

Abstract
Fission yeast Cut5/Rad4 plays a unique role in the genome maintenance as it is required for replication, replication checkpoint, and normal UV sensitivity. It is unknown, however, how Cut5 protein is linked to other checkpoint proteins, and what part it plays in replication and UV sensitivity. Here we report that Cut5 interacts with a novel checkpoint protein Crb2 and that this interaction is needed for normal genome maintenance. The carboxyl terminus of Crb2 resembles yeast Rad9 and human 53BP1 and BRCA1. Crb2 is required for checkpoint arrests induced by irradiation and polymerase mutations, but not for those induced by inhibited nucleotide supply. Upon UV damage, Crb2 is transiently modified, probably phosphorylated, with a similar timing of phosphorylation in Chk1 kinase, which is reported to restrain Cdc2 activation. Crb2 modification requires other damage-sensing checkpoint proteins but not Chk1, suggesting that Crb2 acts at the upstream of Chk1. The modified Crb2 exists as a slowly sedimenting form, whereas Crb2 in undamaged cells is in a rapidly sedimenting structure. Cut5 and Crb2 interact with Chk1 in a two-hybrid system. Moreover, moderate overexpression of Chk1 suppresses the phenotypes of cut5 and crb2 mutants. Cut5, Crb2, and Chk1 thus may form a checkpoint sensor-transmitter pathway to arrest the cell cycle.
Keywords: UV damage, hydroxyurea, DNA polymerase, phosphorylation, two-hybrid screen
The eukaryotic genome is maintained by a number of controlling elements. Weinert and Hartwell (1988) identified a checkpoint gene RAD9 in the budding yeast Saccharomyces cerevisiae, which monitors the damage of DNA on UV irradiation, and delays the progression of cell cycle. In the absence of the RAD9 function, cells containing the damaged DNAs enter fatal cell division without a delay. A number of checkpoint genes involved in DNA damage, replication, and spindle defects have been identified (e.g., Elledge 1996; Kitazono and Matsumoto 1997). The word checkpoint in the present paper is used as synonymous to a surveillance mechanism that blocks or delays cell-cycle transitions (Nasmyth 1996). Cell-cycle regulators are also important elements in maintenance of the genome. When the mitotic cyclin gene, cdc13+, is lost in the fission yeast Schizosaccharomyces pombe, cells are unable to enter mitosis but perform repeated DNA replications, leading to the formation of polyploidy giant nucleus (Hayles et al. 1994). Similar phenotypes can be produced by overproducing Rum1, the inhibitor for CDK/cyclin B (Moreno and Nurse 1994) or constitutively activating Cdc18, an essential replication initiation factor (Kelly et al. 1993; Nishitani and Nurse 1995). In mammalian cells, the genome is maintained by tumor-suppressing proteins such as p53 and RB. p53 is a transcription factor, activating transcription of certain inhibitors against CDK kinase on DNA damage, leading to the arrest of cells in the G1 phase (e.g., El-Deiry et al. 1993). If p53 is lost, the damage checkpoint in the G1 phase no longer seems to be functional. These various gene functions contribute to the quality control of genome.
The fission yeast cut5+ gene is uniquely implicated in the genome maintenance as it is essential for DNA replication, replication checkpoint control, and normal UV sensitivity (Saka et al. 1994a;b). Temperature-sensitive cut5 mutants at the restrictive temperature (36°C) block DNA replication but enter mitosis, producing the cut phenotype (Saka and Yanagida 1993), which strikingly differs from the arrest phenotype of temperature sensitive cdc mutations in DNA polymerase or ligase, or ribonucleotide reductase (RNR) (Nurse et al. 1976; Gordon and Fantes 1986). The cell-cycle arrest of these replication-defective cdc mutants is caused by the replication checkpoint control that also operates in the presence of hydroxyurea (HU), an inhibitor of RNR. The double-mutants between cut5 and polymerase or ligase mutants also displayed no retardation in cell division at 36°C (Saka et al. 1994a;b). These results showed that Cut5 was required for the replication checkpoint arrest induced by HU or mutations in the replication enzymes. In S. pombe, the HU-induced replication checkpoint arrest is also abolished in hus and certain rad mutants, rad1, rad3, rad9, rad17, and rad26 (Al-Khodairy and Carr 1992; Enoch et al. 1992; Rowley et al. 1992; Sheldrick and Carr 1993; Ford et al. 1994; Griffiths et al. 1995; Bentley et al. 1996; Lieberman et al. 1996; Kostrub et al. 1997).
In addition to the phenotypes described above, cut5 mutants are sensitive to UV irradiation at the permissive temperature. Consistent with the phenotype, isolation of the cut5+ gene (Saka and Yanagida 1993) indicated that it was identical to rad4+ (Duck et al. 1976; Fenech et al. 1991). All cut5/rad4 mutants were found to be UV sensitive (Duck et al. 1976; Saka and Yanagida 1993; Saka et al. 1994a). Intriguingly, however, the UV- or 4NQO-induced checkpoint control was retained in cut5 mutants even at 36°C (Saka et al. 1994a,b; F. Esashi, Y. Saka, and M. Yanagida, unpubl.).
Cut5 may sense the replication defect and generate a signal to delay the cell cycle. Multiple phenotypes of cut5 in replication and damage, however, are intriguing, and the actual molecular role of Cut5 in maintaining the genome is unknown. In this study, we addressed the question of what kind of proteins interact with Cut5. Identification of the interacting proteins should shed light on how Cut5 protein functions within cells. Cut5 is a 74-kD nuclear protein consisting of several regions (Saka et al. 1994a). The amino-terminal 100 amino acids long motif repeats twice, and is similar to the regions found in the human repair protein XRCC1 (Thompson et al. 1990), the oncoprotein Ect1 (Miki et al. 1993) and the yeast protein Rev1 (Larimer et al. 1989). This region may serve as the site for protein–protein interaction. The S. cerevisiae Dpb11 (Araki et al. 1995), a suppressor for mutations in an essential subunit of DNA Pol II (epsilon) has a weak resemblance to Cut5.
