

Abstract
Schizosaccharomyces pombe cells have a well-defined pattern of polarized growth at the cell ends during interphase and divide symmetrically into two equal-sized daughter cells. We identified a gene, pom1, that provides positional information for both growth and division in S. pombe. pom1 mutants form functioning growth zones and division septa but show several abnormalities: (1) After division, cells initiate growth with equal frequencies from either the old or the new end; (2) most cells never switch to bipolar growth but instead grow exclusively at the randomly chosen end; (3) some cells mislocalize their growth axis altogether, leading to the formation of angled and branched cells; and (4) many cells misplace and/or misorient their septa, leading to asymmetric cell division. pom1 encodes a putative protein kinase that is concentrated at the new cell end during interphase, at both cell ends during mitosis, and at the septation site after mitosis. Small amounts of Pom1p are also found at the old cell end during interphase and associated with the actin ring during mitosis. Pom1p localization to the cell ends is independent of actin but requires microtubules and Tea1p. pom1 mutations are synthetically lethal with several other mutations that affect cytokinesis and/or the actin or microtubule cytoskeleton. Thus, Pom1p may position the growth and cytokinesis machineries by interaction with both the actin and microtubule cytoskeletons.
Keywords: S. pombe, morphogenesis, polarity, actin, microtubules, cell division
The generation of a specific cell shape and three-dimensional organization is crucial for cell function in both unicellular and multicellular organisms. A central aspect of cellular morphogenesis is polarization of the cell along an appropriate axis. Polarization depends on positional information within the cell and is critical for both cell division and development (Strome 1993; Lehmann 1995; Drubin and Nelson 1996). The mechanisms that generate and maintain cell polarity are not yet well understood, but they presumably involve positional signals from within or outside the cell, signal transduction elements, and the cytoskeleton. For example, in the budding yeast Saccharomyces cerevisiae, polarization of the cytoskeleton toward the bud site involves both cortical positional markers and GTPase signaling modules that communicate the positional information to the cytoskeleton (Pringle et al. 1995; Drubin and Nelson 1996).
In the fission yeast Schizosaccharomyces pombe, positional information is required both for polarized growth at the cell ends and for proper positioning of the centrally located plane of cell division. S. pombe cells grow as cylindrical rods of constant diameter by elongating at the cell ends. After cell division, both daughter cells initiate polarized growth by elongating exclusively at the end that existed in the mother cell prior to division (the old end). In early G2 phase, the end generated by the preceding cell division (the new end) also begins to grow. This transition from unipolar to bipolar growth is known as new end take off (NETO; Mitchison and Nurse 1985). After mitosis, cytokinesis and septum formation occur at a site that is normally midway between the cell ends. The sites of cell growth and division are reflected by the distribution of the actin cytoskeleton (Marks and Hyams 1985; Marks et al. 1986). During interphase, F-actin patches are concentrated at the growing cell end(s). At mitosis, cell elongation ceases, and actin redistributes to the cell center to form a cortical ring, and later adjacent patches, in preparation for division. After division, actin relocates to the old ends of the two daughter cells, where polarized growth is reinitiated.
The microtubule cytoskeleton also appears to be involved in the positioning of polarized growth in S. pombe: Mutations in tubulin genes or treatment with microtubule-disrupting drugs leads to the formation of bent and/or branched cells (Umesono et al. 1983; Hiraoka et al. 1984; Yaffe et al. 1996). During interphase, cytoplasmic microtubules are organized longitudinally in the cell, with many appearing to run from end to end (Hagan and Hyams 1988). At the onset of mitosis, these microtubules are replaced by spindle microtubules, and, after nuclear division, new cytoplasmic microtubules are formed from two microtubule-organizing centers at the cell center.
Recent genetic analyses of morphogenesis in S. pombe have identified several interesting types of mutants (Snell and Nurse 1993, 1994; Nurse 1994; Verde et al. 1995). For example, the ban and tea mutants are defective in the orientation of growth polarization; they can still polarize their growth machinery, but they form bent and/or T-shaped cells. Recently, Mata and Nurse (1997) have reported that Tea1p is directed by microtubules to the two cell ends, where it may act as a positional marker for the growth machinery. In contrast, the products of the mid1 (or dmf1) and pos genes are involved in determining the site of cell division. Mutants defective in these genes place septa at seemingly random positions and orientations, rather than at the cell center (Chang et al. 1996; Edamatsu and Toyoshima 1996; Sohrmann et al. 1996). Mid1p localizes to the nucleus during interphase but forms a central ring at the cell cortex at mitosis; this ring might direct the cell division machinery to the cell center (Sohrmann et al. 1996).
In this report, we describe a novel gene, pom1, that encodes a predicted protein kinase that is involved in selecting the sites for both polarized growth and cell division in S. pombe.
Results
Isolation of morphogenetic mutants
To identify S. pombe mutants with defects in morphogenesis, 7100 individual clones of mutagenized cells were screened by fluorescence microscopy as described in Materials and Methods. Forty mutants were identified that had interesting morphogenetic phenotypes caused by single mutations. Some of these mutants had phenotypes similar to those of previously described mutants, such as the formation of round, bent, or branched cells (see introductory section). Among these, six mutants that formed T-shaped cells all proved to contain mutations in the previously described tea1 and tea2 genes (data not shown). Other mutants showed novel alterations of cell shape, septum formation, and/or cell-wall deposition. One mutant harbored a recessive mutation that we named pom1-1 (polarity misplaced); it showed defects in the positioning and orientation of division septa, as well as a few angled and T-shaped cells (Fig. 1A). The abnormal septum localization in the pom1-1 mutant resembled that of mid1 mutants (Chang et al. 1996; Sohrmann et al. 1996), but pom1-1 segregated independently of mid1 as well as of various other mutations affecting morphogenesis or cytokinesis, including tea1, tea2, ban1, ban2, ban3, and ban4 (data not shown). The remainder of this report describes our analysis of the pom1 gene and its product.
Figure 1.

(A) Abnormalities in cell shape and cell division in pom1-1 mutant cells. Strain JB100 cells growing exponentially at 30°C were stained with Calcofluor and bisBenzimide to visualize septa and DNA. (Asterisks) Cells with branched or angled growth; (arrows) cells with septa in abnormal positions and/or orientations; (arrowheads) cells with longitudinal or irregular septa. (B–D) Abnormal septum placement in pom1-Δ1 mutant cells. (B) Cells of wild-type strain 972 (left panels) and pom1-Δ1 strain JB110 (right panels) growing exponentially at 36°C were stained with Calcofluor and bisBenzimide (upper panels) and with rhodamine–phalloidin to visualize F-actin (lower panels). (a, b, and c) Cells with different degrees of septum misplacement, as used for the quantitative analysis in C. (C) Septation patterns were scored in >300 cells from each culture shown in B. (Left) Septum position was scored relative to the long axis of the cell as divided into five equal compartments (see examples in B). (Right) Septum angle was scored relative to the long axis of the cell. Septa with angles of 80° to 90° were designated as orthogonal and septa with smaller angles as tilted. Septa that looked very aberrant (e.g., consisting of several sheets) or were placed longitudinally in the cell (see examples in A) were scored as irregular. (D) JB110 cells growing exponentially at 30°C were treated with FITC-conjugated lectin to stain their cell walls, then washed and grown for 45 min before staining with Calcofluor (left). The cell ends that had grown during the 45 min after labeling with lectin can be identified by their unlabeled surface (right) and are marked with asterisks (left).
Cloning of pom1, a novel protein kinase gene
To clone the gene defined by the pom1-1 mutation, we exploited the synthetic lethality of the pom1-1 dmf1-6 and pom1-1 cdc14-118 double mutants (see below and Materials and Methods). Several plasmids with overlapping inserts rescued the viability of both double mutants as well as the morphological phenotypes of the pom1-1 single mutant. Partial sequencing revealed that the inserts were derived from a region near cdc25 on chromosome I that had been sequenced as part of the S. pombe genome project (cosmid c2F7; EMBL/GenBank/DDBJ accession no. Z50142). Crossing of pom1-1 (JB99) and cdc25-22 (JB20) strains showed that the two genes are linked (26 wild-type segregants among 460 segregants analyzed), suggesting that the isolated plasmids indeed contained pom1 and not a multicopy suppressor. Subcloning experiments showed that ORF SPAC2F7.03c was responsible for the rescue of pom1-1 (data not shown). This ORF was deleted by use of a PCR-generated fragment containing the ura4+ marker (see Materials and Methods). The phenotype of the resulting pom1-Δ1 mutant was indistinguishable from that of pom1-1 (see below), and pom1-Δ1 did not complement pom1-1 in a diploid strain (data not shown). Thus, SPAC2F7.03c is pom1.
pom1 encodes a predicted protein of 1087 amino acids with a putative protein kinase catalytic domain near its carboxyl terminus (Fig. 2). This domain shows all 12 of the major conserved protein kinase subdomains (Hanks and Hunter 1995), including an ATP-binding site (amino acids 705–728). No other significant sequence motifs have been detected in Pom1p. pom1 does not contain any consensus splicing sites, suggesting that it has no introns, a conclusion supported by the sequences of partial cDNA clones (see Materials and Methods). The closest Pom1p homolog found in the databases was another predicted S. pombe protein kinase (accession no. Q09815; SPAC16C9.07). The two proteins are 55% identical in their kinase domains but otherwise share only a short region of homology immediately upstream of the kinase domains (33% identity between amino acids 592–698 of Pom1p and the corresponding region in the SPAC16C9.07 product). Other protein kinases with strong homology to Pom1p are rat Dyrk (Kentrup et al. 1996), human Dyrk2 and Dyrk3 (accession nos. Y13493 and Y12735), the Caenorhabditis elegans F49E11.1 product (accession no. Z70308), S. cerevisiae Yak1p (Garrett and Broach 1989), and the Drosophila and human Minibrains (Tejedor et al. 1995; Smith et al. 1997), which show 43% to 52% identity to Pom1p in their kinase domains. These proteins belong to an emerging novel family of protein kinases with potential dual specificity (Kentrup et al. 1996). Minibrain is involved in postembryonic neurogenesis and is implicated in learning defects associated with Down Syndrome (Tejedor et al. 1995; Smith et al. 1997; and references cited therein). Outside the kinase domains, only Dyrk2, Dyrk3, the F49E11.1 product, and Minibrain show any homology to Pom1p (31% to 47% identity over 29–73 amino acids immediately upstream of the kinase domain). Notably, no homology other than to the kinase domain was found with any protein predicted by the S. cerevisiae genome sequence.
