

Abstract
The multisubunit eukaryotic translation initiation factor (eIF) 4F recruits 40S ribosomal subunits to the 5′ end of mRNA. The eIF4F subunit eIF4E interacts directly with the mRNA 5′ cap structure. Assembly of the eIF4F complex is inhibited by a family of repressor polypeptides, the eIF4E-binding proteins (4E-BPs). Binding of the 4E-BPs to eIF4E is regulated by phosphorylation: Hypophosphorylated 4E-BP isoforms interact strongly with eIF4E, whereas hyperphosphorylated isoforms do not. 4E-BP1 is hypophosphorylated in quiescent cells, but is hyperphosphorylated on multiple sites following exposure to a variety of extracellular stimuli. The PI3-kinase/Akt pathway and the kinase FRAP/mTOR signal to 4E-BP1. FRAP/mTOR has been reported to phosphorylate 4E-BP1 directly in vitro. However, it is not known if FRAP/mTOR is responsible for the phosphorylation of all 4E-BP1 sites, nor which sites must be phosphorylated to release 4E-BP1 from eIF4E. To address these questions, a recombinant FRAP/mTOR protein and a FRAP/mTOR immunoprecipitate were utilized in in vitro kinase assays to phosphorylate 4E-BP1. Phosphopeptide mapping of the in vitro-labeled protein yielded two 4E-BP1 phosphopeptides that comigrated with phosphopeptides produced in vivo. Mass spectrometry analysis indicated that these peptides contain phosphorylated Thr-37 and Thr-46. Thr-37 and Thr-46 are efficiently phosphorylated in vitro by FRAP/mTOR when 4E-BP1 is bound to eIF4E. However, phosphorylation at these sites was not associated with a loss of eIF4E binding. Phosphorylated Thr-37 and Thr-46 are detected in all phosphorylated in vivo 4E-BP1 isoforms, including those that interact with eIF4E. Finally, mutational analysis demonstrated that phosphorylation of Thr-37/Thr-46 is required for subsequent phosphorylation of several carboxy-terminal serum-sensitive sites. Taken together, our results suggest that 4E-BP1 phosphorylation by FRAP/mTOR on Thr-37 and Thr-46 is a priming event for subsequent phosphorylation of the carboxy-terminal serum-sensitive sites.
Keywords: Phosphorylation regulation, translation initiation, rapamycin, signal transduction
Regulation of translation plays an important role in controlling cell growth and proliferation (Conlon and Raff 1999). An increase in translation rates is necessary for entry into and transit through the G1 phase of the cell cycle (Brooks 1977). Deregulation of translation is associated with aberrant growth, tumorigenicity, and apoptosis (Sonenberg 1993; Polunovsky et al. 1996). In eukaryotes, the predominant step of translational regulation is the recruitment of the small ribosomal subunit to mRNA. This occurs through the binding of eIF4E to the mRNA 5′ cap structure (m7GpppX, where X is any nucleotide). eIF4E is a component of the tripartite eIF4F (eukaryotic translation initiation factor 4F) complex, along with two other subunits, eIF4G and eIF4A. eIF4A is an RNA helicase posited to unwind mRNA secondary structure. eIF4G serves as a modular scaffolding protein that binds eIF4E, eIF4A, eIF3, and the poly(A)-binding protein, and that bridges the 40S ribosome and the mRNA (for review, see Merrick and Hershey 1996; Hentze 1997).
Cap-dependent translation is regulated in part by the eIF4E-binding proteins (4E-BPs; also known as PHAS, for phosphorylated heat- and acid-stable), a family of three small (10–12 kD) acidic proteins that compete with eIF4G for binding to a common binding site on eIF4E. Both the 4E-BPs and eIF4G interact with eIF4E through an eIF4E-binding motif (YXXXXLφ, where X is any amino acid and φ is a hydrophobic residue). Consistent with this observation, overexpression of 4E-BPs in mammalian cells, or addition of 4E-BPs to translation extracts, results in the inhibition of cap-dependent, but not cap-independent, translation (Pause et al. 1994).
Binding of the 4E-BPs to eIF4E is reversible and is dependent on the phosphorylation status of 4E-BP. Hypophosphorylated 4E-BP1 interacts strongly with eIF4E, whereas hyperphosphorylation of 4E-BP1 drastically decreases its binding to eIF4E. Exposure of cells to various extracellular stimuli (such as hormones, mitogens, growth factors, cytokines, and G-protein-coupled receptor agonists) induces an increase in 4E-BP1 phosphorylation that coincides (in most cases) with an increase in translation rates. Conversely, nutrient or growth factor deprivation results in 4E-BP1 dephosphorylation, an increase in eIF4E binding, and a concomitant decrease in cap-dependent translation (for review, see Kleijn et al. 1998; Sonenberg and Gingras 1998; Raught and Gingras 1999). Infection with certain viruses, such as encephalomyocarditis virus (EMCV), also causes a decrease in 4E-BP1 phosphorylation that coincides with the shut-off of host protein synthesis (Gingras et al. 1996).
The pathway leading to 4E-BP1 phosphorylation involves the phosphoinositide-3 (PI3) kinase and its downstream effector, the serine–threonine kinase Akt (for review, see Sonenberg and Gingras 1998; Raught and Gingras 1999). The role of PI3 kinase in 4E-BP1 phosphorylation was demonstrated in several ways; for example, treatment of cells with LY294002 and wortmannin, two potent PI3-kinase inhibitors, prevents 4E-BP1 hyperphosphorylation following hormone or growth factor stimulation. Conversely, expression of an activated form of the catalytic subunit of PI3 kinase increases 4E-BP1 phosphorylation (von Manteuffel et al. 1996; Gingras et al. 1998). Akt (also known as protein kinase B), which prevents cell death in many cell systems (for review, see Franke et al. 1997; Downward 1998) was also shown to affect 4E-BP1 phosphorylation. Overexpression of a constitutively active, membrane-targeted form of Akt induces 4E-BP1 phosphorylation on the same sites that are phosphorylated in vivo after serum stimulation (Gingras et al. 1998; Kohn et al. 1998). Conversely, overexpression of a kinase-dead Akt acts in a dominant-negative fashion to prevent insulin-induced 4E-BP1 phosphorylation (Gingras et al. 1998). Expression of the activated Akt mutant confers wortmannin but not rapamycin resistance to 4E-BP1 phosphorylation (Gingras et al. 1998), indicating that the rapamycin-sensitive signaling pathway component lies downstream of Akt.
The rapamycin-sensitive component in the 4E-BP1 phosphorylation pathway is FRAP/mTOR (FKBP12-rapamycin associated protein/mammalian target of rapamycin), also known as RAFT1 (rapamycin and 12-kD FK506 binding protein target 1), a member of the PIK (phosphoinositide kinase-related) family of kinases. The PIK family also includes kinases such as DNA-PK and ATM (for review, see Hoekstra 1997). FRAP/mTOR is the target of the immunosuppressive drug rapamycin that, in a complex with the immunophilin FKBP-12, binds to FRAP/mTOR to inhibit its function. FRAP/mTOR is the mammalian homolog of the yeast TOR proteins, which regulate G1 progression, and which modulate translation in response to nutrient availability (for review, see Thomas and Hall 1997). Recent results indicate that regulation of translation by FRAP/mTOR in response to nutrient availability also operates in mammals: Amino acid deprivation decreases the phosphorylation of 4E-BP1, likely through modulation of FRAP/mTOR activity (Fox et al. 1998; Hara et al. 1998; Wang et al. 1998; Xu et al. 1998). Overexpression of a rapamycin-resistant form of FRAP/mTOR confers rapamycin resistance to 4E-BP1 phosphorylation. Immunoprecipitates of FRAP/mTOR from rat brain, or from cells transfected with a tagged form of mTOR, contain a kinase activity capable of phosphorylating 4E-BP1. Immunoprecipitates of a catalytically inactive FRAP/mTOR mutant from mammalian cells fail to phosphorylate 4E-BP1, indicating that FRAP kinase activity is required for 4E-BP1 phosphorylation. Similarly, addition of a complex of FKBP12–rapamycin to the kinase reaction abrogates phosphorylation of 4E-BP1, indicating that the kinase activity present in the FRAP immunoprecipitates is sensitive to rapamycin (Brunn et al. 1997a,b; Burnett et al. 1998). There is disagreement, however, as to the number and identity of the sites phosphorylated by FRAP/mTOR. Brunn et al. (1997a) reported the phosphorylation of five 4E-BP1 Ser/Thr–Pro sites by a FRAP/mTOR immunoprecipitate in vitro, and concluded that FRAP/mTOR was responsible for the phosphorylation of all in vivo 4E-BP1 phosphorylation sites (Fadden et al. 1997). However, Burnett et al. (1998) subsequently reported that only two 4E-BP1 sites, threonines 37 and 46 (the numbering throughout this paper is according to the human 4E-BP1 protein), were phosphorylated by FRAP/mTOR. When threonines 37 and 46 were mutated to alanines, binding to eIF4E was reported to be constitutive (Burnett et al. 1998). These authors concluded that phosphorylation of Thr-37 and Thr-46 leads to the dissociation of 4E-BP1 from eIF4E. Another report demonstrated however that 4E-BP1 phosphorylated on Thr-37 is associated with eIF4E (Fadden et al. 1997).
4E-BP1 is phosphorylated in vivo on multiple residues (Fadden et al. 1997; Heesom et al. 1998). However, the order of phosphate addition and the functional significance of each phosphorylation event regarding the interaction of 4E-BP1 with eIF4E is unknown. The relative sensitivity of each site to different stimuli and pharmacological inhibitors also remains unknown. For example, one report indicates that all of the phosphorylation sites are sensitive to serum and rapamycin (Fadden et al. 1997), whereas other groups have observed a differential sensitivity for different subsets of sites (von Manteuffel et al. 1997; Gingras et al. 1998).
Here, we show that the phosphorylation of 4E-BP1 by both a FRAP/mTOR immunoprecipitate and a baculovirus-expressed FRAP/mTOR protein occurs on two sites only, Thr-37 and Thr-46. FRAP/mTOR is demonstrated to phosphorylate 4E-BP1 in an eIF4E/4E-BP1 complex. Thr-37 and Thr-46 are phosphorylated to a high degree in serum-starved cells, in which most of the 4E-BP1 is complexed to eIF4E. Although phosphorylation of Thr-37 and Thr-46 by FRAP is not sufficient to abolish the eIF4E/4E-BP1 interaction, mutational analysis indicates that it serves as a priming event for subsequent phosphorylation of other Ser/Thr–Pro sites in 4E-BP1.
Results
Both recombinant FRAP/mTOR and a FRAP/mTOR immune complex phosphorylate 4E-BP1
Phosphopeptide mapping has proven to be a powerful method in the study of the sensitivity of specific phosphorylation sites to various stimuli (see, e.g., Fleurent et al. 1997; von Manteuffel et al. 1997; Burnett et al. 1998; Gingras et al. 1998). This technique is also applied to the identification of putative kinases by comparing the pattern of the phosphopeptides generated in vitro with those phosphorylated in vivo. Here it was used to better define the role of FRAP/mTOR in 4E-BP1 phosphorylation. A rat brain FRAP/mTOR immunoprecipitate and a purified baculovirus-expressed Flag-tagged FRAP/mTOR fusion protein were used in an in vitro kinase assay with recombinant human 4E-BP1 as a substrate. 32P-Labeled 4E-BP1 was separated via SDS-PAGE, then transferred to nitrocellulose membranes, which were subjected to autoradiography. 4E-BP1 was readily phosphorylated by both preparations of FRAP/mTOR (data not shown). The membrane piece bearing the labeled 4E-BP1 protein was excised and incubated in a trypsin/chymotrypsin mixture, and two-dimensional phosphopeptide mapping was performed on the liberated peptides. The map prepared from 4E-BP1 phosphorylated by the rat brain FRAP/mTOR immunoprecipitate yielded two major phosphopeptides (indicated as 1 and 2; Fig. 1A). In parallel, serum-stimulated 293 cells were labeled in vivo with [32P]orthophosphate, and endogenous 4E-BP1 was subjected to tryptic/chymotryptic mapping (Fig. 1B). Mixing of the in vitro- and the in vivo-labeled products demonstrated that the two phosphopeptides generated by FRAP/mTOR labeling comigrate with the two most prominent in vivo phosphopeptides (Fig. 1C). The same major phosphopeptides were observed in 4E-BP1 phosphorylated by a recombinant baculovirus-expressed FRAP/mTOR (Fig. 1D), consistent with the notion that 4E-BP1 phosphorylation on these sites is catalyzed by FRAP/mTOR itself, and not by a contaminating kinase in the immunoprecipitate.
Figure 1.
