

Abstract
The non-receptor tyrosine kinase Src is a critical regulator of cytoskeletal contraction, cell adhesion, and migration. In normal cells, Src activity is stringently controlled by Csk-dependent phosphorylation of Src(Y530), and by Cullin-5-dependent ubiquitinylation, which affects active Src(pY419) exclusively, leading to its degradation by the proteosome. Previous work has shown that Src activity is also limited by Cdk5, a proline-directed kinase, which has been shown to phosphorylate Src(S75). Here we show that this phosphorylation promotes the ubiquitin-dependent degradation of Src, thus restricting the availability of active Src. We demonstrate that Src(S75) phosphorylation occurs in vivo in epithelial cells, and like ubiquitinylation, is associated only with active Src. Preventing Cdk5-dependent phosphorylation of Src(S75), by site-specific mutation of S75 or by Cdk5 inhibition or suppression, increases Src(Y419) phosphorylation and kinase activity, resulting in Src-dependent cytoskeletal changes. In transfected cells, ubiquitinylation of Src(S75A) is about 35% that of wild-type Src-V5, and its half-life is approximately 2.5-fold greater. Cdk5 suppression leads to a comparable decrease in the ubiquitinylation of endogenous Src and a similar increase in Src stability. Together, these findings demonstrate that Cdk5-dependent phosphorylation of Src(S75) is a physiologically significant mechanism of regulating intracellular Src activity.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-011-0638-1) contains supplementary material, which is available to authorized users.
Keywords: Cdk5, Src, Signal transduction, Ubiquitination, Kinase activity
Introduction
Src is a non-receptor protein tyrosine kinase with a key role in regulating cell-to-matrix adhesion, migration, and junctional stability [1]. Thus, precise regulation of Src activity is critical for normal cell growth. Inactive Src is locked in a “closed” configuration, in which a phosphorylated tyrosine near the C terminus of Src (Y530 in mammalian Src; Y527 in chicken) interacts with the SH2 domain in the N-terminal region and the SH3 domain interacts with a linker region between the SH2 and kinase domains [2]. These intramolecular interactions restrict access to the kinase domain [2]. Dephosphorylation of Y530 is followed by autophosphorylation at Y419, locking Src in an “open,” active configuration [2, 3]. Active Src may be deactivated by rephosphorylation of Y530 by C-terminal Src kinase (Csk) [3, 4, 5]; alternatively, active Src is reported to be irreversibly destroyed by Cullin-5-dependent ubiquitinylation and proteosomal degradation [6, 7]. Only active Src is subject to ubiquitinylation, raising the possibility that active Src itself may initiate the process leading to its own ubiquitinylation [6]. Phosphorylation of active Src by Csk protects Src from irreversible proteolytic degradation and maintains an inactive pool of Src protein that can be rapidly activated when needed. Defects in these regulatory mechanisms result in over-activation of Src and contribute to cell transformation [3, 7, 8].
Src can also be phosphorylated at S75 [9, 10], a site in the “unique” region at the Src N-terminus, but the cellular function and physiological significance of this phosphorylation has remained obscure. S75 is not present in other members of the Src kinase family, but is evolutionarily conserved in Src, suggesting that it may be functionally important. Phosphorylation of this and other Ser/Thr sites in the unique region was first observed in mitotic cells, where it is caused by the mitotic kinase, Cdk1 [9, 11, 12]. However, mitosis-independent phosphorylation of this site was later observed in neurons and in certain tumor cell lines [10]. This phosphorylation was shown to be due to Cdk5 [10], a widely distributed proline-directed kinase with a substrate specificity similar to Cdk1.
Cdk5 is an atypical member of the cyclin-dependent kinase family, which is not involved in cell cycle control [13]. Cdk5 activation requires one of two specific activating proteins, p35 or p39, rather than a member of the cyclin family. Although Cdk5 is most prominently expressed in neuronal cells, it also regulates a variety of physiological functions in non-neuronal cells [14]. Previous studies of Cdk5 in fibroblasts [15], keratinocytes [16], corneal epithelial cells [17], and lens epithelial cells [18, 19, 20] have shown that Cdk5 activity strengthens cell attachment to the extracellular matrix [15, 20]. The effects of Cdk5 on cell adhesion have been linked to a Cdk5-dependent suppression of Src activity, leading to increased Rho activation, myosin phosphorylation, and stabilization of stress fibers [20], and to Cdk5-dependent phosphorylation of talin, which is necessary for stabilizing nascent focal adhesions [15]. The present study was undertaken to explore the mechanism by which Cdk5 regulates Src activity and to test whether Cdk5-dependent phosphorylation of Src(S75) is involved.
Materials and methods
Cell culture and transfection
Human lens epithelial cells (FHL124) were cultured and transfected with Lipofectamine (Invitrogen, Carlsbad, CA) as described previously [19] and incubated 24–48 h before use. For suppression of Cdk5 or Cullin-5 expression, FHL124 cells were transfected with 160 nM of ON-TARGET plus SMARTpool siRNA: Cdk5 [L-003239-00-0005,Thermo Scientific Dharmacon, Chicago, IL, USA)]; Cullin-5 (h,SIH200914D SA Biosciences, Frederick, MD, USA); or with ON-TARGET plus siCONTROL Non-targeting pool, [D-001810-10, Thermo Scientific Dharmacon, Chicago, IL, USA)], and were harvested after 72 h, as indicated. Cdk5 was also suppressed using a small hairpin plasmid targeted to the Cdk5 sequence, 5′-GGG GAG CTG AAA TTG GCT GAT-3′ [21].
Antibodies
The following antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA): Cdk5 polyclonal (C-8), Cdk5 monoclonal (J-3), Src monoclonal (B-12), Src polyclonal (SRC 2), and ubiquitin polyclonal (FL-76). Antibodies for Src(pY416), non-pY419 Src, Src(pY530), GAPDH, and GFP were purchased from Cell Signaling Technology (Beverly, MA, USA). Cullin-5 (ab33053) and Src (pS75) rabbit antibody (ab79308) were produced by Abcam Ltd (Cambridge, UK). V5 monoclonal antibody was from Invitrogen. Anti-rabbit & anti-mouse IgG, horseradish peroxidase-linked secondary antibodies were from Amersham (Louisville, CO, USA).
