

Abstract
Transcription is regulated by the state of phosphorylation of a heptapeptide repeat known as the carboxy-terminal domain (CTD) present in the largest subunit of RNA polymerase II (RNAPII). RNAPII that associates with transcription initiation complexes contains an unphosphorylated CTD, whereas the elongating polymerase has a phosphorylated CTD. Transcription factor IIH has a kinase activity specific for the CTD that is stimulated by the formation of a transcription initiation complex. Here, we report the isolation of a cDNA clone encoding a 150-kD polypeptide, which, together with RNAPII, reconstitutes a highly specific CTD phosphatase activity. Functional analysis demonstrates that the CTD phosphatase allows recycling of RNAPII. The phosphatase dephosphorylates the CTD allowing efficient incorporation of RNAPII into transcription initiation complexes, which results in increased transcription. The CTD phosphatase was found to be active in ternary elongation complexes. Moreover, the phosphatase stimulates elongation by RNAPII; however, this function is independent of its catalytic activity.
Keywords: Transcription, RNA polymerase II, CTD phosphatase, phosphorylation
Purified eukaryotic RNA polymerases are unable to initiate promoter-specific transcription (Zawel and Reinberg 1995). A search for factors that would confer promoter specificity to RNA polymerase II (RNAPII) led to the discovery of the general transcription factors (GTFs; Zawel and Reinberg 1993; Hampsey 1998).
Stable association of RNAPII with promoter sequences requires TFIID (or TBP), TFIIB, and TFIIF (for review see Orphanides et al. 1996). However, the RNAPII transcription system is unique because, after the polymerase has stably associated with promoter sequences, two additional factors, TFIIE and TFIIH, are necessary for transcription (Flores et al. 1990; for review, see Orphanides et al. 1996). This requirement is likely related to a unique structure found at the carboxyl terminus of the largest subunit of RNAPII known as the carboxy-terminal domain (CTD) (Koleske and Young 1995; Dahmus 1996). This conserved structure consists of multiple tandem repeats of the heptapeptide Tyr-Ser-Pro-Thr-Ser-Pro-Ser, which serves as a substrate for a number of protein kinases (Dahmus 1996). At least two forms of RNAPII have been detected in cells. The most abundant form contains a phosphorylated CTD (RNAPIIO). A second form contains an unphosphorylated CTD and is known as RNAPIIA (Dahmus 1994). The phosphorylation of the CTD has been correlated with function. It was found that the nonphosphorylated form of RNAPII is recruited to the initiation complex (Laybourn and Dahmus 1989; Lu et al. 1991), whereas the elongating polymerase is found with a phosphorylated CTD (Bartholomew et al. 1986; Laybourn and Dahmus 1989; Zawel and Reinberg 1993; O’Brien et al. 1994). TFIIH contains a CTD kinase activity (Feaver et al. 1991; Lu et al. 1992; Serizawa et al. 1992) and this activity is efficient after RNAPII has associated with promoter sequences (Lu et al. 1992). These findings led to the hypothesis that RNAPII cycles during transcription (Laybourn and Dahmus 1989; Zawel et al. 1993; Dahmus 1996). However, studies aimed at identifying the enzyme that recycles RNAPII (i.e., the CTD phosphatase) have been unsuccessful.
Dahmus and co-workers were the first to identify a CTD-specific phosphatase activity in HeLa cell extracts (Chesnut et al. 1992). Subsequently, Chambers and Dahmus (1994) partially purified the CTD phosphatase and reported that it was a type-2C protein phosphatase (resistant to okadaic acid and requiring Mg2+ for activity) with specificity for phosphoserine and phosphothreonine residues. CTD phosphatase activity was correlated with a predominant polypeptide of 205 kD and a minor polypeptide of 150 kD. In a subsequent study, Dahmus and co-workers (Chambers et al. 1995), showed that CTD phosphatase activity is regulated by two GTFs. The large subunit of TFIIF (RAP74) stimulates the CTD phosphatase activity, whereas TFIIB inhibits TFIIF-mediated stimulation. Importantly, the CTD phosphatase interacts functionally with RNAPII independently of the CTD. These findings were followed by a report by Chambers and Kane (1996) demonstrating a CTD phosphatase activity in yeast with properties similar to those described for the human activity. The yeast CTD phosphatase activity was stimulated by TFIIF. However, reconstitution of CTD phosphatase activity apparently requires an additional component. Nonetheless, Chambers and Kane (1996) demonstrated that one component of the yeast phosphatase activity is a polypeptide of ∼100 kD. Subsequent studies by Kane, Greenblatt, and associates isolated an essential yeast gene encoding the 100-kD component of the yeast CTD phosphatase, denoted FCP1 (Archambault et al. 1997). These studies also reported a human homolog. However, no conserved phosphatase motifs were found in the yeast or human sequences. In agreement with the earlier biochemical studies of Dahmus and co-workers (Chambers et al. 1995) demonstrating a functional interaction between the CTD phosphatase and RAP74, FCP1 was found to interact with the large subunit of TFIIF. These studies were extended by Dahmus, Greenblatt, and co-workers, who isolated human cDNAs encoding FCP1 using a yeast two-hybrid screen with RAP74 as the bait (Archambault et al. 1998). Two forms of FCP1 were described, FCP1a, which encodes a 842-amino-acid polypeptide and a shorter splicing variant denoted FCP1b. Unfortunately, it was not determined whether the isolated human cDNA clones encode an active CTD phosphatase or an active subunit of the CTD phosphatase. Nonetheless, antibodies raised against a fragment of FCP1a immunoreacted with the HeLa-derived 150-kD polypeptide identified initially by Chambers and Dahmus (1994). Studies with the CTD phosphatase culminated in a recent report by Dahmus and co-workers demonstrating that the human activity could be reconstituted with the 150-kD polypeptide isolated from a polyacrylamide–gel, highly purified RNAPIIO, and bacterially produced RAP74 (Marshall et al. 1998). Moreover, these studies revealed that CTD phosphatase activity is regulated by an activator: The HIV-encoded transactivator TAT was found to down-regulate its phosphatase activity.
The studies described above provide a clear demonstration of the existence of an evolutionarily conserved CTD phosphatase and have shed light on its regulation by other transcription factors. These studies did not, however, address the function of the phosphatase in the transcription cycle, or whether the CTD phosphatase promotes the recycling of RNAPII. In the studies described here, we have purified a CTD phosphatase from human cells. Using ion-trap mass spectrometry to determine peptide sequences from the purified polypeptide, we designed oligonucleotides that were used to isolate a cDNA encoding the CTD phosphatase. We provide evidence that the polypeptide encoded by the human cDNA is catalytically active and, more importantly, that it recycles RNAPII. Sequence comparison between the phosphatase isolated here and FCP1a indicates that FCP1a represents an amino-terminally truncated protein that is devoid of catalytic activity. We referred to the catalytically active CTD phosphatase as FCP1, whereas FCP1a denotes the truncated form of the polypeptide isolated previously, and FCP1b, a splicing variant of FCP1. Furthermore, we demonstrate that FCP1 functions as a positive elongation factor. This function is independent of CTD phosphatase activity.
Results
Purification of a CTD phosphatase from HeLa cells
A CTD-phosphatase activity was purified from HeLa cell nuclear extracts as described in Materials and Methods and summarized in Figure 1A. The substrate used during purification was RNAPII containing a phosphorylated CTD. Dephosphorylation of the CTD alters the migration of the largest subunit of RNAPII (Rpb1) on SDS-PAGE, which can be detected by Western blots with antibodies against Rpb1 (see Fig. 1B, and below).
Figure 1.

Purification of FCP1. (A) Purification scheme used to isolate FCP1 from HeLa cell nuclear extracts. (B) Silver stain of a 12% (wt/vol) SDS–polyacrylamide gel containing aliquots of fractions [input (I) and column fractions 4–31] derived from the Superose 12 gel-filtration column. (Bottom) Western blot of CTD phosphatase reactions, input, and column fractions 4–13. Other fractions were assayed, but they displayed no activity (data not shown). The low level of the IIo band recovered in the Western blot (lanes 10–13, and see below) is the result of a poor transfer of the phosphorylated Rpb1 subunit to the membrane. (C) p150 and RNAPII are sufficient to reconstitute CTD phosphatase activity. (Top) Silver-stain of a 12% (wt/vol) SDS–polyacrylamide gel containing polypeptides recovered after the denaturation/renaturation procedure. (M) Molecular weight markers; (I) 1% of the input phenyl–Sepharose fraction; (p55, p77, and p150) 10% of the excised polypeptide; (polII) RNAPII0; (RAP74) recombinant RAP74. (Bottom) Western blot of the CTD phosphatase reaction performed with input, p150, p77, and p55 polypeptides in the absence or presence of RAP74(+). (D) Analysis of the electrophoretic mobility of native FCP1 compared with FCP1 and FCP1a that were translated in vitro. Recombinant nontagged polypeptides were produced as indicated in the text. After electrophoresis, the gel was Western blotted and probed with anti-FCP1 antibodies.