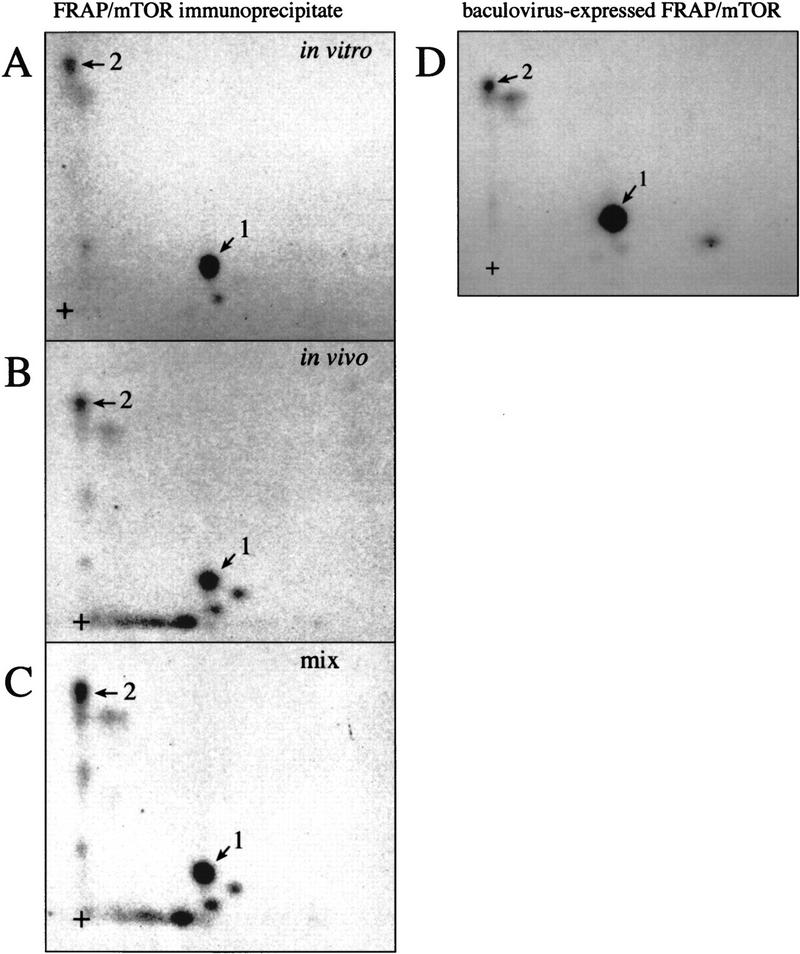
FRAP/mTOR phosphorylates 4E-BP1 in vitro on two sites that are also phosphorylated in vivo. (A) FRAP/mTOR immunoprecipitated from rat brain was utilized in an in vitro kinase assay with human 4E-BP1 (expressed as a GST fusion protein, which was then cleaved and HPLC purified) as a substrate. Phosphopeptide mapping was performed on phosphorylated 4E-BP1. The two most intense phosphopeptides are numbered 1 and 2. (+) The origin of migration. (B) Tryptic/chymotryptic map of 4E-BP1 phosphorylated in 293 cells. The positions of phosphopeptides that comigrate with the FRAP/mTOR phosphorylated peptides 1 and 2 are indicated. (C) An equal number of cpm from the in vitro- and the in vivo-phosphorylated 4E-BP1 were combined and phosphopeptide mapping was performed. (D) Tryptic/chymotryptic map of 4E-BP1 phosphorylated in vitro with purified FRAP/mTOR produced using the baculovirus system.
It should be noted that it remains possible that an endogenous kinase from the insect cells coprecipitates with FRAP/mTOR during the purification procedure; this possibility would be excluded by the use of bacterially-expressed FRAP/mTOR. However, attempts to express FRAP/mTOR in bacteria have been unsuccessful in our laboratory and others. The peptide of lower intensity observed in Figure 1A next to peptide 1 was obtained in several experiments and appears to comigrate with an in vivo peptide. However, unlike peptides 1 and 2, its intensity decreases drastically if the FRAP/mTOR immunoprecipitate is washed at higher stringency, or if recombinant 4E-BP1 is incubated for shorter times with the FRAP/mTOR immunoprecipitate. Also, phosphorylation of this peptide was extremely weak in the 4E-BP1 sample phosphorylated with baculovirus-expressed FRAP/mTOR. Thus, phosphorylation at this residue may arise as a consequence of a contaminating kinase activity in our preparations of FRAP/mTOR. Other peptides are visible in some experiments, but also appear to be due to a contaminating kinase activity. Because the immunoprecipitated material is more readily available, FRAP/mTOR immunoprecipitates were used in all subsequent experiments.
Identification of in vivo 4E-BP1 phosphorylation sites
To identify in vivo 4E-BP1 phosphorylation sites, 293 cells were grown to confluence, then starved of serum. A portion of the cells was then incubated with [32P]orthophosphate, stimulated with serum, and lysed. The remainder of the cells was treated in the same manner, without the labeling step. 4E-BP1 was then immunoprecipitated and the labeled and unlabeled immunoprecipitates were mixed, separated by SDS-PAGE, and transferred onto a nitrocellulose membrane. After autoradiography, the protein was subjected to two-dimensional phosphopeptide mapping (a representative map is shown in Fig. 2A). Peptides 1 and 2, which comigrated with the in vitro FRAP/mTOR phosphorylated 4E-BP1 peptides, as well as peptides 3–5, which migrated in the same area as peptide 2, were scraped from the two-dimensional phosphopeptide map and eluted. Phosphopeptides were identified by capillary liquid chromatography–electrospray ionization tandem mass spectrometry (LC-MS/MS). The tandem mass spectrum for phosphopeptide 1 corresponds to 44-STTPGGTR-51, with the phosphorylation site being Thr-46 (Fig. 2B). Peptide 2 was identified as 21-VVLGDGVQLPPGDYSTTPGGTLF-43, with the phosphorylation site being Thr-37 (data not shown). Peptides 3–5, whose intensities varied from experiment to experiment, contain both phospho-Thr-37 and phospho-Thr-46 and are derived from partially digested products (data not shown). The sequence of each identified phosphopeptide and the position of the phosphorylated amino acids are shown in Figure 2C. In agreement with these findings, all of these peptides contained only phosphothreonine, as detected by phosphoaminoacid analysis (data not shown). Threonines 37 and 46 are located in the middle of 4E-BP1, immediately amino-terminal to the eIF4E-binding site (Fig. 2D).
Figure 2.
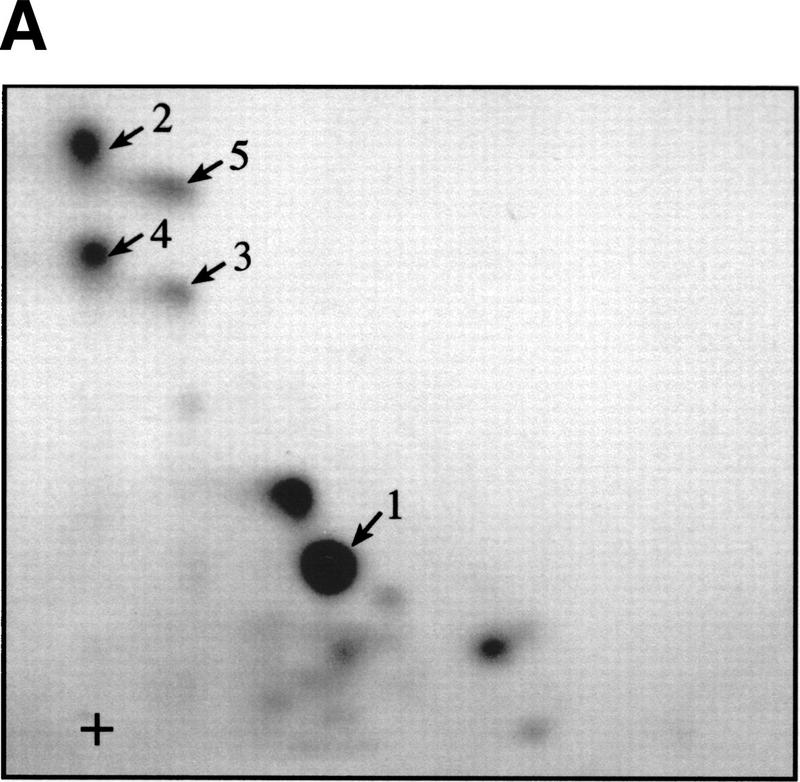



Thr-37 and Thr-46 are phosphorylated in vivo on phosphopeptides that comigrate with FRAP/mTOR phosphorylated peptides. 4E-BP1 immunoprecipitated from 2 × 108 cells (1/20 labeled with [32P]orthophosphate) was separated by SDS-15% PAGE, electroblotted onto a nitrocellulose membrane, and proteolytically cleaved with trypsin/chymotrypsin as described in Materials and Methods. (A) Tryptic/chymotryptic two-dimensional phosphopeptide map of 4E-BP1. Phosphopeptides (labeled 1–5) were isolated from the plates and identified by mass spectrometry. (B) Tandem mass spectrum of m/z 428.9 revealing the sequence and phosphorylation site of the phosphopeptide contained in spot 1 of the map shown in A. The position of the phosphorylated threonine residue (Thr-46) is shown. (C) Identification of the phosphorylated peptides in spots 1–5 from the phosphopeptide map shown in A. Phosphorylated amino acids are in boldface type; the amino acid amino terminal to the cleavage site for trypsin or chymotrypsin is indicated in parentheses. (D) The positions of Thr-37 and Thr-46 in 4E-BP1, relative to the eIF4E-binding site.
FRAP/mTOR phosphorylates Thr-37 and Thr-46
To confirm that the in vivo phosphorylation sites were the same as those phosphorylated in vitro by FRAP/mTOR, histidine-tagged mutants of 4E-BP1 containing either a Thr-37–Ala or a Thr-46–Ala mutation were phosphorylated by a FRAP/mTOR immunoprecipitate. Quantitatively, both mutant proteins were phosphorylated to approximately half the extent of wild-type 4E-BP1 (data not shown). Tryptic/chymotryptic mapping of the mutants was also performed. Whereas the map of the wild-type protein shows the presence of the two phosphopeptides containing Thr-37 and Thr-46, the map of the Thr-37–Ala mutant contains only phosphorylated Thr-46 (cf. Fig. 3, B and A). Similarly, the map from the Thr-46–Ala mutant confirms Thr-46 as a target for FRAP/mTOR (Fig. 3C). When both sites are mutated to alanines, or when the region containing both threonines (amino acids 34–52 in human 4E-BP1) is deleted, the phosphorylation of 4E-BP1 by FRAP/mTOR is almost abolished (these mutants are labeled to <5% of the levels observed for the wild-type protein; data not shown). Thus, Thr-37 and Thr-46 are the two primary amino acids phosphorylated by FRAP/mTOR.
Figure 3.
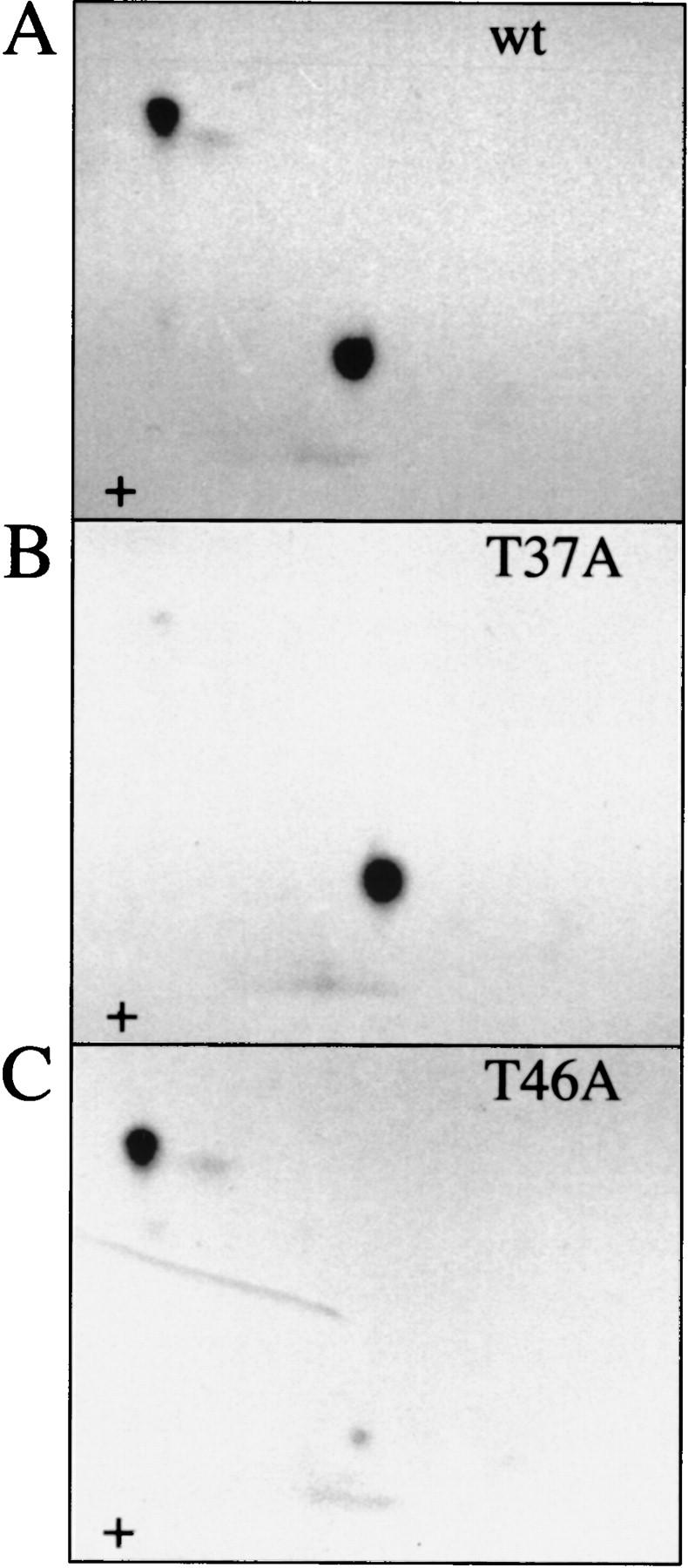
In vitro kinase assay of Thr-37–Ala and Thr-46–Ala mutant proteins confirm that they are the primary FRAP/mTOR phosphorylation sites. Histidine-tagged murine 4E-BP1 was phosphorylated in vitro in an immune-complex kinase assay with FRAP/mTOR, and tryptic/chymotryptic maps were performed. (A) Wild type; (B) Thr-37–Ala; (C) Thr-46–Ala.