Immunoprecipitation and immunoblotting
Cells were lysed in 50 mM Tris (pH 7.5), 150 mM NaCl, and 1% Triton X-100 plus Complete™ protease inhibitor (Roche Diagnostics) and protein tyrosine and serine/threonine phosphatase inhibitors (Upstate). Whole-cell lysate containing 20 μg of protein was used for immunoblotting analysis and 0.5 mg was used for immunoprecipitation. Co-immunoprecipitated proteins were eluted by boiling for 5 min in a 1x SDS sample buffer containing 5% (v/v) 2-mercaptoethanol. Proteins were resolved on a NuPage 4–12% BisTris gel (Invitrogen) and immunoblotted with the indicated antibodies, as described previously [22]. Immunoreactive bands were detected using ECL + regents (Amersham) followed by chemiluminescence on X-ray film.
Immunofluorescence
Transiently Src-V5 or S75A-V5 transfected cells were plated and allowed to spread for 2 h on chamber slides coated with 10 μg/ml fibronectin (Sigma, St Louis, MO, USA). Cells were fixed with 4% paraformaldehyde for 10 min, rinsed with phosphate buffered saline (PBS), and blocked with 5% normal goat serum for 1 h. Blocked specimens were incubated with anti-V5 antibody overnight, followed by anti-mouse IgG secondary antibody conjugated with Alexa Fluor 488 (Invitrogen). A 568-nm laser was used for the excitation of rhodamine phalloidin (Invitrogen). Stained sections were photographed using a Zeiss Axioplan II microscope equipped with a CCD camera.
cDNA constructs
Full-length mouse Src cDNA (Catalog #21-114, Upstate Biotechnology, Lake Placid, NY, USA) was cloned into pENTR/D-TOPO vector (Invitrogen) and sequenced in both directions to ensure accurate duplication. The pENTR/D-TOPO vector and Gateway destination vector, pcDNA6.2/cLumio-DEST (Invitrogen), were then combined according to the manufacturer’s protocol to generate a V5-tagged Src clone (Src-V5) carrying both the V5 epitope and Lumio tag at the C-terminus. S75A, S75E, and Y530F amino acid replacements were generated in Src-V5 by site-directed mutation using the QuickChange™ kit (Stratagene, La Jolla, CA, USA). The point mutations were verified by DNA sequencing.
To generate a non-suppressible Cdk5 rescue plasmid, mutations were made to the shRNA binding site (5′-GGG GAG CTG AAA TTG GCT GAT-3′) [21] of GFP-Cdk5 [23] using the Quik Change II Site-Directed Mutagenesis Kit (Stratagene). Mutagenesis was performed to mutate three nucleotides to achieve a neutral change in amino acid coding sequence. Forward oligonucleotide: 5′-CAGGAAT GGC GAG CTC AAA TTA GCT GAT TTTGG-3′. Reverse oligonucleotide: 5′-CCAAA ATC AGC TAA TTT GAG CTC GCC ATT CCT G-3′. The resulting mutated sequence is GGC GAA CTC AAG TTA GCA GAT (underlined nucleotides were changed).
In vitro kinase assays
For in vitro kinase assays of Src-V5 proteins, cells were transfected with Src-V5, Src(S75A)-V5, Src(S75E)-V5, and Src(Y530F)-V5, respectively. After 24 h, proteins were extracted and immunoprecipitated with anti-V5 antibody and protein-G Sepharose beads. Following immunoprecipitation, the beads were washed three times with kinase buffer (50 mM HEPES, 10 mM MgCl2, and 1 mM MnCl2). In vitro kinase reactions were performed by suspending immunoprecipitated proteins on Sepharose beads in 100 μl of kinase buffer containing final concentrations of 300 μM ATP, 10 μCi of [γ-32P]ATP, and 20 μg Src synthetic peptide substrate (Millipore, 17-111). The reaction was incubated at 30°C for 30 min, and stopped by adding 50 μl of 1.5% phosphoric acid solution. Then, 30 μl of reaction supernatant was spotted on P81 cellulose phosphate paper (Whatman), and washed four times with 200 μl of 0.5% phosphoric acid. The radioactivity captured on the P81 paper was quantified in a scintillation counter (Bioscan).
35S-Methionine pulse-chase experiments
Cells were washed with methionine-free DMEM (Gibco BRL) supplemented with 10% dialyzed fetal calf serum and then incubated for 2 h with the wash medium containing 500 μCi/ml 35S methionine (ICN Biomedicals Inc., Costa. Mesa, CA, USA). Cells were subsequently washed and incubated in complete medium. For experiments involving Src-V5 and Src(S75A)-V5, cells were transfected with the appropriate plasmids 24 h before pulse-labeling. At the indicated chase times, cells were lysed in RIPA buffer and lysates prepared as described above. Lysates were precleared with protein A/G Plus-agarose beads (Santa Cruz Biotechnology) for 4 h and adjusted to equal protein concentrations (1.0 mg/ml). Endogenous Src or Src-V5 were immunoprecipitated and resolved on a NuPage 4–12% BisTris gel (Invitrogen), and [35S]-labeled immunoprecipitated proteins were detected by autoradiography. The same nitrocellulose membranes were immunoblotted using rabbit antibody to Src or to V5 (Invitrogen) to detect the corresponding radiolabeled proteins. To correct for variations in transfection efficiency, radiolabeled Src-V5 and Src(S75A)-V5 were quantified by densitometry at each time point and normalized against the immunoblot of V5. A log-linear plot of the percentage of endogenous Src, Src-V5, or Src(S75A)-V5 remaining at each time point was used to determine the half-life.
Pharmacological inhibitors
The Cdk5 inhibitors, olomoucine and roscovitine (Calbiochem, San Diego, CA, USA), were used at a concentrations of 15 and 10 μM, respectively. The proteosome inhibitor MG132, (Biosciences, EMD Chemicals INC., USA) was used at a concentration of 10 μM. The Src inhibitor, PP1 (Enzo Life Sciences Farmingdale, NY, USA) and its inactive homolog, PP3 (Calbiochem), were used at a concentration of 10 μM.
Analysis of Src ubiquitinylation
Transiently transfected cells with Src-V5 or Src(S75A)-V5 were treated with 10 μM proteasome inhibitor, MG132 10 h. Lysates were harvested in RIPA buffer and immunoprecipitated with monoclonal mouse anti-ubiquitin antibody. Immunoprecipitates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted as described above using anti-V5 rabbit polyclonal antibody.