Figure 2.

Predicted sequence of Pom1p (accession no. Z50142). The kinase domain (amino acids 699–995) is shown in boldface type.
Mislocalization and misorientation of septa in pom1 mutant cells
pom1-Δ1 mutants (strains JB109 and JB110) are viable at temperatures from 20°C to 36°C and showed no obvious defects in mating, meiosis, or sporulation; they did, however, have generation times ∼20% longer than those of wild type (data not shown). As with the pom1-1 mutant, the most obvious phenotype of pom1-Δ1 cells was frequent misplacement and/or misorientation of the septum (Fig. 1B,C). The misplaced septa colocalized with equivalently misplaced actin rings (Fig. 1B), as expected from the role of actin in septation. To investigate the positioning of the misplaced septa relative to the cell ends, we visualized the growing ends and septa simultaneously (see Materials and Methods). This revealed a strong bias in septum positioning: Among 122 cells with asymmetrically positioned septa, 119 had the septum positioned closer to the nongrowing (or less extensively growing) end (as illustrated in Fig. 1D). This bias was also evident in time-lapse studies (Fig. 3, 43 to 263 min).
Figure 3.

Time-lapse analysis of the growth of a pom1-1 strain. JB100 cells growing at room temperature in YE medium were observed by DIC microscopy (see Materials and Methods). The times in minutes since the beginning of observation are indicated. Individual cells are numbered for reference in the text. (Arrows) Growing cell ends.
In contrast to the septa, nuclei appeared to be positioned normally at the cell center during interphase, and to divide normally, in pom1 mutant cells (Figs. 1A,B and 5C). Time-lapse observations (Fig. 3) suggested that the mislocalized and misoriented septa are generally functional for cell division, a conclusion supported by the observations that the percentages of cells with visible septa or with actin rings in the pom1 mutants are similar to those in wild type (Fig. 4B and data not shown). However, anucleate cells were not observed in the pom1 mutants, suggesting that septa forming anucleate compartments (e.g., Fig. 1B, cell c) may not be cleaved.
Figure 4.

Abnormal polarization of growth in pom1-Δ1 strains. (A) Strains 972 (wild type) and JB110 (pom1-Δ1) were grown at 36°C to 5 × 106 cells/ml, then stained with rhodamine–phalloidin to visualize F-actin organization. Strains JB20 (cdc25-22) and JB120 (pom1-Δ1 cdc25-22) were grown to the same titer at 25°C, then shifted to 36°C for 3 hr before staining. (B) Cells from the populations shown in A were scored for unipolar or bipolar actin localization, as were cells of strains SP622 (cdc10–V50) and JB121 (pom1-Δ1 cdc10–V50) (grown as described for the cdc25-22 strains in A). For strains 972 and JB110, the percentages of cells with an actin ring were also scored. In the other strains, these percentages were <5%. At least 250 cells of each strain were evaluated. (C) Strains SP622, JB121, JB20, and JB120 were grown at 25°C to 5 × 106 cells/ml, shifted to 36°C for 4 hr, and stained with Calcofluor. (D) Cells (⩾250) from each population shown in C were scored for the presence or absence of new-end growth.
The time-lapse studies also showed that the asymmetric cell divisions can lead to a pronounced asynchrony in the subsequent cell divisions of the unequal-sized daughter cells (e.g., Fig. 3, cells 1 and 2). On average, however, pom1 mutant cells are only ∼10% shorter than wild-type cells at cell division (data not shown), suggesting that cell size regulation is largely normal.
Mislocalization of polarized growth in pom1 mutant cells
Wild-type cells grow initially at one end but later (after NETO; see introductory section) grow at both ends. In contrast, cell-surface staining (Fig. 1D) and time-lapse observations (Fig. 3, arrows) suggested that pom1 mutant cells grow only at one end until septum formation. To explore further the apparent defect in the switch to bipolar growth, we examined the organization of actin in pom1 mutant cells. Long wild-type cells consistently have F-actin patches concentrated at both cell ends (Fig. 4A,B). In contrast, long pom1 mutant cells showed predominantly unipolar actin staining (Fig. 4A,B). To ask whether the switch to bipolar growth was blocked or merely delayed in the cell cycle, we used a temperature-sensitive cdc25 mutation. The cdc25 single mutant becomes arrested in G2 after NETO (Mitchison and Nurse 1985), as reflected by the localization of actin to both ends of the cells (Fig. 4A,B). In contrast, a pom1 cdc25 double mutant showed unipolar actin localization (Fig. 4A,B), suggesting that the switch to bipolar growth is permanently blocked by the loss of Pom1p function.
The time-lapse studies revealed another interesting aspect of the pom1 mutant phenotype. In wild-type cells, it is always the old end that initiates growth after cell division. In contrast, in pom1 mutant cells, either the old end (Fig. 3, cell 6, 263–312 min) or the new end (Fig. 3, cells 5, 7, and 8, 346–487 min) could initiate growth. These few examples also suggest that there is no necessary correlation between the ends selected for growth by the two daughter cells generated by one cell division.
To explore further this apparent defect in the selection of the growth pole, we stained cdc10 and pom1 cdc10 mutant cells with Calcofluor, which reveals the division scars as weakly stained regions of the cell wall (Mitchison and Nurse 1985). At restrictive temperature, cdc10 mutants arrest in G1 prior to NETO (Mitchison and Nurse 1985); thus, actin polarization (Fig. 4B) and cell growth occur only at the old end, and the most recent division scar is seen as a dark hemisphere at the new cell end (Fig. 4C,D). The pom1 cdc10 double mutant also polarized to just one end (Fig. 4B); however, that end could be either the old end (division scar thus seen at the new end) or the new end (division scar thus displaced from the new end by growth at that end), in approximately equal frequency (Fig. 4C,D). Similar results were obtained with cdc25 and pom1 cdc25 mutant strains. As noted above, the cdc25 single mutant undergoes NETO, so that the division scar is invariably displaced from the new end (Fig. 4C). In contrast, the pom1 cdc25 double mutant, which grows only at one end (see above), could grow either at the old end (Fig. 4C) or the new end (not shown), in approximately equal frequency (Fig. 4D). Thus, cells lacking Pom1p appear to pick one end at random for growth after cell division and then, because of the defect in switching to bipolar growth, grow exclusively at this end until the next cell division.
Although most pom1 mutant cells picked one or the other end for growth after division, about 5% of cells mislocalized their growth axis altogether and formed angled or T-shaped cells (Figs. 1A and 5A,B). This phenotype was more dramatic in a pom1 cdc11 double mutant. At restrictive temperature, the cdc11 mutation causes a defect in cytokinesis, but the nuclear and growth cycles continue (Nurse et al. 1976; Mitchison and Nurse 1985). cdc11 mutant cells grow at both ends after each mitosis, as reflected by the actin cytoskeleton, which forms rings during mitosis and cycles back to the cell ends during interphase (Marks et al. 1986). Presumably, after each mitosis, cdc11 mutant cells must decide where to reinitiate growth. In the absence of Pom1p, these decisions are often aberrant, leading to the formation of cells that are branched and often wildly so (Fig. 5A,B). Staining for actin showed only one or two apparent growth zones per cell (data not shown), suggesting that the branching is caused by mislocalization of polarity in successive cell cycles and not by simultaneous activation of multiple growing ends. This interpretation is supported by the increase over time in the percentage of branched cells in the pom1 cdc11 mutant (Fig. 5B); no such increase in cell branching over time was observed in pom1 cdc10 or pom1 cdc25 double mutants (Fig. 4A,C, and data not shown), in which repeated mitotic cycles did not occur.
Figure 5.

Mislocalization of growth poles and of microtubule-organizing centers in pom1-Δ1 strains. (A) Cells of strains JB110 (pom1-Δ1), JB22 (cdc11-136), and JB122 (pom1-Δ1 cdc11-136) were grown at 25°C to 5 × 106 cells/ml, shifted to 36°C for 4.5 hr, and stained with Calcofluor. (B) 600 cells from each population shown in A were scored for the extent of cell branching as a function of time after the shift to 36°C. The “branched” cell population in the pom1-Δ1 single mutant consisted mainly (∼90%) of angled cells (see A, cell 1); the rest were T-shaped cells, the majority of which (∼90%) had a division scar at each end (see A, cell 2). The cdc11-136 single mutant showed <1% cell branching at all times. (C) Cells of strains 972 and JB110 were grown at 30°C to 1 × 107 cells/ml, then fixed and stained with anti-tubulin antibodies and with bisBenzimide to visualize DNA. Some cells are numbered for reference in the text. (Arrowhead and arrows) Post-anaphase microtubule-organizing centers.
Microtubule organization in pom1 mutant cells
Mutations or drugs that compromise microtubule function also cause bending and branching of S. pombe cells (see introductory section). Therefore, we examined the microtubule cytoskeleton in pom1-Δ1 cells. The mutant cells displayed apparently normal arrays of interphase cytoplasmic microtubules (Fig. 5C, cells labeled 1) and normal mitotic spindles (Fig. 5C, cell 2). However, the mutant cells differed from wild-type cells in the placement of the post-anaphase microtubule-organizing centers, which nucleate the new cytoplasmic microtubules. In wild-type cells, these structures appear in the center of the cell (Fig. 5C, arrowhead; Hagan and Hyams 1988), but in pom1 mutants they are frequently displaced from the center (Fig. 5C, arrows). The displaced microtubule-organizing centers coincide with the mislocalized septa as visualized by differential-interference contrast (DIC) microscopy (data not shown).