An initial investigation for searching Cut5-interacting proteins by the two-hybrid screen method (Fields and Song 1989) indicated that the GAL4 DNA-binding domain (GAL4D) fused to full-length Cut5 activated transcription without the activation domain. Therefore, Cut5 was divided into several regions and each of the regions was used as bait. We identified a novel gene product Crb2, which interacts specifically with the amino-terminal region of Cut5. The deletion of the crb2+ gene profoundly affects checkpoint control induced by UV as well as DNA polymerase mutations. Crb2 is modified on DNA damage, and appears to mediate the signal for arrest of the cell cycle through Chk1, a protein kinase which restrains the activation of Cdc2 kinase by regulating phosphorylation of Tyr15 (Walworth et al. 1993; Walworth and Bernards 1996; O’Connell et al. 1997; Rhind et al. 1997).
Results
Mutations of cut5/rad4 in the conserved amino-terminus
To locate an essential region in Cut5, we determined mutation sites of cut5. The mutant gene was amplified from three cut5/rad4 alleles [rad4-116; Duck et al. (1976); cut5-580, Hirano et al. (1986); cut5-T401, Samejima et al. (1993)] by PCR and sequenced. Surprisingly, all three mutations contained the same substitution at the 45th codon (from ACG to ATG), leading to the amino acid change T45M (Fig. 1A). An additional alteration (resulting in K62Q) was present in cut5-T401. The T45M mutation should create a new EcoT22I site (ATGCAT, Fig. 1B). This was verified by Southern hybridization (Fig. 1B, right), which produced expected sizes of the EcoT22I fragments for the mutant genomic DNAs. The T45M mutation resided in the middle of the first amino-terminal repeat R1 (consensus is indicated in the bottom of panel A; Saka et al. 1994b). T45 is present within the conserved stretch (consensus, VTHLIA), hereafter designated as the TH domain.
Figure 1.
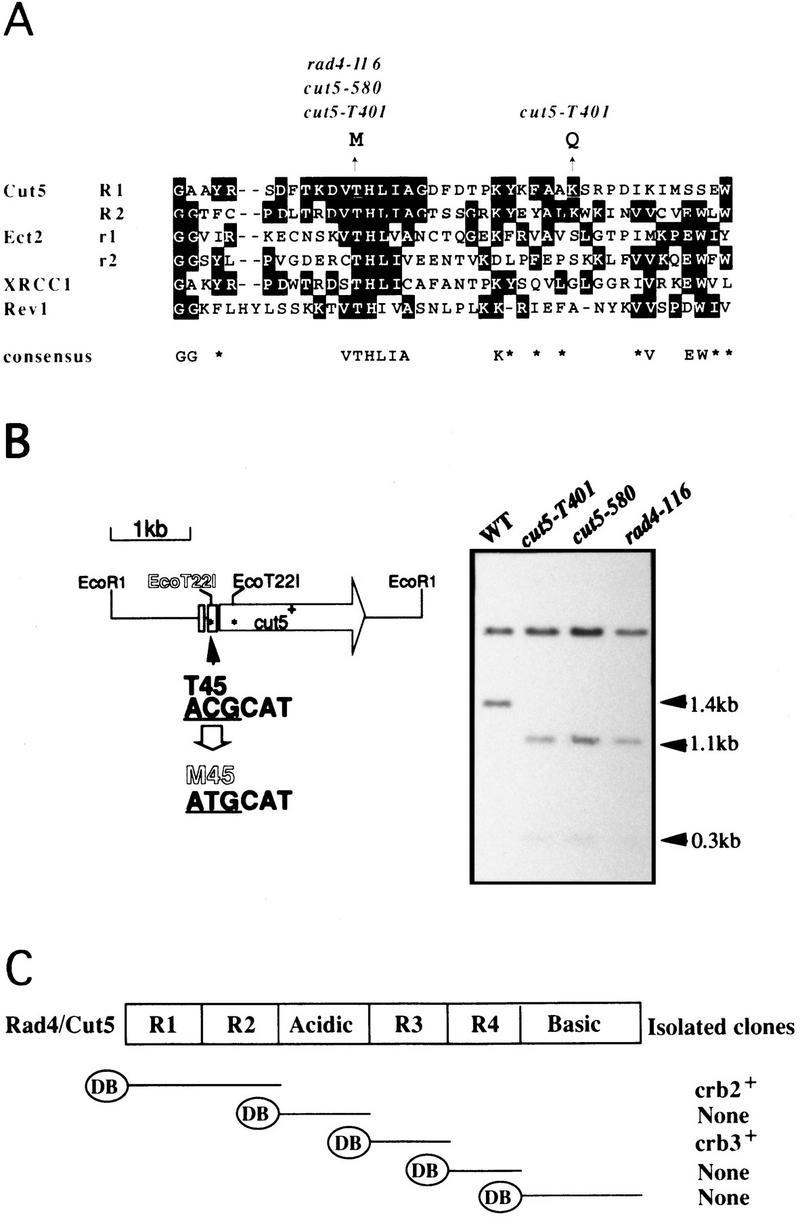
cut5/rad4 mutations reside in the highly conserved T45. (A) Mutation sites for rad4-116, cut5-580, and cut5-T401 were determined to be the identical T45M, residing in the R1 repeat. The amino-terminal regions R1 and R2 are similar to oncoprotein Ect2 (Miki et al. 1993), DNA repair proteins XRCC1 (Thompson et al. 1990), and Rev1 (Larimer et al. 1989). The consensus is indicated below. (B) The cut5/rad4 mutation created a new site for EcoT22I (left). This was verified by hybridization probed with EcoRI fragment containing the cut5+ gene (right). Genomic DNAs from wild-type and mutant DNAs digested with EcoT22I and EcoRI gave rise to the fragments with the expected sizes. (C) Construction of bait for two-hybrid screening. Each of five DNA fragments was placed under GAL4DB. The S. pombe cDNA library associated with the GAL4 activation domain was employed for screening. Crb2 and Crb3 cDNAs were obtained when the amino-terminal R1R2 and R3 were used as bait, respectively.
