons are shown as boxes, and the nucleotides are numbered starting from the ATG translation initiation codon. The enzymatic active site pentapeptide domain QACRG is indicated. The differential splicing events that produce two messages encoding the short or long form of caspase-2 are indicated. The position of the probe used for the genomic Southern blot analysis is shown. (B) RT–PCR analysis of spleen mRNA for caspase-2-deficient and wild-type mice. Sequences amplified are indicated at the top. A 300-bp fragment from actin mRNA was amplified as a control, and the caspase-2 cDNA sequence was amplified from nucleotide 901 to 1079 within the active site or from nucleotide 181 to 598 upstream of the active site. The positive control is a fragment amplified from a cloned caspase-2 cDNA; the negative control is a PCR reaction performed without the addition of template cDNA. The sizes of the amplified fragments are indicated at right. (C,D) Immunoblot analysis of tissues from wild-type and mutant mice. Immunoblot analysis of adult (C) (strain 72) and E11.5 (D) (strain 511) mouse tissues using a rabbit polyclonal antibody raised against the full-length recombinant caspase-2 protein. This antibody cross-reacts with two other unrelated proteins (44 and 33 kD).
We analyzed caspase-2 gene expression in the mutant mice by reverse transcription of mRNA followed by polymerase chain reaction (RT–PCR). No caspase-2 message could be detected in spleen mRNA from homozygous null mice using a primer complementary to the deleted caspase-2 active site exon and a carboxy-terminal primer (bases 901–1079) (Fig. 1B). Less than 1/10 of the amino-terminal PCR product present in wild-type mice (bases 181–598) could be detected in mutant mice (Fig. 1B). On Western blot using a polyclonal antibody recognizing the amino-terminus of caspase-2, we could not detect the pro-caspase-2 protein (46 kD) in any tissue tested in homozygous mutants, including whole embryos from embryonic day 11.5 (E11.5) (Fig. 1C,D). These data indicate that we have generated a null allele of caspase-2. caspase-2 is expressed at high levels in embryonic tissues and the adult spleen and thymus. Low levels of caspase-2 protein were observed in adult heart, lung, and brain (Fig. 1C). The protein was barely detectable in kidney and undetectable in the adult liver. This pattern of expression therefore points to a role for caspase-2 during development and in immune cells during postnatal life.
Caspase-2 is required for female germ cell death
To assess the consequences of a lack of caspase-2 function, we first examined survival rates in the female germ line as a paradigm of development-related cell death. During mammalian fetal development, over one-half to two-thirds of the ovarian germ cells produced through clonal expansion undergo apoptosis. These waves of massive germ-cell death in mice occur in the later stages of fetal life and persist through day 3 postpartum (P3), at which time the remaining oocytes are enclosed by granulosa cells to form primordial follicles (Ratts et al. 1995; Tilly 1996; Tilly et al. 1997). Using a quantitative RT–PCR/Southern blotting-based technique (Rambhatla et al. 1995), we determined that caspase-2 mRNA in oocytes was maternally derived, possessed a functional poly(A) tail, and was present at relatively high levels (∼12,600 mRNA copies/oocyte). These findings, coupled with the fact that the rodent ovary expresses almost exclusively caspase-2L mRNA (Flaws et al. 1995) (Fig. 3B, below), led us to speculate that an important role exists for this caspase in germ-cell demise. Gross histological analysis of ovaries of wild-type and caspase-2-deficient female mice at P4, the day immediately following cessation of germ-cell death when the maximal number of primordial follicles are present, revealed a normal architecture (data not shown). However, the number of newly formed primordial follicles containing oocytes was significantly higher in caspase-2-null females when compared with wild-type siblings (Fig. 2A), suggesting that fetal germ-cell attrition was attenuated in the absence of caspase-2. To confirm that female germ cells do in fact require caspase-2 to undergo apoptosis, we next employed a clinically relevant paradigm of oocyte death, that being exposure of germ cells to anticancer drugs. Germ-cell death induced by doxorubicin can be inhibited by the general caspase inhibitor ZVAD-FMK (benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone) (Perez et al. 1997). Treatment of oocytes harvested from young adult wild-type female mice with therapeutic levels of doxorubicin (adriamycin, 14-hydroxydaunomycin; 200 nm), in vitro, resulted in budding and fragmentation in over two-thirds of the germ cells cultured for 24 hr (Fig. 2B,C). However, caspase-2-deficient oocytes exhibited almost complete resistance to apoptosis induced by the chemotherapeutic drug (Fig. 2C), collectively supporting a central role for caspase-2 in mediating both normal and pathophysiological apoptosis in this cell lineage.
Figure 3.


Cell death of facial motor neurons during development. (A) Decreased number of neurons in the facial motor nuclei of E19.5 and newborn caspase-2-deficient mice. Coronal serial 5-μm sections were stained with cresyl violet, and the numbers of neurons in every fourth section were recorded. Only neurons with a nucleus were counted. The data are presented as the mean and s.e.m. from 8 wild-type and 10 mutant nuclei from both strains of mutant mice. The data from E19.5 and newborn mice were identical and therefore pooled. Significant differences were assessed using a two-tailed Student’s t-test at a 95% confidence interval. (*) P < 0.0001 vs. wild-type). (B) PCR analysis of caspase-2 isoforms in ovary and brain. Primers complementary to regions in exons VIII and X of caspase-2 cDNA were used to amplify caspase-2L message (167 bp) or caspase-2S message containing exon IX (228 bp).
Figure 2.