Genetic interactions of pom1
To gain additional insights into the function of Pom1p, we investigated the possible genetic interactions (synthetic lethality, suppression, or epistasis) between pom1-Δ1 and other mutations affecting cytoskeletal function and/or cytokinesis (by use of strains indicated in Table 1, below). Interestingly, we observed synthetic lethal interactions between pom1-Δ1 and mutations affecting both the actin and microtubule cytoskeletons. In particular, pom1-Δ1 and the cold-sensitive act1-48 mutation (McCollum et al. 1996) were synthetically lethal at all temperatures tested (18°C to 36°C). In addition, pom1-Δ1 cdc3-6 (profilin; Balasubramanian et al. 1994) and pom1-Δ1 cdc8-110 (tropomyosin; Balasubramanian et al. 1992) double mutants grew slowly at 23°C and failed to grow at 30°C, temperatures at which the cdc3-6 and cdc8-110 single mutants grew well. Similarly, a pom1-Δ1 nda3–KM311 (β-tubulin; Hiraoka et al. 1984) double mutant failed to grow at 28°C, a temperature at which the cold-sensitive nda3–KM311 single mutant grew well. In addition, a pom1-Δ1 ban5-3 (α2-tubulin; Adachi et al. 1986; Yaffe et al. 1996) double mutant grew very slowly at 30°C and 36°C, temperatures at which the ban5-3 single mutant grew well. pom1-Δ1 also showed a temperature-sensitive synthetic lethality with the cytokinesis mutations cdc14-118 (Nurse et al. 1976; Fankhauser and Simanis 1993) and dmf1-6 (an allele of mid1; Sohrmann et al. 1996). A pom1-Δ1 tea1Δ double mutant was viable at temperatures from 23°C to 36°C but showed an additive phenotype of strong cell branching (like the tea1Δ single mutant; Mata and Nurse 1997) and misplaced septa (like the pom1-Δ1 single mutant; see above). No obvious genetic interactions were seen between pom1-Δ1 and arp3-c1 (actin-related protein; McCollum et al. 1996).
Table 1.
S. pombe strains used in this study
| Strain
|
Genotype
|
Reference/Source
|
|---|---|---|
| 972 | wild-type h− | Leupold (1970) |
| JB12 | leu1-32 h+ | J. Kohli, |
| JB16 | ade6–M210/ade6–M216 ura4–D18/ura4–D18 h−/h+ | this studya |
| JB20 | cdc25-22 h− | Thuriaux et al. (1980) |
| SP622 | cdc10–V50 ura4–D18 h− | Reymond et al. (1992) |
| JB22 | cdc11-136 leu1-32 h− | Nurse et al. (1976) |
| JB23 | cdc14-118 leu1-32 h− | Nurse et al. (1976) |
| JB24 | cdc3-6 h− | Nurse et al. (1976) |
| JB26 | cdc8-110 h− | Nurse et al. (1976) |
| JB30 | nda3–KM311 leu1-32 h− | Umesono et al. (1983) |
| JB31 | tea1-1 leu1-32 ura4–D18 h+ | Snell and Nurse (1994) |
| JB32 | ban5-3bade6–M210 leu1-32 h− | Verde et al. (1995) |
| JB40 | dmf1-6 leu1-32 h− | Sohrmann et al. (1996) |
| JB50 | tea1Δ leu1-32 ura4-D18 h− | Mata and Nurse (1997) |
| KGY1010 | act1-48 leu1-32 lys1-131 ura4–D18 h− | McCollum et al. (1996) |
| YDM188 | arp3–c1 ade6–M210 leu1-32 ura4–D18 h− | McCollum et al. (1996) |
| JB99 | pom1-1 h+ | see text |
| JB100 | pom1-1 leu1-32 h− | see text |
| JB101 | pom1-1 cdc14-118 leu1-32 h− | JB23 × JB99 |
| JB102 | pom1-1 dmf1-6 leu1-32 h− | JB40 × JB99 |
| JB109 | pom1-Δ1 ade6–M216 ura4–D18 h+ | see text |
| JB110 | pom1-Δ1 ura4–D18 h− | 972 × JB109 |
| JB111 | pom1–3HA h− | see text |
| JB112 | pom1–3HA h+ | see text |
| JB113 | pom1–3HA cdc10–V50 h− | SP622 × JB112 |
| JB114 | pom1–3HA cdc25-22 h− | JB20 × JB112 |
| JB115 | pom1–GFPS65T h− | see text |
| JB120 | pom1-Δ1 cdc25-22 ura4–D18 h− | JB20 × JB109 |
| JB121 | pom1-Δ1 cdc10–V50 ura4–D18 h− | SP622 × JB109 |
| JB122 | pom1-Δ1 cdc11-136 ura4–D18 h− | JB22 × JB109 |
| JB130 | pom1–3HA nda3–KM311 h− | JB30 × JB112 |
| JB131 | pom1–3HA tea1-1 h− | JB31 × JB111 |
| JB132 | pom1–3HA ban5-3b h− | JB32 × JB112 |
Derived by mating two haploid strains obtained from J. Kohli.
ban5-3 is an allele of atb2 (Adachi et al. 1986; Yaffe et al. 1996), one of the two genes for α-tubulin in S. pombe.
Localization of Pom1p
We used three methods to investigate the intracellular localization of Pom1p. First, we generated polyclonal antibodies specific for Pom1p (see Materials and Methods). In addition, we tagged the 3′ end of pom1 (under its own promoter and in its normal genomic location) with sequences encoding either a triple hemagglutinin epitope (3HA) or green fluorescent protein (GFP) (see Materials and Methods). The growth rates and cell morphologies of strains containing the tagged genes were indistinguishable from those of wild-type strains (data not shown), indicating that the tagged genes are functional. Immunofluorescence staining of wild-type cells with the Pom1p-specific antibodies, immunofluorescence staining of pom1–3HA cells with HA-specific antibodies, and observations on pom1–GFP cells all revealed the same patterns of Pom1p localization, as described below. The failure of pom1-Δ1 cells to stain with the Pom1p-specific antibodies (Fig. 6B) further confirmed the specificity of the staining. Because the tagged strains yielded stronger and more reproducible Pom1p localization signals than did the Pom1p-specific antibodies, we used these strains for most of the studies described below.
Figure 6.
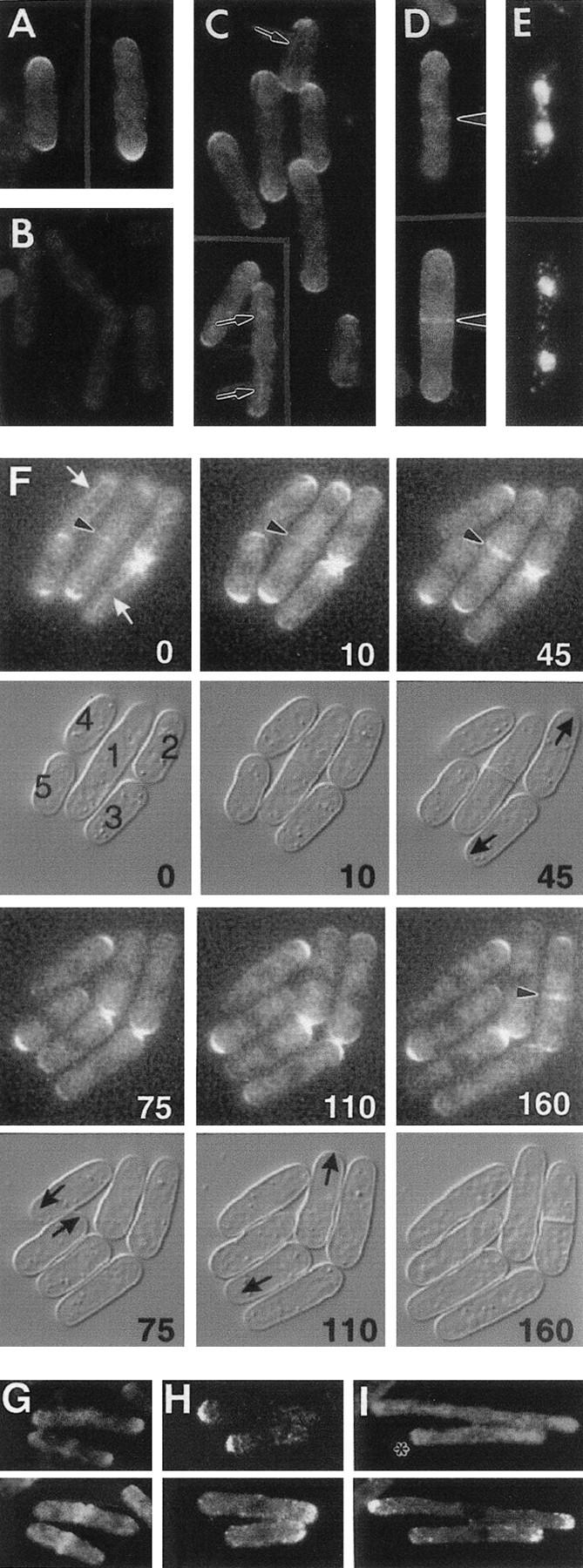
Localization of Pom1p. (A–E) Immunolocalization. All strains were grown at 30°C. (A,B) Wild-type strain 972 (A) and pom1-Δ1 strain JB110 (B) were stained with antibodies to Pom1p. (C–E) The pom1–3HA strain JB111 was stained with HA-specific antibodies (C) or double-stained with HA-specific antibodies (D) and bisBenzimide to visualize the DNA (E). Arrows and arrowheads indicate structures discussed in the text. (F) Time-lapse study of Pom1p–GFP localization through the cell-cycle. Strain JB115 was grown at room temperature in EMM medium and observed by fluorescence and DIC microscopy (see Materials and Methods). The times in minutes since the beginning of observation are indicated. Individual cells are numbered for reference in the text. (White arrows) Filamentous structures; (arrowheads) central Pom1p; (black arrows) growing ends. (G–I) Comparison of Pom1p and actin localization in wild-type and mutant strains. Cells expressing Pom1p–3HA were double-stained with antibodies to HA (top) and actin (bottom). (G) Strain JB111 (wild-type except for pom1–3HA) was grown at 30°C to exponential phase before staining. (H,I) cdc10-V50 strain JB113 (H) and cdc25-22 strain JB114 (I) were grown at 25°C to 5 × 106 cells/ml, then shifted to 36°C for 3 hr before staining.