Fractionation of the CTD phosphatase activity on a gel-filtration Superose 12 column, the last step of purification demonstrates that the activity coeluted with an apparent native mass of ∼300 kD (data not shown) comigrating with a polypeptide of ∼150 kD (p150), as judged by SDS-PAGE (Fig. 1B). In agreement with the gel-filtration results, CTD phosphatase activity could be recovered from p150 excised from a SDS–polyacrylamide gel (Fig. 1C). In this experiment, phosphatase activity was demonstrated with p150 isolated from a denaturing gel and highly purified RNAPII; the activity, however, was stimulated by the addition of bacterially purified RAP74. p150 was not active with CTD-fusion proteins or with Rpb1 (data not shown). Other than RNAPII, we found no other substrate for the phosphatase (data not shown). Thus, we conclude that we have purified a 150-kD polypeptide that, together with RNAPII, reconstitutes a CTD phosphatase.
FCP1 is related to FCP1a, a component of a human and yeast CTD phosphatase
Ion-trap mass spectrometry analysis of p150 revealed that it is related to the previously characterized CTD phosphatase component FCP1a (data not shown; see Fig. 2). However, because the polypeptide derived from the cDNA encoding human FCP1a has a faster mobility on SDS-PAGE than the native phosphatase (Fig. 1D) and, more importantly, because we could not demonstrate enzymatic activity using recombinant FCP1a (see below), we attempted to isolate the cDNA encoding the CTD phosphatase.
Figure 2.

The amino acid sequence of FCP1. The 5′- and 3′-untranslated cDNA sequences as shown above and below, respectively. (Red) Amino acids unique to FCP1, not found in the reported human FCP1a amino acid sequence. Red letters throughout the sequence also denote discrepancies between FCP1 and FCP1a. (○) Initiation methionine assigned for FCP1a. (Purple) Conserved DXDX(T/V) motif (underlined) and the hydrophobic residues preceding the conserved motif present in phosphatases acting on phosphate esters (Collet et al. 1998). Sequences determined by ion-trap mass spectrometry analysis are boxed. (Green) Cysteine/histidine residues of a putative zinc-binding domain. (Yellow) Amino acids that share homology with carboxy-terminal residues of acid tyrosine phosphatase.
On the basis of the peptide sequences derived from p150 and the sequence reported for the human and yeast FCP1a (Archambault et al. 1997, 1998), a human EST was obtained from which oligonucleotides were derived to screen a human cDNA library (see Materials and Methods). The complete amino acid sequence of the longer cDNA clone is shown in Figure 2. The sequence contains all of the p150-derived peptides and is highly related to FCP1a (Fig. 2). However, the open reading frame encodes a polypeptide of 961 amino acids, which is larger than FCP1a at the amino terminus by 119 amino acids (Fig. 2). The in vitro-translated untagged polypeptide derived from the cDNA clone comigrates with the polypeptide purified from human cells on SDS-PAGE (Fig. 1D). The major difference between human FCP1a and FCP1 is due to a different assignment of the translation initiation codon.
Analysis of the amino acid sequence revealed some putative important motifs. A putative zinc-binding motif is present near the amino terminus (Fig 2). More importantly, the motif DXDX(T/V), preceded by four hydrophobic residues, identified previously as belonging to a large hydrolase family comprising several phosphatases or phosphomutases (Collet et al. 1998), is also present within the sequence of FCP1 (Fig. 2). A domain of unknown function present in many tyrosine phosphatases (Denu and Dixon 1995) is also present in FCP1. Thus, FCP1 has signature sequences that are present in other phosphatases.
The amino-terminal domain of FCP1 is necessary for catalytic activity
In light of the results described above demonstrating that the cDNA encoding FCP1 contains 119 amino acids at its amino-terminus, that are absent in FCP1a, we investigated the function of these residues in the phosphatase activity.
In vitro-translated, amino-terminally His-tagged FCP1 and truncated proteins (see Fig. 3A) were analyzed for their ability to dephosphorylate the CTD of RNAPII in the presence of RAP74. The proteins were immunoprecipitated from the lysates with monoclonal antibodies against the His tag, washed extensively with a buffer containing high salt (for details, see Materials and Methods), and analyzed for CTD phosphatase activity (Fig. 3B). The analysis revealed that the full-length 961-amino-acid protein FCP1 was active, but that deletion of the amino-terminal 119 amino acids, FCP1a, compromised phosphatase activity (Fig 3B, cf. lanes 1 and 2).
Figure 3.
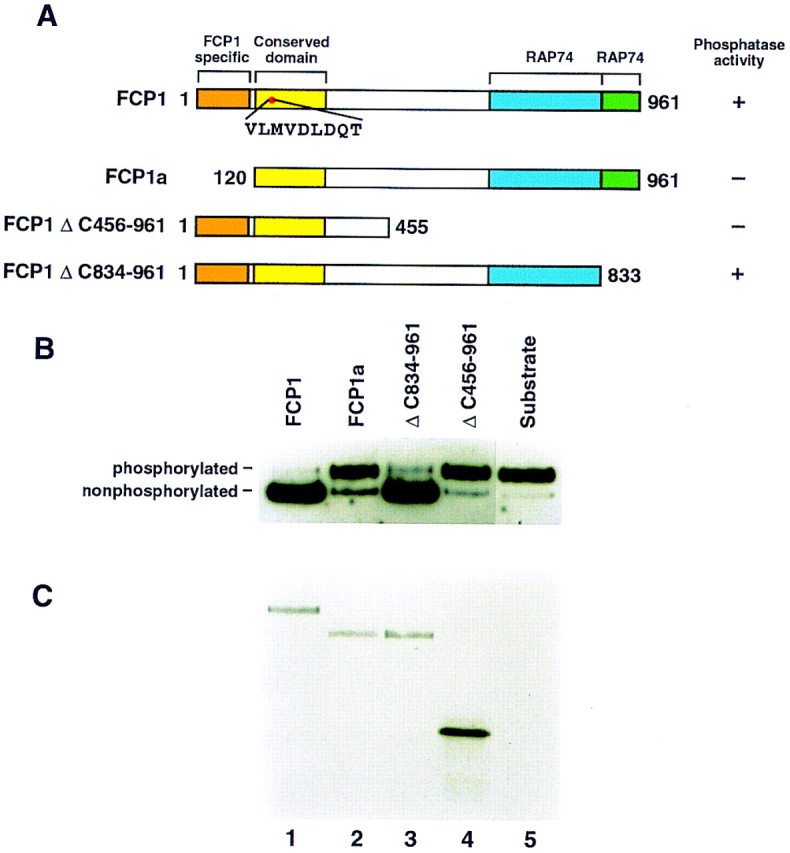
(A) Schematic diagram of amino-terminally His-tagged FCP1 constructs. (Top) Schematic (conserved domain) representation of FCP1. The FCP1-specific motif, absent in FCP1a, is indicated. The domain conserved between different CTD phosphatases, that contains the phosphatase motif DXDX(T/V). RAP74-interaction domains, according to studies described by others (Archambault et al. 1997), are also depicted. (Right) Summary of the CTD phosphatase activity of the different constructs. Numbers on either side of the proteins indicate the amino-terminal and carboxy-terminal amino acids of each FCP1 construct. (B) CTD phosphatase activity of different His-tagged FCP1 truncated proteins. In vitro-translated amino-terminally His-tagged FCP1 proteins were immunoprecipitated with antibodies against the His tag as described in Materials and Methods. Immunoprecipitates were subjected to CTD phosphatase reactions and analyzed by Western blot as described in Materials and Methods. (Top) phosphatase activity of FCP1 (lane 1), FCP1a (lane 2), ΔC834-961 (lane 3), ΔC456-961 (lane 4), and antibody alone (lane 5). (Bottom) Autoradiograph of the Western blot denoting the in vitro-translated [35S]methionine-labeled amino-terminally His-tagged proteins.
In attempts to define the minimum sequence required for the phosphatase activity, we analyzed different carboxy-terminal truncations. A carboxy-terminal deletion of 128 amino acids, which includes one of the RAP74-binding domains (Archambault et al. 1997, 1998), was dispensable for activity (Fig. 3B, lane 3). However, the removal of the second RAP74-binding domain (Archambault et al. 1997) was detrimental for activity (Fig. 4B, lane 4). Unfortunately, we have not been able to obtain viable recombinant baculoviruses carrying FCP1 with mutations in the conserved phosphatase domain (D to A changes at residues 188 and 190, see Fig. 2). However, recent studies performed with yeast FCP1 have demonstrated that residues in the conserved phosphatase domain are necessary for enzymatic activity (J. Greenblatt, pers. comm.).
Figure 4.

(A) Silver stain of SDS–polyacrylamide gels showing the purity of the factors used in the functional transcription assays below. Baculovirus-expressed FCP1 (rFCP1) and FCP1a (rFCP1a) are shown by Coomassie blue staining. Recombinant GTFs (lanes 1–4) were purified as described by Maldonado et al. (1996). The two forms of RNAPII (lane 5) were purified from HeLa cells as described by Lu et al. (1991). A silver-stained gel containing the phosphorylated RNAPII used in the experiment described below is shown in Fig. 1. TFIIH was isolated from HeLa cells by use of an immunopurification protocol described by LeRoy et al. (1998). (B) The effect of FCP1 on transcription reconstituted with RNAPIIA and RNAPIIO. Preinitiation complexes were formed with either RNAPIIA (top) or RNAPIIO (bottom) in the presence of rFCP1 or rFCP1a as shown in the panel and by use of the protocol described in Materials and Methods. Reactions indicated as Pre PIC received rFCP1 or rFCP1a at the beginning of the reaction, and factors and DNA were incubated for 20 min. This incubation was followed by the addition of nucleotides and further incubation for 35 min. Reactions labeled as Post PIC received FCP1 in the second incubation together with the nucleotides. Transcripts were resolved on an 8 m urea–6% (wt/vol) polyacrylamide gel. (C) Quantitative representation of the effect of FCP1 on transcription reconstituted with RNAPIIO. Data were obtained from two different experiments, one of which is shown in B. (Inset) Comparison of the effect of FCP1 on transcription reconstituted with the phosphorylated and nonphosphorylated forms of RNAPII. Additionally, a comparison of the extent of transcription observed in reactions reconstituted with RNAPIIO and FCP1 to reactions reconstituted with RNAPIIA is shown.