Results
Cdk5 is a specific regulator of Src activity
Our previous studies had shown that pharmacological inhibition of Cdk5 using olomoucine increased Src activity approximately 50% in epithelial cells of the cornea and lens [17, 20]. To confirm the specificity of this effect, we tested alternative methods of inhibiting Cdk5. An alternative pharmacological inhibitor, roscovitine, like olomoucine, increased Src activity (Src(pY419)/total Src) 51.6 ± 10.2% (p < 0.05) (Fig. 1a). Transfecting lens epithelial cells with dominant negative Cdk5 (D144 N) [24] had a similar effect, increasing Src activity 70.6 ± 11.6% (p < 0.05) (Fig. 1b). In addition, suppressing endogenous Cdk5 expression with siRNA, which reduced Cdk5 expression by 90%, also increased endogenous Src activity by 74.0 ± 14.3% (p < 0.05) compared to the control, non-targeting siRNA pool (Fig. 1c). Cdk5 suppression with a small hairpin plasmid, which targets a different region of the Cdk5 mRNA led to a 72.7 ± 10.3% (p < 0.05) increase in Src(pY419). This increase was reversed by expression of a rescue plasmid, which can not be suppressed by the small hairpin plasmid (Fig. 1d), thus verifying that the increase in Src activity was solely due to the loss of Cdk5. The high degree of specificity provided by the dominant negative Cdk5 protein, the siRNA oligonucleotides, the small hairpin plasmid, and the rescue plasmid confirms that endogenous Src activity is specifically regulated by Cdk5.
Fig. 1.

Inhibiting Cdk5 increases Src activity. a Human lens epithelial cells were incubated with or without roscovitine. Cells were lysed and immunoblotted with antibodies against Src(pY419), Src, Cdk5, and GAPDH. b Cells were transfected with dominant negative Cdk5(D144 N), Cdk5, or a GFP control plasmid, and immunoblotted as in a. c Cells were transfected with siRNA oligonucleotides to suppress endogenous Cdk5 (siCdk5), or with non-targeting oligonucleotides (siControl). Cell lysates were immunoblotted as in a. d Cells were transfected with siRNA-resistant Cdk5 rescue plasmid (rCdk5), a small hairpin plasmid to suppress endogenous Cdk5 (shCdk5), or shCdk5 plus rescue plasmid Cdk5, or were mock-transfected (Mock). Cell lysates were immunoblotted as in a
Src-V5 and Src-V5 mutants are properly regulated and suggest a role of Cdk5-dependent S75 phosphorylation
To develop tools to investigate the mechanism of Cdk5-dependent regulation of Src, we constructed V5-tagged expression plasmids bearing V5-Lumio tags at the C-terminus. Since Cdk5 was previously implicated in phosphorylation of Src(S75) [10], site-specific mutations were generated at this site. To test whether the corresponding Src-V5 fusion proteins are properly regulated in the cell, the kinase activity of all constructs was examined by in vitro kinase assay using a peptide substrate, and the results were normalized to amount of V5-tagged protein, as determined by immunoblotting (Fig. 2a). The results indicated that the activity of Src-V5, was 4.53 ± 0. 10% (p < 0.05) that of the constitutively active construct, Src(Y530F)-V5. Since previous studies have indicated that approximately 5–10% of endogenous Src is active under normal physiological conditions [25], Src-V5 appears to be normally regulated with approximately 5% in the active configuration. Interestingly, the kinase assay also indicated that the activity of Src(S75E)-V5 was approximately equal to that of Src-V5, indicating that the presence of the phospho-mimetic amino acid does not affect Src activation. Thus, Src(S75E)-V5, like the wild-type Src-V5, appears to be mostly in the inactive configuration. In contrast, the activity of Src(S75A)-V5, which lacks a phosphorylatable serine at amino acid 75, was 1.96 times greater than Src-V5 (p < 0.05).
Fig. 2.
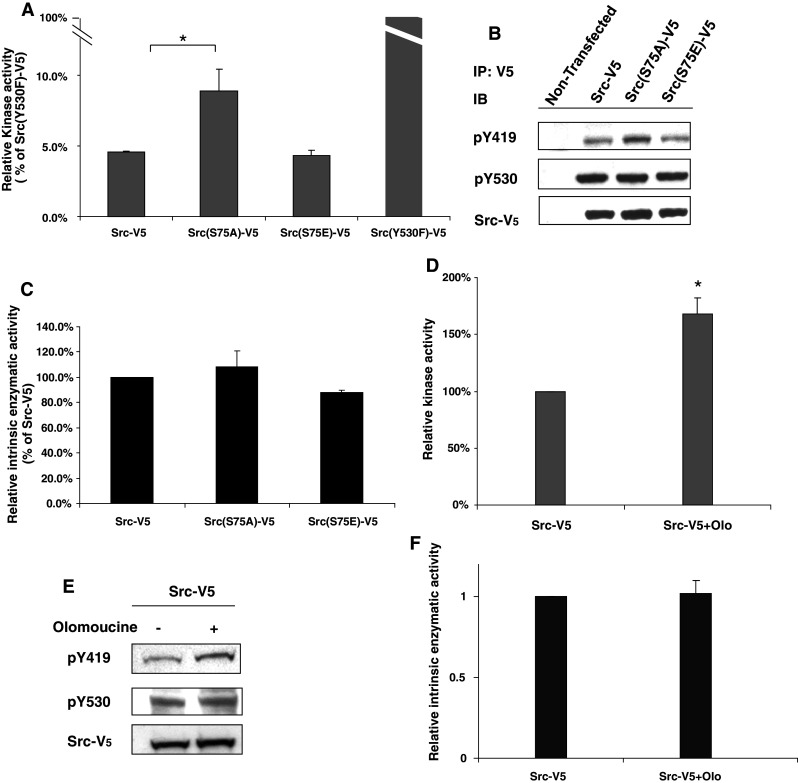
Kinase assay and Y419 phosphorylation of Src-V5 constructs. a Cells were transfected with Src-V5, Src(S75A)-V5, Src(S75E)-V5, and Src(Y530F))-V5, and cell extracts were immunoprecipitated with V5 antibody. Cell extract from non-transfected cells was similarly immunoprecipitated as a control. The kinase activity of the immunoprecipitated proteins was examined by in vitro kinase assay using a peptide substrate and the results were normalized to amount of V5-tagged protein present, as determined by immunoblotting (see panel b). Kinase activity of the non-transfected cells was insignificant (less than 10% of Src-V5), but is not plotted, as no V5 was detected. The relative kinase activity of the Src-V5 constructs is shown as a percentage of Src(Y530F)-V5. Statistical significance was evaluated using one-way ANOVA (*p < 0.05, n = 3). b Immunoprecipitated proteins assayed in a were immunoblotted to detect Src(pY419), Src(pY530), and V5. c The ratio of kinase activity to pY419 of Src-V5, Src(S75A)-V5, and Src(S75E)-V5 was plotted as a percentage of Src-V5. d A single flask of cells expressing Src-V5 was evenly divided into two dishes, which were incubated overnight in the presence or absence of olomoucine. Cell extracts were treated as in a. Kinase activity in the presence of olomoucine is plotted as a percentage of control. e Immunoprecipitated proteins assayed in d were immunoblotted to detect Src(pY419), Src(pY530), and V5. f The ratio of kinase activity to pY419 of Src-V5 in the presence of olomoucine was plotted as a percentage of Src-V5 in the absence of the inhibitor
We next examined the phosphorylation status of each of the V5-tagged Src constructs by immunoblotting with phospho-specific antibodies for Src(pY419) and Src(pY530). The results demonstrate that all three constructs are indeed phosphorylated at both sites (Fig. 2b). Moreover, the greater enzymatic activity of Src(S75A)-V5 is paralleled by a comparable increase in Y419 phosphorylation. As a result, the ratio of kinase activity to Y419 phosphorylation of Src(S75A) is not significantly different from that of Src-V5 and Src(S75E)-V5 (p > 0.05) (Fig. 2c). Thus, as expected, the increased activity of Src(S75A)-V5 is due to an increase in the amount of Y419 phosphorylated Src, not to any other effect of the mutation on kinase activity [i.e., the amount of substrate phosphorylated per mole of Src(pY419)]. Because the ratio of pY419/substrate phosphorylated remains constant for all three Src constructs, pY419 provides an accurate measure of their kinase activity.