Pom1p was observed at both cell ends in most cells (Fig. 6A,C,F), although one end usually had a stronger signal than the other. In some cells, Pom1p could also be observed in association with filaments (possibly microtubules; see below) running along the cell (Fig. 6C, arrows; Fig. 6F, white arrows). In addition, Pom1p was observed at the center of cells undergoing division (Fig. 6D,F, arrowheads). This signal was very faint in cells early in nuclear division (Fig. 6D,E, upper cell; Fig. 6F, cell 1, 0 min) but became much stronger as nuclear division and septation were completed (Fig. 6D,E, lower cell; Fig. 6F, cell 1, 10–45 min). The central Pom1p signal always spanned the full cell diameter (i.e., it did not appear to contract) until after cell division, when it marked the new ends of both daughter cells (Fig. 6F, cell 1, 45 and 75 min).
The cell cycle dynamics of Pom1p localization were revealed most clearly by time-lapse studies of living pom1–GFP cells. In addition to the progressive changes in Pom1p signal at the cell center (see above), it was apparent that the new ends of cells had a high concentration of Pom1p at the time of birth and through much, if not all, of interphase (Fig. 6F, cell 1 daughters, 75–160 min; cells 2 and 3, 0–110 min). (Note that cells 4 and 5 were apparently nonsister cells that had happened to settle into the medium with their old ends adjacent.) The concentration of Pom1p seemed to be high and approximately equivalent at the two cell ends during the early stages of septum formation (Fig. 6F, cell 1, 10 min; cell 2, 110–160 min) and then decreased while the concentration at the cell center increased as cell division proceeded (Fig. 6F, cell 1, 10–75 min).
Actin independence of Pom1p localization
Although Pom1p and actin both localize to the cell ends and to the cell center, their patterns of localization through the cell cycle differ, as shown by comparison of the Pom1p localization described above to the previously described distribution of actin (see introductory section) and by double-staining experiments (Fig. 6G–I). In wild-type cells, when actin relocalized from the cell ends to the cell center in preparation for division, Pom1p was still found predominantly at the cell ends, although a small amount of Pom1p colocalized with the central actin (Fig. 6G). In interphase cells, there was typically a negative correlation between the amount of Pom1p and the amount of actin at a particular end, which reflects, at least in part, the high concentration of Pom1p at the new end (Fig. 6F) and the high concentration of actin at the old end (Marks and Hyams 1985) in recently divided cells. This negative correlation was also apparent in cdc10 and cdc25 mutant backgrounds. In cdc10–V50 cells, where actin was concentrated only at the old end, Pom1p was found almost exclusively at the new end (Fig. 6H). In cdc25-22 cells, where actin was typically concentrated at both ends (Fig. 4A,B; Fig. 6I), only small amounts of Pom1p were typically seen at either end (Fig. 6I). In some cdc25-22 cells, particular ends had lower concentrations of actin; such ends typically also had more distinct concentrations of Pom1p (Fig. 6I, asterisk).
These results suggested that Pom1p localization does not depend on actin. To test this hypothesis, we treated cells with the inhibitor latrunculin-A (LAT-A), which quickly and completely disrupts the S. cerevisiae actin cytoskeleton (Ayscough et al. 1997). S. pombe cells treated for 3 hr with LAT-A had no remaining visible actin structures (Fig. 7A, right), but Pom1p was still localized to the cell ends (Fig. 7A, left), indicating that Pom1p localization to the cell ends is indeed independent of actin. In contrast, Pom1p was not observed at the cell centers in LAT-A-treated cells, suggesting that actin is required (directly or indirectly) for localization of Pom1p to the division site. In some LAT-A-treated cells, small amounts of Pom1p were also observed in association with filament-like structures (Fig. 7A, arrows), as noted also in untreated cells (Fig. 6C,F). Thus, these structures do not appear to be actin filaments.
Figure 7.

Effects of cytoskeletal inhibitors and mutations on Pom1p localization. All strains expressed Pom1p-3HA and were stained with antibodies to HA. Arrows and arrowheads indicate structures discussed in the text. (A) Strain JB111 (wild type) was grown at 30°C to 2 × 106 cells/ml, and LAT-A was added to 100 μm. After 3 hr, cells were stained separately for Pom1p and for F-actin (using rhodamine–phalloidin). (B) Strain JB111 was grown at 30°C to 5 × 106 cells/ml and TBZ was added to 100 μg/ml (right panels). A control culture (left panels) received no TBZ. After 1 hr, aliquots of cells from each culture were double-stained for Pom1p (top panels) and DNA (middle panels) or stained separately for microtubules (bottom panels). (C) Strain JB130 (nda3-KM311; left panel) was grown at 32°C to 5 × 106 cells/ml and shifted to 20°C for 1.5 hr before staining for Pom1p. Strain JB132 (ban5-3; right panel) was grown at 25°C to 5 × 106 cells/ml and shifted to 36°C for 3 hr before staining for Pom1p. (D) Strain JB131 (tea1-1) was grown at 25°C to 5 × 106 cells/ml, shifted to 36°C for 3.5 hr, and stained for Pom1p. (E) Strains 972 (wild type) and JB110 (pom1-Δ1) growing exponentially at 30°C were fixed and stained with antibodies to Tea1p.
Microtubule dependence of Pom1p localization
To ask whether Pom1p localization depends on the microtubule cytoskeleton, we depolymerized microtubules using thiabendazole (TBZ; Umesono et al. 1983). Treatment of cells with TBZ for ⩾60 min caused a drastic shortening of microtubules and essentially complete loss of Pom1p from the cell ends (Fig. 7B), and a significant decrease in the Pom1p staining of cell ends could be detected within 10 min of TBZ addition (data not shown). Thus, the polar localization of Pom1p appears to depend directly or indirectly on microtubules. In contrast, some TBZ-treated cells still showed Pom1p staining at the cell centers (Fig. 7B, right, arrowhead), indicating either that this aspect of Pom1p localization is microtubule independent or that the short residual microtubules near the cell nucleus are sufficient for it. In some TBZ-treated cells, Pom1p staining was observed along short filaments (Fig. 7B, right, arrows) that were probably residual spindle microtubules, as judged from their colocalization with the nuclear DNA. This observation suggests that Pom1p has some affinity for microtubules and that the filaments seen in untreated cells (Figs. 6, C and F, and 7, A and B, left, arrows) may represent microtubules. Indeed, the positions of some of these Pom1p-stained filaments suggested that they might be spindles (Fig. 7B, left, arrows), raising the possibility that a fraction of Pom1p localizes to the spindle during mitosis.
To confirm that the loss of Pom1p localization from the cell ends in TBZ-treated cells was caused by disassembly of microtubules and not by some other effect of the drug, we also examined conditional nda3 and atb2 mutants. These tubulin mutations caused a more nearly complete loss of microtubules than did TBZ treatment (data not shown). As expected, both mutants failed to localize Pom1p to the cell ends (Fig. 7C); in addition, the mutant cells did not show detectable Pom1p at the cell center (see Discussion).
Relationship between Pom1p and Tea1p localization
Like pom1 mutants, tea1 mutants form branched cells (Snell and Nurse 1994; Verde et al. 1995), and Tea1p also localizes to the cell ends in a microtubule-dependent manner (Mata and Nurse 1997). Thus, we examined the relationship between Pom1p and Tea1p localization. In a tea1 mutant, Pom1p was not observed at the cell ends at either 25°C (data not shown) or 36°C (Fig. 7D), although some cells did show weak Pom1p signals along filaments (Fig. 7D, arrow) and/or at the cell center (Fig. 7D, arrowheads). In contrast, Tea1p localization appeared normal in a pom1-Δ1 strain (Fig. 7E). Thus, Tea1p localization does not require Pom1p, but at least some aspects of Pom1p localization depend directly or indirectly on Tea1p.
Discussion
This report describes a novel S. pombe gene, pom1, whose product is required for the proper positioning of growth zones and division septa but not for polarized growth or division per se. Although the available data do not yet allow a comprehensive model of Pom1p function, there are intriguing clues to its role in each of the processes in which it is implicated.
Role of Pom1p in distinguishing the old from the new cell end
An important problem in S. pombe morphogenesis is how a newborn cell distinguishes its old end (which will grow immediately) from its new end (which will grow only after a delay). Pom1p is the first protein shown to play a critical role in this process: In pom1 mutants, newborn cells initiate growth from either end with equal frequency. Strikingly, in newborn wild-type cells, Pom1p is highly concentrated at the new end and virtually absent from the old end. This suggests that the high concentration of Pom1p may mark the new end as such and provide a negative signal for assembly of the growth machinery at that end. However, a negative signal from Pom1p cannot be the only mechanism determining the selection of a growth pole, because wild-type cells faithfully begin growth from the old end and not from the sides of the cell, and even pom1 mutant cells usually begin growth from one end or the other.