FRAP/mTOR phosphorylates a 4E-BP1/eIF4E complex
Hypophosphorylated 4E-BPs interact strongly with eIF4E, and one or more kinases are posited to induce the disruption of the eIF4E/4E-BP complex by phosphorylation of the 4E-BPs. Thus, the in vivo substrate for a 4E-BP kinase should be a 4E-BP/eIF4E complex, and a physiological 4E-BP kinase(s) should be able to phosphorylate 4E-BP when bound to eIF4E. To determine whether the FRAP/mTOR immunoprecipitate can phosphorylate a 4E-BP1/eIF4E complex in vitro, an equimolar quantity (Fig. 4A, lane 2) or a twofold molar excess of eIF4E (lane 3) was preincubated with 4E-BP1, then a kinase assay was performed. As a control for the specificity of the kinase reaction, 4E-BP1 was also phosphorylated under the same conditions with the MAP kinase ERK2. ERK2 was reported previously to phosphorylate 4E-BP1 preferentially on Ser65 (Haystead et al. 1994), but is unable to phosphorylate 4E-BP1 complexed with eIF4E (Lin et al. 1995). The presence of eIF4E did not reduce FRAP/mTOR phosphorylation of 4E-BP1 (Fig. 4A, cf. lanes 2 and 3 to lane 1). In contrast, although ERK2 readily phosphorylated free 4E-BP1, it failed to phosphorylate 4E-BP1 in the presence of a twofold molar excess of eIF4E (cf. lanes 6 and 4). eIF4E prevented phosphorylation of the wild-type 4E-BP1 by ERK2, but not of a 4E-BP1 mutant lacking the eIF4E binding site (Fig. 4A, lanes 7 and 8), indicating that the inhibition by eIF4E is a direct consequence of its interaction with 4E-BP1. eIF4E was not a substrate for FRAP/mTOR, nor for ERK2 (Fig. 4A). 4E-BP1 phosphorylated by FRAP/mTOR in the presence (Fig. 4B) or absence (Fig. 4C) of eIF4E was analyzed by two-dimensional phosphopeptide mapping. In both cases, Thr-37 and Thr-46 were the primary phosphorylation sites. Phosphorylation of the 4E-BP1/eIF4E complex by a FRAP/mTOR immunoprecipitate is consistent with it (or an associated kinase) being a physiological 4E-BP1 kinase.
Figure 4.


FRAP/mTOR phosphorylates 4E-BP1 complexed with eIF4E. (A) 4E-BP1 was pre-incubated without eIF4E, with an equimolar quantity or with a twofold molar excess of eIF4E, and subjected to a kinase assay using FRAP/mTOR immunoprecipitated from rat brain (lanes 1–3) or recombinant ERK2 (lanes 4–8). Phosphorylated proteins were separated via SDS-15%-PAGE, transferred to nitrocellulose membranes, and subjected to autoradiography. The substrates used were as follows. (Lanes 1–6) A thrombin-cleaved and HPLC-purified GST–4E-BP1; (lane 7) an uncleaved GST–4E-BP1 wild-type protein; (lane 8) an uncleaved GST–4E-BP1 mutant lacking the eIF4E-binding site. (B) Tryptic/chymotryptic map of 4E-BP1 alone (A, lane 1). (C) Tryptic/chymotryptic map of 4E-BP1 complexed with a twofold molar excess eIF4E (A, lane 3).
FRAP/mTOR phosphorylation of 4E-BP1 does not disrupt the 4E-BP1/eIF4E complex
After establishing that FRAP/mTOR can phosphorylate 4E-BP1 complexed with eIF4E, it was important to determine whether phosphorylation of Thr-37/Thr-46 disrupts the 4E-BP1/eIF4E complex. A 4E-BP1/eIF4E complex was phosphorylated by FRAP/mTOR, then incubated with a cap analog (m7GDP) coupled to agarose beads, washed, and subjected to SDS-PAGE. As demonstrated in Figure 4, the addition of eIF4E did not affect the phosphorylation of 4E-BP1 by FRAP/mTOR (Fig. 5A, bottom panel, cf. lanes 1 and 2). Importantly, most of the 32P-labeled 4E-BP1 (∼65%) was retained on the m7GDP beads in the presence of eIF4E (Fig. 5A, cf. lane 2, top and lane 2, bottom panel). A portion of 4E-BP1 (∼35%) was found in the unbound fraction after the m7GDP-agarose pull-down (cf. middle and bottom panels, lane 2). This procedure does not quantitatively sequester eIF4E either, however, as ∼20% of the eIF4E is also present in the unbound fraction (data not shown). 4E-BP1 in the bound and unbound fractions was analyzed by two-dimensional phosphopeptide mapping. Similar levels of phospho-Thr-37 and phospho-Thr-46 were observed in each case (Fig. 5B), demonstrating that the phosphorylation of 4E-BP1 on Thr-37 and Thr-46 does not disrupt the 4E-BP1/eIF4E complex. It cannot be ruled out, however, that only monophosphorylated 4E-BP1 molecules interact with eIF4E and that the proteins phosphorylated on both residues do not bind. Nevertheless, more phosphorylated 4E-BP1 remains bound to eIF4E than dissociates after incubation with FRAP/mTOR. This indicates that the kinase activity present in the FRAP/mTOR immunoprecipitate is insufficient to disrupt the 4E-BP1:eIF4E complex. Consistent with this result, a deletion of the region encompassing Thr-37 and Thr-46 (amino acids 34–50) binds to eIF4E to the same extent as the wild-type protein, indicating that these amino acids are not involved in mediating eIF4E binding (data not shown). Furthermore, a bacterially expressed double mutant of 4E-BP1 Thr-37–Glu/Thr-46–Glu (designed to mimic phosphorylation at these residues) was also without effect on eIF4E binding (data not shown).
Figure 5.
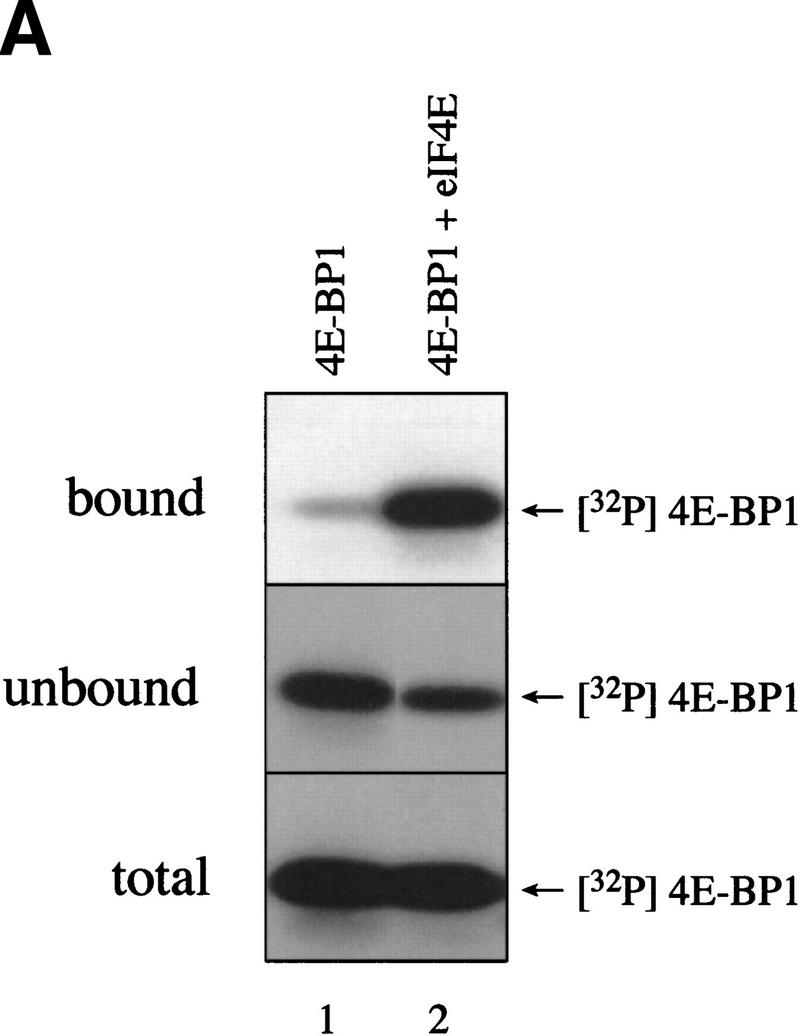
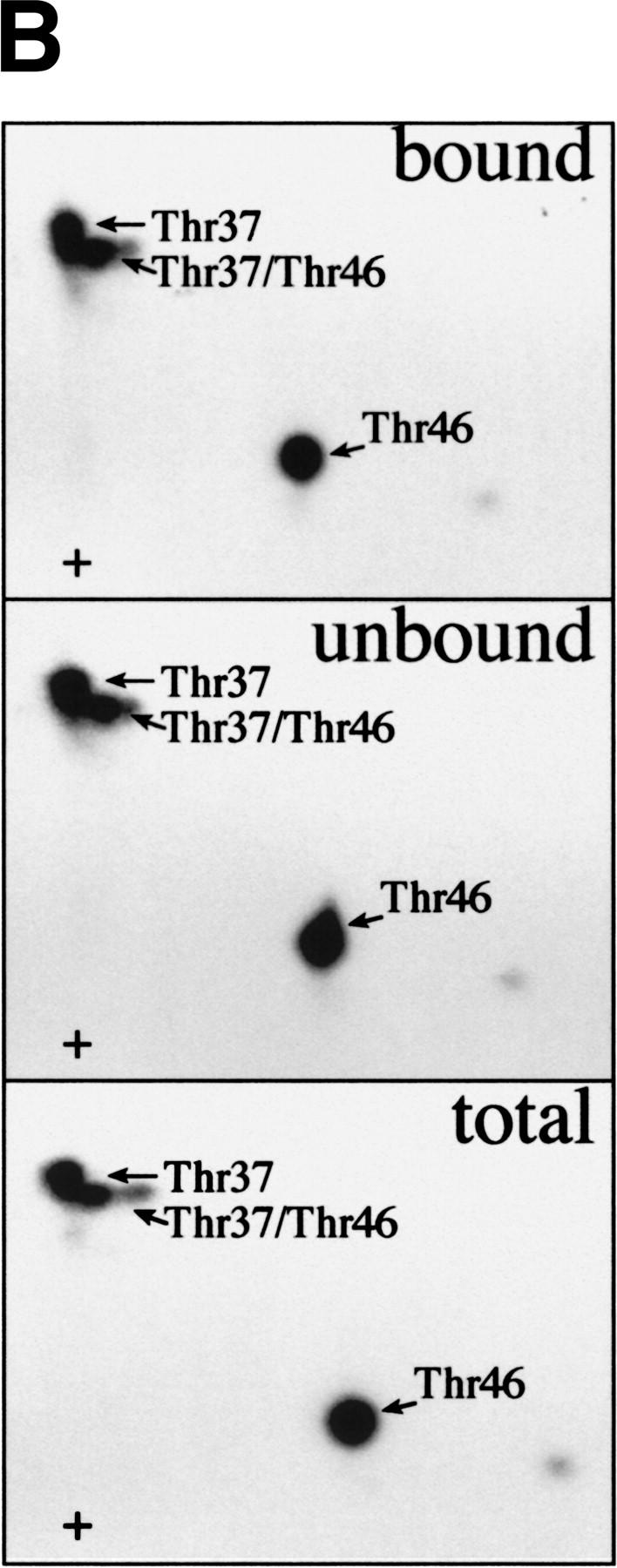
FRAP/mTOR phosphorylation of Thr-37 and Thr-46 does not disrupt the 4E-BP1/eIF4E complex. (A) 4E-BP1 alone, or a 4E-BP1/eIF4E complex, was incubated with FRAP/mTOR, as in Fig. 4. The kinase reaction was stopped and one-half of the material was immediately subjected to SDS-PAGE (bottom). The remaining material was incubated with m7GDP-agarose beads to purify eIF4E and associated proteins. The bound (top) and unbound (middle) fractions were analyzed by SDS-PAGE. (B) Phosphorylated 4E-BP1 (A, lane 2; bound, unbound, and total) was analyzed by two-dimensional phosphopeptide mapping.
Thr-37 and Thr-46 are phosphorylated to a high stoichiometry in serum-starved cells and are detected in all phosphorylated isoforms in vivo
Multiple sites on 4E-BP1 are phosphorylated following the addition of serum to serum-starved cells. However, 4E-BP1 is phosphorylated to a significant extent even in 293 cells deprived of serum. To map the phosphoaminoacids present in serum-starved cells, starved or stimulated 293 cells were labeled with [32P]orthophosphate in vivo, and phosphopeptide maps were generated from immunoprecipitated 4E-BP1. Phosphopeptides containing phospho-Thr-37 and -Thr-46 are present at relatively high levels in serum-starved cells, and their total quantity increased only marginally following serum-stimulation relative to the quantity of protein present, in contrast to the serum-induced phosphopeptides a–d (Fig. 6, cf. panels A and B; phosphopeptides a–d are very sensitive to minor variations in serum treatment. Depending on the confluency and passage number of the cells, the time span of serum-starvation and stimulation and even the batch of serum, the quantity of these phosphopeptides vary; see, e.g., Figs. 1B, 2A, and 6B).
Figure 6.