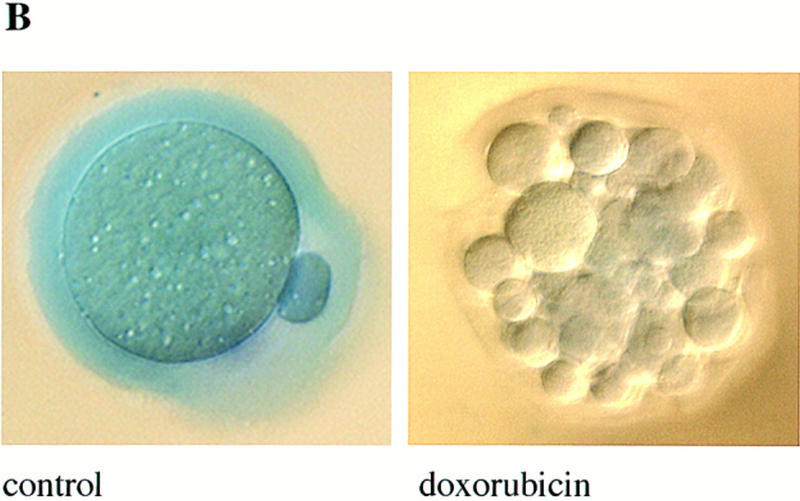

Germ cells from caspase-2-deficient mice are resistant to developmental and doxorubicin-induced apoptosis. (A) Number of follicles in the neonatal females. Ovaries from 4-day-old females were sectioned and stained with hematoxylin–pycric methyl blue and the number of follicles determined (mean ± s.e.m., n = 4 mice for each group; [(*) P < 0.05 vs. wild-type)]. (B) Morphological changes in wild-type oocytes treated with doxorubicin. Nomarski photomicroscopy of oocytes (magnification, 400×), untreated and treated with 200 nm doxorubicin. (C) Apoptosis in normal and caspase-2-deficient oocytes treated with doxorubicin. Oocytes were collected and cultured without (control) or with 200 nm doxorubicin for 24 hr. Oocytes with an apoptotic morphology were counted. The number of oocytes analyzed in each group were 76 (control +/+); 101 (control −/−); 108 (doxorubicin +/+); 152 (doxorubicin −/−). Both mutant mouse lines were used with the same outcome and results were pooled. The graph indicates the mean ± s.e.m. (*) P < 0.0001 vs. all other group).
In addition to the robust expression of the caspase-2 gene noted in oocytes, blastocysts contained abundant levels of caspase-2 mRNA (∼16,000 copies/blastocyst). To test for the functional requirement of caspase-2 in apoptosis in the preimplantation embryo, blastocysts were cultured in vitro in the absence or presence of staurosporine (100 μm) or doxorubicin (100 nm) for 16 hr, and the extent of apoptosis was assessed by Hoechst staining and fluorescence microscopy. Chromatin condensation and pyknotic nuclei were observed in 60%–80% of the cells of wild-type blastocysts treated with either staurosporine or doxorubicin (data not shown). Caspase-2-deficient blastocysts were equally sensitive to apoptosis induced by either of the two compounds, indicating that caspase-2 is dispensable for apoptosis at this stage of embryonic development. The possibility that other caspase family members may compensate for the loss of caspase-2 function in blastocysts is supported by the finding that at least another member of this protease family, caspase-3, is expressed in blastocysts (Jurisicova et al. 1998).
Accelerated cell death of facial motor neurons during development
caspase-2 is highly expressed in the developing brain between embryonic day 9 and 16 and thus it may play a role in the extensive cell death that takes place in the embryonic nervous system. We first investigated the structural integrity of the brain at embryonic day 16.5 (E16.5), 19.5 (E19.5), and at birth. Histological analysis of coronal serial sections revealed no gross abnormalities in caspase-2-mutant mice. We therefore looked for more subtle differences by counting the neurons in specific nuclei and ganglia. Neurons were counted on every fourth section throughout the brain of E16.5, E19.5, and newborn animals. The number of neurons in the vestibular, geniculate, nodose, and superior cervical ganglia were not significantly different in caspase-2-deficient and wild-type mice and were in accordance with expected numbers for normal mice (data not shown). We next examined the role of caspase-2 in the well-characterized physiological death of facial motor neurons. In the mouse facial nucleus, cell death occurs predominantly during the last 4 days of gestation and the first 3 days following birth (Ashwell 1983; Michaelidis 1996). Interestingly, the number of motor neurons in the facial nuclei of late embryonic and newborn caspase-2 mutant mice amounted to only 73% of that in the wild-type animals (P < 0.0001) (Fig. 3A), suggesting that the caspase-2 locus encodes an activity that can partially protect against facial motor neuron death. This reduction in the number of facial neurons in caspase-2-deficient mice appears to be caused by accelerated cell death and is not because of a decrease in neuron formation, as the number of facial motor neurons in wild-type and mutant mice was the same at E16.5, preceding the period of physiological cell death. However, the number of facial motor neurons in the wild-type (2376 ± 266) and mutant (2359 ± 206) mice at postnatal day 7 (P7) to P9 were the same. These data suggest that programmed cell death of facial motor neurons during development is accelerated in caspase-2 mutant mice but, eventually, the same number of neurons die. This unexpected result indicates that caspase-2 is involved not only as a positive effector of cell death (e.g., oocytes) but that in some cells it may also serve to delay apoptosis. We have shown previously that transcripts encoding both the long and short forms of caspase-2 are present in the embryonic brain (Wang et al. 1994). caspase-2L message is present at high levels in the brain at E15 but decreases dramatically to much lower levels in adult mice. caspase-2S message is present at low levels (∼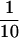 of the levels of the long form) in the embryonic brain, but its levels appear to be held constant so that the ratio of the long-to-short form of caspase-2 in adult brain is ∼1:1. Similar observations have been previously reported by Kumar et al. (1997). We disrupted both isoforms in our mutant mice and, therefore, may have offset the balance between these positive and negative regulators of cell death, resulting in decreased facial motor neuron survival. By comparison, the message levels for caspase-2S in the ovary are essentially below detectable limits (Flaws et al. 1995) (Fig. 3B), and thus disruption of caspase-2 in this tissue likely leads to enhanced survival through specific deletion of the long isoform. However, further investigation is needed to unequivocally establish the role of caspase-2S in neuronal survival.
of the levels of the long form) in the embryonic brain, but its levels appear to be held constant so that the ratio of the long-to-short form of caspase-2 in adult brain is ∼1:1. Similar observations have been previously reported by Kumar et al. (1997). We disrupted both isoforms in our mutant mice and, therefore, may have offset the balance between these positive and negative regulators of cell death, resulting in decreased facial motor neuron survival. By comparison, the message levels for caspase-2S in the ovary are essentially below detectable limits (Flaws et al. 1995) (Fig. 3B), and thus disruption of caspase-2 in this tissue likely leads to enhanced survival through specific deletion of the long isoform. However, further investigation is needed to unequivocally establish the role of caspase-2S in neuronal survival.