Role of Pom1p in defining the ends of the cell
About 5% of pom1 mutant cells show a defect in orientation of the growth axis relative to the ends and sides of the cell, leading to the formation of angled and T-shaped cells. Similar aberrations are more frequent in the tea1 and tea2 mutants (Verde et al. 1995; Mata and Nurse 1997), suggesting that the cytoplasmic microtubule/Tea1p (and perhaps Tea2p) system (Mata and Nurse 1997) plays the primary role in defining the ends of the cell and that Pom1p plays a secondary role, perhaps as one of several factors communicating positional information from the cytoplasmic microtubule/Tea protein system to the actin cytoskeleton (see also below). In this regard, the extensively branched cells produced by the cdc11 pom1 double mutant are of interest. As a cdc11 mutant reorganizes growth poles after each mitosis and failed cytokinesis (Mitchison and Nurse 1985; Marks et al. 1986), the extensive branching of the cdc11 pom1 double mutant may reflect primarily the inappropriate resumption of growth from “new ends” (actually the centers of the cells because of the failure of cytokinesis) after mitosis in the absence of Pom1p and only secondarily the difficulties of Pom1p-deficient cells in orienting the growth axis relative to the ends and sides of the cell. Consistent with this interpretation, cdc10 pom1 and cdc25 pom1 double mutants, which do not undergo repeated cycles of mitosis and failed cytokinesis, do not branch upon extended arrest, although the cells are less straight than those of cdc10 and cdc25 single mutants. Taken together, the results suggest that a major role of Pom1p is in proper polarity re-establishment after mitosis.
Role of Pom1p in NETO and the function of bipolar growth
During interphase, Pom1p is required for the initiation of a second growth pole at the new end (NETO): pom1 mutant cells continue to grow at the one randomly selected end until division. It is possible that NETO is normally triggered by a loss of the negative signal hypothesized to prevent growth at the new end immediately after division; although the concentration of Pom1p at the new end seems to remain high until well after NETO, the protein might be inactivated at the appropriate stage in the cell cycle. However, in this case, one would expect a pom1 mutant to initiate bipolar growth immediately after cell division. Thus, it seems more likely that Pom1p plays a positive role in the establishment of bipolar growth; for example, a change in Pom1p substrate specificity at the appropriate cell cycle stage might simultaneously eliminate the negative signal and produce a positive signal for recruitment of the growth machinery to the new end. Further studies will be needed to discriminate among these and other possible models.
Whatever the actual role of Pom1p in NETO and the establishment of bipolar growth, the viability of the pom1-Δ1 mutant demonstrates that NETO is not essential for viability or cell-cycle progression. This conclusion is supported by observations on other mutants (tea1, orb2, and ban2) with NETO defects (Verde et al. 1995). However, the switch to bipolar growth may provide for more efficient cell elongation. On average, pom1 mutant cells are about 10% shorter than wild-type cells during septation. Moreover, pom1 cdc25 double-mutant cells are clearly shorter on average than cdc25 single-mutant cells (Fig. 4A,C). In addition, bipolar growth may contribute to the cell’s ability to distinguish its ends from its sides: It was striking that most T-shaped pom1 mutant cells had a division scar at each of the original cell ends (such as would result from old end growth, without NETO, followed by division), suggesting that ends that had not grown were not recognized efficiently as appropriate sites for polarized growth.
Role of Pom1p in the positioning and orientation of division planes
The phenotypes of pom1 mutants and of the other mutants (mid1 and pos1, pos2, and pos3) with misplaced division sites suggest some general conclusions about the mechanisms governing the positioning and orientation of division planes in S. pombe. First, all of the mutants produce septa that are misoriented (i.e., not orthogonal to the long axis of the cell) as well as misplaced (Chang et al. 1996; Edamatsu and Toyoshima 1996; Sohrmann et al. 1996; this study); this suggests that the mechanisms for positioning and orienting division planes are related. Second, at least in the mid1 and pom1 mutants, the post-anaphase microtubule-organizing centers are also misplaced (Chang et al. 1996; this study), suggesting that their placement is governed by the same signal(s) that govern the placement of division sites. Finally, in both mid1 and pom1 mutants (this is less clear for the pos mutants; see Edamatsu and Toyoshima 1996), septa that would form anucleate cells do not seem to be cleaved (Chang et al. 1996; this study), raising the possibility of a checkpoint that monitors whether there is a nucleus on either side of the septation site (Chang et al. 1996).
Despite such insights, the mechanisms controlling septum placement in S. pombe remain poorly understood. Among several possible models, the most attractive at present is that the position of the nucleus determines the position of the division site (Chang and Nurse 1996). In this regard, it is of interest that Pom1p was not detected in or associated with the nucleus (except for the possible association of some Pom1p with spindle microtubules). However, Pom1p might be involved in receiving and/or transmitting a signal (perhaps carried by Mid1p; see introductory section and Sohrmann et al. 1996) that emanates from the nucleus. Whether or not the signal for positioning the division site originates in the nucleus, Pom1p might be involved either in directing components of the cytokinesis machinery to the cell center or in anchoring them there. Consistent with these possibilities, a faint Pom1p signal was detected at the center of cells at the beginning of nuclear division, which is approximately the time at which the medial actin ring also appears. However, this ring constricts during septation (Jochová et al. 1991; McCollum et al. 1995; Kitayama et al. 1997), whereas the centrally located Pom1p signal spans the full cell diameter both during and after septum formation. Thus, Pom1p is unlikely to be a structural component of the medial actin ring.
It is also striking that in all of the known mutants with misplaced septa, the nuclei themselves are positioned normally at the cell center during interphase and appear to divide normally. It is possible that in all of these mutants, the hypothetical signal either is not sent by the nucleus or cannot be received by the cell cortex, and that in the absence of a signal, the division machinery is free to assemble at an abnormal position. However, these observations also might indicate that the positional information for division does not in fact come from the nucleus.
Another challenge for the nuclear-signal model is posed by the observation that pom1 mutants preferentially place their septa closer to the nongrowing end of the cell. It is not yet clear whether such a bias also exists in the other mutants with defects in septum placement. This bias could be reconciled with the model by supposing that the cortical site is marked by the nucleus during interphase and then gets displaced from the cell center by the continuing unipolar growth of the pom1 mutant cells. In this case, Pom1p might actually play no direct role in selection of the division site, and the septum-localization defect would be merely an indirect effect of the failure of NETO. (The movement of Pom1p to the cell center might in this case be only a step toward the marking of the new end after division.) However, early marking of the cortical site is difficult to reconcile with the behavior of other mutants such as wee1, tea1, orb2, and ban2, which divide symmetrically despite growing mainly from one end (Mitchison and Nurse 1985; Verde et al. 1995; Chang and Nurse 1996). Moreover, if Pom1p plays no direct role in selection of the division site, it is hard to understand why septum orientation as well as septum placement is perturbed in pom1 mutants and why pom1 mutations show genetic interactions with mutations affecting cytokinesis.
Interactions of Pom1p with the actin and microtubule cytoskeletons
In S. pombe, actin is required for polarized growth and the cytoplasmic microtubules are involved in the positioning of such growth (see introductory section). Thus, a possible role for Pom1p is in the signal chain that communicates positional information from the microtubules to the actin cytoskeleton. Consistent with this hypothesis, Pom1p requires microtubules, but not actin, for localization to the cell ends, and some Pom1p may localize along microtubules. Moreover, pom1 mutations show synthetic-lethal interactions both with tubulin mutations and with actin cytoskeleton mutations. Pom1p appears to be a protein kinase, and it might function by phosphorylating one or more components of the actin cytoskeleton. However, this general view of Pom1p function faces the difficulty that although Pom1p and actin both localize to the same regions of the cell, they do not do so synchronously. Indeed, at the cell ends, the concentration of actin is typically high when that of Pom1p is low, and vice versa. There are at least three possible explanations for these observations. First, the level of Pom1p protein kinase activity may not always simply reflect the abundance of the protein. Second, as noted above, the specificity of Pom1p may be altered in response to cell cycle signals, so that it could provide both positive and negative signals to the actin cytoskeleton at different times. Finally, as the localization of Pom1p is dynamic, it could be marking sites at one time that are used only later; for example, the Pom1p that accumulates transiently at the old end prior to septation may mark this site for actin assembly in the next cell cycle.
Examination of events at the cell center also does not reveal a simple story. A faint Pom1p signal is observed at about the same time that the actin ring forms, but it is not clear that the Pom1p is present in time to direct actin to that site. Subsequently, actin patches become highly concentrated at the cell center before the major increase in Pom1p concentration at that site. Moreover, in pom1 mutants, actin rings form and actin patches become concentrated at the septation sites, even when these are misplaced. Finally, it is not clear whether microtubules, actin, or both are involved in localizing Pom1p to the cell center. In addition to the cytoplasmic and spindle pole body-associated microtubules present earlier in the cell cycle (Hagan and Hyams 1988; Ding et al. 1997), a microtubule ring colocalizes with the medial actin ring during nuclear division (Pichová et al. 1995). We did not detect Pom1p at the cell center in tubulin mutant cells, but some TBZ-treated cells still showed central Pom1p localization. This localization might have been attributable to residual microtubules in the TBZ-treated cells; alternatively, the failure of central Pom1p localization in the mutants might be an indirect effect of cell cycle blockage resulting from the loss of microtubules. We also did not detect central Pom1p staining in LAT-A-treated cells; it is not yet clear whether this reflects a direct or indirect effect of the loss of actin function.
Functional relationship of Pom1p and Tea1p
Like Pom1p, Tea1p requires microtubules for localization to the cell ends (Mata and Nurse 1997). However, the detailed patterns of Pom1p and Tea1p localization differ. Moreover, although Tea1p requires continuous microtubule function to maintain its localization at the cell ends (Mata and Nurse 1997), Pom1p delocalizes and relocalizes only slowly in response to microtubule disruption and recovery, respectively (see Results; J. Bähler, unpubl.), and it remains concentrated at the cell ends when the cytoplasmic microtubules disassemble at the time of spindle formation. In addition, Tea1p, unlike Pom1p, localizes to the tips of microtubules and directly affects microtubule organization (Mata and Nurse 1997). Thus, the mechanisms for delivery and/or retention of Pom1p and Tea1p to the cell ends may differ.