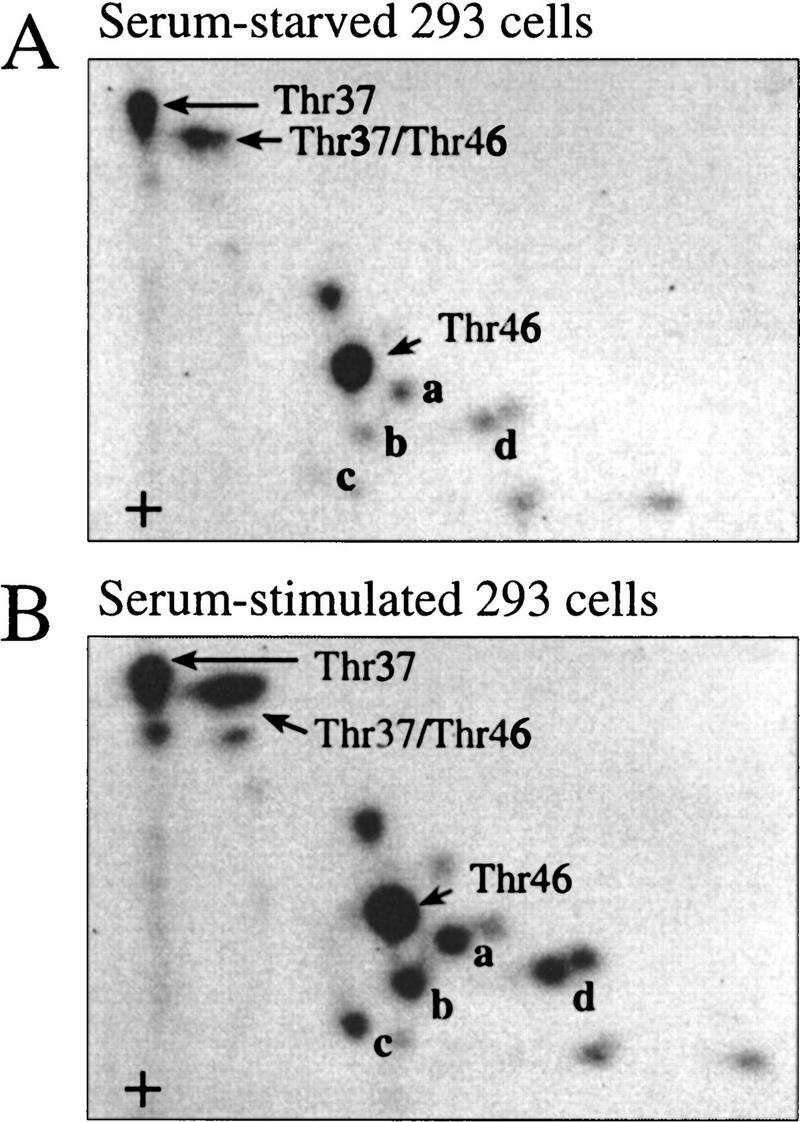
Phosphorylated Thr-37 and Thr-46 are present at a high stoichiometry in serum-starved 293 cells. 293 cells were grown to confluence and serum starved for 30 hr. 32P-Labeling was performed for 4 hr and phosphopeptide maps were generated from starved cells (A) or cells stimulated for 30 min with 10% dialyzed FCS (B). Phosphopeptides containing phospho-Thr-37 or -Thr-46 are identified. Serum-induced phosphopeptides are labeled a–d.
An increase in 4E-BP1 phosphorylation is accompanied by a decrease in its electrophoretic mobility. To determine the order of phosphate addition, each of the forms separated by SDS-PAGE was mapped. As previously observed (e.g., Beretta et al. 1996; Gingras et al. 1996; von Manteuffel et al. 1997; Gingras et al. 1998), multiple isoforms of human 4E-BP1 incorporate 32P in vivo (3–5 distinct bands, depending on the cell type and resolution of the gel; in Fig. 7A, inset, three bands were separated). Tryptic maps from the slowest (Fig. 7A) and the fastest (Fig. 7B) migrating isoform are shown. Phosphopeptides containing Thr-37 and Thr-46 are present in both 4E-BP1 isoforms (cf. panels B and A). This observation is not restricted to human cells, because phospho-Thr-37 and phospho-Thr-46 are also observed in the fastest migrating phosphorylated 4E-BP1 isoforms from Rat1a cells (data not shown). Thus, phosphorylation of Thr-37 and Thr-46 appears to be an early event in a sequential process of 4E-BP1 phosphorylation. Phosphorylated Thr-37 and Thr-46 are detected in isoforms that bind eIF4E as well as in isoforms that do not bind eIF4E (see, e.g., von Manteuffel et al. 1996). These data also show that phosphorylation of Thr-37 and Thr-46 does not cause a shift in electrophoretic mobility upon 4E-BP1 hyperphosphorylation.
Figure 7.
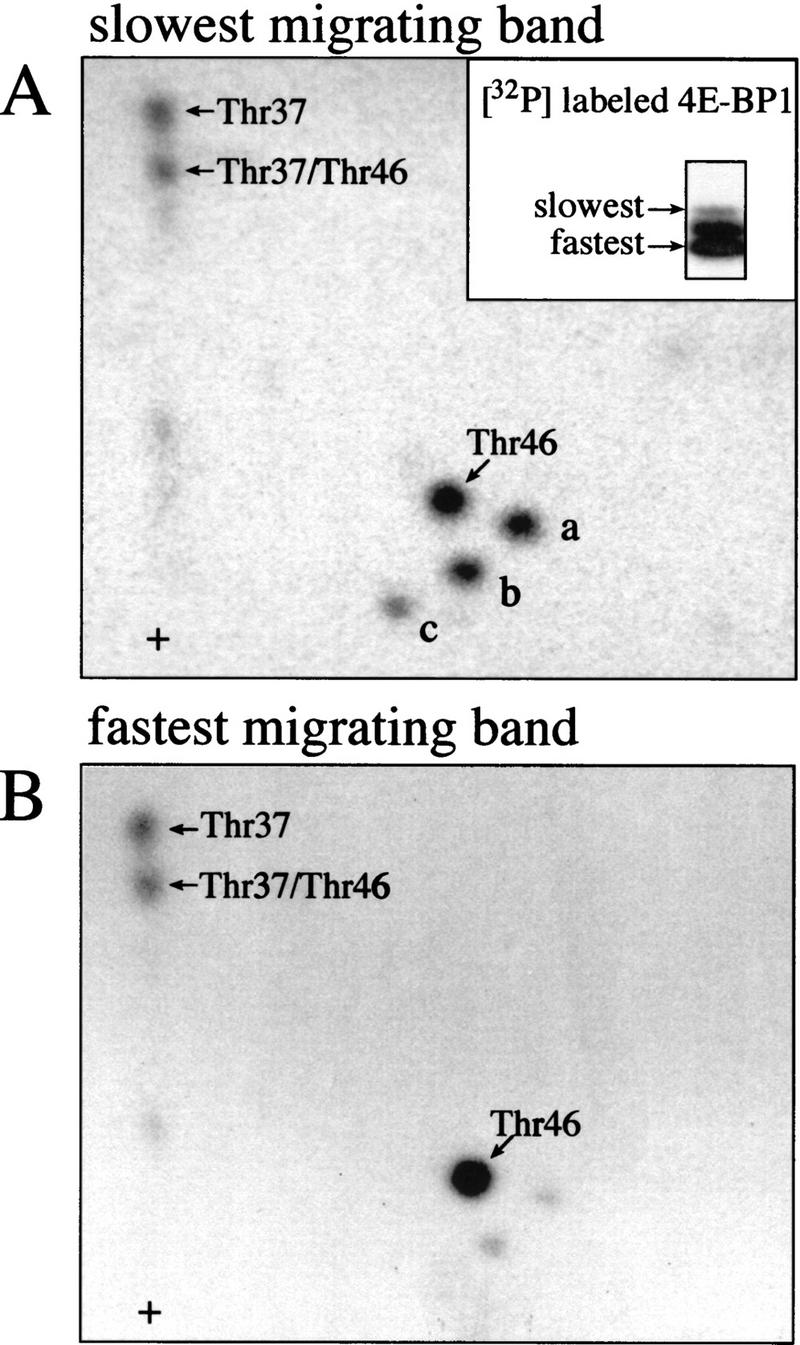
Phosphorylated Thr-37 and Thr-46 are present in the fastest migrating phosphorylated 4E-BP1 isoform. 293 cells were serum starved for 30 hr, incubated with [32P]orthophosphate, stimulated with 10% dialyzed FCS, and extracts were subjected to SDS-PAGE and autoradiography (A, inset). The slowest (A) and fastest-migrating (B) 4E-BP1 isoforms were isolated and phosphopeptide maps were performed.
To confirm that phospho-Thr-37 and -Thr-46 are present in all phosphorylated isoforms and that their phosphorylation status is only marginally affected by serum addition, phosphospecific antibodies to Thr-37 and Thr-46 were developed. The sequences surrounding Thr-37 and Thr-46 are almost identical (Fig. 2D), thus the phosphospecific antibody that was generated against a peptide containing phosphorylated Thr-37 (anti-phospho-Thr-37) does not discriminate between phosphorylated Thr-37 and phosphorylated Thr-46 (data not shown). To confirm the specificity of the phosphospecific antibody, hemagglutinin-tagged wild-type 4E-BP1 and a 4E-BP1 mutant in which Thr-37 and Thr-46 were mutated to alanines were transfected into 293T cells. The phosphospecific antibody detected the wild-type 4E-BP1 protein, but failed to detect the Thr-37–Ala/Thr-46–Ala mutant, although both proteins were expressed to a similar level (Fig. 8A, cf. lanes 1 and 2 with lanes 3 and 4). Also, the phosphospecific antibody recognizes the phosphorylated peptide containing Thr-37, but not the unphosphorylated peptide (data not shown), indicating that the phosphate group is an essential part of the antibody recognition motif.
Figure 8.


Phosphospecific antibodies confirm the relative lack of serum sensitivity of Thr-37 and Thr-46. (A) 293 cells were transfected with wild-type HA–4E-BP1 (wt), HA–4E-BP1 Thr-37–Ala/Thr-46–Ala (mut), or vector alone (vec). Cell extracts were prepared and analyzed by Western blotting with either the phosphospecific antibody to Thr-37 and Thr-46 or with an anti-HA antibody. (B) A time course of serum stimulation (after 30 hr of starvation) of 293 cells was performed for the indicated times. Extracts were prepared for Western blotting (see Materials and Methods) and duplicate blots were performed. Phosphospecific antibody to Thr-37 and Thr-46 (see Materials and Methods; top) and an anti-4E-BP1 antibody raised against the entire protein (bottom) were used to probe the membranes.
Following serum stimulation of 293 cells, duplicate blots were generated, and probed either with a polyclonal anti-4E-BP1 antiserum or with the phosphospecific antibody. The electrophoretic mobility of a significant portion of 4E-BP1 is retarded following serum stimulation (Fig. 8B, bottom panel). The phosphospecific antibody to Thr-37/Thr-46 recognizes all 4E-BP1 isoforms separated by SDS-PAGE (Fig. 8B, top). Using a secondary antibody coupled to 125I, the amount of Thr-37/Thr-46 in all isoforms was quantified. The intensity of the signal obtained for phosphorylated Thr-37 and Thr-46 at the time of peak phosphorylation (time = 30 min; normalized for total expression) is ∼1.7-fold greater than the signal obtained from lysates of serum-starved cells. In other experiments, this increase was between 1.3- and 1.8-fold (data not shown). Thus, as observed for the in vivo 32P-labeling, phosphorylation of Thr-37 and Thr-46 is only marginally responsive to serum stimulation. These data are also consistent with reports demonstrating that FRAP activity is only marginally enhanced in response to insulin stimulation (Scott et al. 1998).
Thr-37 and Thr-46 phosphorylation is sensitive to rapamycin and LY294002 under starvation conditions
The response of Thr-37 and Thr-46 phosphorylation to inhibitors of PI3 kinase or FRAP/mTOR was also examined. Rapamycin, LY294002, or wortmannin treatment of 293 cells prior to serum stimulation prevents the serum-induced hyperphosphorylation of 4E-BP1 (von Manteuffel et al. 1996). On two-dimensional phosphopeptide maps, treatment with rapamycin and wortmannin causes the disappearance of a subset of phosphopeptides, whereas others are relatively rapamycin and wortmannin resistant (von Manteuffel et al. 1997; Gingras et al. 1998). Interestingly, the most rapamycin-, LY294002-, and wortmannin-resistant phosphopeptides are those containing phosphorylated Thr-37 and Thr-46 (see von Manteuffel et al. 1997; Gingras et al. 1998). This result is puzzling, as FRAP/mTOR itself is rapamycin sensitive and is also inhibited, at least in vitro, by wortmannin and LY294002 (Brunn et al. 1996). To address this apparent discrepancy, starved 293 cells were incubated with rapamycin, and half of the cells were serum stimulated in the presence of rapamycin, while the other half was not serum-stimulated. Thr-37 and Thr-46 phosphorylation decreased drastically and rapidly following rapamycin treatment in the absence of subsequent serum stimulation (Fig. 9, top, cf. lane 1 with lanes 2–5). However, when the cells were stimulated with serum subsequent to rapamycin treatment, this effect was much less pronounced (top panel, lanes 6–10). The expression level of 4E-BP1 protein was not affected by rapamycin treatment (Fig. 9, bottom). Pretreatment with the PI3 kinase inhibitor LY294002 produced the same effect as rapamycin (data not shown). The extent of dephosphorylation was calculated to be ∼sixfold in the absence of subsequent stimulation and 1.5–2-fold in the presence of serum. Thus, consistent with Thr-37 and Thr-46 being phosphorylated by FRAP/mTOR, their phosphorylation is sensitive to rapamycin and LY294002. This sensitivity is reduced, however, by ∼threefold in the presence of serum. This difference will be addressed in the Discussion.
Figure 9.

Sensitivity of Thr-37 and Thr-46 phosphorylation to rapamycin. Serum-starved 293 cells were incubated with rapamycin (20 ng/ml). One set of plates was lysed immediately (lanes 1–5); the other was serum stimulated for 30 min prior to lysis (lanes 6–10). Extracts were heat treated and analyzed by Western blotting, as described in Materials and Methods. The membranes were incubated with either an anti-phosphospecific antibody (top) or with an anti-4E-BP1 antibody (bottom).