Isolation of Crb2
For the two-hybrid screening method (Fields and Song 1989), five constructs (Fig. 1C) were made as bait, each of which contained a region of Cut5 fused to the GAL4DB. GAL4DB–R1R2 thus contained the R1 and R2 region, and so on. More than 1 × 106 S. pombe cDNA clones were screened for each plasmid. Two identical clones (designated crb2+, cut5-repeat binding) were obtained by the bait of R1R2, whereas eight identical clones (crb3+) were isolated by the bait of R3. Combinations of R1R2 and Crb2, and of R3 and Crb3, produced β-galactosidase activities (Fig. 2A), respectively, as strong as the control interaction (p53 and T-antigen). Other control combinations showed low activities. The interaction of Cut5 with Crb2 or Crb3 was specific as seen by pairwise combinations (Fig. 2B; β-galactosidase positives are blue). No other positive clone was obtained by the screen with the other baits.
Figure 2.

Crb2 and Crb3 interact with Cut5. (A) β-Galactosidase activities were measured for different combinations. (DB) GAL4 DNA-binding domain; (AD) GAL4 activation domain; (R1R2) the R1R2 region of Cut5; (R3) the central R3 region of Cut5; (T-ag) large T-antigen; (p53) human tumor suppressor. (B) Pairwise combinations were examined. The interactions between Crb2 and R1R2, and between Crb3 and R3, are positive (blue). (C) Binding of Crb2 and Crb3 with Cut5 in vitro. The reticulolysate translation system was employed to synthesize luciferase, Crb2, and Crb3 by use of [35S]methionine (left). GST–R1R2 and GST–R3 made in bacterial cells were purified by association with glutathion-beads. Beads were then incubated with labeled luciferase, Crb2 or Crb3, followed by washing of the beads. 35S-Labeled proteins bound to beads were analyzed by SDS-PAGE and autoradiography (right). (D) The mutant R1R2 DNA containing M45H instead of T45H failed to interact with Crb2 in the two-hybrid method at 26°C and 36°C.
Interaction of Crb2 with Cut5
An in vitro-binding assay described below supported a direct interaction between Crb2 and Cut5. The cDNAs of Crb2 and Crb3 were expressed under the T7 promoter by the in vitro reticulocyte lysate system by use of [35S]methionine for labeling Crb2 and Crb3 (Fig. 2C, left; luciferase was also labeled as control). To examine binding, GST–R1R2 and GST–R3 fusion proteins produced in Escherichia coli and purified by glutathione–agarose beads were incubated with 35S-labeled proteins. The beads were washed, and bound labeled proteins were analyzed by SDS–PAGE and autoradiography (Fig. 2C; right). Radiolabeled Crb2 and Crb3 were detected to be bound to GST–R1R2 and R3, respectively, but luciferase was not. A Crb2 fragment cleaved during incubation was also efficiently bound to GST–R1R2. Another control with the beads containing only GST showed that neither bound Crb2 nor Crb3.
To examine whether mutant Cut5T45M retains the ability to interact with Crb2, the mutant R1R2 was isolated and fused to the GAL4DB. Two-hybrid interaction between Crb2 and mutant R1R2 was completely abolished at both 26°C and 36°C (Fig. 2D). Interaction of Cut5 with Crb2, at least at the level of the two-hybrid system, thus requires the TH domain.
Crb2 resembles Rad9 and p53-binding protein
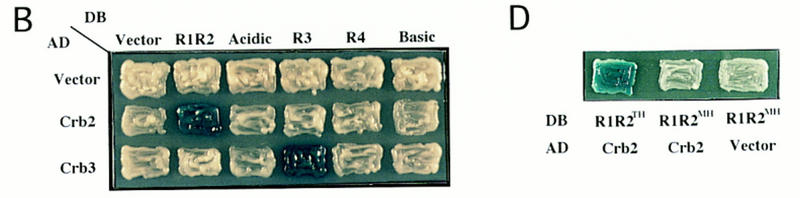
An S. pombe cosmid that contained the genomic crb2+ gene was isolated with cDNA as the probe. Hybridization to an ordered cosmid bank indicated that crb2+ was located near nda2+ in the left arm of chromosome II (Mizukami et al. 1993). The 9-kb PstI fragment containing crb2+ was subcloned from the cosmid (the map shown in Fig. 3A). Plasmid carrying crb2+ (pFE15) fully suppressed the UV sensitivity of cut5-T401 at 26°C, and this suppression was employed for subcloning of crb2+ (+ indicates suppression). The same plasmid, however, did not suppress the temperature sensitive phenotype of cut5-T401 at 36°C (data not shown).
Figure 3.
Crb2 suppressing the UV sensitivity of cut5 resembles yeast Rad9 and human 53BP1 and BRCA1. (A) A cosmid containing crb2+ was obtained by the probe of Crb2 cDNA. Plasmid carrying crb2+ was subcloned by its ability to suppress the UV phenotype of cut5 mutant (indicated by +). pFE15 contained the full-length crb2+ gene. pFE14 was used for gene disruption. The accession no. for Crb2 is D86478. (B,C) The carboxyl terminus of Crb2 resembles that of yeast Rad9 and human 53BP1. Identical amino acids are boxed. (D) The BRCT motifs present in Cut5, Crb2, BRCA1, 53BP1, and Rad9 are shown (based on Bork et al. 1997). Thick characters represent identical or similar amino acids.
Sequencing showed that crb2+ encodes a 778 amino acid protein (predicted molecular mass of 87.5 kD; database accession no. D86478). The near amino-terminal region is required for interacting with Cut5 (data not shown). Database search revealed the highest scores for Rad9 of S. cerevisiae (Weinert and Hartwell 1988) and human 53BP1, which interacts with p53 (Iwabuchi et al. 1994). The carboxy-terminal region of Crb2 is ∼30% and 25% identical to that of Rad9 and 53BP1, respectively. Crb2 is also similar to the carboxyl terminus of BRCA1, a mammary cancer gene, and other repair and cancer-related genes (Koonin et al. 1996; Callebaut and Mornon 1997). A common domain termed BRCT was proposed from detailed sequence analyses (Bork et al. 1997); a number of damage-responsive proteins including Cut5 also contained the BRCT motif (Fig. 3D). The BRCT-containing domain in BRCA1 is implicated in transcriptional activation.
Crb3 is a WD repeat containing protein
The nucleotide sequence of Crb3 cDNA has been determined (database accession no. D45883): crb3+ encodes a protein with the WD repeats (Neer et al. 1994) partly resembling β-transducin (Fig. 4). The predicted sequence contains 446 amino acids (calculated molecular mass of, 49.5 kD) with 6 WD repeats. It is an essential protein for viability and may be implicated in the G1/S progression (T. Matsusaka and M. Yanagida, unpubl.). Structure and functional analyses of the crb3+ gene will be described elsewhere.