FCP1 recycles RNAPII
Next, we analyzed the effect of the CTD phosphatase on transcription using a highly purified reconstituted system (Fig. 4A). We began our analysis by comparing the level of transcription observed when the system was reconstituted with highly purified factors (Fig. 4A) using the phosphorylated and nonphosphorylated forms of RNAPII. We found that the phosphorylated form of RNAPII was compromised in its ability to direct promoter-dependent transcription under the conditions used (Fig. 4B; see Materials and Methods), because of a poor incorporation of the phosphorylated RNAPII into transcription initiation complexes (see below). This defect was specific to the phosphorylation of RNAPII and to the assay used, as the phosphorylated RNAPII was active in a promoter-independent transcription assay (data not shown, see below), and promoter-dependent transcription was detected with an equivalent amount of the nonphosphorylated form of RNAPII (Fig. 4B). More importantly, the phosphorylated form of RNAPII could be reactivated to function in the promoter-dependent assay by the addition of a baculovirus-expressed highly purified FCP1 (Fig. 4A, lane 7, and B). FCP1 had little effect on reactions reconstituted with the nonphosphorylated form of RNAPII (Fig. 4B, see also C, inset). Reactivation of RNAPII requires FCP1 catalytic activity, as the polypeptide lacking the amino-terminal 119 amino acids (Fig 4A, lane 8), which is compromised in its enzymatic activity (Fig. 3), had a reduced effect (Fig. 4B, C, and see below). Importantly, reactivation of the phosphorylated form of RNAPII was precluded when the RNAPII was preincubated with the GTFs and DNA (Fig. 4B,C). This result is likely to be, in part, a manifestation of TFIIB, as previous studies have shown that TFIIB down-regulates phosphatase activity by preventing TFIIF-mediated stimulation of phosphatase activity (Chambers et al. 1995).
The overall level of transcription obtained with the phosphorylated form of RNAPII in the presence of FCP1 under reactivation conditions was approximately one-half the activity observed with an equivalent amount of the nonphosphorylated form of RNAPII (Fig. 4C). This result may reflect a requirement for an additional factor or FCP1 activity maybe most efficient prior to RNAPII disengagement from transcription elongation complexes (see below).
The studies described above establish that FCP1 functions to reactivate the phosphorylated form of RNAPII, but they do not address directly the phosphorylation state of the RNAPII in the transcription complexes. We analyzed CTD phosphorylation directly within the preinitiation complex using a cross-linking assay. In previous work, we demonstrated that Rpb1 can be specifically labeled within the preinitiation complex by the incorporation of a phenyl-azide photoactivatible cross-linking agent through the phosphate at position −4 together with an adjacent radioactive phosphate (for details, see Materials and Methods; Lagrange et al. 1996; Kim et al. 1997). We implemented this approach using DNA attached to beads to analyze the phosphorylation of the CTD in the transcription initiation complex. Under the conditions described above (Fig. 4B), with the phosphorylated form of RNAPII to form transcription initiation complexes, we observed the following: (1) When FCP1 was added to the assay together with all the factors required for transcription, prior to the establishment of a transcription initiation complex, Rpb1 in the complex was nonphosphorylated (Fig. 5, lane 5). (2) When the reaction was performed with an impaired phosphatase (lane 6) or in the absence of CTD phosphatase (lane 4), a phosphorylated Rpb1 was recovered in the transcription complex. The reduced levels of phosphorylated RNAPII recovered in the complex, with respect to the nonphosphorylated form of RNAPII, is consistent with the functional data demonstrating that phosphorylated RNAPII is impaired in its ability to direct promoter-dependent transcription. The recovery of a labeled TAG–Rpb1 requires the formation of a stable transcription initiation complex as it was resistant to sarkosyl rinsing, conditions that we established previously to remove proteins bound nonspecifically (Zawel et al. 1995) and, more importantly, detection of TAG–Rpb1 was dependent on the factors necessary to establish an initiation complex such as TBP (Fig. 5, lanes 1–3), TFIIB, or TFIIF (data not shown). Thus, we conclude that FCP1 dephosphorylates RNAPII, resulting in a form of the enzyme that is capable of efficiently forming transcription initiation-competent complexes. The reduced transcription activity observed with the phosphorylated form of RNAPII correlates with a poor association of phosphorylated RNAPII with transcription complexes. This result is in perfect agreement with earlier studies with gel mobility shift assays to measure complex formation using phosphorylated and nonphosphorylated forms of RNAPII (Lu et al. 1991).
Figure 5.
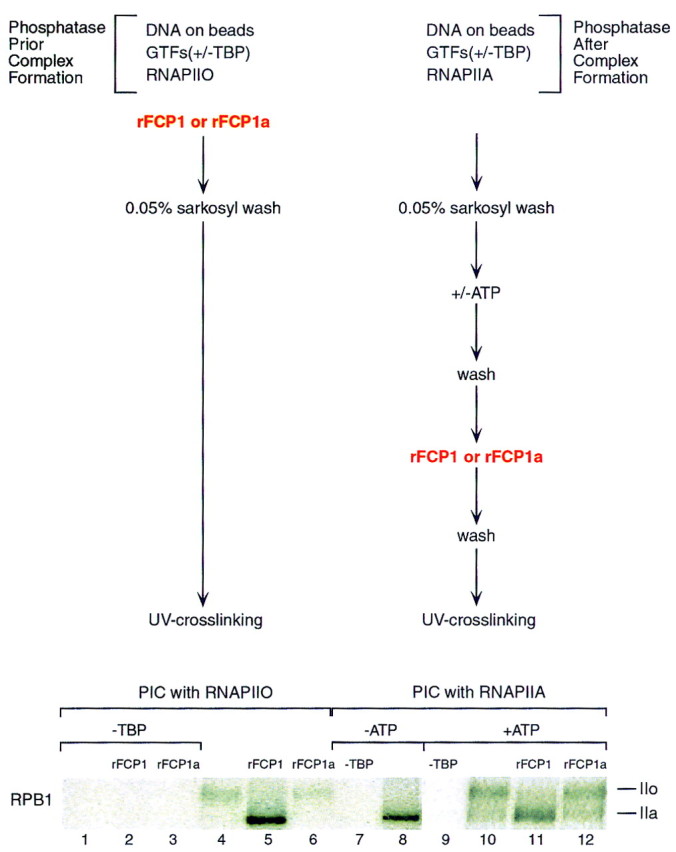
UV cross-linking of Rpb1 in rFCP1- or rFCP1a-treated transcription initiation complexes. (Top) Schematic representation of the protocols followed. (Bottom) Autoradiograph denoting the appearance of labeled Rpb1 (phosphorylated, IIo; nonphosphorylated, IIa) after UV cross-linking performed as described above and in Materials and Methods. Briefly, preinitiation complexes (PIC) were formed with RNAPIIO in the presence or absence of rFCP1 or rFCP1a (left, lanes 1–6). PICs were also formed with RNAPIIA (right, lanes 7,8) and were treated with ATP to phosphorylate the CTD of RNAPII within the preinitiation complex and prior to incubation with rFCP1 or rFCP1a (right, lanes 9–12). TBP was omitted during PIC formation to analyze the specificity of the UV cross-linked Rpb1 (lanes 1,2,3,7,9).
FCP1 dephosphorylates the CTD of RNAPII in transcription elongation complexes
An important issue that we addressed was whether FCP1 was active within transcription elongation complexes. Transcription initiation complexes were formed as above, but on beads and with the nonphosphorylated form of RNAPII (Fig. 5, lanes 7–12). Addition of ATP allowed efficient phosphorylation of the CTD, as evidenced by the recovery of a TAG–Rpb1 migrating slower than the RNAPII used to form the initial complex on SDS-PAGE (Fig. 5, cf. lane 8 with 10). CTD phosphorylation was mediated by TFIIH, because in the absence of highly purified TFIIH (see Fig. 4A, lane 6) no phosphorylation was observed (data not shown). Addition of FCP1 subsequent to preinitiation complex formation and CTD phosphorylation (see Fig. 5, top) resulted in the dephosphorylation of Rpb1 (lane 11). These results suggest that FCP1 is active in dephosphorylating the CTD in a transcription initiation complex; however, this observation should be taken cautiously as TFIIH-mediated phosphorylation of the CTD prior to initiation of transcription may have promoted an abnormal conformation of the transcription complex (see Discussion).