Thr-37 and Thr-46 phosphorylation is a priming step required for subsequent phosphorylation of the serum-sensitive sites
In view of the results described above, the biological significance of Thr-37 and Thr-46 phosphorylation remained elusive. To address this issue, constructs encoding hemagglutinin (HA)-tagged wild-type or point mutants of 4E-BP1 (Thr-37–Ala, Thr-46–Ala, Thr-37–Ala/Thr-46–Ala, and Thr-37–Glu/Thr-46–Glu) were transfected into 293T cells, and the pattern of phosphorylation was examined (Peptide maps prepared from 293T cells are identical to those prepared from 293 cells; data not shown). One set of transfected cells was used for 32P-labeling and another served for quantitation of protein by Western blotting. HA–4E-BP1 from 32P-labeled samples was immunoprecipitated using a monoclonal anti-HA antibody, and proteins were separated by SDS-PAGE. Incorporation of 32P was monitored (a representative autoradiograph is shown in Fig. 10A) and normalized to expression levels (a representative Western blot is shown in Fig. 10B). All of the mutants are capable of binding to eIF4E, as eIF4E was coimmunoprecipitated to approximately the same extent in all lanes (Fig. 10A). Coprecipitation of eIF4E with the HA-tagged proteins was confirmed by Western blotting (not shown), consistent with the results obtained with bacterially expressed 4E-BP1 mutants. Unexpectedly, mutation of either Thr-37 or Thr-46 to alanine caused a 10- to 20-fold decrease in 32P incorporation into HA–4E-BP1 (Fig. 10C, cf. lanes 2 and 3 with lane 1). Mutation of both Thr-37 and Thr-46 to alanine almost abolished 4E-BP1 phosphorylation (cf. lane 4 and lane 1), whereas replacement of the two threonines by glutamic acids had a slightly less severe effect (lane 5). The dramatic effect observed for the mutants is much greater than would be expected by abolishing Thr-37 or Thr-46 phosphorylation alone, as these sites account for ∼30% each of the total phosphorylation on 4E-BP1 (data not shown). The remaining phosphoamino acids represent ∼40% of the total 4E-BP1 phosphorylation. Thus, mutation of the Thr-37 and/or Thr-46 appears to prevent phosphorylation of the other sites.
Figure 10.
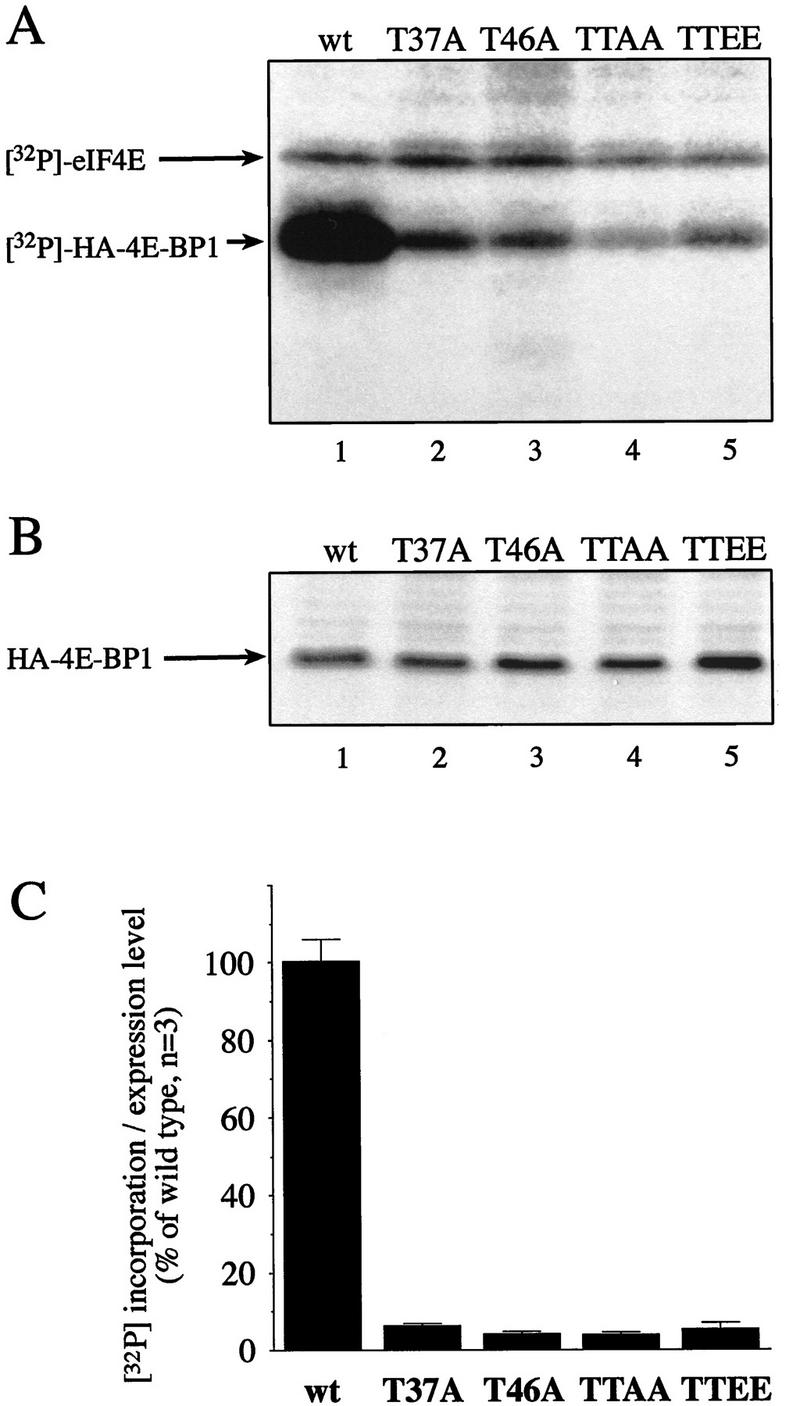
Mutation of Thr-37 or Thr-46 drastically reduces 4E-BP1 phosphorylation in vivo. 4E-BP1 wild type (wt) and mutants Thr-37–Ala (T37A), Thr-46–Ala (T46A), Thr-37–Ala/Thr-46–Ala (TTAA), and Thr-37–Glu/Thr-46–Glu (TTEE) in a pcDNA3-3HA vector were transfected into 293T cells (six plates/construct). Three transfected plates were subjected to 32P-labeling, and HA-tagged 4E-BP1 was immunoprecipitated using an anti-HA antibody (HA.11). Precipitated proteins were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and autoradiographed. The intensity of the HA–4E-BP1 bands was quantified by PhosphorImager. The three other plates were used to analyze the expression levels of the HA-tagged proteins by Western blotting, using anti-HA as the primary antibody and 125I-coupled anti-mouse IgG as the secondary antibody. Expression level was quantified by PhosphorImager. The average of the 32P incorporation into the mutant proteins was normalized to the expression level of each mutant. (A) A representative autoradiograph is shown; arrows indicate 32P-labeled HA–4E-BP1 and coprecipitated eIF4E. (B) A representative Western blot showing the levels of the mutants; this Western blot was developed by chemiluminescence, which produces a brighter signal. (C) Incorporation of 32P into each mutant, normalized to the expression level. The incorporation in the wild-type protein is set to 100%.
To test the hypothesis that Thr-37 and Thr-46 mutation prevents the phosphorylation of other sites, phosphopeptide maps were generated from the mutant and wild-type proteins. Because the mutant proteins were phosphorylated to a much lower extent than the wild-type protein (Fig. 10), an equivalent quantity of radioactivity was loaded on each plate. Thus, ∼10–20 times less material was employed for the wild-type map than for those of the various mutants (Fig. 11, upper left). The characteristic pattern of 4E-BP1 phosphorylation was observed for the wild-type HA-tagged protein (Fig. 11A). The map generated from the Thr-37–Ala mutant shows a similar pattern, with the exception of the disappearance of peptides containing Thr-37 (Fig. 11B; the other peptides migrating in this area result from partial chymotryptic cleavage of the peptide containing Thr-37 and Thr-46, and thus contain phospho-Thr-46). Mutation of Thr-46 has a more pronounced effect on phosphorylation of the other sites. With the exception of two phosphopeptides whose intensity does not change, phosphorylation on all other sites is dramatically reduced as compared to the wild-type protein. It is noteworthy that Thr-37 phosphorylation is also reduced severalfold (Fig. 11C) in the Thr-46–Ala mutant. Thus, Thr-46 appears to play a key role in modulating the phosphorylation of the other sites, including Thr-37. The map generated from the double mutant Thr-37–Ala/Thr-46–Ala showed, as expected, the disappearance of the phosphopeptides containing Thr-37 and Thr-46, but also an extensive decrease in phosphorylation of the remaining sites (Fig. 11D). Strikingly, however, replacement of the Thr-37 and Thr-46 residues by glutamic acids partially restored phosphorylation on the remaining sites, although the signals from Thr-37 and Thr-46 phosphorylation were absent (Fig. 11E). We therefore conclude that mutating threonines 37 and 46 to glutamic acid partially mimics phosphorylated Thr-37 and Thr-46. It should be emphasized, however, that the phosphorylation of the serum-sensitive sites is not restored to wild-type levels in the Thr-37–Glu/Thr-46–Glu mutant (in Fig. 11, an equal number of counts was loaded; thus, ∼10 times less material from the wild-type sample was used). These results demonstrate that phosphorylation on Thr-37 and Thr-46 is a prerequisite for the subsequent phosphorylation of the serum-stimulated sites on 4E-BP1, as illustrated in the model in Figure 11F.
Figure 11.
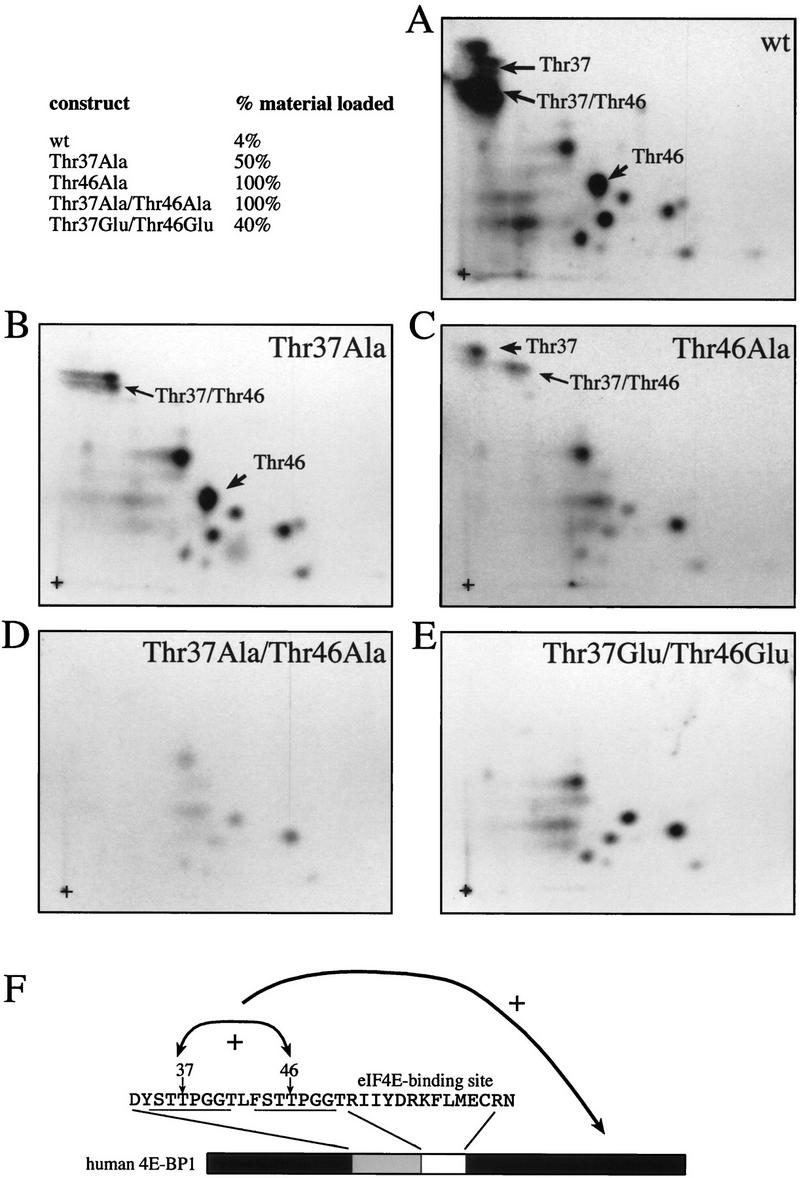
Effects of mutations of Thr-37 and/or Thr-46 on other phosphorylation sites. (A–E) Phosphopeptide maps were performed on HA-tagged 4E-BP1 proteins from Fig. 10. (F) Thr-37 and Thr-46 affect 4E-BP1 phosphorylation. The arrows indicate (1) a mutually positive effect of Thr-37 and Thr-46 phosphorylation, and (2) a positive effect of Thr-37 and Thr-46 phosphorylation on the phosphorylation of the serum-sensitive sites. The fraction of samples from Fig. 10 loaded onto each map is shown (upper left). An equivalent quantity in cpm was loaded for each mutant.