Finally, we examined the effect of Cdk5 inhibition on the kinase activity of Src-V5. In vitro kinase assay indicated that kinase activity was increased by 68.2 ± 14.3% in the presence of the inhibitor, which was paralleled by an equivalent increase in the amount of Src(pY419) (Fig. 2d, e). Thus, Cdk5 inhibition, like Src(S75) mutation, produced no significant change in the ratio of kinase activity to Src(pY419) (Fig. 2f). Together, these data demonstrate that the C-terminal V5 tag does not interfere with Csk-dependent regulation of Src and indicate that the effects of Src(S75) mutation and Cdk5 inhibition on Src activity are simply due to changes in Src(pY419) phosphorylation, and not to some unexpected, independent mechanism.
An intact phosphorylation site at Src(S75) is essential for Cdk5-dependent regulation of Src
The observation that Cdk5 inhibition and the Src(S75A) mutation both increased the amount of Src(pY419), suggested that Src(S75) might be required for Cdk5-dependent regulation of Src activity. To test this possibility, transfected cells expressing Src-V5 or Src(S75A)-V5 were incubated overnight in the absence or presence of the potent Cdk5 inhibitor, olomoucine, and the activity of the V5-tagged Src kinases (pY419/total V5-protein) was examined by immunoprecipitation and immunoblotting. The results again showed that the activity of the Src(S75A)-V5 mutant was 2.5 ± 0.3 times greater than that of a comparable amount of wild-type Src-V5 (Fig. 3), indicating that Cdk5 inhibition and the absence of a phosphorylatable amino acid at S75 have similar effects on Src activity. However, Cdk5 inhibition produced no additional increase in the activity of Src(S75A)-V5 mutant, indicating that Cdk5-dependent regulation of Src activity requires an intact phosphorylation site at S75.
Fig. 3.

Effect of Cdk5 on Src activity requires an intact S75 site. a Cells transfected with Src-V5 or Src(S75A)-V5 were incubated for 24 h with or without 15 μM olomoucine overnight. Cell lysates were immunoprecipitated with V5 antibody, and immunoblotted with antibodies against V5 and Src(pY419). b The ratio of Src(pY419)/Src-V5 was quantified by densitometric scanning of immunoblots and plotted as a percentage of Src-V5 in the untreated cells. Statistical significance was evaluated using one-way ANOVA (*p < 0.05, n = 3)
Src(S75A) mimics the physiological effects of Cdk5 inhibition
We previously showed that inhibiting Cdk5 blocks the Rho-dependent pathway of myosin phosphorylation, leading to a loss of central stress fibers, and demonstrated that this effect is mediated by Src through p190RhoGAP [20]. If the absence of phosphorylation at Src(S75) is responsible for these Src-dependent effects of Cdk5 inhibition, we would expect Src(S75A) to produce similar changes in cytoskeletal organization. To test this, we examined stress fiber formation in cells expression equivalent levels of Src-V5 or Src(S75A)-V5 (as judged by fluorescence intensity). To avoid blocking Src activity during the early phase of cell-spreading, when Src is required for focal adhesion formation and cell attachment, we allowed cells to spread for 60 min before adding PP1 or its inactive homolog, PP3. As shown in Fig. 4, 96.3% (52/54) of Src-V5-transfected cells revealed well-formed central stress fibers. In contrast, only 12.5% (6/48) of cells expressing an equivalent amount of Src(S75A)-V5 contained central stress fibers (Fig. 4). F-actin in most Src(S75A)-V5 transfected cells was either entirely absent or restricted to the cell periphery (Fig. 4). A Chi-square test indicated that the difference in central stress fiber formation and cytoskeletal architecture was statistically significant (p < 0.001, χ2 = 36.37). To test whether the changes in the actin cytoskeleton were dependent on Src activity, we examined the effect of the Src inhibitor PP1 on central stress fiber formation in cells expressing Src(S75A)-V5. In the presence of PP1, 80.3% (49/61) of Src(S75A)-V5 transfected cells contained central stress fibers (p < 0.001, χ2 = 65. 38), indicating that the cytoskeletal changes produced by Src(S75A)-V5 are due to increased Src activity (Fig. 4). However, the inactive homolog, PP3 did not affect stress fiber organization either in Src-V5 or Src(S75A)-V5 cells. Thus, the physiological effects of Src(S75A)-V5 expression mimic those of Cdk5 inhibition.
Fig. 4.