To expand these results, we analyzed whether FCP1 could dephosphorylate the CTD of RNAPII in arrested ternary elongation complexes. This analysis was important because, in this case, FCP1 activity was measured on complexes containing RNAPII and devoid of any other factors (for details, see Zawel et al. 1995). Transcription complexes were formed as above on DNA containing the adenovirus major late promoter directing transcription of a 392-nucleotide-long G-less cassette that was attached to beads and by use of the nonphosphorylated form of RNAPII. Initiation complexes were provided with the nucleotides necessary for transcription to the end of the cassette. Arrested complexes at the end of the cassette were then washed to remove nucleotides and the GTFs released during transcription. Then, the transcription complexes were exposed to FCP1 (for details, see Fig 6A). The result of this experiment demonstrates a conversion from the nonphosphorylated to the phosphorylated form of RNAPII upon transcription; the RNAPII in the preinitiation complex was nonphosphorylated whereas the RNAPII in the stalled ternary complex was phosphorylated (Fig. 6B, cf. lane 1 with lane 2). FCP1 treatment of the ternary complex resulted in the conversion of RNAPII to the dephosphorylated form (lane 4). Dephosphorylation requires active FCP1, as incubation with FCP1a (lane 5) or buffer (lane 3) was without effect. Thus, we concluded that FCP1 can dephosphorylate the CTD in a ternary stalled elongation complex. Importantly, the conversion of the elongating RNAPII to the dephosphorylated form did not promote the release of RNAPII from the template DNA.
Figure 6.

(A) Schematic representation of the protocol used to analyze the effect of FCP1 on ternary complexes. Preinitiation complexes were assembled on immobilized G-less templates with GTFs and RNAPIIA as described in Materials and Methods. Complexes were provided with the ribonucleoside triphosphates necessary for initiation and elongation to the end of the G-less cassette. Elongation complexes were stalled at nucleotide 392 (t0) by nucleotide starvation (G-less). The ternary complexes were washed with BC500 and 0.5% (wt/vol) Sarkosyl and were subsequently equilibrated with BC100 prior to CTD phosphatase treatment. The effect of the CTD phosphatase on these ternary complexes was analyzed in B. Stalled complexes were also analyzed for their ability to resume elongation. Ternary complexes were incubated for 20 min with CTD phosphatase and/or factors TFIIS and TFIIF, as indicated in the figure. Then, ternary complexes were supplemented with all four nucleotides (A/C/U/G) and chased for 4 min. (B) FCP1 can dephosphorylate RNAPIIO in stalled ternary complexes. Western blot of the CTD phosphatase assay performed with the ternary complex. (Lanes 1,2) Phosphorylation state of Rpb1 in the preinitiation complexes (PIC) and t0 complexes, respectively. The low intensity of the phosphorylated Rpb1 detected in the Western blot is due to poor transfer of this polypeptide. (C) Functional analysis of the ternary complexes after treatment with different elongation factors and/or CTD phosphatase. Ternary complexes were chased for 4 min in the presence of different factors as indicated. Products were separated on an 8 m urea–5% (wt/vol) polyacrylamide gel. (Lane 1) Products prior to the addition of factors and chase; the substrate provided to the experiments described in the remaining lanes. On the left of the panel, the 392 nucleotide (t0) and 977 run-off (RO) transcripts are indicated.
FCP1 stimulates elongation by RNAPII independent of the phosphatase activity
The assay described above allowed us to analyze the effect of FCP1 and CTD phosphorylation on elongation. The arrested ternary complexes were allowed to resume elongation by the addition of the four ribonucleoside triphosphates. Under the conditions of the assay (using transcription complexes attached to beads that were washed extensively to remove the factors released during production of the 392-nucleotide RNA; Fig. 6C, lane 1), few of the arrested complexes resumed elongation, and those that did failed to produce the 977-nucleotide run-off transcript after a 4-min chase (lane 2). The same phenomenon was observed after FCP1 treatment; however, the presence of FCP1 appeared to stimulate the production of longer transcripts (cf. lanes 2 and 6). Pretreatment of the arrested complex with the elongation factor TFIIS increased the number of complexes that escaped arrest. However, during the 4-min chase, the complexes did not reach full size (lane 3). The same phenomenon occurred when the complexes were pretreated with FCP1 and TFIIS, but, again, the RNAs were longer than those observed in the absence of FCP1 (cf. lane 3 with lane 10). TFIIF, which affects the rate of elongation (Flores et al. 1989; Price et al. 1989; Bengal et al. 1991), allowed the few complexes that escaped arrest to reach full size (lane 4). The addition of TFIIF and FCP1 apparently stimulated the amount of full-length transcript (cf. lanes 4 and 8). The combination of both TFIIS and TFIIF resulted in almost complete release from arrest and allowed the production of the 977 run-off product (lane 5). The addition of FCP1 together with TFIIS and TFIIF stimulated the production of the full-length transcripts (lane 12). We interpreted these results to suggest that FCP1 stimulates elongation by RNAPII. Importantly, the effect observed was not dependent on the catalytic activity of the CTD phosphatase, as FCP1a had a similar effect as FCP1 (lanes 7, 9, 11, and 13). The effect observed was specific to the protein, as BSA, or proteins derived from mock-infected cells or mammalian capping enzyme purified from baculovirus infected cells were without effect (data not shown).
We analyzed the effect of FCP1 on RNAPII elongation directly. Arrested complexes were formed as above and treated with TFIIF and TFIIS (Fig. 7). One-third of the complexes were also treated with FCP1 or FCP1a. Then, allowed to resume elongation for different periods of time. After 30 s of elongation, the complexes containing FCP1 and FCP1a generated transcripts longer than the complexes containing only TFIIF and TFIIS (cf. lane 2 with lanes 3 and 4). Moreover, the complexes containing FCP1 or FCP1a started to accumulate full-length run-off product after 2 min of elongation (lanes 9 and 10), whereas in reactions lacking FCP1 and FCP1a, the full-length product was visible at later time points (data not shown). We concluded that FCP1 stimulates elongation by RNAPII, and this effect is independent of its phosphatase activity.
Figure 7.

The effect of FCP1 on elongation. Arrested elongating complexes were formed as in Fig. 6A, treated with rFCP1 (lanes 3,6,9) or rFCP1a (lanes 4,7,10) in the presence of TFIIF and TFIIS. Then, the transcripts were chased by the addition of all four nucleotides for different time points as indicated. (Lane 1) Transcripts before the addition of factors and the chase.
Discussion
In these studies, we purified a polypeptide from human cells that, together with purified human RNAPII, reconstitutes a highly specific CTD phosphatase activity. We have termed this activity FCP1. FCP1-mediated CTD phosphatase activity was stimulated by RAP74, the largest subunit of TFIIF. FCP1 activity resides in a single polypeptide of ∼150 kD that eluted from a gel-filtration column with an apparent mass of 300 kD. Thus, we concluded that FCP1 probably exists as a homodimer in solution. We demonstrated that FCP1 activity resides in the biochemically purified polypeptide by microsequencing the isolated protein and isolating a cDNA clone containing all the peptides derived from the native protein. More importantly, expression of the isolated cDNA demonstrated that it encodes an active polypeptide. Previous two-hybrid studies with RAP74 as the bait isolated a human cDNA clone, denoted FCP1a, that is related to FCP1 (Archambault et al. 1998). Our studies demonstrate, however, that FCP1a encodes a truncated polypeptide missing 119 amino acids at the amino terminus that are important for catalytic activity. In the previous studies, the initiation codon was incorrectly assigned (Archambault et al. 1997, 1998).
The primary sequence of FCP1 revealed a motif common to a family of phosphatases that act on phosphate esters (Collet et al. 1998). This phosphatase motif is present within residues that are conserved among different putative CTD phosphatases and other proteins in various databases (Archambault et al. 1997, 1998). Unfortunately, we were unable to obtain viable baculoviruses carrying mutations in this domain; however, studies performed with mutant versions of the yeast derived CTD phosphatase have demonstrated this domain to be important for enzymatic activity and viability (J. Greenblatt, M. Kobor, J. Archambault, and W. Lester, pers. comm.). Additionally, we uncovered a domain that is conserved in tyrosine phosphatases and a putative zinc-binding motif at the amino terminus of FCP1. Because the phosphatase activity required RNAPII and could not be reconstituted with the isolated CTD (H. Cho and D. Reinberg, unpubl., Chambers and Dahmus 1994) or with isolated Rpb1, the largest subunit of RNAPII (H. Cho and D. Reinberg, unpubl.), we cannot conclude that FCP1 encodes a CTD phosphatase. The results allow us to conclude that FCP1, together with highly purified RNAPII, reconstitutes CTD phosphatase activity. We have not ruled out the possibility that an additional phosphatase subunit residing in core RNAPII is also required. However, our results demonstrate that if FCP1 is not the CTD phosphatase, it is an important positive effector of CTD phosphatase activity.