Figure 4.

The amino acid sequence of Crb3 protein. Six WD repeats are present in Crb3 protein, which contains 446 amino acids. (A) Location of the WD repeats is schematized; (B) the repeats are aligned. Identical amino acids are boxed. The accession no. for the database is D45883.
Identification and localization of Crb2
Rabbit antibodies against Crb2 were affinity-purified for detection of Crb2 in S. pombe extracts. Diffused immunoblot bands were obtained at the position of ∼100–110 kD (Fig. 5A, lane 2), the intensity of which increased in cells carrying plasmid pCRB2 (lane 3), but which were absent in Δcrb2 strain (lane 1). These diffuse bands may be caused by post-translational modification (see below).
Figure 5.
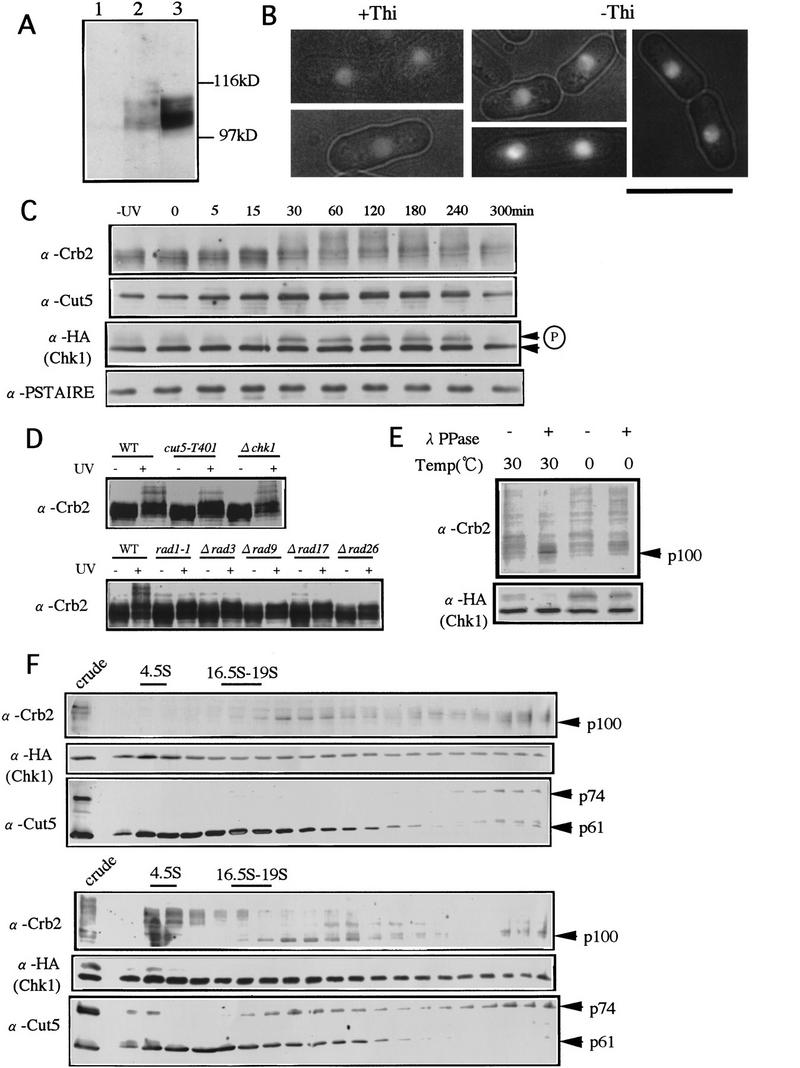
Crb2, a putative nuclear phosphoprotein, alters sedimentation profile upon UV damage. (A) Polyclonal antibodies made against the fusion protein of Crb2 were used for detecting Crb2 in S. pombe extracts. The diffused 100–110-kD band was detected (lane 2), the intensity of which increased in cells carrying pCRB2 (lane 3). No band detected in Δcrb2 (lane 1). (B) GFP-tagged Crb2 was placed under a mild inducible promoter REP41. Cells cultured in the presence of thiamine (+Thi, the promoter repressed) or the absence (−Thi, derepressed) were observed. Nuclear fluorescence of cells in +Thi was weak, and photographically enhanced. The contour of cell shape was produced with the merged exposures of fluorescence and transmission light source. Bar, 10 μm. (C) S. pombe cells integrated with HA-tagged chk1+ (Walworth and Bernards 1996) were irradiated with UV (100 J/m2). Extracts were prepared and immunoblotted by use of antibodies against Crb2, Cut5, HA, and Cdc2 (PSTAIRE). Modification of the Crb2 band occurred ∼30 min and was clearly seen 60 min after irradiation. The upper band of Chk1, intensified from 30 to 240 min, represents phosphorylation (Walworth and Bernards 1996). (D) Immunoblot patterns of wild type (WT), cut5-T401, Δchk1, rad1-1, Δrad3, Δrad9, Δrad17, and rad26 extracts before (−) and after (+) UV irradiation with anti-crb2 antibodies are shown. Cells irradiated by UV (100 J/m2) were cultured at 26°C for 1 hr. The upper band was seen in wild-type and Δchk1 but not in other mutant extracts. (E) Extracts of the UV-irradiated cells were treated with (+) or without (−) λ protein phosphatase at 0°C or 30°C. Upper modified bands of Crb2 and Chk1 were diminished, and the lower bands at 100 and 56 kD, respectively, were intensified after treatment with phosphatase at 30°C. (F) Centrifugation patterns of nonirradiated (top) and UV irradiated (bottom) cell extracts run in a 15–40% sucrose gradient at 40,000 rpm for 12 hr. Fractions collected were immunoblotted with antibodies against Crb2, HA (Chk1), and Cut5. After irradiation, the upper bands of Crb2 and Chk1 were clearly seen in the top, slowly sedimenting fractions. A small population of Cut5 was also shifted to the slowly sedimenting fractions. The positions of the control markers (4.5S and 16.5-19S) are indicated by the bars. The immunoblot patterns of crude extracts are shown in left lanes.