Discussion
In this report we describe that Thr-37 and Thr-46 are phosphorylated by FRAP/mTOR in a 4E-BP1/eIF4E complex and that this phosphorylation does not result in the disruption of the complex. Thr-37 and Thr-46 are phosphorylated to a high stoichiometry under serum-starvation conditions, and their phosphorylation state does not significantly increase following serum stimulation. 32P incorporation into 4E-BP1 in vivo is drastically reduced when either Thr-37 or Thr-46 is mutated to alanine. Phosphopeptide mapping revealed that mutation of Thr-37 or Thr-46, which are located amino-terminal to the eIF4E-binding site, affects phosphorylation of all 4E-BP1 sites, including the serum-inducible residues. This effect is relieved to some extent by the mutation of Thr-37 and Thr-46 to glutamic acid, which partially mimics the phosphorylated threonines (Fig. 11E). Taken together, these data indicate that phosphorylation of Thr-37 and Thr-46 in vivo is a prerequisite for phosphorylation of serum sensitive 4E-BP1 sites. Several phosphorylation sites were previously reported to be serum sensitive; these include Thr-37, Thr-46, Ser-65, Thr-70, Ser-83 (all lying in the consensus S/T-P, Fadden et al. 1997), and Ser-112 (a site preceded by acidic residues, Heesom et al. 1998). In 293 cells, as in Rat1a cells, however, we have observed that the most serum-sensitive sites are Ser-65 and Thr-70 (A.-C. Gingras, B. Raught, and S.P. Gygi, unpubl.). It remains possible that in different cell types the sensitivity of the various phosphorylation sites to extracellular stimuli differs.
Based on our results, we propose a two-step model for 4E-BP1 phosphorylation (Fig. 12). FRAP/mTOR (or a tightly associated kinase) first phosphorylates Thr-37 and Thr-46 on 4E-BP1 complexed with eIF4E. This phosphorylation event appears to be relatively independent of extracellular stimuli. The second step, which is activated by extracellular stimuli, is the phosphorylation of the sites located carboxy-terminal to the eIF4E-binding site, including Ser-65 and Thr-70 (A.-C. Gingras, B. Raught, and S.P. Gygi, unpubl.). The second phosphorylation step results in release from eIF4E and stimulation of translation.
Figure 12.

A two-step model for the phosphorylation of 4E-BP1. FRAP/mTOR phosphorylates 4E-BP1 (in a 4E-BP1/eIF4E complex) on Thr-37 and Thr-46. Phosphorylation of Thr-37 and Thr-46 is a prerequisite for an unidentified serum-sensitive kinase (acting downstream of Akt) to phosphorylate 4E-BP1 on the serum-sensitive sites. The latter phosphorylation triggers the release of 4E-BP1 from eIF4E. A direct link between Akt and FRAP/mTOR remains to be proven.
Phosphorylation of proteins on multiple residues often occurs in a sequential manner. In some instances, the recognition site of a kinase contains a phosphorylated residue. This is the case for glycogen synthase kinase-3 (GSK-3), which phosphorylates serines or threonines lying in the consensus SXXXPS, where PS is a phosphoserine (Fiol et al. 1990). Other kinases, including casein kinase I, also exhibit a preference for phosphorylated amino acids in the vicinity of their target site (Flotow and Roach 1991). Priming phosphorylation is, however, not restricted to the kinase recognition site. For example, phosphorylation may induce a conformational change in the substrate to render a site more easily accessible to a kinase or create a docking site for another protein, similar to the SH2/phosphotyrosine interaction. In this regard, 14-3-3 proteins, which recognize phosphoserines, serve as chaperones, inhibitors, or adaptors to modulate the function of their binding partners (Muslin et al. 1996; Yaffe et al. 1997).
Many kinases are able to phosphorylate 4E-BP1 in vitro, including several different MAP kinases (ERK2, JNK, p38), protein kinase C, and casein kinase II (for review, see Lawrence et al. 1997; see also A.-C. Gingras, unpubl.). Other PIK family members, such as ATM and ATR, can also phosphorylate 4E-BP1 (Banin et al. 1998; Sarkaria et al. 1998). 4E-BP1 is mostly unstructured in solution (Fletcher and Wagner 1998; Fletcher et al. 1998). Thus, it is not surprising that it is a good kinase substrate. Upon binding to eIF4E, however, a localized structure is induced in the 4E-BP1/eIF4E contact region (Fletcher and Wagner 1998; Marcotrigiano et al. 1999). This induced fit appears to mask some phosphorylation sites, as ERK2 is unable to phosphorylate 4E-BP1 in a 4E-BP1/eIF4E complex. As previously mentioned, a 4E-BP1/eIF4E complex is most likely the true physiological substrate. Therefore, using a complex of 4E-BP1/eIF4E in in vitro kinase assays is more likely to identify an in vivo kinase. Contrary to ERK2, FRAP/mTOR phosphorylates 4E-BP1 to the same extent, and on the same sites, regardless of whether 4E-BP1 is free in solution or bound to eIF4E.
The cocrystal structure of eIF4E complexed with a 4E-BP1 peptide (amino acids 51–67; amino acids 65–67 were not visible in the density map) was recently determined (Marcotrigiano et al. 1999). 4E-BP1 binds to a phylogenetically conserved region on the convex surface of eIF4E. Similar results were obtained by nuclear magnetic resonance (NMR), using both the full-length protein as well as a fragment of 4E-BP1. Although chemical shifts induced by the peptide and the full-length 4E-BP1 were largely overlapping, the full-length 4E-BP1 induced shifts in additional eIF4E residues (Matsuo et al. 1997). This effect appears to be due to the amino-terminal extension of full-length 4E-BP1, which may include the Thr-46 phosphorylation site. Acidic residues flank the region on eIF4E in contact with the 4E-BP1 peptide [Glu-140 and Asp-143 are predicted to be close to 4E-BP1 Thr-46 (J. Marcotrigiano, pers. comm.); Glu-70 is predicted to be in the vicinity of Ser-65; (J. Marcotrigiano, A.-C. Gingras, N. Sonenberg, and S.K. Burley, in prep.)]. Thus, it is conceivable that the mechanism leading to 4E-BP1 release from eIF4E involves electrostatic repulsion between the negatively charged phosphates on 4E-BP1 and the acidic residues on eIF4E (Marcotrigiano et al. 1997). In this regard, phosphorylation of 4E-BP1 at Thr-37 and Thr-46 could theoretically decrease the affinity of 4E-BP1 for eIF4E, although this was not detected in the m7GDP–agarose-binding experiments (which are only qualitative; a better stoichiometry of 4E-BP1 phosphorylation by FRAP/mTOR is required to perform quantitative measurements of affinity). Alternatively, it is possible that phosphorylation of Thr-37 and Thr-46 alters the conformation of the 4E-BP1/eIF4E complex to allow access to the other phosphorylation sites on 4E-BP1. A third possibility is that phosphorylation of 4E-BP1 by FRAP/mTOR creates a docking site for a different kinase or for an adaptor molecule that recruits a kinase.
The two sites phosphorylated by FRAP/mTOR in vitro are the least rapamycin sensitive in vivo. Also, under conditions of serum starvation, Thr-37 and Thr-46 are phosphorylated to a high stoichiometry, relative to the other sites. Furthermore, in cells treated with rapamycin and subsequently stimulated with serum, phosphorylation at these sites is only slightly affected (von Manteuffel et al. 1997; Gingras et al. 1998, this paper). Only when starved cells were treated with rapamycin was a rapid reduction in the phosphorylation of Thr-37 and Thr-46 observed. Several hypotheses, which are not mutually exclusive, could explain this observation: (1) FRAP is inhibited by rapamycin, even in the presence of serum, but another rapamycin-insensitive/serum-sensitive kinase (for example a MAP kinase) is responsible for phosphorylating 4E-BP1 under these conditions; (2) Some residual FRAP kinase activity is retained in the presence of rapamycin but is inhibited in the absence of serum; (3) A phosphatase active in the absence of serum, but inactive in the presence of serum, is responsible for the effect. In the latter case, if the phosphatase is rapidly inactivated following serum stimulation, then even a low FRAP/mTOR kinase activity could be sufficient to induce 4E-BP1 phosphorylation. However, when cells are serum deprived, the phosphatase activity may predominate. In the absence of rapamycin, FRAP/mTOR is active and this is sufficient to phosphorylate Thr-37 and Thr-46. When rapamycin is added in the absence of serum, however, the balance may change: The phosphatase could remain active, and the activity of FRAP/mTOR is inhibited, so a net decrease in the phosphorylation of Thr-37 and Thr-46 is observed.
Thr-37 and Thr-46 phosphorylation is only moderately increased (1.3- to 1.7-fold) following serum stimulation. This modest serum sensitivity is comparable to that reported for FRAP/mTOR activation by insulin (Scott and Lawrence 1998; Scott et al. 1998). There is no evidence at present that Thr-37 and Thr-46 phosphorylation occurs downstream of Akt, as opposed to the phosphorylation of the serum-sensitive sites (Gingras et al. 1998). The induction of phosphorylation by serum on the sites carboxy-terminal to the eIF4E-binding motif is far more pronounced (see Fig. 6), although an exact quantification was not possible in this study. The most prominent serum-induced sites have been mapped to Ser-65 and Thr-70 (A.-C. Gingras, B. Raught, and S.P. Gygi, unpubl.). Because the serum-responsive sites on 4E-BP1 are also the most rapamycin sensitive, it is highly likely that FRAP/mTOR is involved in their phosphorylation, although probably not directly. It remains possible that the rapamycin-induced dephosphorylation of Thr-37 and Thr-46 prevents the subsequent phosphorylation of the serum-sensitive sites following serum stimulation. This is, however, unlikely because phosphorylation on Thr37 and Thr46 is restored with rapid kinetics to almost maximal levels following addition of serum, whereas the serum-sensitive sites remain dephosphorylated. It is also important to mention that the relatively high level of phosphorylated Thr-37 and Thr-46 in the rapamycin-treated, serum-stimulated cells (Fig. 9) is not sufficient to induce phosphorylation on the serum-sensitive sites. Thus, FRAP/mTOR likely impacts on the phosphorylation of the serum-sensitive sites either by stimulating their phosphorylation following serum stimulation or by inducing their dephosphorylation in the presence of rapamycin. In this regard, an attractive hypothesis as to the function of FRAP/mTOR is that it serves as a gatekeeper, somewhat similar to the mechanisms controlling the cell cycle at the restriction point. FRAP/mTOR could alter the phosphorylation of the serum-sensitive sites of 4E-BP1 through the modulation of the activity of a kinase or a phosphatase.
In Saccharomyces cerevisiae, a nutrient-induced, rapamycin-sensitive association between the phosphatases PP2A or Sit4 (the yeast homolog of protein phosphatase 6) and a protein named TAP42 was reported. This association was postulated to modulate the activity or substrate specificity of the phosphatases, or to target them to specific substrates. Because of the rapamycin-sensitive nature of this interaction, it was postulated that the phosphatases and TAP42 were downstream components in the TOR signaling pathway. In mammals, a homolog of TAP42, α4, associates with mammalian PP2A, as well as with protein phosphatases 4 and 6 (Chen et al. 1998). However, regulation of the association and activity of these proteins by FRAP/mTOR remains controversial. A recent report indicates that PP2A can partially dephosphorylate a FRAP/mTOR-phosphorylated 4E-BP1 substrate in vitro and that the dephosphorylation can be prevented by preincubation of TAP42 with PP2A (Nanahoshi et al. 1998). Thus, it is possible that phosphatases play an active role in FRAP/mTOR signaling to 4E-BP1.
Our data are consistent with FRAP/mTOR being a physiological 4E-BP1 kinase, as was suggested previously (Brunn et al. 1997a,b; Burnett et al. 1998). However, there are discrepancies between our results and the data reported previously. Brunn et al. (1997a) found that FRAP/mTOR phosphorylates five sites (Thr-37, Thr-46, Ser-65, Thr-70, and Ser-83) on 4E-BP1. Using the same antibody used by Brunn et al. (as well as baculovirus-expressed FRAP/mTOR), we obtained significant and reproducible phosphorylation only on Thr-37 and Thr-46. We also detected other phosphopeptides in some of our kinase assays, but phosphorylation of these phosphopeptides was not reproducible. These phosphopeptides were labeled only to a very low stoichiometry as compared to Thr-37 and Thr-46, and, as mentioned previously, washing the FRAP/mTOR immunoprecipitate with a stringent buffer decreased their phosphorylation even further. We believe that the differences between our results and those of Brunn et al. are probably due to the washing conditions of the immunoprecipitate and times of incubation (see Materials and Methods). It is possible that a kinase that is tightly associated with FRAP/mTOR is responsible for the phosphorylation reported for the other residues on 4E-BP1. Our data are largely in agreement with those of Burnett et al. (1998), which defined Thr-37 and Thr-46 as the two sites phosphorylated by FRAP/mTOR on 4E-BP1. However, Burnett et al. (1998) reported that, in vitro, phosphorylation of 4E-BP1 at Thr-37 and Thr-46 by FRAP/mTOR decreases binding to eIF4E. We did not observe such a decrease, as we clearly detected Thr-37 and Thr-46 in the fractions bound to eIF4E. However, no quantitative measurement of the eIF4E/4E-BP1 interaction was performed, so it remains possible that the strength of this interaction is decreased by phosphorylation on Thr-37 and Thr-46. Burnett et al. (1998) also reported that mutants of Thr-37 and Thr-46 expressed in 293 cells bind constitutively to eIF4E. We obtained the same result (A.-C. Gingras, unpubl.), but explain it by the fact that Thr-37 and Thr-46 are necessary for the phosphorylation of the serum-inducible phosphorylation sites, which lead to 4E-BP1 dissociation from eIF4E.
It is of the utmost importance to determine which kinases phosphorylate the serum-sensitive sites of 4E-BP1. It is highly likely that Ser-65 and Thr-70, the most serum-sensitive sites, are phosphorylated by a proline-directed kinase. However, because of the lack of sensitivity to the MEK inhibitor PD98059, it is unlikely that ERKs are involved in mediating the phosphorylation on the serum-sensitive sites. Other MAP kinase family members (such as JNK and p38), which can phosphorylate 4E-BP1 in vitro, are not activated by the same stimuli that cause 4E-BP1 phosphorylation. Other proline-directed kinases, such as the cyclin-dependent kinases, are not likely to be involved in phosphorylating 4E-BP1 in vivo, because they are mostly nuclear, whereas 4E-BP1 is present mainly in the cytoplasm. One approach to identify these kinases, based on our results, is to use a 4E-BP1/eIF4E complex prephosphorylated at Thr-37 and Thr-46 as a substrate in a biochemical purification scheme.