Interestingly, Pom1p requires Tea1p for localization to the cell ends, whereas Tea1p localization is independent of Pom1p. This suggests that Pom1p acts downstream of Tea1p at the cell ends. In this case, tea1 mutants should show the same abnormalities as do pom1 mutants. Indeed, there are striking similarities in the mutant phenotypes, such as the formation of T-shaped cells and the NETO defects (Verde et al. 1995; Mata and Nurse 1997). Moreover, some tea1 cells seem to initiate growth at the new end after cytokinesis (J. Mata and P. Nurse, pers. comm.). However, Tea1p does not appear to function exclusively through Pom1p, as tea1 mutants have the additional phenotypes of abnormal microtubule organization and more prevalent cell branching (Mata and Nurse 1997). Conversely, Pom1p appears to have some functions that are independent of Tea1p, as Pom1p localizes to the cell center (although, interestingly, not to high levels) in the absence of Tea1p, and tea1 mutants are not defective in septum placement. Moreover, a tea1 pom1 double mutant shows an additive phenotype, consistent with the hypothesis that Tea1p and Pom1p act in part in a common pathway, but that each protein has additional independent function(s).
Conclusions
Pom1p is involved in providing positional information both for polarized growth and for cell division in S. pombe. Both processes involve assembly of actin and associated proteins at defined sites, and Pom1p may transmit positional information from the microtubule to the actin cytoskeleton. However, at present the exact role of Pom1p in directing the organization of the actin cytoskeleton is not clear; it may involve negative signals, positive signals, or both. It will be important to identify proteins that interact with Pom1p, either as regulators or as substrates, and to examine the role and cell cycle regulation of the putative protein kinase activity. As signaling pathways involving protein kinases may also be important in the organization of the actin and/or microtubule cytoskeletons in other eukaryotes, elucidation of the mechanisms of Pom1p function should be of general interest for an understanding of eukaryotic cellular morphogenesis.
Materials and methods
Strains, growth conditions, and inhibitor methods
The S. pombe strains used are listed in Table 1; all are isogenic to 972 (Leupold 1970). Standard growth media were used (Moreno et al. 1991); except where noted, cells for experiments were grown at 30°C in EMM minimal medium. Growth rates were determined both in EMM and in YE rich liquid medium at 23°C, 30°C, and 36°C; exponentially growing cultures (<5 × 106 cells/ml) were diluted twofold, and the times required for return to the original absorbances were measured. For experiments with microtubule and actin inhibitors, cells were grown to early exponential phase (<3 × 106 cells/ml). TBZ (Sigma; final concentration 20–100 μg/ml) or LAT-A (Molecular Probes; final concentration 100 μm) was then added, and the culture was incubated further before examination.
Genetic and molecular biology methods
Standard genetic and recombinant DNA methods (Moreno et al. 1991; Sambrook et al. 1989) were used except where noted. Yeast transformations were performed by use of a lithium acetate method (Keeney and Boeke 1994). DNA was prepared from bacteria and isolated from agarose gels by use of Qiagen kits and from yeast cells as described by Hoffman and Winston (1987). DNA was sequenced by the UNC–Chapel Hill Automated Sequencing Facility. Oligonucleotide primers were obtained from Integrated DNA Technologies.
Mutant screen
Strain 972 was grown in EMM medium to stationary phase and mutagenized with 300 μm nitrosoguanidine (Sigma) for 60 min (∼10% survival) as described by Moreno et al. (1991). Individual colonies of mutagenized cells were picked into 20-μl aliquots of water in the wells of microtiter plates. Cells were then transferred with a multiprong device onto two YE plates and two plates of YE containing 6% ethanol (Jimenez and Oballe 1994); one plate of each type was incubated at 25°C and one at 36°C. All plates contained 2 μg/ml phloxin B, which accumulates in dead cells (Kohli et al. 1977). Growth of the clones was scored after three days. Cells were then transferred with the multiprong device from the YE + ethanol plates at 36°C to microtiter plates with 50 μl of water in each well, and 10-μl aliquots of the clonal cell suspensions were then spotted with a multipipettor onto gelatin-coated slides, stained with Calcofluor, and examined by fluorescence microscopy (Zahner et al. 1996). Clones that showed aberrations in cell shape, cell division, and/or Calcofluor staining pattern were recovered for further analysis from the YE plates that had been kept at 25°C. The original pom1-1 mutant was backcrossed four times to wild-type strains JB12 and 972, yielding strains JB99 and JB100, before detailed characterization.
Cloning, deletion, and tagging of pom1
To clone pom1, we exploited the observations that the pom1-1 dmf1-6 and pom1-1 cdc14-118 double mutants are viable (although slow growing) at 23°C but inviable at 30°C. Double mutants that also carried leu1-32 (strains JB101 and JB102) were grown at 23°C and transformed with a S. pombe genomic DNA library (a gift from P. Young, Queens University, Ontario, Canada) constructed in plasmid pWH5 (Wright et al. 1986). Leu+ transformants were grown for 12 hr at 23°C and then shifted to 30°C for three days. Plasmid DNA was recovered from three transformants that showed plasmid-dependent rescue. These plasmids had identical or overlapping inserts as judged from restriction digests, and all three complemented both of the double mutants as well as the pom1-1 single mutant (strain JB100) upon retransformation. The ORF responsible for complementation of pom1-1 was identified by gel purification of different restriction fragments of the original inserts and cotransformation of these fragments into strains JB101 and JB102 along with plasmid pREP3 (Maundrell 1993). (Many transformants selected for the pREP3 selectable marker also have the cotransformed fragment integrated in the genome.) In addition, cDNA clones containing partial pom1 sequences (codons 696–1087) were recovered from two different libraries (Fikes et al. 1990; Kelly et al. 1993) by colony hybridization.
pom1 was deleted by use of a PCR-generated fragment (Bähler et al. 1998). The S. pombe ura4+ gene was amplified by use of plasmid KS–ura4 (which contains ura4+ as a 1.8-kb HindIII fragment in the HindIII site of pBluescript KS−; a gift from S. Parisi, University of Bern, Switzerland) as template, a forward primer that contained 76 nucleotides from the 5′-end of pom1 (from position −55 to +21 relative to the start of the ORF) and the 24 nucleotides of the M13 forward primer, and a reverse primer that contained 76 nucleotides from the 3′-end of pom1 (the last five codons of the ORF and the 61 nucleotides immediately downstream) and the 24 nucleotides of the M13 reverse primer. The PCR product was purified by phenol extraction and transformed into diploid strain JB16 under selection for Ura+. Genomic DNAs from 16 transformants were checked by PCR for integration of ura4+ at the pom1 locus, using the forward primer (see above) together with a primer corresponding to nucleotides 78–105 downstream of the pom1 stop codon. Six transformants produced a PCR fragment of the expected size (1.9 kb). Tetrads were dissected from four of these transformants. Most tetrads yielded four viable spores, and all of the four-spored tetrads showed a 2:2 segregation of Ura+:Ura− that correlated with the presence of the pom1 phenotype and the PCR product diagnostic for correct integration. Strain JB109 is one of these segregants; its deletion allele was designated pom1-Δ1 and used in further analyses. The success of the deletion construction was also confirmed by Southern blotting (data not shown).
Pom1p was tagged at its carboxyl terminus with GFP carrying the S65T mutation (Heim et al. 1995) or with a 3HA epitope by direct chromosomal integration of PCR-generated fragments (Bähler et al. 1998). Sequences encoding each tag together with a G418-resistance marker were amplified with plasmids pFA6a–GFP(S65T)–kanMX6 and pFA6a–3HA–kanMX6 as templates. The two primers had 80-bp tails corresponding to the regions just upstream and 200–280 bp downstream of the pom1 stop codon. The PCR fragments were then used to transform strain 972. Transformants were selected on YE plates containing 100 μg/ml G418 (Geneticin; Life Technologies), and both junctions were checked for correct integration by PCR using primers hybridizing to the kanamycin marker and primers hybridizing 2400 bp upstream or 400 bp downstream of the pom1 stop codon. For each construct, one strain showing a PCR reaction diagnostic for correct integration was analyzed by crossing to strain JB12 and dissecting tetrads. This revealed a 2:2 segregation of G418 resistance in both cases, indicating that each marker had integrated at a single site in the genome. Strains JB111, JB112, and JB115 are segregants from these crosses.
Generation of Pom1p-specific antibodies
A 878-bp fragment of pom1 (codons 283–575) with BclI and EcoRI sites at the upstream end and BclI and XhoI sites at the downstream end was PCR amplified by use of the Expand System with primers 5′-CTATTCCTCCTAATATGATCAATGAATTCGATCACCAACATCCTAAAGCAAATATATC-3′ and 5′-CATTTGAAGACTCGAGAGTGATCAAAGGCTGTAATGATTCACTCGTATCAG-3′ (restriction sites underlined). The PCR product was digested with EcoRI and XhoI or with BclI and cloned into the corresponding sites of plasmids pMAL–c2 (New England Biolabs) and pGEX3X (Pharmacia), respectively, to generate fusions of Pom1p to MalE and glutathione S-transferase (GST). The Pom1p fusion proteins were expressed in Escherichia coli strain DH5α (Life Technologies). Attempts to purify the GST fusion protein with glutathione–agarose beads resulted in extensive degradation. Therefore, full-length GST–Pom1p was obtained by separation of whole-cell extracts by SDS-PAGE. The gel slices containing the fusion protein were used to inject two rabbits by use of standard procedures (Cocalico Biologicals). Antibodies were affinity purified as described previously (Pringle et al. 1991) by use of nitrocellulose strips containing MalE-Pom1p and elution with 4.5 m MgCl2.