Jellyfish green fluorescence protein (GFP) was tagged to the amino-terminus of Crb2 under a moderate promoter REP41 and used to determine intracellular localization of Crb2 (Figure 5B). The nuclear chromatin region (verified by double stain with DAPI; data not shown) showed green fluorescence when the promoter was repressed in the presence of thiamine (+Thi). When Crb2 was mildly overproduced in the absence of thiamine (−Thi), the same nuclear region was intensely fluorescent. Hence, Crb2 appears to be a nuclear chromatin protein like Cut5 (Saka et al. 1994). We have been unsuccessful in detecting the stable complex formation between Cut5 and Crb2 in S. pombe extracts, however. Immunoprecipitation by anti-cut5 antibodies was performed under different buffer conditions. Cut5 was clearly detected in the precipitates, whereas Crb2 was not (data not shown). Conversely, Crb2 was immunoprecipitated by anti-crb2 antibodies, but Cut5 was not detected in the precipitates.
Hypermodification of Crb2 upon irradiation
We examined whether Crb2 was further modified after cells were UV irradiated (100 J/m2) at 26°C. Additional upper bands (apparent molecular mass of 120–150 kD) became visible, greatly intensified after 1–2 hr, and diminished after 4–5 hr (Fig. 5C, top). This transient band-shift clearly indicated the occurrence of strong post-translational modification in Crb2 after irradiation. The intensity of 74-kD Cut5 increased (approximately two-fold) after irradiation (Fig. 5C; second panel).
Chk1 kinase has been reported to be transiently phosphorylated in UV irradiated cells (Walworth and Bernards 1996). We compared the timing of Crb2 band-shift with that of Chk1 phosphorylation by immunoblot with anti-HA antibodies and a strain integrated with the HA-tagged chk1+ gene (a kind gift of Dr. N. Walworth, Robert Wood Johnson Medical School, Piscataway, NJ; this strain was also used to obtain data for Crb2 modification described above). The upper phosphorylated band of Chk1 was seen at approximately the same timing (Fig. 5C, third panel) as Crb2 modification. Cdc2 detected by anti-PSTAIRE antibodies was shown as control (Fig. 5C, bottom panel). UV irradiation thus induced a change in Crb2 during the arrest of cell cycle (see below).
We examined whether this 120- to 150-kD super-band shift required the presence of other damage checkpoint genes. Extracts of wild type, cut5-T401, Δchk1, rad1-1, Δrad3, Δrad9, Δrad17, and Δrad26 were prepared before (−) and 1 hr after (+) UV irradiation at 26°C and immunoblotted with anti-crb2 antibodies (Fig. 5D). The 120- to 150-kD band shift of Crb2 after irradiation was observed in Δchk1 deleted cells and also in cut5-T401 (the level in cut5 somewhat reduced) but not in other checkpoint mutants, suggesting that the damage-sensing checkpoint genes, Rad1, Rad3, Rad9, Rad17, and Rad26, were needed to modify Crb2 upon irradiation.
We wanted to know whether the band shift of Crb2 was produced by phosphorylation, and performed phosphatase treatment of UV irradiated cell extracts as described in Walworth and Bernards (1996) at 30°C and 0°C. The intensity of the unmodified sharp 100-kD band clearly increased after the phosphatase treatment at 30°C (Fig. 5E, second lane). The control Chk1 was also dephosphorylated after phosphatase treatment. The upper 110-kD and 120- to 150-kD bands of Crb2 thus formed at least partly because of phosphorylation. Crb2 in nonirradiating cells thus appeared to be already phosphorylated and this phosphorylation was greatly enhanced further following UV irradiation. This hypermodification of Crb2 is not caused by the cell-cycle arrest, because it did not occur in G2-arrested cdc25 mutant cells (data not shown). UV-induced DNA damage is probably the direct cause of this hypermodification.
To know whether Crb2 and Cut5 were present in monomeric or oligomeric complexes, sucrose gradient centrifugation of S. pombe extracts was run to determine the sedimentation profiles of Crb2 and Cut5 (Fig. 5F). Cell extracts were first centrifuged at 14,000 rpm for 20 min, and the supernatants were overlaid on the top of a 15%–40% linear gradient, followed by centrifugation at 40,000 rpm for 12 hr. A broad peak ranging from 20S to the near bottom of the centrifugal tube (>50S) was obtained by immunoblot with anti-crb2 antibodies (Fig. 5F, top panel, α-Crb2), suggesting that Crb2 was associated with relatively large heterogenously sized particles in extracts. For Cut5, the native 74-kD band was also present in the heavy fractions, whereas the cleaved 61-kD band (Saka et al. 1974) sedimented much more slowly (α-Cut5). The cleavage of 74-kD Cut5 occurs during extract preparations and centrifugations, and is difficult to control; the cleaved 61-kD Cut5 band is enriched in slowly sedimenting fractions. Chk1 kinase sedimented very broadly from the top to the bottom of sucrose gradient (α-HA). Fractions of Crb2, Cut5, and Chk1 thus sedimented as large structures though they did not form any cosedimenting peak.
Sedimentation profiles in cell extracts prepared 1 hr after UV irradiation (100 J/m2) differed strikingly from those in nonirradiated cells. The modified forms of Crb2 sedimented very slowly at ∼3–5S (Fig. 5F, bottom panel, α-Crb2), and the upper band of Chk1 was also present in the slowly sedimenting fractions. Cut5 showed a broad profile, with a small fraction also present in the slowly sedimenting fractions. These results strongly suggested that the modified populations of Cut5, Crb2, and Chk1 were dissociated from the larger complexes after UV irradiation. We did immunoprecipitation of the upper slowly-sedimenting fractions after UV irradiation using antibodies against Crb2, Cut5, and Chk1, and could detect respective immunoprecipitated proteins, but were not able to detect coimmunoprecipitation with others under the conditions employed (data not shown).
crb2 null is sensitive to UV and HU
The genomic copy of crb2+ was disrupted (Fig. 6A, Materials and Methods). Resulting heterozygous diploids produced all four spores that were viable in each tetrad, while Ura+ and Ura− segregated 2 : 2. The crb2+ gene is thus not essential for viability. The gene disruption was confirmed by genomic Southern hybridization of haploid segregants (Fig. 6B) and the lack of protein confirmed by immunoblotting (Fig. 5A, lane 1). Haploid crb2 null cells (designated Δcrb2) divide normally as do wild type at both 26°C and 36°C in minimal or rich culture medium, with a slightly longer generation time (3.4 hr) than wild type (2.9 hr at 26°C in YPD). Δcrb2 cells showed no anomaly in DNA content as determined by FACScan analysis (data not shown).