Src(S75A) reduces the formation of central stress fibers. Transfected cells expressing Src-V5 or Src(S75A)-V5 were replated on fibronectin-coated chamber slides for 1 h. Cells were then incubated for 2 h in absence or presence of PP1 or PP3. Representative fluorescence images of V5-expressing cells (upper images; green channel), phalloidin-stained actin cytoskeleton (middle images; red channel), and DAPI-stained nuclei (lower images; blue channel) are shown. Well-formed central stress fibers are present in 96.3% of Src-V5 cells at the selected fluorescence intensity. In contrast, 87.5% of cells transfected with Src(S75A)-V5 at comparable fluorescence intensity either showed no central polymerized actin (long arrow) or had peripheral actin fibers (short arrow). PP1 did not disturb central stress fiber formation in Src-V5 cells, but rescued the disrupting effect of Src(S75A)-V5 on central stress fibers. PP3 did not affect stress fiber organization either in Src-V5 or Src(S75A)-V5 cells (arrows). This experiment is representative of three experiments with similar results. Scale bar = 20 μm
Cdk5-dependent phosphorylation of Src(S75) occurs in cells
The above results suggested that the effects of Cdk5 on Src activity involve Cdk5-dependent phosphorylation at Src(S75). Since Src-V5 shows the same response to Cdk5 inhibition as endogenous Src (see Figs. 1, 3), we first used this tagged fusion protein to test for S75 phosphorylation using a phospho-specific antibody raised against a Src(pS75) peptide. This antibody detected a single band of 60 kDa in Src-V5 transfected cells; this band was not seen in Src(S75A)-V5 transfected cells, indicating that the antibody is specific for Src(pS75) (Fig. 5a). As additional confirmation of the specificity of the antibody, we tested the ability of the phosphorylated antigenic peptide and its non-phosphorylated counterpart to block immunoreactivity to endogenous Src(pS75) in Cos7 cells (Fig. 5b). The antibody detected an endogenous protein of the correct molecular weight (approximately 60 kDa), which was completely blocked by the phosphorylated, but not the non-phosphorylated, peptide (Fig. 5b). We next tested whether S75 phosphorylation of Src-V5 in transfected Cos7 cells (Fig. 5c) and endogenous Src of lens epithelial cells (Fig. 5d) were Cdk5-dependent. When transfected cells expressing Src-V5 were treated with the Cdk5 inhibitor olomoucine, Src(pS75) was no longer detected (Fig. 5c). Similarly, suppression of Cdk5 expression with siRNA oligonucleotides inhibited phosphorylation of endogenous Src(pS75) (Fig. 5d). Thus, Src(S75) is phosphorylated in a Cdk5-dependent manner.
Fig. 5.

Cdk5-dependent phosphorylation of Src(S75) occurs in cells. a Human lens epithelial cells were transfected with Src-V5 or Src(S75A)-V5. Cell lysates were immunoprecipitated with V5 antibody and immunoblotted with phospho-specific antibody to Src(pS75). The phospho-specific antibody detected Src(pS75) in cells transfected with Src-V5, but not in those transfected with Src(S75A)-V5. b Cos7 cells were transfected with Src-V5 or Src(S75A)-V5, and immunoblotted to detect endogenous Src(pS75) in the presence or absence of the phosphorylated antigenic peptide (PP) or its non-phosphorylated counterpart (NP). The phosphorylated peptide, but not the non-phosphorylated peptide, completely blocked immunoreactivity. c Cos7 cells were transfected with Src-V5 or Src(S75A)-V5 and incubated for a total of 24 h with or without 15 μM olomoucine during the final 16 h. Cell lysates were immunoprecipitated with V5 antibody then immunoblotted to detect Src(pS75) and V5. The antibody detected significant Src(pS75) in Src-V5 transfected cells, but only trace amounts in olomoucine treated Src-V5 cells and Src(S75A)-V5 cells. d Lens epithelial cells were transfected with siRNA to suppress Cdk5 (siCdk5) or with non-targeting siRNA oligonucleotides (non-targeting). Cell lysates were immunoblotted with antibodies against Src(pS75), Src, Cdk5, and GAPDH. Src(pS75) expression was inhibited in siCdk5-transfected cells. e Cell lysates were immunoprecipitated with antibody specific for either inactive Src (Non-pY419) or active Src (pY419). The supernatant fractions and immunoprecipitated fractions were immunoblotted with antibodies against Src, Non-pY419, Src(pY419), and Src(pS75). This experiment is representative of three experiments with similar results
The availability of the Src(pS75) phospho-specific antibody also allowed us to determine whether both the active and inactive conformations of Src are subject to phosphorylation at S75. Immunoprecipitation with an antibody specific for inactive Src (non-pY419 antibody) precipitated the majority of Src protein, but all detectable Src(pY419) and Src(pS75) remained in the supernatant fraction. In a parallel experiment, phospho-specific Src(pY419) antibody was used to immunoprecipitate active Src(pY419). In this case, all detectable Src(pY419) and Src(pS75) were found in the immunoprecipitate (Fig. 5e). These findings indicate that only active Src is phosphorylated on S75.
Regulation of Src activity by Cdk5 requires Cullin-5
Since the results indicated that Cdk5 affects Src activity via S75 phosphorylation, we next considered possible mechanisms for this effect. Src activity is regulated by reversible interconversion of the active and inactive forms and by irreversible degradation of the active form:
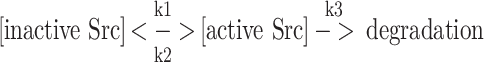 |
In principle, Src(S75) phosphorylation might limit Src activity by increasing Cullin-5-dependent ubiquitinylation of active Src or by shifting the balance of phosphorylation and dephosphorylation to favor the inactive form [i.e., reducing k1, the rate of phosphatase-dependent Src(pY530) dephosphorylation, or increasing k2, the rate of Csk-dependent Y530 re-phosphorylation]. To examine these possibilities, we first tested whether inhibiting Cdk5 would alter the activity of constitutively active Src(Y530F)-V5, which can not be phosphorylated at Y530. We expected that this mutant would be resistant to any effects that were mediated by altering the phosphorylation/dephosphorylation of Y530. A single flask of Src(Y530F)-V5 transfected cells was evenly divided into two dishes to ensure that the transfection efficiency of olomoucine treated cells and control cells was the same; then one dish was incubated overnight with olomoucine to inhibit Cdk5. The results showed that both the Y419 phosphorylation and the total amount of Src(Y530F)-V5 increased significantly (Fig. 6a), indicating that Cdk5 does not exert its effect on Src activity by regulating phosphorylation/dephosphorylation at Y530. We also noted that the expression of Src(Y530F)-V5 was always lower (32.3 ± 3.8%; p < 0.05, n = 4) than that of Src-V5 when equal amounts of DNA were used for transfection. This is not unexpected, since Src-V5 is largely in the inactive configuration and is therefore degraded more slowly [6, 26].
Fig. 6.