To further our investigation on the role of caspase-2 in motor neuron death, we evaluated motor neuron degeneration caused by early postnatal axotomy (Snider 1992; Michaelidis et al.1996). The facial nerve of P3 mice was unilaterally transected, and at P7, the number of facial motor neurons was determined in two mice of each genotype. As expected, the majority of the neurons on the transected side were lost and most of the remaining cells showed signs of atrophy. There was no difference in cell survival between caspase-2-deficient mice (mean for two animals of 35.5% survival) versus wild-type (36.6% survival) or heterozygous (32.5% survival). Therefore, caspase-2 protects motor neurons against naturally occurring cell death during embryonic development but this action is lost in postnatal life.
Caspase-2 is not required for the death of sympathetic neurons induced by trophic factor withdrawal
Sympathetic neurons from newborn animals die by apoptosis when cultured in the absence of nerve growth factor (NGF) (Edward et al. 1991; Snider et al. 1992; Deckwerth et al. 1993; Michaelidis et al. 1996). Previous reports have suggested a role for caspase-2 in the death of sympathetic neurons induced by NGF withdrawal. Pro-caspase-2 is apparently activated after NGF removal (Deshmukh et al. 1996) and antisense caspase-2 oligonucleotides protect sympathetic neurons subjected to NGF deprivation (Troy et al. 1997). In light of these results, we examined neuronal cultures from caspase-2-deficient mice. Sympathetic neurons from the superior cervical ganglia (SCG) were maintained in culture in the presence of NGF for 4 days. At that time, no difference was observed in the morphology and number of neurons recovered from caspase-2-null and wild-type mice. SCG neuronal cultures were deprived of NGF for 24 and 30 hr and viability was assessed by counting neurons with bright-phase cell bodies. Surprisingly, caspase-2-deficient sympathetic neurons were not protected from NGF withdrawal but died slightly faster than wild-type neurons (Fig. 4).
Figure 4.

Death of sympathetic neurons deprived of NGF. Neurons from the superior cervical ganglia were cultured in the absence of NGF and surviving neurons were counted after the indicated period of time. After 48 hr NGF deprivation 0%–5% of the cells survived in all cultures. Neurons from individual newborn mice were cultured independently in two wells. The first category (+/−) represents data from one wild-type and three heterozygous mice (n = 7 wells); the second category (−/−) represents data from two caspase-2 mutant littermates (n = 4 wells). The error bars represent the s.e.m.. P = 0.15 at 24 hr considered not significant.P = 0.03 (*) at 30 hr is considered significant.
Ischemic brain injury in caspase-2-deficient mice
Recent reports have indicated that inhibition of caspases can prevent brain damage induced by ischemia in a mouse model of stroke (Friedlander et al. 1997b; Hara et al. 1997). We assessed the role of caspase-2 in mediating brain injury after permanent focal ischemia caused by middle artery occlusion. Neurological symptoms and infarct volume were determined 24 hr after occlusion in wild-type and caspase-2-deficient littermates (Fig. 5A). Inactivation of caspase-2 had no effect on neurological deficits or tissue damage after ischemia. Furthermore, immunoblot analysis revealed that the amount of pro-caspase-2 in the forebrain of wild-type mice remained unchanged after either permanent or transient artery occlusion (Fig. 5B), indicating that caspase-2 is not up-regulated or processed during ischemia. We also monitored neurological damage after transient ischemia with MCA filament occlusion for 2 hr and reperfusion for 18 hr and obtained similar results. Therefore caspase-2 is not an essential effector of cell death caused by forebrain ischemia.
Figure 5.


Role of caspase-2 in ischemic injury. (A) Brain damage as measured by infarct TTC staining of infarct area 24 hr after permanent focal ischemia in wild-type and caspase-2-deficient mice. Infarct area was determined in each of five coronal sections (2 mm) from anterior (2 mm from anterior pole) to posterior (10 mm from anterior pole) as percent of TTC (○) +/+ (n = 6); (•) −/− (n = 8). (B) Immunoblot of caspase-2 from brain hemisphere on the ischemic side (I) or contralateral side (C) after either transient (3 hr ischemia; 6 hr reperfusion) or permanent (24 hr ischemia) occlusion. Ten micrograms of protein was loaded on the gel per lane. Samples from thymus tissues are shown at left as a comparison for the amount of caspase-2 present in brain and thymus.
Motor neuron degeneration in an amyotrophic lateral sclerosis mouse model carrying a caspase-2 mutation
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by the loss of motor neurons in the brain, brain stem, and spinal cord, ultimately resulting in paralysis and death. Although most cases of ALS are sporadic, the disease also exists in a familial form. A subset of inherited cases is caused by mutations in the Cu/Zn superoxide dismutase (SOD-1) gene. Transgenic mice expressing mutant SOD-1 genes develop progressive motor weaknesses similar to human ALS (Gurney 1994; Gurney et al. 1994; Tu et al. 1996). The exact mechanisms of the disease are uncertain; however, overexpression of Bcl-2 delays the onset of neuronal loss (Kostic et al. 1997), whereas neuron-specific expression of a dominant-negative caspase-1 mutant protein slows down the progression of the disease (Friedlander et al. 1997a). We studied the role of caspase-2 in the death of motor neurons in ALS by crossing a SOD(G93/A) transgenic male (Gurney et al. 1994) with a caspase-2-deficient female. Two male offspring carrying the SOD-1 (G93/A) transgene and heterozygous for caspase-2 mutation were crossed with four wild-type and four caspase-2-deficient females. We selected offspring carrying the SOD(G93/A) transgene by PCR and followed the course of motor control loss using the following criteria: tremors, posturing one or more legs when held by the tail, weakness of grip in front paws, slowness in walking, paralysis, and mortality. The mice were sacrificed when unable to right themselves in 30 sec when on their sides. The onset of the disease was scored at the first appearance of symptoms (usually tremors or leg posturing) in days. caspase-2 mutant mice carrying the SOD(G93/A) transgene developed ALS-like symptoms with similar time course and characteristics as that of wild-type/SOD mice (Table 1). Therefore, caspase-2 is dispensable for neuronal death caused by overexpression of SOD(G93/A) gene.