Figure 6.

Deletion of crb2+ is sensitive to UV and HU. (A) Gene disruption of crb2+. Resulting crb2::ura4+ was used for transformation after linearization with EcoRI. (B) Disrupted Ura+ segregants were viable. 4.7-kb EcoRI fragment was detected in the disrupted cells when probed with the crb2+ DNA. S, hypersensitive to UV or HU. (C) The deletion strain of crb2+ (Δcrb2) is hypersensitive to UV. This UV sensitivity was suppressed by pCRB2 and pCHK1, but not by pCUT5. (D) Colony of Δcrb2 was scarcely formed on YPD containing 6 mm HU at 26°C. (WT) wild type; (cdc2-3w) a HU sensitive cdc2 allele (Enoch et al. 1992).
We found that Δcrb2 deletion was both UV and HU sensitive (Fig. 6B,C,D). It was more UV sensitive than cut5 (Saka et al. 1993). This UV hypersensitivity of Δcrb2 was suppressed by plasmid pCRB2 but not by pCUT5 (Fig. 7C). Δcrb2 was also HU sensitive and its sensitivity was similar to that of cdc2-3w (Fig 6D).
Figure 7.
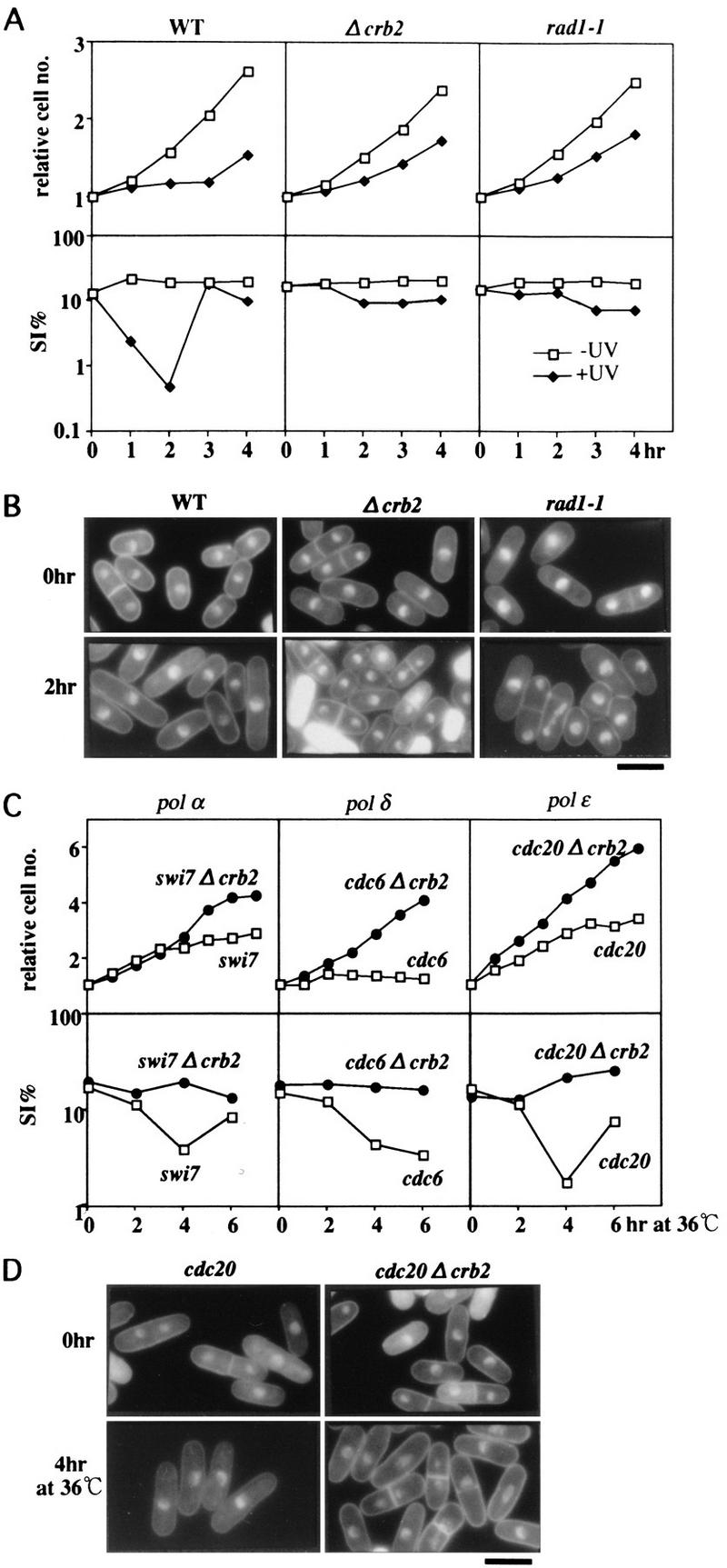
Damage- and RC mutation-induced checkpoint abolished in Δcrb2. (A) Wild-type (WT), Δcrb2, and rad1-1 cells were irradiated by UV (+UV, 100 J/m2) at 26°C, and the cell number and the septation index (SI%) were measured. The control of no irradiation (−UV) is also shown. (B) Cells 0 and 2 hr after irradiation were stained by DAPI. Bar, 10 μm. (C) temperature sensitive strains swi7, cdc6, and cdc20, and double mutants swi7-Δcrb2, cdc6-Δcrb2, and cdc20-Δcrb2 were grown at 26°C and then transferred to 36°C. The cell number and the SI% were measured. (D) Single cdc20 and double cdc20 Δcrb2 cultured at 36°C for 0 and 4 hr were stained by DAPI. Bar, 10 μm.