Materials and methods
Plasmids and antibodies
The human 4E-BP1 coding sequence cloned into the cytomegalovirus-based vector pACTAG-2 was used as a template for PCR mutagenesis, which was used to mutate Thr-37 and Thr-46 to either Ala or Glu. To generate double mutants Thr-37–Ala/Thr-46–Ala and Thr-37–Glu/Thr-46–Glu, either Thr-46–Ala or Thr-46–Glu cloned into pACTAG-2 was used as template to mutate Thr-37. Similarly, the deletion of amino acids 34–50 and the deletion in the eIF4E-binding site (amino acids 54–63) were generated by PCR mutagenesis. The products were inserted in-frame into pACTAG-2 and sequenced in their entirety. Alternatively, the mutated sequences were inserted in-frame in the vector pGEX-6p1 (Pharmacia) for protein expression in bacteria. GST-2T–4E-BP1 wild-type was described previously (Pause et al. 1994). It contains almost the entire coding region of human 4E-BP1 (lacking the first 6 amino acids) fused in-frame in the vector pGEX-2T (Pharmacia; contains a thrombin cleavage site). Murine 4E-BP1 wild type was cloned in-frame into pQE-30 vector (Qiagen), which contains an amino-terminal 6-histidine tag (His). PCR mutagenesis was utilized to mutate Thr-36 (corresponding to human Thr-37) or Thr-45 (corresponding to human Thr-46) to alanines. The constructs were sequenced in their entirety.
The rabbit polyclonal antisera 11208 and 11209 against human 4E-BP1 have been described previously (Gingras et al. 1996, 1998). Antiserum 11208 was used for Western blotting analysis and 11209 for immunoprecipitation. The antibody to FRAP/mTOR was described previously (Brunn et al. 1997b). The anti-HA mouse monoclonal antibody HA.11 was purchased from BAbCO. The phosphospecific antibodies directed against Thr-37 and Thr-46 sites in 4E-BP1 were produced by immunizing New Zealand white rabbits with the following synthetic peptides coupled to keyhole limpet hemocyanin: Thr-37(P), PGDYSTT*PGGTLFC, and Thr-46(P), GTLFSTT*PGGTRIIC. Enzyme-linked immunosorbent assay (ELISA), using the phosphopeptide and corresponding nonphosphopeptide, was used to identify the best-responding rabbits. IgG was purified using protein-A–Sepharose. Antibodies reactive with the nonphosphopeptide were removed by adsorption to a nonphosphopeptide affinity column. Antibodies that flowed through this column were next passed over a column of immobilized phosphopeptide; after washing, antibodies were eluted at low pH and dialyzed. The resulting antibodies were characterized by ELISA against the phosphopeptide and nonphosphopeptide to determine the extent of phosphospecificity and by Western blots against whole-cell extracts and purified 4E-BP1 proteins. The antibody directed against phospho-Thr-37, however, cross-reacts with phospho-Thr-46, because of the high sequence identity surrounding the two sites. Similarly, the anti-phospho-Thr-46 antibody cross-reacts with phosphorylated Thr-37.
Protein expression and purification
An amino-terminal deletant (NΔ27) of eIF4E, which behaves essentially as the wild-type protein, was bacterially expressed and purified by m7GDP chromatography (Marcotrigiano et al. 1997). Glutathione S-transferase (GST)-fused 4E-BP1 proteins were expressed and purified from DH10β Escherichia coli essentially according to the manufacturer’s (Pharmacia) instructions. Specifically, bacteria were grown to an OD of 0.8 and induced for 2 hr at 37°C with 0.5 mm IPTG. Bacteria were pelleted, resuspended in PBS containing 1 mm PMSF, and lysed by sonication. After centrifugation (Sorvall SS34 for 1 hr at 15,000 rpm), the cleared lysate was incubated for 15 min at 20°C with glutathione–Sepharose beads (1 ml beads per liter of starting culture). Beads were pelleted and rinsed three times with PBS containing 1 mm PMSF and 1% Triton X-100. The elution was performed in 20 mm Tris-HCl (pH 8.5) containing 10 mm reduced glutathione. Proteins were dialyzed against 20 mm HEPES–KOH (pH 7.4), 74 mm KCl, and 1 mm DTT, and quantified by SDS-PAGE and Coomassie staining, as compared to a BSA standard. Alternatively, the construct pGEX-2T–4E-BP1 wild type was expressed and bound to the glutathione beads as above and digested with thrombin on the beads, according to the manufacturer’s instructions. The cleaved 4E-BP1 protein was then purified by reverse-phase HPLC on a C4 column, as described (J. Marcotrigiano, A.-C. Gingras, N. Sonenberg, and S.K. Burley, in prep.). Histidine-tagged 4E-BP1 proteins were expressed and purified from M15pREP4 cells according to the manufacturer’s (Qiagen) instructions, eluted with imidazole, and dialyzed against 20 mm HEPES–KOH (pH 7.4), 74 mm KCl, and 1 mm DTT. Epitope-tagged FRAP was purified from two-liter baculovirus infections using Sf9 host cells (Invitrogen) grown in 10-liter fermentors for infection. Infection conditions for large-scale cultures ranged from 24–48 hr. The baculovirus construct, lysis conditions, and chromatography methods were described previously (Brown et al. 1995).
Cell culture and transient transfections
Human embryonic kidney 293 cells (ATCC CRL 1573) and human embryonic kidney 293T cells (overexpressing the SV40 large T antigen; ATCC CRL 11268) were maintained in Dulbecco’s modified Eagle medium (DMEM) containing 10% FCS. For transient transfection of 293T cells, 1 × 106 cells were plated onto a 100-mm dish 24 hr prior to transfection of 15 μg of pACTAG-2–4E-BP1 DNA, using a modified calcium phosphate technique (Chen and Okayama 1988). Cells were either harvested or labeled 48 hr post-transfection.
Extract preparation and Western blotting
Cells were rinsed twice with cold buffer A (20 mm Tris-HCl at pH 7.5, 100 mm KCl, 20 mm β-glycerolphosphate, 1 mm DTT, 0.25 mm Na3VO4, 10 mm NaF, 1 mm EDTA, 1 mm EGTA, 10 nm okadaic acid, 1 mm PMSF) and scraped into a minimal volume of the same buffer. Lysis was performed by three freeze–thaw cycles. Cell debris was pelleted by centrifugation, and the protein concentration in the supernatant was measured using the Bio-Rad assay. For analysis of endogenous 4E-BP1, 75 μg of total cell extract was incubated at 100°C for 7 min to enrich for 4E-BP1, which is heat stable (Hu et al. 1994; Pause et al. 1994). Samples were then incubated on ice for 5 min, and precipitated material was removed by centrifugation (microcentrifuge, 13,000 rpm for 5 min). Laemmli sample buffer was added to the supernatant, which was then subjected to SDS-15% PAGE. For analysis of transfected HA–4E-BP1, cells were lysed by three freeze–thaw cycles, and 50 μg of protein was analyzed by SDS-15% PAGE. For qualitative analysis, Western blotting and chemiluminescence detection were performed as described, using either the anti-4E-BP1 11208 antiserum (1:1500) or the phosphospecific anti-Thr-37/Thr-46 antibody (1:1000). For quantitative analysis of the endogenous proteins, membranes were first incubated with the anti-4E-BP1 11208 antiserum (1:1500) or the phosphospecific anti-Thr-37/Thr-46 antibody (1:1000), followed by incubation with [125I]-coupled protein A. For the quantitation of overexpressed HA-tagged 4E-BP1, the anti-HA (HA.11) monoclonal antibody (0.5 μg/ml) was utilized.
Metabolic labeling and immunoprecipitation
32P-Labeling was performed essentially as described (Gingras et al. 1998). In small scale experiments, 100- or 150-mm dishes of 293 cells or 293T cells were 32P-labeled and processed under the same conditions described in Gingras et al. (1998), but using nitrocellulose membranes instead of PVDF membranes for the transfer. For large-scale experiments designed to identify the phosphorylation sites, ten 150-mm plates of confluent 293 cells (∼2 × 108 cells) were starved for 30 hr and incubated for 3.5 hr in phosphate-free DMEM (GIBCO) containing 1 mCi/ml of [32P]orthophosphate (DuPont NEN; 3000 mCi/mmole; total volume of 7.5 ml). Dialyzed FCS (15%; GIBCO) was then added for 40 min, and the cells were rinsed and lysed as described (Gingras et al. 1998). In parallel, 200 × 150-mm plates of confluent 293 cells (∼4 × 109 cells) were starved for 33 hr and stimulated for 40 min with 15% FCS (GIBCO). Cells were rinsed and extracts were prepared in the same manner as for the 32P-labeled extracts. The extract (at this point, the cold and hot extracts were processed separately) was precleared by incubation with protein A beads (50 μl of packed beads per 108 cells) with end-over-end rotation at 4°C for 2 hr. The supernatant was transferred to a fresh tube, together with 25 μl of 11209 antisera bound to 25 μl of packed protein A–Sepharose beads per 108 cells, and incubated end-over-end for 4 hr at 4°C. Beads were spun and washed three times (with 100 times the bead volume) in lysis buffer, two times in RIPA buffer, and two times in LiCl solution (200 mm LiCl, 1 mm DTT). Immunoprecipitates from the 32P-labeled and cold samples were eluted in Laemmli sample buffer and the elutions were combined. Immunoprecipitated material was subjected to SDS–15% PAGE and transferred to 0.2-μm pore size nitrocellulose (Schleicher & Schuell), which were dried and autoradiographed. Radioactive bands corresponding to 4E-BP1 were excised and Cerenkov counted.
Phosphopeptide maps and phosphoaminoacid analysis
Tryptic/chymotryptic digestion of 4E-BP1 immobilized on the nitrocellulose membranes was performed essentially as described (van der Geer et al. 1994; Gingras et al. 1998). For the large-scale experiment, the sample was divided into three tubes for digestion and mapping. The digest was performed using a 200:1 mixture of N–tosyl–l–phenylalanine chloromethyl ketone-treated trypsin and chymotrypsin (1 μg per sample, Worthington) for 10 hr, followed by the addition of 0.5 μg of enzyme mix three times for 1 hr each. The sample was then lyophilized (Speed-vac, Savant), resuspended in 500 μl of water, lyophilized again, resuspended in 250 μl of water, lyophilized a third time, resuspended in 250 μl pH 1.9 buffer [2.5% (vol/vol) formic acid, 7.8% (vol/vol) glacial acetic acid], and lyophilized a fourth time. For two-dimensional phosphopeptide mapping, the first dimension (electrophoresis) was performed in pH 1.9 buffer using an HTLE 7000 apparatus (CBS Scientific); the second dimension was performed in phosphochromatography buffer [37.5% (vol/vol) n-butanol, 25% (vol/vol) pyridine, 7.5% (vol/vol) glacial acetic acid]. Plastic-backed cellulose-coated TLC plates (Kodak; 20 × 20 cm) were employed. The plates were dried and subjected to autoradiography.
To determine which phosphoamino acid is phosphorylated in the spots of interest, the cellulose was scraped off. Peptides were eluted from the cellulose with pH 1.9 buffer, using a spin filter (Bio101). The material was lyophilized, resuspended in 100 μl 6n HCl and incubated at 110°C for 60 min in a screw-cap tube. The sample was lyophilized and rinsed several times with water (debris was removed by centrifugation before the last lyophilization). Two-dimensional phosphoamino acid analysis mapping was performed essentially as described (van der Geer et al. 1994).
Mass spectrometry
Spots containing radioactivity were scraped from the cellulose plates and prepared for analysis by LC-MS/MS as described (Watts et al. 1994). The system used was adapted from previous reports (Gygi et al. 1999). In brief, a capillary HPLC column (50 μm i.d., 12 cm long) packed in-house with C18 material was coupled via a homemade μESI interface to a triple quadrupole mass spectrometer (TSQ 7000; Finnigan Mat, San Jose, CA). A pressurisable bomb was used to load 5 μl from a solution containing the phosphopeptide onto the capillary column. The column was then reattached to the HPLC gradient delivery system (Michrom, Auburn, CA). A gradient for column elution was developed over 30 min, and a precolumn flow splitter reduced the flow from 50 μl/min to ∼150 nl/min.
The eluted peptides were ionized by electrospray ionization, detected, and the specific peptide ions were automatically selected and fragmented by a triple quadrupole mass spectrometer (Gygi et al. 1999). To achieve this, the mass spectrometer switched between the MS mode (for peptide mass identification) and the MS/MS mode (for peptide characterization and sequencing). The selected peptides were fragmented by a process called collision-induced dissociation (CID) to generate a tandem mass spectrum (MS/MS spectrum or CID mass spectrum), which contained the sequence information for a single peptide. Individual CID mass spectra were then searched with the computer program Sequest (Eng et al. 1994) against the known sequence of 4E-BP1. The search was modified to allow for the potential increase in mass to serine, threonine, and tyrosine by phosphorylation (Yates et al. 1995). All phosphopeptides matched by computer searching algorithms were manually verified.
Kinase assays
FRAP/mTOR immunoprecipitation from rat brain was carried out essentially as described (Brunn et al. 1997a) with the following modification: The immunoprecipitated material was washed three times with lysis buffer, three times with lysis buffer containing 400 mm KCl, and twice with kinase buffer (Brunn et al. 1997a). FRAP/mTOR kinase assays were performed essentially as described (Brunn et al. 1997a) with either FRAP/mTOR immunoprecipitated from rat brain (Brunn et al. 1997a) or with baculovirus-expressed, FLAG-tagged FRAP/mTOR. FRAP/mTOR kinase reactions were incubated for 20–40 min at 30°C, with gentle agitation. The ERK2 assay was performed for 10 min using recombinant ERK2 (NEB), according to the manufacturer’s instructions.