Table 1.
Effect of caspase-2 inactivation on motor control degeneration in SOD (G93/A) mice
|
caspase-2 genotype
|
+/+
|
+/−
|
−/−
|
|
|---|---|---|---|---|
| Onset | 112 ± 4.4 | 102 ± 2.2 | 99.1 ± 1.0 | |
| (n = 7) | (n = 26) | (n = 15) | (P = 0.04) | |
| Length | 27.7 ± 2.9 | 29.1 ± 2.6 | 30.3 ± 4.2 | |
| (n = 7) | (n = 26) | (n = 15) | (P = 0.70) | |
| Mortality | 140 ± 5.8 | 131 ± 2.4 | 129 ± 2.5 | |
| (n = 7) | (n = 26) | (n = 15) | (P = 0.06) |
Disease onset, length, and mortality listed as mean ± s.e.m. in days for SOD(G93/A) transgenic mice carrying deletion at the caspase-2 locus. All mice are hemizygous for SOD(G93/A) transgene. The caspase-2 genotype is indicated: (+/+) Wild type; (+/−) heterozygous; (−/−) homozygous for caspase-2 deletion. P values are indicated at right for comparison between caspase-2+/+ and caspase-2−/−groups.
B lymphoblasts lacking caspase-2 are deficient for granzyme B but not Fas-induced apoptosis
In addition to the ovary and embryonic brain, caspase-2 is highly expressed in the spleen. We therefore examined the integrity of apoptotic pathways in caspase-2−/− B lymphoblasts. Cytotoxic T lymphocytes (CTL) initiate apoptosis in target cells through the Fas (Kagi et al. 1994) and granzyme pathways (Shi et al. 1992; Heusel et al. 1994). The release from CTL granules of the serine protease granzyme B (GB), along with the protein perforin, induces apoptosis by directly or indirectly activating members of the caspase family (Shi et al. 1996). Furthermore, it has been reported that GB is able to cleave caspase-2 in vitro (Harvey et al. 1996). We found that treatment of HeLa cells with GB and perforin, but not perforin alone, leads to cleavage of the pro-caspase-2 (46 kD) into fragments of apparent molecular masses of 33 and 18 kD (Fig. 6A). This suggests that caspase-2 is activated when cells are killed by GB (Li et al. 1997). We therefore examined the sensitivity of caspase-2-deficient B lymphoblasts to GB and perforin. B lymphoblasts from caspase-2-deficient mice were more resistant to GB than wild-type cells (Fig. 6B). We also investigated the sensitivity of thymocytes as well as B and T lymphoblast apoptosis induced by anti-Fas antibody (Jo-2), doxorubicin, etoposide, γ-irradiation, or staurosporine. These treatments have all been shown to cause early processing of caspase-2 (Harvey et al. 1997; Li et al. 1997). Caspase-2-deficient and wild-type cells were equally sensitive to all of these treatments (data not shown), suggesting that in the absence of caspase-2, these agents can trigger apoptosis through alternate pathways.
Figure 6.


(A) Caspase-2 is processed in cells during apoptosis mediated by GB. Immunoblot of caspase-2 from HeLa cells untreated (lane 1), treated with 0.2 μg/ml perforin (lane 2), or 0.2 μg/ml perforin and 1 μg/ml GB (lane 3) for 2 hr. (B) caspase-2-deficient B lymphoblasts are partially resistant to apoptosis induced by GB. Splenocytes were treated with 10 μg/ml lipopolysaccharide (LPS) for 3 days. The resulting activated B lymphocytes (lymphoblasts) were cultured in the presence of perforin (0.2 μg/ml) and the indicated concentrations of GB/perforin for 3 hr. Cell death was measured by counting the number of cells with condensed chromatin or fragmented nuclei after Hoechst dye staining. Each experimental point represents a percentage calculated from at least 200 counted cells. The experiment was repeated three times with similar results. Mice from strain 72 were used.
Discussion
In view of its high expression between embryonic days 8 and 16 in various tissues when extensive cell death takes place, caspase-2 was a strong candidate as an effector of programmed cell death during development. Mice carrying a null mutation for caspase-2, however, develop normally and are devoid of severe phenotypic abnormalities. Thorough analysis of the rate of cell death in tissues expressing caspase-2 revealed that this protease is an essential inducer of apoptosis in female germ cells and involved in mediating apoptosis induced by granzyme B. Surprisingly, in some neuronal populations, caspase-2 functions to delay cell death.
Oocytes lacking caspase-2 are resistant to apoptosis
Caspase-2 appears to be an important mediator of the death of germ cells that occurs in the fetal ovary to establish the germ cell pool of the female. Inactivation of caspase-2 causes a significant increase in the number of primordial follicles in the postnatal ovary. Oocytes are very sensitive to the widely used chemotherapeutic drug doxorubicin, which causes germ cell depletion in women undergoing cancer treatment. Oocytes collected from caspase-2-deficient females were almost completely resistant to the effects of doxorubicin. Caspase-2 is therefore an essential effector of programmed cell death in female germ cells.