UV damage checkpoint is abolished in Δcrb2
We examined whether UV-induced damage checkpoint was retained in Δcrb2 cells. When wild-type cells were irradiated with UV (100 J/m2 at 26°C), their mitotic entry was greatly delayed, and cell number increase was arrested (Fig. 7A, WT). The septation index (SI) decreased for ∼2 hr because of the activation of damage checkpoint control (Al-Khodairy and Carr 1992; Rowley et al. 1992). The damage checkpoint mutant rad1-1, however, did not show such delay nor a decrease in SI. We found that Δcrb2 also lost the delay, basicly showing the identical damage phenotype to that of rad1-1 (Fig. 7A).
UV-irradiated wild-type cells are known to be elongated with the single nucleus (Fig. 6B, 2 hr after irradiation). In sharp contrast, after irradiation Δcrb2 and rad1-1 cells were divided and the septated cells, with occasional cut phenotype, were frequently seen. The deletion of crb2+ thus led to loss of the cell-cycle arrest in response to UV irradiation. The UV sensitivity of the double mutant cut5 Δcrb2 was identical to Δcrb2, and its UV-induced checkpoint control was also abolished (data not shown).
Polymerase mutant-induced arrest abolished in Δcrb2
We wanted to know whether the cell-cycle arrest phenotypes of polymerase mutants at 36°C were maintained in the absence of crb2+. Double-mutants were constructed by crossing of Δcrb2 with cdc6-121, swi7-H4, or cdc20-M10 (Singh and Klar 1993; Francesconi et al. 1995; Murakami and Okayama 1995). These single polymerase mutants (α, δ, and ε) were arrested at 36°C and the SI decreased (Fig. 7C). The double-mutants, however, were not arrested at 36°C, and the level of SI remained the same after the temperature-shift. The double-mutant cells frequently displayed the cut phenotype at 36°C for 4 hr (Fig. 7D, right), whereas single mutant cells were elongated with the single nucleus (left). Checkpoint control was thus lost if the crb2+ gene was disrupted in the polymerase mutants at 36°C. The phenotype of the other double-mutant with cdc17-K42 (defective in DNA ligase) was also examined. The arrest phenotype was abolished in the double-mutant (data not shown). We concluded from these results that Crb2 was required for the cell-cycle arrest induced by defects in the replication machinery enzymes.
Nucleotide pool-dependent replication checkpoint retained in Δcrb2
We then determined whether the HU-induced arrest was maintained in Δcrb2. In sharp contrast to the results with polymerase mutations, Δcrb2 cells were arrested in the presence of HU (+HU) at 36°C (Fig. 8A). Δcrb2 cells in the presence of HU were elongated with the single nucleus as wild-type cells, and did not show the cut phenotype (right panel). The SI index was strikingly lowered (data not shown). Similar results were obtained at 26°C (data not shown). The HU-induced cell-cycle arrest was thus completely retained in Δcrb2.
Figure 8.

HU- and cdc22-induced checkpoint occurs in Δcrb2. (A) Wild type (WT) and Δcrb2 were cultured at 36°C in the presence (+) or the absence (−) of HU, and their cell numbers were counted (left). Cells were stained by DAPI (right). The cell number of wild type and Δcrb2 did not increase in the presence of HU. Cell elongation and single nuclear phenotype was observed for both wild-type and Δcrb2 strains in the presence of HU. Bar, 10 μm. (B) Single mutants Δcrb2 and temperature sensitive cdc22, and the double mutant cdc22 Δcrb2 were cultured at 36°C. The double mutant showed the cdc phenotype and no cut phenotype. Cells were stained by DAPI after 4 hr at 36°C. Both cdc22 and cdc22 Δcrb2 showed the checkpoint arrest at 36°C. Bar, 10 μm.
Basically the same result was obtained for the double mutant Δcrb2 temperature sensitive cdc22. The cdc22+ gene encodes the large subunit of ribonucleotide reductase (Gordon and Fantes 1986; Fernandez-Sarabia et al. 1993), and is known to be directly inhibited by HU. The double mutant Δcrb2 cdc22 grew normally at 26°C, and was arrested at 36°C, displaying the identical arrest phenotypes of HU-added Δcrb2 or single cdc22 mutant cells (Fig. 8B). These results established that the cell-cycle arrest induced by HU or cdc22 mutation did not require Crb2.
Interaction of crb2+ with other checkpoint genes
We examined whether the UV sensitivity of Δcrb2 can be suppressed by overexpression of other checkpoint genes. For this purpose, plasmids carrying crb2+, cut5+, rad1+, rad3+, rad9+, rad17+, rad26+, or chk1+/rad27+ (Al-Khodairy and Carr 1992; Rowley et al. 1992; Sheldrick and Carr 1993; Al-Khodairy et al. 1994) placed at the downstream of the moderate promoter REP41 (a generous gift of Dr. A.M. Carr, Medical Research Council Cell Mutation Unit, University of Sussex, Brighton, UK, except for the first two strains constructed in the present study) were introduced into Δcrb2, cut5-T401 and Δchk1 strains, and the UV-sensitive phenotype of transformants was investigated. The results are summarized in Table 1. None of the damage-sensing checkpoint genes (rad1+, rad3+, rad9+, rad17+, rad26+) could suppress the UV sensitivity of Δcrb2, cut5-T401, and Δchk1. In contrast, moderate overexpression of Chk1 kinase, a checkpoint signal transmitter, suppressed the UV phenotype of Δcrb2 (see Fig. 6C) and cut5-T401. Moreover, pCRB2 suppressed the UV sensitivity of cut5, but pCUT5 suppressed neither Δcrb2 nor Δchk1. These results suggested that Crb2 played an upstream role for Chk1 and acted at the downstream of, or parallel to, other checkpoint gene products in the UV damage-signaling pathway (see Discussion).
Table 1.
UV sensitivity of Δcrb2 suppressed by plasmid pCHK1 carrying the chk1+/rad27+ gene
| Plasmids
|
||||
|---|---|---|---|---|
| Mutant
|
pCRB2
|
pCUT5
|
pCHK1
|
pRAD1 pRAD3 pRAD9 pRAD17 pRAD26
|
| Δcrb2 | + | − | + | − |
| cut5–T401 | + | + | + | − |
| Δchk1 | − | − | + | − |
(+) Normal sensitivity; (−) hypersensitivity; (Δcrb2, Δchk1) deletion mutants of crb2+ and chk1+, respectively.