Chromatography on m7GDP–agarose
4E-BP1 and eIF4E proteins were incubated for 1 hr with m7GDP coupled to agarose adipic resin in a total volume of 50 μl (5–30 μl of packed beads per reaction, beads were prepared according to Edery et al. 1988). Beads were then spun in a microcentrifuge (3000 rpm, 30 sec), washed 3 times with 20 volumes of buffer containing 75 mm KCl, 20 mm HEPES–KOH (pH 7.4), and resuspended in Laemmli sample buffer. Samples were analyzed by SDS-PAGE and subjected to autoradiography.
Acknowledgments
We thank Joe Marcotrigiano for providing the purified eIF4E NΔ27 protein and the purified cleaved GST–4E-BP1 proteins, Eric J. Brown and Stuart L. Schreiber for the baculovirus-flag FRAP construct, Chantal Binda and Colin Lister for excellent technical assistance, Yanick Matteau for help with tissue culture, and Hiroaki Imataka for the pcDNA3-3HA vector. We thank William B.C. Merrick, Eric J. Brown, Michel L. Tremblay, Francis Poulin, Stéphane Pyronnet and Mathieu Miron for helpful discussions and comments on the manuscript. This work was supported by grants from the National Cancer Institute of Canada and the Howard Hughes Medical Institute (HHMI) to N.S. and from the National Science Foundation Science and Technology Center for Molecular Biotechnology to R.A. N.S. is a Medical Research Council (MRC) of Canada Distinguished Scientist and a HHMI International Scholar. A.-C.G. and B.R. were supported by a doctoral award and a post-doctoral fellowship, respectively, from the MRC of Canada. S.P.G. is supported by a National Institutes of Health grant T32HG00035-3 and a grant from Oxford Glycosciences.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL nsonen@med.mcgill.ca; FAX (514) 398-1287.
References
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Beretta L, Svitkin YV, Sonenberg N. Rapamycin stimulates viral protein synthesis and augments the shutoff of host protein synthesis upon picornavirus infection. J Virol. 1996;70:8993–8996. doi: 10.1128/jvi.70.12.8993-8996.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks RF. Continuous protein synthesis is required to maintain the probability of entry into S phase. Cell. 1977;12:311–317. doi: 10.1016/0092-8674(77)90209-4. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–446. doi: 10.1038/377441a0. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC, Jr, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J. 1996;15:5256–5267. [PMC free article] [PubMed] [Google Scholar]
- Brunn GJ, Fadden P, Haystead TAJ, Lawrence JC., Jr The mammalian target of rapamycin phosphorylates sites having a (Ser/Thr)-Pro motif and is activated by antibodies to a region near its COOH terminus. J Biol Chem. 1997a;272:32547–32550. doi: 10.1074/jbc.272.51.32547. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC, Jr, Abraham RT. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997b;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CA, Okayama H. Calcium phosphate-mediated gene transfer: A highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- Chen J, Peterson RT, Schreiber SL. Alpha 4 associates with protein phosphatases 2A, 4, and 6. Biochem Biophys Res Commun. 1998;247:827–832. doi: 10.1006/bbrc.1998.8792. [DOI] [PubMed] [Google Scholar]
- Conlon I, Raff M. Size control in animal development. Cell. 1999;96:235–244. doi: 10.1016/s0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- Edery I, Altmann M, Sonenberg N. High-level synthesis in Escherichia coli of functional cap-binding eukaryotic initiation factor eIF-4E and affinity purification using a simplified cap-analog resin. Gene. 1988;74:517–525. doi: 10.1016/0378-1119(88)90184-9. [DOI] [PubMed] [Google Scholar]
- Eng J, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Fadden P, Haystead TA, Lawrence JC., Jr Identification of phosphorylation sites in the translational regulator, PHAS-I, that are controlled by insulin and rapamycin in rat adipocytes. J Biol Chem. 1997;272:10240–10247. doi: 10.1074/jbc.272.15.10240. [DOI] [PubMed] [Google Scholar]
- Fiol CJ, Wang A, Roeske RW, Roach PJ. Ordered multisite protein phosphorylation. Analysis of glycogen synthase kinase 3 action using model peptide substrates. J Biol Chem. 1990;265:6061–6065. [PubMed] [Google Scholar]
- Fletcher CM, Wagner G. The interaction of eIF4E with 4E-BP1 is an induced fit to a completely disordered protein. Protein Sci. 1998;7:1639–1642. doi: 10.1002/pro.5560070720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher CM, McGuire AM, Gingras A-C, Li H, Matsuo H, Sonenberg N, Wagner G. 4E binding proteins inhibit the translation factor eIF4E without folded structure. Biochemistry. 1998;37:9–15. doi: 10.1021/bi972494r. [DOI] [PubMed] [Google Scholar]
- Fleurent M, Gingras A-C, Sonenberg N, Meloche S. Angiotensin II stimulates phosphorylation of the translational repressor 4E-binding protein 1 by a mitogen-activated protein kinase-independent mechanism. J Biol Chem. 1997;272:4006–4012. doi: 10.1074/jbc.272.7.4006. [DOI] [PubMed] [Google Scholar]
- Flotow H, Roach PJ. Role of acidic residues as substrate determinants for casein kinase I. J Biol Chem. 1991;266:3724–3727. [PubMed] [Google Scholar]
- Fox HL, Pham PT, Kimball SR, Jefferson LS, Lynch CJ. Amino acid effects on translational repressor 4E-BP1 are mediated primarily by L-leucine in isolated adipocytes. Am J Physiol. 1998;275:C1232–C1238. doi: 10.1152/ajpcell.1998.275.5.C1232. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC. PI3K: Downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Gingras A-C, Svitkin Y, Belsham GJ, Pause A, Sonenberg N. Activation of the translational suppressor 4E-BP1 following infection with encephalomyocarditis virus and poliovirus. Proc Natl Acad Sci. 1996;93:5578–5583. doi: 10.1073/pnas.93.11.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A-C, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes & Dev. 1998;12:502–513. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- Haystead TA, Haystead CM, Hu C, Lin TA, Lawrence JC., Jr Phosphorylation of PHAS-I by mitogen-activated protein (MAP) kinase. Identification of a site phosphorylated by MAP kinase in vitro and in response to insulin in rat adipocytes. J Biol Chem. 1994;269:23185–23191. [PubMed] [Google Scholar]
- Heesom KJ, Avison MB, Diggle TA, Denton RM. Insulin-stimulated kinase from rat fat cells that phosphorylates initiation factor-4E binding protein 1 on the rapamycin-insensitive site (serine-111) Biochem J. 1998;336:39–48. doi: 10.1042/bj3360039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW. eIF4G: A multipurpose ribosome adapter? Science. 1997;275:500–501. doi: 10.1126/science.275.5299.500. [DOI] [PubMed] [Google Scholar]
- Hoekstra MF. Responses to DNA damage and regulation of cell cycle checkpoints by the ATM protein kinase family. Curr Opin Genet Dev. 1997;7:170–175. doi: 10.1016/s0959-437x(97)80125-6. [DOI] [PubMed] [Google Scholar]
- Hu C, Pang S, Kong X, Velleca M, Lawrence JC., Jr Molecular cloning and tissue distribution of PHAS-I, an intracellular target for insulin and growth factors. Proc Natl Acad Sci. 1994;91:3730–3734. doi: 10.1073/pnas.91.9.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleijn M, Scheper GC, Voorma HO, Thomas AAM. Regulation of translation initiation factors by signal transduction. Eur J Biochem. 1998;253:531–544. doi: 10.1046/j.1432-1327.1998.2530531.x. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Barthel A, Kovacina KS, Boge A, Wallach B, Summers SA, Birnbaum MJ, Scott PH, Lawrence JC, Jr, Roth RA. Construction and characterization of a conditionally active version of the serine/threonine kinase Akt. J Biol Chem. 1998;273:11937–11943. doi: 10.1074/jbc.273.19.11937. [DOI] [PubMed] [Google Scholar]
- Lawrence JC, Jr, Fadden P, Haystead TA, Lin TA. PHAS proteins as mediators of the actions of insulin, growth factors and cAMP on protein synthesis and cell proliferation. Adv Enzyme Regul. 1997;37:239–267. doi: 10.1016/s0065-2571(96)00016-7. [DOI] [PubMed] [Google Scholar]
- Lin TA, Kong X, Saltiel AR, Blackshear PJ, Lawrence JC., Jr Control of PHAS-I by insulin in 3T3-L1 adipocytes. Synthesis, degradation, and phosphorylation by a rapamycin-sensitive and mitogen-activated protein kinase-independent pathway. J Biol Chem. 1995;270:18531–18538. doi: 10.1074/jbc.270.31.18531. [DOI] [PubMed] [Google Scholar]
- Marcotrigiano, J., A.-C. Gingras, N. Sonenberg, and S.K. Burley. 1999. Cap–dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol. Cell. (in press). [DOI] [PubMed]
- Matsuo H, Li H, McGuire AM, Fletcher CM, Gingras A-C, Sonenberg N, Wagner G. Structure of translation factor eIF4E bound to m7GDP and interaction with 4E-binding protein. Nat Struct Biol. 1997;4:717–724. doi: 10.1038/nsb0997-717. [DOI] [PubMed] [Google Scholar]
- Merrick WC, Hershey JWB. The pathway and mechanism of eukaryotic protein synthesis. In: Hershey JWB, Mathews M, Sonenberg N, editors. Translational control. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 31–69. [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Nanahoshi M, Nishiuma T, Tsujishita Y, Hara K, Inui S, Sakaguchi N, Yonezawa K. Regulation of protein phosphatase 2A catalytic activity by alpha4 protein and its yeast homolog TAP42. Biochem Biophys Res Commun. 1998;251:520–526. doi: 10.1006/bbrc.1998.9493. [DOI] [PubMed] [Google Scholar]
- Pause A, Belsham GJ, Gingras A-C, Donzé O, Lin TA, Lawrence JC, Jr, Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- Polunovsky VA, Rosenwald IB, Tan A, White J, Chiang L, Sonenberg N, Bitterman PB. Translational control of programmed cell death: Eukaryotic translation initiation factor 4E blocks apoptosis in growth-factor-restricted fibroblasts with physiologically expressed or deregulated Myc. Mol Cell Biol. 1996;16:6573–6581. doi: 10.1128/mcb.16.11.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raught B, Gingras A-C. eIF4E is regulated at multiple levels. Int J Biochem Cell Biol. 1999;31:43–57. doi: 10.1016/s1357-2725(98)00131-9. [DOI] [PubMed] [Google Scholar]
- Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998;58:4375–4382. [PubMed] [Google Scholar]
- Scott PH, Lawrence JC., Jr Attenuation of mammalian Target of Rapaycin activity by increased cAMP in 3T3-L1 cells. J Biol Chem. 1998;272:34496–34501. doi: 10.1074/jbc.273.51.34496. [DOI] [PubMed] [Google Scholar]
- Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC., Jr Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci. 1998;95:7772–7777. doi: 10.1073/pnas.95.13.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N. Translation factors as effectors of cell growth and tumorigenesis. Curr Opin Cell Biol. 1993;5:955–960. doi: 10.1016/0955-0674(93)90076-3. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Gingras A-C. The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr Opin Cell Biol. 1998;10:268–275. doi: 10.1016/s0955-0674(98)80150-6. [DOI] [PubMed] [Google Scholar]
- Thomas G, Hall MN. TOR signalling and control of cell growth. Curr Opin Cell Biol. 1997;9:782–787. doi: 10.1016/s0955-0674(97)80078-6. [DOI] [PubMed] [Google Scholar]
- van der Geer P, Luo K, Sefton BM, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis on cellulose thin-layer plates. In: Celis JE, editor. Cell biology: A laboratory handbook. San Diego, CA: Academic Press; 1994. pp. 422–448. [Google Scholar]
- von Manteuffel SR, Gingras A-C, Ming XF, Sonenberg N, Thomas G. 4E-BP1 phosphorylation is mediated by the FRAP-p70s6k pathway and is independent of mitogen-activated protein kinase. Proc Natl Acad Sci. 1996;93:4076–4080. doi: 10.1073/pnas.93.9.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Manteuffel SR, Dennis PB, Pullen N, Gingras A-C, Sonenberg N, Thomas G. The insulin-induced signalling pathway leading to S6 and initiation factor 4E binding protein 1 phosphorylation bifurcates at a rapamycin-sensitive point immediately upstream of p70s6k. Mol Cell Biol. 1997;17:5426–5436. doi: 10.1128/mcb.17.9.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Campbell LE, Miller CM, Proud CG. Amino acid availability regulates p70 S6 kinase and multiple translation factors. Biochem J. 1998;334:261–267. doi: 10.1042/bj3340261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JD, Affolter M, Krebs DL, Wange RL, Samelson LE, Aebersold R. Identification by electrospray ionization mass spectrometry of the sites of tyrosine phosphorylation induced in activated Jurkat T cells on the protein tyrosine kinase ZAP-70. J Biol Chem. 1994;269:29520–29529. [PubMed] [Google Scholar]
- Xu G, Kwon G, Marshall CA, Lin TA, Lawrence JC, Jr, McDaniel ML. Branched-chain amino acids are essential in the regulation of PHAS-I and p70 S6 kinase by pancreatic beta-cells. A possible role in protein translation and mitogenic signaling. J Biol Chem. 1998;273:28178–28184. doi: 10.1074/jbc.273.43.28178. [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- Yates D, Jr, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]