Microscopy
Cells were observed by fluorescence microscopy using a Nikon Microphot SA microscope with a 60× Plan-apo objective. F-actin was stained with rhodamine-conjugated phalloidin (Sigma) as described by Balasubramanian et al. (1997). To visualize actin together with septa and nuclear DNA, cells were stained with rhodamine–phalloidin as described by Alfa et al. (1993) with the addition of 50 μg/ml Calcofluor (Sigma) to the staining solution. Cells were then examined in mounting medium containing 0.05 μg/ml bisBenzimide (Sigma). Birth scars were visualized by staining with Calcofluor as described by Verde et al. (1995). To visualize growth zones by use of FITC-conjugated Bandeiraea simplicifolia lectin (May and Mitchison 1986), cells grown in EMM medium were resuspended in EMM plus 5 μg/ml FITC-lectin (Sigma, L-9381) for 5 min, washed twice in EMM, and incubated further in EMM in the dark for 30–60 min. Cells were then counterstained with Calcofluor (1 mg/ml in ice-cold 1% NaCl) and observed.
To visualize 3HA-tagged Pom1p by immunofluorescence, exponentially growing cells were fixed with 3.8% formaldehyde for 30 min in EMM medium, washed three times with PEM (100 mm Na-PIPES at pH 6.9, 1 mm EGTA, 1 mm MgSO4) (Hagan and Hyams 1988), and digested for 15 min at 30°C in PEM plus 1 m sorbitol (PEMS) containing 0.5 mg/ml yeast lytic enzyme (ICN Biochemicals) and 0.1 mg/ml lysing enzymes (Sigma, L2773). Cells were washed once in PEMS plus 1% Triton X-100 and three times in PEM, resuspended in PEMBAL (PEM plus 0.1 m l-lysine, 1% BSA, 0.1% sodium azide), and shaken gently for 30 min. Cells were then applied to multiwell slides as described previously (Pringle et al. 1991) and incubated for 16 hr at room temperature with the monoclonal HA11 antibody (Berkeley Antibody Co.) diluted 1:200 in PEMBAL. After three 10-min washes with PEMBAL, FITC-tagged anti-mouse-IgG secondary antibodies (Jackson ImmunoResearch) diluted 1:200 in PEMBAL were applied for 4 hr at room temperature. Cells were again washed three times with PEMBAL and then observed in mounting medium as described above. For double staining of Pom1p and actin, cells were treated with −20°C methanol and acetone for 6 min and 30 sec, respectively, before incubation with the primary antibodies. Goat antibodies to S. cerevisiae actin (a gift from J. Cooper, Washington University, St. Louis, MO) were diluted 1:100 in PEMBAL, applied to the cells together with the HA11 antibody, and detected by use of TRITC-tagged anti-goat-IgG antibodies (Jackson ImmunoResearch) diluted 1:200 in PEMBAL and applied to the cells together with the FITC-tagged anti-mouse-IgG antibodies. Photographs of double-stained cells were taken using a Zeiss laser-scanning confocal microscope with a 63× objective and were analyzed by Zeiss and Adobe Photoshop software.
Immunofluorescence with anti-Pom1p antibodies followed the same protocol as just described. Affinity-purified antibodies were diluted 1:20 to 1:50 in PEMBAL and detected by use of FITC-conjugated anti-rabbit-IgG secondary antibodies (Jackson ImmunoResearch) diluted 1:200 in PEMBAL. To visualize Tea1p, cells were fixed with −70°C methanol for 30 min, stained with anti-Tea1p antibodies (Mata and Nurse 1997) diluted 1:30 in PEMBAL, and detected with anti-rabbit-IgG antibodies as described above. To stain microtubules with the monoclonal anti-tubulin antibody TAT1 (Woods et al. 1989), cells were fixed using a combination of formaldehyde and glutaraldehyde and then treated with sodium borohydride as described previously (Hagan and Hyams 1988). TAT1 was diluted 1:10 in PEMBAL, and FITC-tagged anti-mouse-IgG secondary antibodies were used as described above.
To observe GFP-tagged Pom1p in living cells, cells were grown to early exponential phase (<5 × 106 cells/ml), mounted on a thin layer of EMM containing 25% gelatin, sealed under a coverslip with Valap (1:1:1 vaseline/lanolin/paraffin), and observed at room temperature using a Nikon FXA microscope equipped with a Hamamatsu cooled-CCD camera and an Apo 60X/1.4 n.a. objective in combination with a 2× Optivar (Salmon et al. 1994). Images were collected every 1–5 min by a 3-sec exposure of 490-nm light and analyzed using the Universal Metamorph Imaging System. For some time-lapse series of the GFP-tagged cells, digitally enhanced DIC images were recorded in parallel (Yeh et al. 1995; Shaw et al. 1997). Time-lapse studies of non-GFP-tagged cells growing on YE medium were recorded by DIC with the same procedure.
Acknowledgments
We thank F. Chang, J. Cooper, B. Edgar, J. Fikes, K. Gull, J. Kohli, M. Longtine, J. Mata, D. McCollum, A. McKenzie, P. Nurse, S. Parisi, P. Philippsen, V. Simanis, A. Wach, and P. Young for gifts of antibodies, plasmids, and strains; J. Adam, E. Yeh, T. Salmon, A. Steever, and J. Wu for assistance with some experiments; J. Mata and P. Nurse for communicating results prior to publication; S. Whitfield for superb photographic services; and O. Al-Awar, E. Bi, D. Brunner, M. Longtine, J. Mata, D. McCollum, P. Nurse, and K. Sawin for valuable discussions and/or critical reading of the manuscript. This work was supported by National Institutes of Health grant GM31006. J.B. was supported by fellowships from the Swiss National Science Foundation and the Ciba-Geigy-Jubiläums-Stiftung.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jpringle@email.unc.edu; FAX (919) 962-0320.
References
- Adachi Y, Toda T, Niwa O, Yanagida M. Differential expressions of essential and nonessential α-tubulin genes in Schizosaccharomyces pombe. Mol Cell Biol. 1986;6:2168–2178. doi: 10.1128/mcb.6.6.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfa C, Fantes P, Hyams J, McLeod M, Warbrick E. Experiments with fission yeast. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. [Google Scholar]
- Ayscough KR, Stryker J, Pokala N, Sanders M, Crews P, Drubin DG. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J Cell Biol. 1997;137:399–416. doi: 10.1083/jcb.137.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler, J., J.-Q. Wu, M.S. Longtine, N.G. Shah, A. McKenzie III, A.B. Steever, A. Wach, P. Philippsen, and J.R. Pringle. 1998. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast (in press). [DOI] [PubMed]
- Balasubramanian MK, Helfman DM, Hemmingsen SM. A new tropomyosin essential for cytokinesis in the fission yeast S. pombe. Nature. 1992;360:84–87. doi: 10.1038/360084a0. [DOI] [PubMed] [Google Scholar]
- Balasubramanian MK, Hirani BR, Burke JD, Gould KL. The Schizosaccharomyces pombe cdc3+ gene encodes a profilin essential for cytokinesis. J Cell Biol. 1994;125:1289–1301. doi: 10.1083/jcb.125.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian MK, McCollum D, Gould KL. Cytokinesis in the fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1997;283:494–506. doi: 10.1016/s0076-6879(97)83039-x. [DOI] [PubMed] [Google Scholar]
- Chang F, Nurse P. How fission yeast fission in the middle. Cell. 1996;84:191–194. doi: 10.1016/s0092-8674(00)80973-3. [DOI] [PubMed] [Google Scholar]
- Chang F, Woollard A, Nurse P. Isolation and characterization of fission yeast mutants defective in the assembly and placement of the contractile actin ring. J Cell Sci. 1996;109:131–142. doi: 10.1242/jcs.109.1.131. [DOI] [PubMed] [Google Scholar]
- Ding R, West RR, Morphew M, Oakley BR, McIntosh JR. The spindle pole body of Schizosaccharomyces pombe enters and leaves the nuclear envelope as the cell cycle proceeds. Mol Biol Cell. 1997;8:1461–1479. doi: 10.1091/mbc.8.8.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Edamatsu M, Toyoshima YY. Isolation and characterization of pos mutants defective in correct positioning of septum in Schizosaccharomyces pombe. Zool Sci. 1996;13:235–239. doi: 10.2108/zsj.13.235. [DOI] [PubMed] [Google Scholar]
- Fankhauser C, Simanis V. The Schizosaccharomyces pombe cdc14 gene is required for septum formation and can also inhibit nuclear division. Mol Biol Cell. 1993;4:531–539. doi: 10.1091/mbc.4.5.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fikes JD, Becker DM, Winston F, Guarente L. Striking conservation of TFIID in Schizosaccharomyces pombe and Saccharomyces cerevisiae. Nature. 1990;346:291–294. doi: 10.1038/346291a0. [DOI] [PubMed] [Google Scholar]
- Garrett S, Broach J. Loss of Ras activity in Saccharomyces cerevisiae is suppressed by disruptions of a new kinase gene, YAK1, whose product may act downstream of the cAMP-dependent protein kinase. Genes & Dev. 1989;3:1336–1348. doi: 10.1101/gad.3.9.1336. [DOI] [PubMed] [Google Scholar]
- Hagan IM, Hyams JS. The use of cell division cycle mutants to investigate the control of microtubule distribution in the fission yeast Schizosaccharomyces pombe. J Cell Sci. 1988;89:343–357. doi: 10.1242/jcs.89.3.343. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Hunter T. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Hiraoka Y, Toda T, Yanagida M. The NDA3 gene of fission yeast encodes β-tubulin: A cold-sensitive nda3 mutation reversibly blocks spindle formation and chromosome movement in mitosis. Cell. 1984;39:349–358. doi: 10.1016/0092-8674(84)90013-8. [DOI] [PubMed] [Google Scholar]