The most important finding of our study concerns with the function of FCP1 in the transcription cycle. Our previous studies (Lu et al. 1991), as well as those of Dahmus and co-workers (Chesnut et al. 1992), demonstrated that the nonphosphorylated form of RNAPII efficiently associates with the transcription initiation complex. Elongation, however, is catalyzed by the phosphorylated form of RNAPII both in isolated nuclei (Cadena and Dahmus 1987) and in vitro (Bartholomew et al. 1986; Payne et al. 1989; Zawel et al. 1995). These findings led to the hypothesis that RNAPII cycles during the transcription cycle and invoke the requirement for a CTD phosphatase. Although CTD phosphatases have been reported in humans (Chambers and Dahmus 1994) and yeast (Chambers and Kane 1996), their role in the transcription cycle has not been addressed. The studies presented here demonstrate that FCP1 recycles RNAPII. In agreement with earlier observations (Lu et al. 1991; Chesnut et al. 1992), we found that the phosphorylated form of RNAPII was impaired in its ability to associate with transcription initiation complexes. Importantly, FCP1 dephosphorylates the CTD of RNAPII allowing efficient incorporation of RNAPII into transcription initiation complexes and resulting in increased transcription. Surprisingly, we observed that RNAPII incorporated into transcription initiation complexes was also susceptible to dephosphorylation by FCP1. Although this finding may suggest that FCP1 is active in initiation complexes, it should be taken cautiously. The transcription initiation complexes were formed with the nonphosphorylated form of RNAPII and phosphorylation was obtained in the initiation complex, in a TFIIH-dependent manner, after treatment with ATP, but prior to transcription initiation. This treatment likely resulted in conformational changes of the initiation complex and the release of TFIIB (Zawel et al. 1995), which previous studies have demonstrated to down-regulate CTD phosphatase activity (Chambers et al. 1995). It is important to emphasize that the FCP1 transcription-stimulatory activity was impaired by the preincubation of GTFs with RNAPII and DNA. Moreover, in the transcription cycle, the CTD is apparently phosphorylated subsequently to transcription initiation (Akoulitchev et al. 1995).
Our studies support the model presented in Figure 8. As demonstrated previously, RNAPII, through its interactions with TFIIF, recognizes the TB complex composed of TBP in association with the TATA motif and TFIIB. Because TFIIF was found to interact with both the phosphorylated and nonphosphorylated forms of RNAPII (Zawel et al. 1995) and FCP1 and to stimulate FCP1 activity (Chambers et al. 1995), its association with RNAPII prior to association with the TB complex may be important in attaining an RNAPII that is fully dephosphorylated. The association of RNAPII with promoter sequences provides the foundation for the entry of TFIIE and allows the association of TFIIH, resulting in the formation of a fully competent transcription initiation complex. During the process of initiation and prior to the formation of a fully competent elongation complex, the CTD becomes phosphorylated. Phosphorylation of the CTD does not affect elongation efficiency (Peng et al. 1998), but allows RNAPII to disengage from the promoter and from transcription initiation factors (Usheva et al. 1992; Zawel et al. 1995). CTD phosphorylation provides a foundation for the association of factors involved in RNA processing such as the capping enzyme (Cho et al. 1997; McCracken et al. 1997a; Yue et al. 1997; Ho et al. 1998), splicing factors (McCracken et al. 1997b), and factors involved in 3′-end formation (Hirose and Manley 1998). Upon transcription of termination/polyadenylation signals, the elongating complex is altered, resulting in the release of RNAPII from the template by an unknown process. It is possible that RNAPII is converted to the nonphosphorylated form prior to, or concomitant with, its release from the DNA template. This possibility is supported by our studies demonstrating that FCP1 is capable of dephosphorylating the CTD of RNAPII not only in solution prior to incorporation into transcription initiation complexes, but also in active ternary elongation complexes stalled as a result of nucleotide starvation. The finding that FCP1 also stimulates elongation by RNAPII independent of its phosphatase activity, suggests that FCP1 may remain associated with RNAPII during elongation. The finding that FCP1 is active in ternary complexes has implications for the mechanism of transcription termination as well as for the down-regulation of RNA processing. Similar to the signal imposed on phosphorylation of the CTD—disengagement of RNAPII from the promoter and from interaction with initiation factors—dephosphorylation of the CTD may result in a signal that releases factors involved in RNA maturation from RNAPII.
Figure 8.
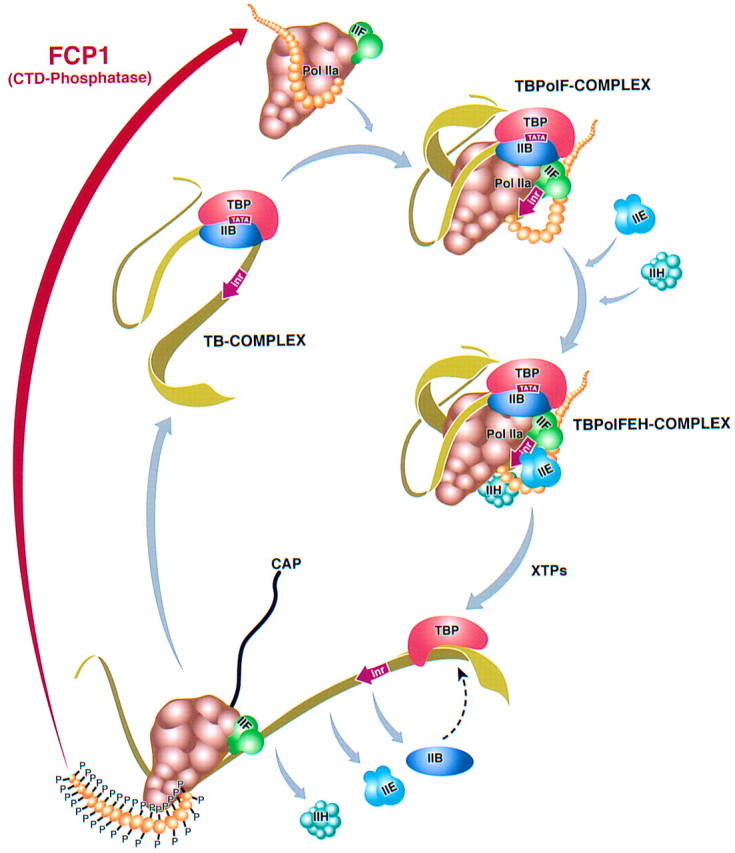
Model illustrating the RNAPII transcription cycle. The TB complex is recognized by the RNAPII–TFIIF complex that was released from ternary complexes and subjected to the action of FCP1. FCP1 dephosphorylates the CTD converting RNAPII to an initiation competent form. After association with promoter sequences (TBPolF complex), TFIIE and TFIIH enter into the transcription cycle generating an initiation competent complex (TBPolFEH). In the presence of the ribonucleoside triphosphates, the transcription initiation complex disassembles with the release of TFIIB, TFIIE, and TFIIH. TBP remains bound to the TATA motif and provides the foundation for the reassociation of TFIIB. The CTD of RNAPII becomes phosphorylated providing a platform for the association of factors involved in RNA processing. The nascent RNA becomes capped. Upon reaching termination sequences, the CTD becomes dephosphorylated by FCP1 within the ternary complex or immediately after the release of RNAPII from the DNA template. TFIIF is associated with the elongation competent form of RNAPII.
Materials and methods
Purification of CTD phosphatase activity from HeLa nuclear extracts
HeLa cell nuclear extract preparation and phosphocellulose chromatography were as described previously (Maldonado et al. 1996). Protein from the 0.1 m phosphocellulose eluate (5.8 grams) was loaded onto a 600-ml DEAE–cellulose column equilibrated with BC100{100 mm KCl containing buffer C [20 mm Tris-HCl (pH 7.9), 0.2 mm EDTA, 10 mm β-mercaptoethanol, 10% (vol/vol) glycerol, 0.2 mm PMSF] }. The column was washed extensively with BC100 until no protein was detected in the eluate. CTD phosphatase activity (2 grams) was step eluted with BC350 (350 mm KCl containing buffer C). Prior to phenyl–Sepharose chromatography, CTD-phosphatase activity was precipitated with 60% (wt/vol) ammonium sulfate. The precipitate (945 mg) was resuspended in buffer D [20 mm Tris-HCl (pH 7.9), 0.2 mm EDTA, 1 mm DTT, 10% (vol/vol) glycerol, and 0.2 mm PMSF] to a concentration of 700 mm ammonium sulfate. Proteins were loaded onto a phenyl–Sepharose HiLoad 26/10 FPLC column (Pharmacia) equilibrated with BD700 (700 mm ammonium sulfate containing buffer D). The column was resolved with a linear gradient from 700 to 0 mm ammonium sulfate in buffer D. Fractions were assayed for CTD phosphatase activity as described below. Fractions containing CTD phosphatase activity were pooled and dialyzed against BD100 (100 mm ammonium sulfate containing buffer D) and loaded onto an S-Sepharose (Sigma) column. The flowthrough (72 mg) was loaded onto a DEAE-5PW HPLC column (21.5 mm ID × 15 cm, Toso Haas) equilibrated with BD100. The column was developed with a 15-column volume linear gradient from BD100 to BD700. Fractions containing CTD phosphatase activity were pooled (12 mg) and fractionated repeatedly on a DEAE- 5PW column. The final active fractions (1.2 mg) were pooled and dialyzed against BC100. The protein fraction was loaded onto a MonoP 5/5 FPLC column equilibrated with BC100 and developed with a 10-column volume linear gradient from BC100 to BC1000. Fractionation on this column was repeated, active fractions were pooled (0.5 μg), and the 150 and 77 kD polypeptides were analyzed by ion-trap mass spectrometry. An aliquot of the Mono
P fraction was analyzed on a Superose 12 3.2/30 gel-filtration column in BC500 (buffer C containing 500 mm KCl) and 0.01% (vol/vol) Triton X-100.