The effect of Cdk5 on Src activity is Cullin-5-dependent. a Cells transfected with a Src-V5 or constitutively active Src(Y530F)-V5 were incubated overnight in the presence or absence of olomoucine. Cells were lysed and immunoblotted with antibodies against Src(pY419), Src(pY530), and V5. b Relative levels of Src and Src(pY419) were quantified and tested for statistical significance one-way ANOVA (*p < 0.05, n = 4). c Cells transfected with a non-targeting siRNA oligonucleotides (non-targeting) or siRNA oligonucleotides to suppress Cullin-5 (siCullin-5) were incubated for a total of 72 h, with or without 15 μM olomoucine during the final 16 h. Cells were lysed and immunoblotted with antibodies against Src(pY419), total Src, Cullin-5, and GAPDH. d Relative levels of total Src and Src(pY419) were quantified by densitometric scanning of immunoblots and plotted as a percentage of the non-targeting control. Statistical significance was evaluated using one-way ANOVA (*p < 0.05, n = 4)
Since the above result implied that Cdk5 affects the rate of degradation of active Src (k3), we next tested whether the effect of Cdk5 on Src activity requires Cullin-5, the E3 ligase that regulates ubiquitinylation of active Src [6, 7]. Cells were incubated with siRNA oligonucleotides to suppress Cullin-5, and Src activity was examined in the presence and absence of olomoucine. Cullin-5 siRNA oligonucleotides reduced Cullin-5 expression by 86.0 ± 6.6% (p < 0.001) and increased Src(pY419) as compared to non-targeting siRNA controls (2.58 ± 0.79-fold; p < 0.05, n = 4); however, inhibiting Cdk5 activity with olomoucine in the presence of Cullin-5 siRNA did not produce any further increase in Src(pY419) (p > 0.05, n = 4) (Fig. 7c, d). Together, the above findings indicate that Cdk5 affects the Cullin-5-dependent pathway of Src regulation, rather than the reversible, phosphatase and Csk-dependent pathway. Consistent with previous reports [7], our data showed that Cullin-5 suppression did not significantly affect total levels of Src, most likely because only 5–10% of Src is active under normal physiological conditions [25].
Fig. 7.

Src(S75A) and Cdk5 suppression reduces ubiquitinylation of Src. Cells were transfected with Src-V5 or Src(S75A)-V5 (a, b), and with non-targeting oligonucleotides or siCdk5 (c, d), then incubated 12 h in medium containing 10 μM MG132. a Cell lysates were immunoprecipitated with ubiquitin antibody, and immunoblotted with V5 antibody to detect ubiquitinylated V5-tagged protein. A typical immunoblot is presented, showing high-molecular-weight ubiquitinylated Src-V5, Src(S75A)-V5, and Src(S75E)-V5. b Relative amounts of ubiquitin incorporation were calculated and plotted as a percentage of Src-V5. c Cell lysates were immunoprecipitated with ubiquitin antibody, and immunoblotted with Src antibody to detect endogenous ubiquitinylated Src protein. d Relative incorporation of ubiquitin into endogenous Src in the presence of Cdk5 siRNA was calculated as a percentage of the non-targeting control. Statistical significance was evaluated using one-way ANOVA (*p < 0.05, n = 3)
Mutation of Src(S75) and Cdk5 suppression alter the ubiquitinylation of Src
The above findings implied that Cdk5-dependent phosphorylation of Src(S75) regulates the rate of Cullin-5-dependent ubiquitinylation and proteosomal degradation of active Src. To test this hypothesis, we inhibited proteosomal degradation with MG132 and measured ubiquitin incorporation into Src-V5, Src(S75A)-V5, and Src(S75E)-V5 by immunoprecipitating the ubiquitinylated proteins and immunoblotting for V5-tagged proteins. Ubiquitinylation of Src(S75A)-V5 was significantly less under these conditions than ubiquitinylation of either Src-V5 or Src(S75E)-V5 (Fig. 8a). Quantification indicated that ubiquitin incorporation into Src(S75A)-V5 was about 35.3 ± 0.64% (p < 0.05) that incorporated into Src-V5 or Src(S75E) (Fig. 8b), even though the amount of Y419 phosphorylated Src(S75A) was greater (Fig. 2b). We next tested the effect of suppressing Cdk5 by siRNA on the ubiquitinylation of endogenous Src. As Fig. 8c and d show, Src ubiquitinylation in the presence of Cdk5 siRNA was 47.4 ± 8.1% (p < 0.05) of control, whereas the Y419 phosphorylation under these conditions was elevated (Fig. 1c). These findings confirm the view that the Src(S75A) mutation and Cdk5 suppression increase Src activity by reducing the ubiquitinylation of active Src.
Fig. 8.

Src(S75A) and Cdk5 inhibition increases the stability of Src. Cells were transfected with Src-V5 or Src(S75A)-V5 (a, b), and with non-targeting oligonucleotides or siCdk5 (c, d), then metabolically labeled with [35S] methionine for 2 h, and chased with nonradioactive medium for 9 h. a Autoradiogram of 35S -labeled proteins immunoprecipitated with V5 antibody from a representative experiment. b The time-dependent decrease in radiolabeled Src-V5 (diamonds) and Src(S75A) (squares) were plotted on a log-linear scale and regression analysis was used to determine the best-fit straight line. Statistical significance at 9 h was evaluated using one-way ANOVA (*p < 0.05, n = 3). c Autoradiogram of 35S -labeled protein immunoprecipitated with antibody to Src shows the time-dependent decrease in endogenous Src protein. d Radiolabeled Src from three experiments was quantified and plotted as shown in b
Cdk5 inhibition and Src(S75) mutation alter the degradation of Src
As an additional confirmation of the effect of phosphorylation at S75 on the stability of active Src, we measured protein expression of Src-V5 and Src(S75A)-V5 by pulse-labeling transfected lens epithelial cells with 35 S-methionine and following the decay of labeled V5-tagged proteins during a 9-h chase period. At 9 h, the remaining Src-V5 was significantly less than Src(S75A)-V5 (56.1 ± 10.0 vs. 83.0 ± 7.1%, p < 0.05; n = 3). The calculated half-life of Src-V5 (based on the best-fit straight line of a log-linear plot during the chase period) was 11.5 h. Under the same conditions, Src(S75A)-V5 was significantly more stable, with a calculated half-life during this period of 38.7 h (Fig. 9a, b). Thus, replacing Src(S75) with a non-phosphorylatable amino acid reduces both ubiquitinylation and degradation of Src.
Fig. 9.