- Hoffman CS, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- Jimenez J, Oballe J. Ethanol-hypersensitive and ethanol-dependent cdc− mutants in Schizosaccharomyces pombe. Mol Gen Genet. 1994;245:86–95. doi: 10.1007/BF00279754. [DOI] [PubMed] [Google Scholar]
- Jochová J, Rupeš I, Streiblová E. F-actin contractile rings in protoplasts of the yeast Schizosaccharomyces. Cell Biol Int Rep. 1991;15:607–610. doi: 10.1016/0309-1651(91)90007-6. [DOI] [PubMed] [Google Scholar]
- Keeney JB, Boeke JD. Efficient targeted integration at leu1-32 and ura4-294 in Schizosaccharomyces pombe. Genetics. 1994;136:849–856. doi: 10.1093/genetics/136.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TJ, Martin GS, Forsburg SL, Stephen RJ, Russo A, Nurse P. The fission yeast cdc18+ gene product couples S phase to START and mitosis. Cell. 1993;74:371–382. doi: 10.1016/0092-8674(93)90427-r. [DOI] [PubMed] [Google Scholar]
- Kentrup H, Becker W, Heukelbach J, Wilmes A, Schürmann A, Huppertz C, Kainulainen H, Joost H-G. Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J Biol Chem. 1996;271:3488–3495. doi: 10.1074/jbc.271.7.3488. [DOI] [PubMed] [Google Scholar]
- Kitayama C, Sugimoto A, Yamamoto M. Type II myosin heavy chain encoded by the myo2 gene composes the contractile ring during cytokinesis in Schizosaccharomyces pombe. J Cell Biol. 1997;137:1309–1319. doi: 10.1083/jcb.137.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli J, Hottinger H, Munz P, Strauss A, Thuriaux P. Genetic mapping in Schizosaccharomyces pombe by mitotic and meiotic analysis and induced haploidization. Genetics. 1977;87:471–489. doi: 10.1093/genetics/87.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann R. Cell–cell signaling, microtubules, and the loss of symmetry in the Drosophila oocyte. Cell. 1995;83:353–356. doi: 10.1016/0092-8674(95)90111-6. [DOI] [PubMed] [Google Scholar]
- Leupold U. Genetical methods for Schizosaccharomyces pombe. Methods Cell Physiol. 1970;4:169–177. [Google Scholar]
- Marks J, Hyams JS. Localization of F-actin through the cell division cycle of S. pombe. Eur J Cell Biol. 1985;39:27–32. [Google Scholar]
- Marks J, Hagan IM, Hyams JS. Growth polarity and cytokinesis in fission yeast: The role of the cytoskeleton. J Cell Sci Suppl. 1986;5:229–241. doi: 10.1242/jcs.1986.supplement_5.15. [DOI] [PubMed] [Google Scholar]
- Mata J, Nurse P. tea1 and the microtubular cytoskeleton are important for generating global spatial order within the fission yeast cell. Cell. 1997;89:939–949. doi: 10.1016/s0092-8674(00)80279-2. [DOI] [PubMed] [Google Scholar]
- Maundrell K. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene. 1993;123:127–130. doi: 10.1016/0378-1119(93)90551-d. [DOI] [PubMed] [Google Scholar]
- May JW, Mitchison JM. Length growth in fission yeast cells measured by two novel techniques. Nature. 1986;322:752–754. [Google Scholar]
- McCollum D, Balasubramanian MK, Pelcher LE, Hemmingsen SM, Gould KL. Schizosaccharomyces pombe cdc4+ gene encodes a novel EF-hand protein essential for cytokinesis. J Cell Biol. 1995;130:651–660. doi: 10.1083/jcb.130.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollum D, Feoktistova A, Morphew M, Balasubramanian M, Gould KL. The Schizosaccharomyces pombe actin-related protein, Arp3, is a component of the cortical actin cytoskeleton and interacts with profilin. EMBO J. 1996;15:6438–6446. [PMC free article] [PubMed] [Google Scholar]
- Mitchison JM, Nurse P. Growth in cell length in the fission yeast Schizosaccharomyces pombe. J Cell Sci. 1985;75:357–376. doi: 10.1242/jcs.75.1.357. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Nurse P. Fission yeast morphogenesis—posing the problems. Mol Biol Cell. 1994;5:613–616. doi: 10.1091/mbc.5.6.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P, Thuriaux P, Nasmyth K. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol Gen Genet. 1976;146:167–178. doi: 10.1007/BF00268085. [DOI] [PubMed] [Google Scholar]
- Pichová A, Kohlwein SD, Yamamoto M. New arrays of cytoplasmic microtubules in the fission yeast Schizosaccharomyces pombe. Protoplasma. 1995;188:252–257. [Google Scholar]
- Pringle JR, Adams AEM, Drubin DG, Haarer BK. Immunofluorescence methods for yeast. Methods Enzymol. 1991;194:565–602. doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Pringle JR, Bi E, Harkins HA, Zahner JE, De Virgilio C, Chant J, Corrado K, Fares H. Establishment of cell polarity in yeast. Cold Spring Harbor Symp Quant Biol. 1995;60:729–744. doi: 10.1101/sqb.1995.060.01.079. [DOI] [PubMed] [Google Scholar]
- Reymond A, Schmidt S, Simanis V. Mutations in the cdc10 start gene of Schizosaccharomyces pombe implicate the region of homology between cdc10 and SWI6 as important for p85cdc10 function. Mol Gen Genet. 1992;234:449–456. doi: 10.1007/BF00538705. [DOI] [PubMed] [Google Scholar]
- Salmon ED, Inoué T, Desai A, Murray AW. High resolution multimode digital imaging system for mitosis studies in vivo and in vitro. Biol Bull. 1994;187:231–232. doi: 10.1086/BBLv187n2p231. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Shaw SL, Yeh E, Bloom K, Salmon ED. Imaging GFP-fusion proteins in Saccharomyces cerevisiae. Curr Biol. 1997;7:701–704. doi: 10.1016/s0960-9822(06)00299-5. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Stevens ME, Sudanagunta SP, Bronson RT, Makhinson M, Watabe AM, O’Dell TJ, Fung J, Weier H-UG, Cheng J-F, Rubin EM. Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down Syndrome. Nature Genetics. 1997;16:28–36. doi: 10.1038/ng0597-28. [DOI] [PubMed] [Google Scholar]
- Snell, V. and P. Nurse. 1993. Investigations into the control of cell form and polarity: The use of morphological mutants in fission yeast. Development (Suppl.) : 289–299. [PubMed]
- ————— Genetic analysis of cell morphogenesis in fission yeast—a role for casein kinase II in the establishment of polarized growth. EMBO J. 1994;13:2066–2074. doi: 10.1002/j.1460-2075.1994.tb06481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohrmann M, Fankhauser C, Brodbeck C, Simanis V. The dmf1/mid1 gene is essential for correct positioning of the division septum in fission yeast. Genes & Dev. 1996;10:2707–2719. doi: 10.1101/gad.10.21.2707. [DOI] [PubMed] [Google Scholar]
- Strome S. Determination of cleavage planes. Cell. 1993;72:3–6. doi: 10.1016/0092-8674(93)90041-n. [DOI] [PubMed] [Google Scholar]
- Tejedor F, Zhu XR, Kaltenbach E, Ackermann A, Baumann A, Canal I, Heisenberg M, Fischbach KF, Pongs O. minibrain: A new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14:287–301. doi: 10.1016/0896-6273(95)90286-4. [DOI] [PubMed] [Google Scholar]
- Thuriaux P, Sipiczki M, Fantes PA. Genetical analysis of a sterile mutant by protoplast fusion in the fission yeast Schizosaccharomyces pombe. J Gen Microbiol. 1980;116:525–528. doi: 10.1099/00221287-116-2-525. [DOI] [PubMed] [Google Scholar]
- Umesono K, Toda T, Hayashi S, Yanagida M. Two cell division cycle genes NDA2 and NDA3 of the fission yeast Schizosaccharomyces pombe control microtubular organization and sensitivity to anti-mitotic benzimidazole compounds. J Mol Biol. 1983;168:271–284. doi: 10.1016/s0022-2836(83)80018-7. [DOI] [PubMed] [Google Scholar]
- Verde F, Mata J, Nurse P. Fission yeast cell morphogenesis: Identification of new genes and analysis of their role during the cell cycle. J Cell Biol. 1995;131:1529–1538. doi: 10.1083/jcb.131.6.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Sherwin T, Sasse R, MacRae TH, Baines AJ, Gull K. Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J Cell Sci. 1989;93:491–500. doi: 10.1242/jcs.93.3.491. [DOI] [PubMed] [Google Scholar]
- Wright A, Maundrell K, Heyer W-D, Beach D, Nurse P. Vectors for the construction of gene banks and the integration of cloned genes in Schizosaccharomyces pombe and Saccharomyces cerevisiae. Plasmid. 1986;15:156–158. doi: 10.1016/0147-619x(86)90051-x. [DOI] [PubMed] [Google Scholar]
- Yaffe MP, Harata D, Verde F, Eddison M, Toda T, Nurse P. Microtubules mediate mitochondrial distribution in fission yeast. Proc Natl Acad Sci. 1996;93:11664–11668. doi: 10.1073/pnas.93.21.11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Skibbens RV, Cheng JW, Salmon ED, Bloom K. Spindle dynamics and cell cycle regulation of dynein in the budding yeast, Saccharomyces cerevisiae. J Cell Biol. 1995;130:687–700. doi: 10.1083/jcb.130.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahner JE, Harkins HA, Pringle JR. Genetic analysis of the bipolar pattern of bud site selection in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:1857–1870. doi: 10.1128/mcb.16.4.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]