Accelerated cell death in caspase-2-deficient neurons
To study the kinetics of cell death in neurons during development, we chose two well-characterized models of physiological death in neurons. We monitored the death of facial motor neurons during development in vivo and the time course of apoptosis in culture of sympathetic neurons deprived of trophic factor. To our surprise, we observed a modest increase in cell-death rate in both models. Caspase-2L is the predominant isoform expressed in most tissues. However, in the brain, the long and short isoforms are present in roughly equal amounts. Overexpression of caspase-2S can prevent cell death in fibroblast cell lines deprived of trophic factor but is unable to directly inhibit caspase-2L (L. Bergeron and J. Yuan, unpubl.). Interestingly, the short isoform appears to be expressed in terminally differentiated tissues such as brain and muscles where it may play a role in survival by acting through mediators of cell death other than caspase-2L. We disrupted both isoforms in our mouse lines. Deletion of caspase-2S in neurons may have tipped the balance towards death. Alternatively, caspase-2 may be involved in the processing of factors that are important for neuronal survival. This result was unexpected as Troy et al. (1997) demonstrated previously that inhibiting caspase-2 with antisense RNA blocked cell death in sympathetic neurons and caspase-2 is apparently activated when sympathetic neurons undergo apoptosis in the absence of NGF as pro-caspase-2 disappeared, whereas the levels of pro-caspase-3 remained constant (Deshmukh et al. 1996). Thus, sympathetic neurons are likely to express other caspases that may act with caspase-2 to regulate their survival. It is possible that antisense caspase-2 may have cross-inhibited other related caspases. These results remind us that extreme caution should be exercised when one tries to draw conclusions from antisense experiments. Alternatively, other caspases may compensate for the loss of caspase-2L by up-regulating their expression when caspase-2 is missing throughout development. These possibities remain to be examined by future experimentation.
We did not observe any difference in the rate of facial motor neuron death after denervation. Furthermore, the extent of neuronal injury induced by ischemia was unaffected in caspase-2-deficient mice. Ischemic injury may induce additional apoptosis inducers in addition to caspase-2. Caspase-2-deficient mice carrying the SOD-1 mutations, which cause progressive motor neuron degeneration similar to ALS, lose motor control at the same rate as that of wild-type/SOD mice. No significant differences were observed in onset or progression of the disease. These results suggest that the function of caspase-2 is not essential for neuronal cell death under pathological conditions.
B lymphoblasts from caspase-2-deficient mice are more resistant to GB but not Fas- or drug-induced death
CTL have evolved to kill their target by directly activating effectors of the cellular death machinery. Thus, GB is able to directly process and activate caspases to induce apoptosis. caspase-2−/− B cells are more resistant to apoptosis induced by GB and perforin than are wild type cells, suggesting that caspase-2 is a downstream target of GB in B cells. Activation of multiple caspases has been observed in cells treated with GB and perforin which include caspase-2, -6, -9 (Srinivasula et al. 1996), -7 (Chinnaiyan and Dixit 1996), and ICE (Shi et al. 1996). Although B cells express at least another caspase, caspase-3 (Krajewski et al. 1997), the functions of caspase-2 and caspase-3 do not seem to be totally redundant in B cells. Because GB can activate both caspase-2 (Harvey 1996) and -3 (Darmon et al. 1995) directly, our result suggests that caspase-2 and -3 may be responsible for cleaving different sets of substrates after being activated by granzyme B. Consistent with this hypothesis, the specificity of caspase-3 is quite different from that of caspase-2, which prefers a hydrophobic residue in P5 position. Also, very few substrates of caspase-2 have been identified, whereas caspase-3 can cleave many proteins (Porter et al. 1997; Talanian et al. 1997; H. Li and J. Yuan, unpubl.).
We observed no difference in the rate of apoptosis induced by triggering of the Fas receptor in thymocytes or B and T lymphoblasts. Multiple caspases have been shown to be activated during the course of Fas-induced apoptosis and deleting one of them does not affect the outcome (Li et al. 1997). Similarly, we observed no difference in apoptosis of caspase-2−/− embryonic fibroblasts induced by TNF-α (20 μg/ml) and CHX (0.2 μg/ml) with most of the cells dying within 48 hr despite the high levels of caspase-2 expressed in wild-type cells (data not shown). Caspase-2 has been shown to physically interact with RAIDD, an adaptor protein containing regions homologous to both the death domain and prodomain of caspase-2 (Duan and Dixit 1997). The death domain of RAIDD interacts with RIP, a death domain containing serine/threonine kinase that is a part of TNF-α death pathway (Grimm et al. 1996). The caspase-2 prodomain homologous region in RAIDD interacts specifically with prodomain of caspase-2 (Duan and Dixit 1997). This result suggests that caspase-2 may be a part of the TNF death complex. Our result, however, indicates that the role of caspase-2 and RAIDD may be redundant with other caspases such as caspase-8 since caspase-2−/− embryonic fibroblasts die normally when induced by TNF-α and CHX.
Conclusion
Targeted disruption of the caspase-2 locus confirms that the protease encoded by this gene serves as both a negative and positive regulator of apoptosis. Caspase-2 is uniquely important for germ-cell apoptosis in the female ovary. Whereas CED-3 appears to be the only caspase required for apoptosis in C. elegans, apoptosis in vertebrates is orchestrated by a family of caspases with complex interactions. The ultimate action of caspase-2 appears to be dependent upon tissue type, cell lineage, developmental stage, differential splicing of the message, and the presence or absence of other caspases.
Materials and methods
Construction of the caspase-2 targeting vector
We isolated the caspase-2 genomic locus by screening a lambda phage FixII 129/sv mouse genomic library (Stratagene, La Jolla, CA) with a 440-bp cDNA probe similar to the Nedd2 sequence described by Kumar et al. (1992). A 20-kb clone was digested with NotI, subcloned into pBSKII (Stratagene), and was designated clone 1N. Clone 1N was further digested with NotI and SalI and subcloned into pBSKII, resulting in subclones 1NS (12.5 kb) and 1S (7 kb). A ClaI–SalI genomic DNA fragment from clone 1NS spanning 10.7 kb was subcloned into pBSKII vector (pJ553) and characterized by restriction site mapping and sequencing of the intron/exon boundaries surrounding the active site of the enzyme. The 3′ SalI site of pJ553 is situated in the last exon of the caspase-2 gene that encodes the carboyxyl terminus of the protein. A replacement-type vector was constructed by inserting the neomycin resistance gene under the control of the phosphoglycerokinase (PGK) promotor in the reverse orientation in place of a 1.65-kb BamHI fragment (Fig. 1A). The resultant targeting vector (pJ556) contained 6.5 kb of genomic DNA from the caspase-2 gene upstream of the PGK-neo insertion and 2.8 kb downstream. We obtained a probe for genomic Southern blot from clone 1S. This genomic fragment includes sequences downstream of our targeting vector from the 3′ end SalI site. We used a 600-bp EcoRI fragment downstream of the targeting vector as a probe for Southern blot analysis (Fig. 1A).