CTD phosphatase assay
RNAPIIO was purified as described previously (Lu et al. 1991). Approximately 100 ng of RNAPIIO was incubated with protein fractions in buffer P [20 mm HEPES (pH 7.5), 10 mm MgCl2, 1 mm DTT, 10% (vol/vol) glycerol and 0.2 mm PMSF] in 20 μl for 30 min at 30°C. An aliquot of each column fraction was dialyzed against buffer C containing 20 mm KCl prior to the assay. Where indicated, bacterially expressed TFIIF or RAP74 (50 ng) was added to the RNAPIIO prior to the addition of the phosphatase fraction. Reactions were terminated by the addition of 6 μl of SDS loading buffer [12% (wt/vol) SDS, 0.6 mm DTT, 300 mm Tris-HCl (pH 6.8) 0.6% (wt/vol) bromophenol blue, and 60% (vol/vol) glycerol] prior to a 2-min boiling. The reaction mixture was loaded onto a 5%–12% (wt/vol) gradient SDS–polyacrylamide gel and transferred to a PVDF membrane (Immobilon, Millipore). The Western blot was blocked with 3% (wt/vol) nonfat powdered milk in TTBS [10 mm Tris-HCl (pH 7.5), 200 mm NaCl, 0.05% (vol/vol) Tween-20] for 30 min at room temperature with gentle shaking. The blots were washed with TTBS buffer briefly and subsequently incubated with anti-GAL4–CTD antiserum (diluted 1:500 in TTBS) for 2 hr at room temperature with gentle shaking. After washing with TTBS, the blots were incubated with peroxidase-conjugated protein A (Bio-Rad; diluted to 1:2000 in TTBS) for 30 min at room temperature. Finally, Western blots were washed and developed by enhanced chemiluminescence according to the manufacturer’s protocol (Boehringer Mannheim).
Renaturation of CTD phosphatase activity
Approximately 60 μg of protein derived from the phenyl–Sepharose fraction was sedimented by a 15-min centrifugation at 12000g after a 10-min incubation with 0.015% (wt/vol) deoxycholate at room temperature and addition of 7.2% (wt/vol) trichloroacetic acid. Precipitates were washed with 80% (vol/vol) ethanol and vacuum dried. Precipitates were resuspended in 10 mm Tris base buffer and loaded onto a 10% (wt/vol) SDS–polyacrylamide gel. Polypeptides in the molecular weight range of 150 and 77 kD, which coeluted with CTD phosphatase activity in the Mono P column, together with a 55-kD polypeptide used as a negative control excised from the gel and eluted at 37°C with gentle shaking in diffusion buffer [50 mm Tris-HCl (pH 7.5), 0.1 mm EDTA, 0.1% (wt/vol) SDS, 5 mm DTT, 150 mm NaCl] for 12 hr. The supernatant was collected and spun through a spin column (Chroma-spin TE-10, Clontech), equilibrated with denaturation buffer [50 mm Tris-HCl (pH 7.9), 1 mm EDTA, 1 mm DTT, 20% (vol/vol) glycerol, 0.1 m KCl, 0.1% (vol/vol) NP40, 6 m guanidine-HCl] and incubated at room temperature for 30 min. Then, the samples were spun again in a spin column equilibrated with equilibration buffer [50 mm Tris-HCl (pH 7.9), 1 mm EDTA, 1 mm DTT, 20% (vol/vol) glycerol, 0.1 m KCl, 10 mm MgCl2, 0.1% (vol/vol) NP40], and samples were assayed for activity after a 15-min renaturation at room temperature.
Ion-trap mass spectrometry and peptide sequencing of p150
Multiple peptide sequences were determined at high sensitivity in a single run by microcapillary reverse-phase chromatography coupled directly to a Finnigan LCQ ion-trap mass spectrometer. To perform this step, an excised Coomassie-stained p150 band from SDS-PAGE was subjected to in-gel reduction, carboxyamidomethylation, and tryptic digestion (Promega). Ten percent of the digestion mixture was pressure loaded onto 5 cm of reverse-phase support (POROS) packed in-house into a 75-μm I.D. column. A gradient of 0% to 50% acetonitrile in 0.5 m acetic acid over 25 min chromatographed peptides directly into the electrospray source of the mass spectrometer. The ion trap was programmed to acquire successive sets of three scan modes consisting of full scan MS over the range of 395–1118 m/z, followed by two data-dependent scans on the most abundant ion in those full scans. These data-dependent scans allowed the automatic acquisition of a high resolution (zoom) scan to determine charge state and exact mass, and MS/MS spectra for the peptide sequence information. MS/MS spectra were acquired with a relative collision energy of 35% and an isolation width of 2.5 daltons. Interpretation of the resulting MS/MS spectra of the peptides was facilitated by the database correlation with the algorithm SEQUEST and by programs developed in the Harvard Microchemistry Facility (Eng et al. 1994; Chittum et al. 1998).
Library screening and molecular cloning of p150
Approximately 5 × 105 plaques from a HeLa cell cDNA library (Clontech) were screened with a DNA fragment that was obtained by 5′ RACE (Clontech) labeled with [α-32P]dCTP by a random primer (Boehringer Mannheim) according to the manufacturer’s protocol. Briefly, the first PCR round was accomplished with the AP1 primer (Clontech), which hybridizes to the adapter sequence of the cDNA library, and a gene-specific primer (CCTGCAGCACCTTCTCTGTGCCGC), which hybridizes to FCP1a cDNA. To enrich further for a gene-specific PCR product, nested PCR was performed with the AP2 primer (Clontech) and a nested gene-specific primer (CTGAGCGGGAAGAGCTGCTCCTC). The resulting PCR fragment was subcloned into the pCR2.1 vector (Invitrogen) for DNA sequence analysis and was also used as a template to generate the radioactive probe for library screening. Two plaques were isolated, and the DNA was extracted. Each cDNA was excised from the λ vector by EcoRI digestion and subcloned into the EcoRI site in pBluescript SK (SK3-1 and SK7-1) which contains amino-terminal truncated FCP1 (see below). Sequences from the 5′ end of the cDNA (SK3-1) were PCR amplified with the T7 primer and a gene-specific primer (GACAACCGGGTGGCTGCACCT) and labeled with [α-32P]dCTP using the random priming method (Boehringer Mannheim) for use as a probe for further library screening. Two independent clones containing additional 5′-sequences were obtained from ∼7 × 105 plaques. Each DNA was extracted and excised from the λ vector by EcoRI digestion and subcloned into the EcoRI site in pBluescript SK(−) (SK4-1 and SK14-1). DNA sequences of both strands were determined. Amino-terminally His-tagged expression vectors were constructed by subcloning of the entire p150 cDNA sequence of SK4-1 (SalI fragment) into the SalI site in pET-28a (+, Novagen). FCP1ΔC269-961 was generated by deletion of an XbaI fragment. FCP1ΔC456-961 was generated by subcloning of a NotI fragment from SK14-1 into pET-28a (+). FCP1a was generated by subcloning of the coding sequences from amino acids 120–961 by PCR from SK3-1 plasmid into TP33.
In vitro translation of nontagged and amino-terminally His-tagged FCP1 constructs
In vitro translation was carried out as recommended by the manufacturer (NEB). Nontagged full-length and amino-terminally truncated (119-amino-acid) FCP1 proteins were generated from 1 μg of SK4-1 and SK3-1, respectively. The DNAs were transcribed by T7 RNA polymerase and translated by rabbit reticulocyte with [35S]methionine in vitro. Equal amounts of the translated products were separated on a 10% (wt/vol) SDS–polyacrylamide gel, which was analyzed by Western blot with antibodies against FCP1. For CTD phosphatase assays, the amino-terminally His-tagged expression vectors (1 μg) were in vitro transcribed and translated as described above. One-third of each reaction mixture was immobilized onto protein G–agarose beads (Boehringer Mannheim) with monoclonal antibodies against the His tag (Clontech). The immunoprecipitates were washed extensively with BC500 and 0.1% (vol/vol) NP40 and subsequently equilibrated with BC100. Each immunoprecipitate was subjected to the CTD phosphatase assay. Western blots of the assay were also autoradiographed to visualize the [35S]methionine-labeled immunoprecipitated proteins.
Recombinant baculoviruses
FCP1 and FCP1a cDNAs were cloned into the pVL1393 baculovirus transfer vector (Invitrogen) at the BamHI and XbaI sites and cotransfected with BaculoGold DNA (Pharmingen) containing AcNPV baculovirus DNA into SF9 insect cells as described by the manufacturer. These cells were maintained in Hink′s Insect Cell media supplemented with 10% FBS and an antibiotic/antimycotic mix (a/a). Recombinant baculoviruses were selected by survival selection and amplified twice. Recombinant proteins were expressed by infection of Hi 5 cells at low cell density of about 1.5 × 106 cells/ml for 72 hr at 30°C. Recombinant proteins were purified by Ni–NTA agarose chromatography.
Antibody production
Polyclonal rabbit antibodies were raised against a multiple antigenic peptide (RLHRNKL VLNVDLDQTTHTTEQHCQQ) of hFCP1 amino acid sequences obtained after the report by Archambault et al. (1997) describing the sequence of yeast FCP1 and its human homolog. After five booster injections, the rabbits were sacrificed, and serum was collected. Antibodies were affinity purified by use of the linear peptide cross-linked to Affiprep 10 according to the manufacturer’s protocol (Bio-Rad).