Proposed model for the targeted degradation of active Src by Cdk5. Src protein exists in both inactive and active configurations. Inactive Src is activated by dephosphorylation of Y530 (k1). Active Src may be deactivated by Csk (k2), which phosphorylates Y530, or destroyed by Cullin-5-dependent ubiquitinylation and proteosomal degradation (k3). Active Src is also subject to phosphorylation at S75 by Cdk5/p35. This phosphorylation promotes the Cullin-5-dependent ubiquitinylation and degradation of Src, thus regulating Src activity by controlling the stability of the active form. Available evidence also suggests a possible auto-regulatory feedback loop in which active Src participates its own ubiquitinylation by phosphorylating Cdk5(Y15) (dotted arrow), thus increasing Cdk5/p35 activity
Since our data indicate that Src(S75) phosphorylation is Cdk5-dependent, we next tested the effect of suppressing Cdk5 on the protein stability of endogenous Src by pulse-labeling cells with 35 S-methionine. At 9 h, the remaining endogenous Src was significantly less when Cdk5 was suppressed by siRNA (60.0 ± 7.1 vs. 81.2 ± 10.4%, p < 0.05; n = 3). We found that endogenous Src is degraded with a calculated half-life of about 11.9 h in lens epithelial cells under normal conditions (Fig. 9c, d). This value is in good agreement with the published values for endogenous Src [6, 26], and with the value of 11.5 h we observed for Src-V5, indicating that the V5-tagged protein is degraded normally. In contrast, when Cdk5 expression was suppressed, the calculated half-life of endogenous Src during the chase period increased to approximately 30.0 h (Fig. 9c, d), a value comparable to that of Src(S75A)-V5. The difference between these two values may be explained by the incomplete suppression of Cdk5. Thus, preventing phosphorylation of Src at S75 by inhibiting Cdk5 or by inserting a non-phosphorylatable amino acid at this position significantly increases the stability of Src.
Discussion
This study demonstrates that Cdk5-dependent phosphorylation of Src(S75) limits endogenous Src activity by promoting the ubiquitinylation and proteosomal degradation of active Src. Since other Src family kinases lack this phosphorylation site, Src may be the only family member whose ubiquitinylation is regulated by Cdk5. The present results also indicate that the effect of Cdk5 on Src activity requires Cullin-5, in agreement with previous studies indicating that Cullin-5 is the primary E3 ubiquitin ligase responsible for in vivo ubiquitinylation of Src [7]. Although Cdk5-dependent phosphorylation of Src(S75) has been known for some time [9, 12], the present study provides the first information about its physiological role. Our data also demonstrate that Cdk5-dependent phosphorylation of Src(S75) is not restricted to neurons and certain tumor cell lines as was originally thought [9, 12], but also occurs in non-transformed, epithelial cell lines. When the closely related kinase Cdk1 is activated during mitosis, it, too, can phosphorylate Src(S75) and would, presumably, also stimulate the ubiquitin-dependent degradation of Src, thus limiting the increase in Src activity that occurs at this time [9, 11, 12].
A schematic diagram of the functional interaction between Cdk5 and Src is proposed in Fig. 9. Activation of Src permits Cdk5-dependent phosphorylation of Src(S75) at a Cdk5 consensus site in the unique, N-terminal region of Src. This phosphorylation promotes the ubiquitinylation of active Src, catalyzed primarily by Cullin-5 [7]. When phosphorylation of Src at S75 is blocked, either by Cdk5 inhibition or by mutation, ubiquitinylation is inhibited and active Src accumulates, increasing the intracellular Src activity. While the present study demonstrates that Src undergoes Cdk5-dependent phosphorylation, our previous work with lens epithelial cells has shown that the reverse is also true: Src is largely responsible for the phosphorylation of Cdk5(Y15) [19]. Since Cdk5(Y15) phosphorylation increases Cdk5/p35 activity [27], these findings suggest a possible regulatory negative feedback loop between Src and Cdk5, in which Src activation promotes its own ubiquitinylation-dependent degradation by phosphorylating Cdk5(Y15) and increasing the activity of Cdk5. Consistent with such a regulatory scheme, we have previously shown that increasing Src activity by inhibiting Cdk5 activity with olomoucine or dominant negative Cdk5, also increases the phosphorylation of Cdk5(Y15) [19].
It is not yet clear whether phosphorylation at Src(S75) constitutes a specific signal for Cullin-5, although this is a possibility. Alternatively, Src(S75) phosphorylation may cause a conformational change that exposes a binding site for Cullin-5 or allows binding to a scaffolding protein needed for efficient ubiquitinylation. Clearly, Src(S75) phosphorylation is not an absolute requirement for ubiquitinylation of active Src, since we observed incorporation of ubiquitin into Src(S75A)-V5, albeit at a reduced rate. This lower residual rate of ubiquitinylation may be due to less efficient ubiquitinylation by Cullin-5, or to another E3 ligase, such as Cbl, which has also been implicated in degradation of Src family kinases [28, 29].
It is worth noting that the simple kinetic model usually used to determine the half life of a protein by the radioactive pulse-chase technique (as in Fig. 8) is not strictly valid for Src, since the degradation machinery targets active Src, rather than the total pool of active and inactive Src (see Supplement 1). As a result, the turnover of the total Src pool is slower than the initial slope of the radioactive decay curve and the calculated half-life would suggest. An additional consequence of the more complex kinetics of Src is that the increase in Src protein seen when the rate of degradation changes depends on the rate of Src(Y530) phosphorylation and dephosphorylation (k1 and k2) as well as the rate of degradation (k3). This may help to explain why we and others [7] have observed little or no change in total Src when the degradation of active Src is inhibited.
Previous work from this laboratory showed that the 50–70% increase in Src activity produced by Cdk5 inhibition is physiologically significant, as it results in Src-dependent inactivation of Rho signaling and loss of central stress fibers [20, 30]. The present data extend these findings by demonstrating that Cdk5 affects Src activity by phosphorylating Src(S75), thus modulating the ubiquitin-dependent degradation of active Src. Because ubiquitinylation leads to proteolysis and the irreversible destruction of Src, phosphorylation of Src(S75) by Cdk5 may be involved in switching cells from states favored by Src activity, such as Rac activation and lamellipodia formation, to states that are antagonized by Src activity, such as Rho activation and stress fiber formation. Such a mechanism may be particularly important for Cdk5-dependent regulation of cell migration [15, 17, 20].
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Dr. John Reddan of Oakland University for the human lens epithelial cell line (FHL124), Drs. Xiao Dong Jiao and J. Fielding Hejtmancik for generously sequencing DNA clones, and Ms. Andrea Elfstrom for technical assistance. This work was supported by the National Eye Institute Intramural Research Program Z01-EY000238-25.
Conflict of interest
None.