Screening of ES cells and generation of caspase-2-deficient mice
J1-ES cells were transfected with 15 μg of SacI linearized pJ556 plasmid by electroporation (400 V, 25 μF; BioRad Gene Pulser, Hercules, CA). Three days after transfection, G418 (200 μg/ml) was added to the medium. Resistant colonies were picked 2 weeks later and expanded. DNA was extracted, digested with EcoRI, and analyzed by Southern blot. The wild-type allele produced a 9.4-kb band. The mutant allele, which has an additional EcoRI site located in the PGK-neo cassette, generated a 3.8-kb band. We verified that a single insertion site at the caspase-2 locus was present in positive clones by Southern blot analysis of BamHI or EcoRI-digested genomic DNA using a neo probe. From screening 656 clones, we obtained eight independent recombinant ES cell lines. Chimeric mice were produced by microinjecting C57BL/6J blastocysts with J1 cells from two individual targeted clones (72 and 511) and transferring these blastocysts into pseudopregnant foster females. Chimeric male progeny were mated with C57BL/6J × DBA2 F1 females and heterozygous progenies were used as founders for two separate lines of mutant mice (72 and 511).
RT–PCR
Poly(A)+ RNA was isolated from mouse tissues using the Fast Tract RNA purification kit (Invitrogen, San Diego, CA) and was reverse transcribed by random priming using Moloney murine leukemia virus reverse transcriptase (New England Biolabs, Beverly, MA). The primers used to amplify caspase-2, named from the base-pair number at the 5′ end, were: forward-901 (5′-TGACAATGCTAACTGTCCAA), reverse-1079 (5′-GTCTCATCTTCATCAACTCC), forward-181 (5′-CTACAGAAGGACATTATC), and reverse-598 (5′-AGCCACGGGGCTGAGATT). The actin primers were forward-650 (5′-GACCTGACAGACTACCTCAT) and reverse-960 (5′-AGACAGCACTGTGTTGGCTA). Primers to detect caspase-2 long and short form were forward-1180(5′-ATGCTAACTGTCCAAGTCTA) and reverse-1390(5′-TCTCATCTTCATCAACTCC). For caspase-2 mRNA analysis from oocytes and embryos, we used a quantitative (RT–PCR)-based assay described by Rambhatla et al. (1995).
Western blot analysis
Mouse tissues were extracted and immediately frozen in liquid nitrogen to be crushed using a mortar and pestle. The resulting tissue powder was resuspended in lysis buffer (0.08 m Tris, 2% SDS, 0.1 m DTT, 10% glycerol, 10 μg/ml PMSF) and 10 μg of protein were loaded on a 12% polyacrylamide gel for electrophoresis. Caspase-2 was detected by immunobloting using a rabbit polyclonal anti-human caspase-2 as previously described (Li et al. 1997). For immunoblot of caspase-2 from GB-treated cells, HeLa cells were incubated with GB and perforin or each agent alone for 2 hr then lysed and processed as described above.
Histological analysis of ovaries
Ovaries were fixed in 4% formalin, 28% ethanol, 2% acetic acid, paraffin-embedded, and serial-sectioned (5 μm) through the entire tissue. Sections were stained with hematoxylin-pycric methyl blue and analyzed for total oocyte-containing follicle numbers, as described (Ratts et al. 1995; Perez et al. 1997).
Collection and culture of oocytes and embryos
Mature metaphase II oocytes were obtained by superovulation of adult female mice with 10 IU of equine chorionic (ECG) followed 48 hr later with 10 IU of human chorionic gonadotropin (HCG). Ovulated eggs were harvested from the oviductal ampullae 16 hr after hCG injection. Embryos were generated from females that were placed with fertile males immediately after hCG injection. Embryos were recovered from the females 17–30 hr after hCG injection and maintained in vitro (Erbach et al. 1994).
Histological analysis of brain
Brains were fixed in formalin, paraffin-embedded, and 5-μm coronal serial sections were obtained throughout the whole brain. Neurons with a visible nucleus and nucleoli were counted in every fourth section. The total number of neurons was obtained by multiplying the counted neurons by four. Four to ten ganglia from each category were examined from both caspase-2 mutant strains. The data is expressed as mean ±s.e.m..
Facial nerve lesion
Transection of the facial nerve was performed as previously described (Michaelidis et al. 1996). Briefly, P3 mice were anesthetized by hypothermia and the right facial nerve was cut by gently pulling on the nerve with forceps. The animals were killed at P7 for histological analysis and neuron counting. The data are expressed as percent of neuron present in the transected side versus the contralateral side. The results represent the mean obtained from two caspase-2-deficient and two wild-type mice of strain 511.
Sympathetic neuron cultures
Primary cultures of sympathetic neurons were established as described previously (Deckwerth 1993) from superior cervical ganglia (SCG) from offspring from caspase-2-heterozygous parents. Both ganglia from a pup were dissected, digested with 1 mg/ml collagenase and 1 mg/ml dispase, dissociated by trituration through a Pasteur pipette, and plated on four poly-ornithine/laminin coated wells in a 24-well plate. The ganglia were suspended in 600 μl of culture medium (MEM supplemented with 10% fetal calf serum) and plated as a drop in the center of the wells. The neurons were allowed to adhere for 45 min and 0.6 ml of culture medium containing 50 ng/ml 2.5S mouse NGF (50 ng/ml) and 3.3 μg/ml aphidicolin was added to each well. The neurons were maintained in culture for 4 days before NGF deprivation. Neurons were deprived of NGF by changing the culture medium with medium containing 0.025% goat anti-2.5S mouse NGF antiserum. Control neurons were maintained in 50 ng/ml NGF. Viability was assessed by counting the neuron with bright-phase appearance on the plate.