Transcription assays
Transcription assays were reconstituted with highly purified Escherichia coli-expressed TBP, TFIIB, TFIIE, TFIIF, and TFIIS, highly purified native RNAPIIA, RNAPIIO, and TFIIH isolated from HeLa cells and baculovirus-expressed recombinant FCP1 (rFCP1) and FCP1a (rFCP1a). All reactions were performed at 25°C.
Preinitiation complexes were assembled on linear DNA (HindIII–SspI) derived from pΔ50ML, which contains the adenovirus major late core promoter driving transcription of a 392-nucleotide G-less cassette. Because our initial trials analyzing differences in transcription by RNAPIIA and RNAPIIO failed, we introduced the following changes. We rationalized that if the interactions between the RNAPII and GTFs were compromised by a rise in salt concentration, the system may become more sensitive to the overall interactions between GTFs and RNAPII, and thus, the state of phosphorylation of the CTD may influence transcription. Transcription was compromised by formation of preinitiation complexes in 30 μl of buffer E [20 mm HEPES (pH 7.8), 10 mm MgCl2, 10 mm DTT, 10 mm ammonium sulfate, 2% (wt/vol) PEG 8000, 60 μm KCl, 40 mm NaCl, 0.01% (vol/vol) NP40, 0.1% (vol/vol) Triton X-100, 20 mm imidazole, 7% glycerol] for 20 min at room temperature in the absence or presence of baculovirus-expressed FCP1 or FCP1a as indicated. After preinitiation complex formation, transcription was allowed to occur for 35 min by addition of 10 μl of elongation buffer [2.4 mm ATP and CTP, 60 μm of UTP (Pharmacia), 5 μCi of [α-32P]UTP (NEN), RNase T1 (30 units; Boehringer Mannheim) in 20 mm HEPES (pH 7.8), 8 mm MgCl2]. The final volume of the reaction mixtures was 40 μl. Reactions were terminated by the addition of 80 μl of stop solution [0.1 m sodium acetate (pH 5.2), 0.2% (wt/vol) SDS, 10 mg/ml yeast tRNA]. Purified transcripts were analyzed on an 8 m urea–6% (wt/vol) polyacrylamide gel and exposed overnight to X-ray film at −80°C. Elongation assays were performed on a linear DNA (HindIII–SspI) fragment of pΔ50ML immobilized on streptavidin-coated magnetic beads (Dynal) through the HindIII site biotinylated with biotinyl–dATP (GIBCO-BRL). Preinitiation complexes were preformed on immobilized beads in buffer F [20 mm HEPES (pH 7.8), 8 mm MgCl2, 10 mm DTT, 10 mm ammonium sulfate, 2% (wt/vol) PEG 8000, 60 mm KCl, 5.7% (vol/vol) glycerol] for 20 min. Transcription was allowed to occur for 35 min with the addition of 600 μm ATP and CTP, 15 μm UTP (Pharmacia), 5 μCi of [α-32P]UTP (NEN) in 20 mm HEPES (pH 7.5), and 8 mm MgCl2. Ternary complexes were arrested at the end of the G-less cassette as a result of nucleotide starvation. Stalled complexes were washed with BC500 and 0.5% (wt/vol) sarkosyl to remove released factors and nucleotides and were subsequently equilibrated with BC100. Prior to resuming elongation, ternary complexes were incubated in 35 μl of 20 mm HEPES (pH 7.8), 9.1 mm MgCl2, 5.6 mm DTT, 34 mm NaCl, 17 mm imidazole, 0.01% (vol/vol) NP40, 0.09% (vol/vol) Triton X-100, 0.86% (vol/vol) glycerol for 20 min with or without additional factors (TFIIF, TFIIS, FCP1, and/or FCP1a) as indicated in the figure. The phosphorylation state of Rpb1 on ternary complexes treated with FCP1 were analyzed from the beads by Western blot. Elongation was resumed by the addition of 5 μl of nucleotides and ammonium sulfate to a final concentration of 600 μm of ATP, CTP, UTP, and GTP and 7.5 mm ammonium sulfate for the time indicated, and terminated by the addition of 80 μl of stop solution as above. Transcripts were analyzed on an 8 m urea–5% (wt/vol) polyacrylamide gel.
UV cross-linking experiments
UV cross-linking was performed as described (Lagrange et al. 1996; Kim et al. 1997) with the following modifications. Preinitiation complexes were formed on a promoter DNA fragment derivatized at position −4 on the nontemplate DNA strand (Kim et al. 1997). The DNA was immobilized on streptavidin-coated magnetic beads. Transcription reactions were performed as described in Figure 6, above. The transcription complexes were washed with transcription buffer [13 mm HEPES (pH 7.9), 5 mm MgCl2, 2.6% (wt/vol) PEG 8000, 12 mm Tris-HCl (pH 7.9), 60 mm KCl, 0.1 mm EDTA, 0.3 mm DTT, 0.1 mm PMSF, 12% (vol/vol) glycerol], and 0.05% (wt/vol) sarkosyl. Preinitiation complexes were UV cross-linked directly (Fig. 5, lanes 1–8) or incubated further at 37°C for 7 min with ATP (1 mm, as indicated in Fig. 5). After incubation, the complexes were washed again with transcription buffer to remove ATP and then incubated with CTD phosphatase in 28 mm HEPES (pH 7.8), 10 mm MgCl2, 2 mm DTT, 40 mm NaCl, 0.01% (vol/vol) Triton X-100, 0.1% (vol/vol) NP40, 20 mm imidazole, and 1% (vol/vol) glycerol for 20 min at room temperature as indicated in the figure. Prior to UV cross-linking, the transcription complexes were washed with buffer C containing 50 mm KCl. All UV cross-linked complexes were processed further as in Kim et al. (1997). UV-crosslinked proteins were resolved on 5%–12% (wt/vol) SDS–polyacrylamide gels. The gels were vacuum dried and analyzed by STORM PhosphorImager.
Acknowledgments
We thank Dr. D. Luse for the gift of TFIIS, Dr. A. Shatkin for capping enzyme, and Drs. Michael Dahmus, Mike Hampsey, and George Orphanides for comments on the manuscript. We also thank Kerry A. Pierce for technical expertise. We are grateful to Drs. J. Greenblatt, M. Kobor, J. Archambault, and W. Lester for communicating umpublished results. This work was supported by a grant to D.R. from the National Institutes of Health (GM-37120) and from the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
The sequence data described in this paper have been submitted to GenBank under accession number AF154115.
Footnotes
E-MAIL reinbedf@umdnj.edu; FAX (732) 235-5294.