References
- 1.Frame MC. Newest findings on the oldest oncogene; how activated Src does it. J Cell Sci. 2004;117:989–998. doi: 10.1242/jcs.01111. [DOI] [PubMed] [Google Scholar]
- 2.Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 3.Brown EJ, Schreiber SL. A signaling pathway to translational control. Cell. 1996;86:517–520. doi: 10.1016/S0092-8674(00)80125-7. [DOI] [PubMed] [Google Scholar]
- 4.Piwnica-Worms H, Saunders KB, Roberts TM, Smith AE, Cheng SH. Tyrosine phosphorylation regulates the biochemical and biological properties of pp60c-src. Cell. 1987;49:75–82. doi: 10.1016/0092-8674(87)90757-4. [DOI] [PubMed] [Google Scholar]
- 5.Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- 6.Hakak Y, Martin GS. Ubiquitin-dependent degradation of active Src. Curr Biol. 1999;9:1039–1042. doi: 10.1016/S0960-9822(99)80453-9. [DOI] [PubMed] [Google Scholar]
- 7.Laszlo GS, Cooper JA. Restriction of Src activity by cullin-5. Curr Biol. 2009;19:157–162. doi: 10.1016/j.cub.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russello SV, Shore SK. Src in human carcinogenesis. Front Biosci. 2004;9:139–144. doi: 10.2741/1138. [DOI] [PubMed] [Google Scholar]
- 9.Kato G, Maeda S. Production of mouse ES cells homozygous for cdk5-phosphorylated site mutation in c-src alleles. J Biochem. 2003;133:563–569. doi: 10.1093/jb/mvg072. [DOI] [PubMed] [Google Scholar]
- 10.Kato G, Maeda S. Neuron-specific cdk5 kinase is responsible for mitosis-independent phosphorylation of c-src at ser75 in human y79 retinoblastoma cells. J Biochem (Tokyo) 1999;126:957–961. doi: 10.1093/oxfordjournals.jbchem.a022540. [DOI] [PubMed] [Google Scholar]
- 11.Chackalaparampil I, Shalloway D. Altered phosphorylation and activation of pp60c-src during fibroblast mitosis. Cell. 1988;52:801–810. doi: 10.1016/0092-8674(88)90422-9. [DOI] [PubMed] [Google Scholar]
- 12.Kato G, Maeda S. Novel phosphorylation at a mitotic site, serine 75, in human pp60c-src from unsynchronized human tumor cells having a spherical morphology. Biochem Biophys Res Commun. 1995;216:619–629. doi: 10.1006/bbrc.1995.2667. [DOI] [PubMed] [Google Scholar]
- 13.Tang D, Lee KY, Qi Z, Matsuura I, Wang JH. Neuronal cdc2-like kinase: From cell cycle to neuronal function. Biochem Cell Biol. 1996;74:419–429. doi: 10.1139/o96-046. [DOI] [PubMed] [Google Scholar]
- 14.Rosales JL, Lee KY. Extraneuronal roles of cyclin-dependent kinase 5. Bioessays. 2006;28:1023–1034. doi: 10.1002/bies.20473. [DOI] [PubMed] [Google Scholar]
- 15.Huang C, Rajfur Z, Yousefi N, Chen Z, Jacobson K, Ginsberg MH. Talin phosphorylation by cdk5 regulates smurf1-mediated talin head ubiquitylation and cell migration. Nat Cell Biol. 2009;11:624–630. doi: 10.1038/ncb1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakano N, Nakao A, Ishidoh K, Tsuboi R, Kominami E, Okumura K, Ogawa H. Cdk5 regulates cell-cell and cell-matrix adhesion in human keratinocytes. Br J Dermatol. 2005;153:37–45. doi: 10.1111/j.1365-2133.2005.06583.x. [DOI] [PubMed] [Google Scholar]
- 17.Gao CY, Stepp MA, Fariss R, Zelenka P. Cdk5 regulates activation and localization of Src during corneal epithelial wound closure. J Cell Sci. 2004;117:4089–4098. doi: 10.1242/jcs.01271. [DOI] [PubMed] [Google Scholar]
- 18.Negash S, Wang HS, Gao C, Ledee D, Zelenka P. Cdk5 regulates cell-matrix and cell-cell adhesion in lens epithelial cells. J Cell Sci. 2002;115:2109–2117. doi: 10.1242/jcs.115.10.2109. [DOI] [PubMed] [Google Scholar]
- 19.Qiao F, Gao CY, Tripathi BK, Zelenka PS (2008) Distinct functions of cdk5(y15) phosphorylation and cdk5 activity in stress fiber formation and organization. Exp Cell Res [DOI] [PubMed]
- 20.Tripathi BK, Zelenka PS (2009) Cdk5-dependent regulation of rho activity, cytoskeletal contraction, and epithelial cell migration via suppression of Src and p190rhogap. Mol Cell Biol [DOI] [PMC free article] [PubMed]
- 21.Alexander K, Yang HS, Hinds PW, Yang HS, Alexander K, Santiago P, Hinds PW. Cellular senescence requires cdk5 repression of rac1 activity. Mol Cell Biol. 2004;24:2808–2819. doi: 10.1128/MCB.24.7.2808-2819.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao CY, Zakeri Z, Zhu Y, He HY, Zelenka PS. Expression of cdk-5, p35, and cdk5-associated kinase activity in the developing rat lens. Develop Genet. 1997;20:267–275. doi: 10.1002/(SICI)1520-6408(1997)20:3<267::AID-DVG9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 23.Gao C, Negash S, Wang HS, Ledee D, Guo H, Russell P, Zelenka P. Cdk5 mediates changes in morphology and promotes apoptosis of astrocytoma cells in response to heat shock. J Cell Sci. 2001;114:1145–1153. doi: 10.1242/jcs.114.6.1145. [DOI] [PubMed] [Google Scholar]
- 24.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 25.Shenoy S, Chackalaparampil I, Bagrodia S, Lin PH, Shalloway D. Role of p34cdc2-mediated phosphorylations in two-step activation of pp60c-src during mitosis. Proc Natl Acad Sci USA. 1992;89:7237–7241. doi: 10.1073/pnas.89.15.7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris KF, Shoji I, Cooper EM, Kumar S, Oda H, Howley PM. Ubiquitin-mediated degradation of active Src tyrosine kinase. Proc Natl Acad Sci USA. 1999;96:13738–13743. doi: 10.1073/pnas.96.24.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM, Gertler FB, Vidal M, Van Etten RA, Tsai LH. Cables links cdk5 and c-abl and facilitates cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–646. doi: 10.1016/S0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 28.Andoniou CE, Lill NL, Thien CB, Lupher ML, Jr, Ota S, Bowtell DD, Scaife RM, Langdon WY, Band H. The Cbl proto-oncogene product negatively regulates the Src-family tyrosine kinase Fyn by enhancing its degradation. Mol Cell Biol. 2000;20:851–867. doi: 10.1128/MCB.20.3.851-867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao N, Ghosh AK, Douillard P, Andoniou CE, Zhou P, Band H. An essential role of ubiquitination in cbl-mediated negative regulation of the Src-family kinase fyn. Signal Transduct. 2002;2:29–39. doi: 10.1002/1615-4061(200205)2:1/2<29::AID-SITA29>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tripathi BK, Zelenka PS. Cdk5: A regulator of epithelial cell adhesion and migration. Cell Adh Migr. 2010;4:333–336. doi: 10.4161/cam.4.3.11131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.