Ischemic mouse model
Experiments were performed as described by Hara et al. (1997), except that occlusion of the middle cerebral artery was continuous for 24 hr. Briefly, mice were anesthetized and the left MCA was occluded with an 8-0 monofilament. Neurological deficits were evaluated and scored as follows: 0, normal; 1, failure to extend right forepaw; 2, circling to the contralateral side; 3, loss of walking. After permanent MCA occlusion, the forebrains were divided into five coronal sections (2 mm) using mouse brain matrix, and the sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC).
SOD mice
Mice carrying the SOD(G93/A) transgene were originally described by Gurney et al. (1994) and were obtained from Jackson Laboratories (Bar Harbor, ME). We crossed one male heterozygous for the SOD(G93/A) insertion with one female caspase-2−/− (strain 511). Two male offspring were subsequently crossed with caspase-2 +/+ and caspase-2−/− females from both strain 511 and 72. Offspring carrying the SOD(G93/A) transgene were observed every second day for neurodegenerative disease progression. The mice were genotyped for the presence of the SOD transgene by PCR using specific primers: Forward SOD 5.3 (5′-GTATTGTTGGGAGGAGGTAGTGAT) and reverse SOD 5.5 (5′-TTCTACAGCTAGCAGGATAACAGAT). Genotyping for caspase-2 mutation was also done by PCR using the following primers: Forward caspase-2 exon VII (5′-CTCACTGGCTACCTAACTTCC) and forward PJK promoter (5′-GCTACCGGTGGATGTGGAATGTG), and reverse intron IX (5′-CCATGCATTGGGAGACACTTAC).
Acknowledgments
We thank Hong Zhu (Harvard Medical School), Lin Wang (Millenium, Cambridge, MA), and Jeanne M. Reeis (Whitehead Institute, Massachusetts Institute of Technology) for excellent technical assistance, Xin Liu (Whitehead Institute, Massachusetts Institute of Technology) for help with neuron population identification, and Vince Cryns and Shari Corin (Cell Biology, Harvard Medical School) for critical reading of the manuscript. We thank John Biggers (Harvard Medical School) for advice with embryo collection and culture, Mark S. Pasternack (Massachusetts General Hospital) for CTL data, and Eugene Johnson and Mohanish Deshmukh (Washington University School of Medicine) for help with SCG neuron cultures. J.Y. was supported in part by grants from the National Institute of Aging, the Amyotrophic Lateral Sclerosis Association, Bristol-Meyers Squibb, and the National Science Foundation (NSF). J.T. was supported by National Institutes of Health (NIH) grants (R01-HD34226 and R01-AG12279) and by the NIH Office of Research on Women’s Health. L.B. was supported by a fellowship from the National Cancer Center. G.P. was supported in part by a grant from the Massachusetts General Hospital Fund for Medical Discovery. K.E.L. was supported by funding from the NSF (MCB 9630370).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jyuan@hms.harvard.edu; FAX (617) 432-4177.
References
- Ashwell KW, Watson CRR. The development of facial motoneurons in the mouse: Neuronal death and the innervation of the facial muscle cells. J Embryol Exp Morphol. 1983;77:117–141. [PubMed] [Google Scholar]
- Chinnaiyan AM, Dixit VM. The cell-death machine. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- Darmon AJ, Nicholson DW, Bleackley RC. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature. 1995;377:446–448. doi: 10.1038/377446a0. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson Jr EM. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1212. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson Jr EM. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan H, Dixit VM. RAIDD is a new ’death’ adaptor molecule. Nature. 1997;385:86–89. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- Edward SN, Tolkovsky AM. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. J Cell Biol. 1991;124:537–546. doi: 10.1083/jcb.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbach GT, Lawitts JA, Papaioannou VE, Biggers JD. Differential growth of the mouse preimplantation embryo in chemically defined media. Biol Reprod. 1994;50:1027–1033. doi: 10.1095/biolreprod50.5.1027. [DOI] [PubMed] [Google Scholar]
- Flaws JA, Kugu K, Trbovich AM, DeSanti A, Tilly KI, Hirshfield AN, Tilly JL. Interleukin-1 beta-converting enzyme-related proteases (IRPs) and mammalian cell death: Dissociation of IRP-induced oligonucleosomal endonuclease activity from morphological apoptosis in granulosa cells of the ovarian follicle. Endocrinology. 1995;136:5042–5053. doi: 10.1210/endo.136.11.7588240. [DOI] [PubMed] [Google Scholar]
- Friedlander RM, Brown RH, Gagliardini V, Wang J, Yuan JY. Inhibition of Ice slows AlS in mice. Nature. 1997a;388:31. doi: 10.1038/40299. [DOI] [PubMed] [Google Scholar]
- Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J. Expression of a dominant negative mutant of ICE in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med. 1997b;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm S, Stanger BZ, Leder P. RIP and FADD: Two ‘death domain‘-containing proteins can induce apoptosis by convergent, but dissociable, pathways. Proc Natl Acad Sci. 1996;93:10923–10927. doi: 10.1073/pnas.93.20.10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. New Eng J Med. 1994;331:1721–1722. doi: 10.1056/NEJM199412223312516. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation [published erratum appears in Science 269(5221): 149. 1995] Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey NL, Trapani JA, Fernandes-Alnemri T, Liwack G, Alnemri ES, Kumar S. Processing of the Nedd2 precursor by ICE-like proteases and granzyme B. Genes Cells. 1996;1:673–685. doi: 10.1046/j.1365-2443.1996.00255.x. [DOI] [PubMed] [Google Scholar]
- Harvey NL, Butt AJ, Kumar S. Functional activation of Nedd2/ICH-1 (caspase-2) is an early process in apoptosis. J Biol Chem. 1997;272:13134–13139. doi: 10.1074/jbc.272.20.13134. [DOI] [PubMed] [Google Scholar]
- Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell. 1994;76:977–987. doi: 10.1016/0092-8674(94)90376-x. [DOI] [PubMed] [Google Scholar]
- Jurisicova A, Varmuza S, MacLusty NJ, Casper RF. Expression of cell death genes during human preimplantation embryo development. J Soc Gynecol Invest. 1998;5:149A. [Google Scholar]
- Kagi D, Vignaux F, Ledermann B, Burki K, Depraetere V, Nagata S, Hengartner H, Golstein P. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. 1994;265:528–530. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: Prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Gascoyne RD, Zapata JM, Krajewska M, Kitada S, Chhanabhai M, Horsman D, Berean K, Piro LD, Fugier-Vivier I, Liu YJ, Wang HG, Reed JC. Immunolocalization of the ICE/Ced-3-family protease, CPP32 (Caspase-3), in non-Hodgkin’s lymphomas, chronic lymphocytic leukemias, and reactive lymph nodes. Blood. 1997;89:3817–3825. [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS-S, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1b converting enzyme. Science. 1995;267:2000–2002. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C-Y, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kumar S. ICE-like proteases in apoptosis. Trends Biochem Sci. 1995a;20:198–202. doi: 10.1016/s0968-0004(00)89007-6. [DOI] [PubMed] [Google Scholar]
- ————— Inhibition of apoptosis by the expression of antisense Nedd2. FEBS Lett. 1995b;368:69–72. doi: 10.1016/0014-5793(95)00602-6. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185:1155–1161. doi: 10.1016/0006-291x(92)91747-e. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1Β-converting enzyme. Genes & Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kinoshita M, Noda M. Origin, expression and possible functions of the two alternatively spliced forms of the mouse Nedd2 mRNA. Cell Death Differ. 1997;4:378–387. [Google Scholar]
- Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, Towne E, Tracey D, Wardwell S, Wei F-Y, Wong W, Kamen R, Seshadri T. Mice deficient in IL-1Β-converting enzyme are defective in production of mature IL-1B and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- Li H, Bergeron L, Cryns V, Pasternack MS, Zhu H, Shi L, Greenberg A, Yuan J. Activation of caspase-2 in apoptosis. J Biol Chem. 1997;272:21010–21017. doi: 10.1074/jbc.272.34.21010. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Green DR. Protease activation during apoptosis: Death by a thousand cuts. Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtmann B, Meyer M, Thoenen H. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron. 1996;17:75–89. doi: 10.1016/s0896-6273(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry NA. Caspases—killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Perez GI, Knudson CM, Leykin L, Korsmeyer SJ, Tilly JL. Apoptosis-associated signaling pathways are required for chemotherapy-mediated female germ cell destruction. Nature Med. 1997;3:1228–1232. doi: 10.1038/nm1197-1228. [DOI] [PubMed] [Google Scholar]
- Porter AG, Ng P, Janicke RU. Death substrates come alive. BioEssays. 1997;19:501–507. doi: 10.1002/bies.950190609. [DOI] [PubMed] [Google Scholar]
- Rambhatla L, Patel B, Dhanasekaran N, Latham KE. Analysis of G-protein-α subunit mRNA abundance in preimplantation mouse embryos using a rapid, quantitative RT–PCR approach. Mol Reprod Dev. 1995;41:314–324. doi: 10.1002/mrd.1080410306. [DOI] [PubMed] [Google Scholar]
- Ratts V, Flaws JA, Kolp R, Sorenson CM, Tilly JL. Ablation of bcl-2 gene expression decreases the number of oocytes and primordial follicles established in the post-natal female mouse gonad. Endocrinology. 1995;136:3665–3668. doi: 10.1210/endo.136.8.7628407. [DOI] [PubMed] [Google Scholar]
- Shi L, Kam CM, Powers JC, Aebersold R, Greenberg AH. Purification of three cytotoxic lymphocyte granule serine proteases that induce apoptosis through distinct substrate and target cell interactions. J Exp Med. 1992;176:1521–1529. doi: 10.1084/jem.176.6.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Chen G, MacDonald G, Bergeron L, Li H, Miura M, Rotello RJ, Miller DK, Li P, Seshadri T, Yuan J, Greenberg A. Activation of an interleukin 1 converting enzyme-dependent apoptosis pathway by granzyme B. Proc Natl Acad Sci. 1996;93:11002–11007. doi: 10.1073/pnas.93.20.11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD, Elliot JL, Yan Q. Axotomy-induced neuronal death during development. J Neurobiol. 1992;23:1231–1246. doi: 10.1002/neu.480230913. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Fernandes-Alnemri T, Zangrilli J, Robertson N, Armstrong RC, Wang L, Trapani JA, Tomaselli KJ, Litwack G, Elnemri ES. The Ced-3/interleukin 1Β converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2α are substrates for the apoptotic mediator CPP32. J Biol Chem. 1996;271:27099–27106. doi: 10.1074/jbc.271.43.27099. [DOI] [PubMed] [Google Scholar]
- Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- Tilly JL. Apoptosis and ovarian function. Rev Reprod. 1996;1:162–172. doi: 10.1530/ror.0.0010162. [DOI] [PubMed] [Google Scholar]
- Tilly JL, Tilly KI, Perez GI. The genes of cell death and cellular susceptibility to apoptosis in the ovary: A hypothesis. Cell Death Differ. 1997;4:180–187. doi: 10.1038/sj.cdd.4400238. [DOI] [PubMed] [Google Scholar]
- Troy CM, Stefanis L, Greene LA, Shelanski ML. Nedd2 is required for apoptosis after trophic factor withdrawal, but not superoxide dismutase (SOD1) downregulation, in sympathetic neurons and PC12 cells. J Neurosci. 1997;17:1911–1918. doi: 10.1523/JNEUROSCI.17-06-01911.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- Yuan JY, Horvitz HR. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1Β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]