References
- Akoulitchev S, Mäkelä TP, Weinberg RA, Reinberg D. Requirement for TFIIH kinase activity in transcription by RNA polymerase II. Nature. 1995;377:557–560. doi: 10.1038/377557a0. [DOI] [PubMed] [Google Scholar]
- Archambault J, Chambers RS, Kobor MS, Ho Y, Cartier M, Bolotin D, Andrews B, Kane CM, Greenblatt J. An essential component of a C-terminal domain phosphatase that interacts with transcription factor IIF in Saccharomyces cerevisiae. Proc Natl Acad Sci. 1997;94:14300–14305. doi: 10.1073/pnas.94.26.14300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambault J, Pan G, Dahmus GK, Cartier M, Marshall N, Zhang S, Dahmus ME, Greenblatt J. FCP1, the RAP74-interacting subunit of a human protein phosphatase that dephosphorylates the carboxyl-terminal domain of RNA polymerase IIO. J Biol Chem. 1998;273:27593–27601. doi: 10.1074/jbc.273.42.27593. [DOI] [PubMed] [Google Scholar]
- Bartholomew B, Dahmus ME, Meares ME. RNA contacts subunits IIo and IIc in HeLa RNA polymerase II transcription complexes. J Biol Chem. 1986;261:14226–14231. [PubMed] [Google Scholar]
- Bengal E, Flores O, Krauskopf A, Reinberg D, Aloni Y. Role of the mammalian transcription factors IIF, IIS, and IIX during elongation by RNA polymerase II. Mol Cell Biol. 1991;11:1195–1206. doi: 10.1128/mcb.11.3.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena DL, Dahmus ME. Messenger RNA synthesis in mammalian cells is catalyzed by the phosphorylated form of RNA polymerase II. J Biol Chem. 1987;262:12468–12474. [PubMed] [Google Scholar]
- Chambers RS, Dahmus ME. Purification and characterization of a phosphatase from HeLa cells which dephosphorylates the C-terminal domain of RNA polymerase II. J Biol Chem. 1994;269:26243–26248. [PubMed] [Google Scholar]
- Chambers RS, Kane CM. Purification and characterization of an RNA polymerase II phosphatase from yeast. J Biol Chem. 1996;271:24498–24504. doi: 10.1074/jbc.271.40.24498. [DOI] [PubMed] [Google Scholar]
- Chambers RS, Wang BQ, Burton ZF, Dahmus ME. The activity of COOH-terminal domain phosphatase is regulated by a docking site on RNA polymerase II and by the general transcription factors IIF and IIB. J Biol Chem. 1995;270:14962–14969. doi: 10.1074/jbc.270.25.14962. [DOI] [PubMed] [Google Scholar]
- Chesnut JD, Stephens JH, Dahmus ME. The interaction of RNA polymerase II with the adenovirus-2 major late promoter is precluded by phosphorylation of the C-terminal domain of subunit IIa. J Biol Chem. 1992;267:10500–10506. [PubMed] [Google Scholar]
- Chittum HS, Lane WS, Carlson BA, Roller PP, Lung FD, Lee BJ, Hatfield DL. Rabbit β-globin is extended beyond its UGA stop codon by multiple suppressions and translational reading groups. Biochemistry. 1998;37:10866–10870. doi: 10.1021/bi981042r. [DOI] [PubMed] [Google Scholar]
- Cho EJ, Takagi T, Moore CR, Buratowski S. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes & Dev. 1997;11:3319–3326. doi: 10.1101/gad.11.24.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet JF, Stroobant V, Pirard M, Delpierre G, Van Schaftingen E. A new class of phosphotransferases phosphorylated on an aspartate residue in an amino-terminal DXDX(T/V) motif. J Biol Chem. 1998;273:14107–14112. doi: 10.1074/jbc.273.23.14107. [DOI] [PubMed] [Google Scholar]
- Dahmus ME. The role of multisite phosphorylation in the regulation of RNA polymerase II activity. Prog Nucleic Acid Res Mol Biol. 1994;48:143–179. doi: 10.1016/s0079-6603(08)60855-7. [DOI] [PubMed] [Google Scholar]
- ————— Reversible phosphorylation of the C-terminal domain of RNA polymerase II. J Biol Chem. 1996;271:19009–19012. doi: 10.1074/jbc.271.32.19009. [DOI] [PubMed] [Google Scholar]
- Denu JM, Dixon JE. A catalytic mechanism for the dual-specific phosphatases. Proc Natl Acad Sci. 1995;92:5910–5914. doi: 10.1073/pnas.92.13.5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormick AL, Yates JRI. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Feaver WJ, Gileadi O, Li Y, Kornberg RD. CTD kinase associated with yeast RNA polymerase II initiation factor b. Cell. 1991;67:1223–1230. doi: 10.1016/0092-8674(91)90298-d. [DOI] [PubMed] [Google Scholar]
- Flores O, Maldonado E, Reinberg D. Factors involved in specific transcription by mammalian RNA polymerase II. Factors IIE and IIF independently interact with RNA polymerase II. J Biol Chem. 1989;264:8913–8921. [PubMed] [Google Scholar]
- Flores O, Ha I, Reinberg D. Factors involved in specific transcription by mammalian RNA polymerase II. J Biol Chem. 1990;265:5629–5634. [PubMed] [Google Scholar]
- Hampsey M. Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol Mol Biol Rev. 1998;62:465–503. doi: 10.1128/mmbr.62.2.465-503.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose Y, Manley JL. RNA polymerase II is an essential mRNA polyadenylation factor. Nature. 1998;395:93–96. doi: 10.1038/25786. [DOI] [PubMed] [Google Scholar]
- Ho CK, Sriskanda V, McCracken S, Bentley D, Schwer B, Shuman S. The guanylyltransferase domain of mammalian mRNA capping enzyme binds to the phosphorylated carboxyl-terminal domain of RNA polymerase II. J Biol Chem. 1998;273:9577–9585. doi: 10.1074/jbc.273.16.9577. [DOI] [PubMed] [Google Scholar]
- Kim TK, Lagrange T, Wang YH, Griffith JD, Reinberg D, Ebright RH. Trajectory of DNA in the RNA polymerase II transcription preinitiation complex. Proc Natl Acad Sci. 1997;94:12268–12273. doi: 10.1073/pnas.94.23.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske AJ, Young RA. The RNA polymerase II holoenzyme and its implications for gene regulation. Trends Biochem Sci. 1995;20:113–116. doi: 10.1016/s0968-0004(00)88977-x. [DOI] [PubMed] [Google Scholar]
- Lagrange T, Kim T-K, Orphanides G, Ebright YW, Ebright RH, Reinberg D. High-resolution mapping of nucleoprotein complexes by site-specific protein-DNA photocrosslinking: Organization of the human TBP-TFIIA-TFIIB-DNA quaternary complex. Proc Natl Acad Sci. 1996;93:10620–10625. doi: 10.1073/pnas.93.20.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laybourn PJ, Dahmus ME. Transcription-dependent structural changes in the C-terminal domain of mammalian RNA polymerase subunit IIa/o. J Biol Chem. 1989;264:6693–6698. [PubMed] [Google Scholar]
- LeRoy G, Drapkin R, Weis L, Reinberg D. Immunoaffinity purification of the human multisubunit transcription factor IIH. J Biol Chem. 1998;273:7134–7140. doi: 10.1074/jbc.273.12.7134. [DOI] [PubMed] [Google Scholar]
- Lu H, Flores O, Weinmann R, Reinberg D. The nonphosphorylated form of RNA polymerase II preferentially associates with the preinitiation complex. Proc Natl Acad Sci. 1991;88:10004–10008. doi: 10.1073/pnas.88.22.10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Zawel L, Fisher L, Egly J-M, Reinberg D. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature. 1992;358:641–645. doi: 10.1038/358641a0. [DOI] [PubMed] [Google Scholar]
- Maldonado E, Drapkin R, Reinberg D. Purification of human RNA polymerase II and general transcription factors. Methods Enzymol. 1996;274:72–100. doi: 10.1016/s0076-6879(96)74009-0. [DOI] [PubMed] [Google Scholar]
- Marshall NF, Dahmus GK, Dahmus ME. Regulation of carboxyl-terminal domain phosphatase by HIV-1 tat protein. J Biol Chem. 1998;273:31726–31730. doi: 10.1074/jbc.273.48.31726. [DOI] [PubMed] [Google Scholar]
- McCracken S, Fong N, Rosonina E, Yankulov K, Brothers G, Siderovski D, Hessel A, Foster S, Shuman S, Bentley DL. 5′-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes & Dev. 1997a;11:3306–3318. doi: 10.1101/gad.11.24.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson SD, Wickens M, Bentley DL. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997b;385:357–361. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- O’Brien T, Hardin S, Greenleaf A, Lis JT. Phosphorylation of RNA polymerase II C-terminal domain and transcriptional elongation. Nature. 1994;370:75–77. doi: 10.1038/370075a0. [DOI] [PubMed] [Google Scholar]
- Orphanides G, Lagrange T, Reinberg D. The general transcription factors of RNA polymerase II. Genes & Dev. 1996;10:2657–2683. doi: 10.1101/gad.10.21.2657. [DOI] [PubMed] [Google Scholar]
- Payne JM, Laybourn PJ, Dahmus ME. The transition of RNA polymerase II from initiation to elongation is associated with phosphorylation of the carboxyl-terminal domain of subunit IIa. J Biol Chem. 1989;264:19621–19629. [PubMed] [Google Scholar]
- Peng J, Liu M, Marion J, Zhu Y, Price DH. RNA polymerase II elongation control. Cold Spring Harbor Symp Quant Biol. 1998;63:365–370. doi: 10.1101/sqb.1998.63.365. [DOI] [PubMed] [Google Scholar]
- Price DH, Sluder AE, Greenleaf AL. Dynamic interaction between a Drosophila transcription factor and RNA polymerase II. Mol Cell Biol. 1989;9:1465–1475. doi: 10.1128/mcb.9.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serizawa H, Conaway RC, Conaway JW. A carboxy-terminal-domain kinase associated with RNA polymerase II transcription factor d from rat liver. Proc Natl Acad Sci. 1992;89:7476–7480. doi: 10.1073/pnas.89.16.7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usheva A, Maldonado E, Goldring A, Lu H, Houbavi C, Reinberg D, Aloni J. Specific interaction between the nonphosphorylated form of RNA polymerase II and the TATA-binding protein. Cell. 1992;69:871–881. doi: 10.1016/0092-8674(92)90297-p. [DOI] [PubMed] [Google Scholar]
- Yue Z, Maldonado E, Pillutla R, Cho H, Reinberg D, Shatkin AJ. Mammalian capping enzyme complements mutant Saccharomyces cerevisiae lacking mRNA guanylyltransferase and selectively binds the elongating form of RNA polymerase II. Proc Natl Acad Sci. 1997;94:12898–12903. doi: 10.1073/pnas.94.24.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L, Reinberg D. Initiation of transcription by RNA polymerase II: A multi-step process. Prog Nucleic Acid Res Mol Biol. 1993;44:67–108. doi: 10.1016/s0079-6603(08)60217-2. [DOI] [PubMed] [Google Scholar]
- ————— Common themes in assembly and function of eukaryotic transcription factors. Annu Rev Biochem. 1995;64:533–561. doi: 10.1146/annurev.bi.64.070195.002533. [DOI] [PubMed] [Google Scholar]
- Zawel L, Lu H, Cisek LJ, Corden JL, Reinberg D. The cycling of RNA polymerase II during transcription. Cold Spring Harb Symp Quant Biol. 1993;58:187–198. doi: 10.1101/sqb.1993.058.01.023. [DOI] [PubMed] [Google Scholar]
- Zawel L, Kumar KP, Reinberg D. Recycling of the general transcription factors during RNA polymerase II transcription. Genes & Dev. 1995;9:1479–1490. doi: 10.1101/gad.9.12.1479. [DOI] [PubMed] [Google Scholar]