Interactions of Chk1 with Crb2 and Cut5
More evidence for genetic interactions among Crb2, Cut5, and Chk1 was obtained in following experiments. HU, as well as UV, sensitivities were examined for Δcrb2 or Δchk1 carrying multicopy vector plasmid (1, 2 in Fig. 9A), pCHK1(3,4), pCRB2 (5,6), or pCUT5 (7,8). These plasmid-borne cells were spotted after dilution, and cultured at 26°C with the addition of HU to the medium (right, HU) or the pretreatment by UV irradiation (100 J/m2, UV, middle, +UV) as control. pCHK1 suppressed the HU-phenotype of Δcrb2, whereas pCRB2 did not suppress that of Δchk1.
Figure 9.

Chk1 interacts with Crb2 and Cut5. (A) Elevated gene dosage of chk1+ suppresses the UV- and HU-sensitive phenotypes of Δcrb2. Each of the four transformant strains of Δcrb2 (top) and Δchk1 (bottom) strains carrying plasmids (1 and 2, vector; 3 and 4, pCHK1; 5 and 6, pCRB2; 7 and 8, pCUT5) were spotted after dilution, and incubated at 26°C (left). Identical cells were also incubated after UV irradiation (middle) or in the presence of HU (right). The plates did not contain thiamine. (B) Δcrb2 null cells carrying plasmid with the chk1+ gene under a moderate inducible promoter REP41 were precultured at 26°C for 18 hr in the presence of thiamine (+Thi; repressed condition) or the absence (−Thi; derepressed condition), and then irradiated by UV and cultured for 4 hr at 26°C. The cell number and the septation index were measured. (C) Phosphorylation of Chk1 does not take place in Δcrb2 on UV irradiation. Three strains (wild type, cut5-T401 and Δcrb2) integrated with chk1+ tagged with the HA antigen (Walworth and Bernards 1996) were irradiated with UV (+UV) and cultured at 26°C for 1 hr. The level of Cdc2 (anti-PSTAIRE) is shown as control. The intense upper band of Chk1 appeared after UV irradiation in the wild type was absent in Δcrb2. (D) Two-hybrid interactions among Chk1, Crb2, and Cut5. (DB) DNA-binding domain; (AD) activation domain; (R1, R2, acidic, R3, R4) regions in Cut5 protein; (T-ag, p53) control T antigen and p53. β-Galactosidase activities were assayed for each pair of two-hybrid interactions.
We wanted to know whether the loss of UV-induced checkpoint arrest in Δcrb2 could be restored by moderate overproduction of Chk1 (Fig. 9B). REP41, a moderate inducible promoter, was derepressed in the absence of thiamine (−Thi). The ability to arrest cells following UV damage was restored in Δcrb2 cells that overexpressed Chk1 (−Thi; shown by a temporal decrease of the SI) but not in cells without overproduction (+Thi), showing that moderate overexpression of Chk1 also suppressed the loss of damage checkpoint. Hence, the UV-induced checkpoint is partly restored in Δcrb2 mutant by overproducing Chk1, suggesting that the gene product other than Crb2 might be able to activate Chk1, but in a way much less efficiently.
We found that phosphorylation of Chk1 after UV irradiation required functional Crb2. The upper band of Chk1 formed 1 hr after UV irradiation at 26°C (+UV) in wild type and cut5 mutant cells but did not in Δcrb2 (Fig. 9C, +UV); the intensity of Chk1 upper band in cut5 mutant was significantly reduced, however. However, after UV irradiation, Crb2 may become essential for cells to survive by sending a signal for Chk1 phosphorylation, that leads to the accumulation of activated Chk1 (Walworth and Bernards 1996).
Moreover, strong two-hybrid interactions existed between Chk1 and Crb2 and between Chk1 and the central region (Acidic R3R4) of Cut5 (Fig. 9D). The degree of interactions were the highest for Chk1 and Crb2, whereas the interaction between Chk1 and the region of Cut5 spanning from the acidic to R3R4 region (acidicR3R4) was high. No two-hybrid interaction was found between Chk1 and R1R2 of Cut5, however. The interaction between Chk1 and R3R4 of Cut5 was relatively weak. Immunoprecipitation experiments, however, did not show the presence of a detectable stable complex. Neither Chk1 and Crb2, nor Chk1 and Cut5, existed as the stable complex in cell extracts.
Discussion
We report in this study (1) identification and characterization of crb2+, an S. pombe checkpoint gene, which is required for both DNA damage and replication machinery checkpoint; and (2) interactions among three checkpoint gene products Cut5, Crb2, and Chk1 for proper maintenance of the genome. Cut5 and Crb2 contain BRCT-motif present in a number of damage-responsive proteins, whereas Chk1 is a protein kinase. Although the stable complex has not been detected in S. pombe extracts, evidence for their putative direct interaction is presented. Functions of Crb2 and Cut5 are distinct, but overlapping. Crb2 is responsive to the DNA damage, whereas Cut5 responds to the impaired precursor nucleotide supply for replication. Both Crb2 and Cut5, however, are required for the checkpoint response to defects in DNA polymerases and ligase. Cut5 and Crb2 may act as sensors that sense damage and/or replication defect, and generate signals to activate Chk1 for regulating Cdc2. Thus, Cut5, Crb2, and Chk1 may form a checkpoint sensor-transmitter pathway to arrest the cell cycle.
The present paper also shows that replication checkpoints present in S. pombe may be categorized into two classes: one caused by the defect in replication machinery (mutations in polymerases and ligase), and the other caused by the inhibition of normal nucleotide supply (HU and cdc22 mutation). Crb2 is required only for the former, but Cut5 is necessary for both (Saka et al. 1994a,b). A possible explanation for this difference is that the mutant polymerase and ligase enzymes may synthesize bad or damaged DNAs that are sensed by Crb2, whereas such bad DNAs are not made in HU-arrested cells. This implies that Crb2 may monitor the damaged DNAs accumulated during the S phase, consistent with the concept that DNA damage causes replication to be blocked (Paulovich and Hartwell 1995).
Chk1 appeared to act at the downstream of Crb2 and Cut5 as a checkpoint signal transmitter. This hypothesis explains a number of results presented in this paper (Table 1; Figs. 5, 6, and 9). Consistent with this hypothesis, Chk1 is required not only for the damage checkpoint (Walworth et al. 1993; Al-Khodairy et al. 1994) but also for the replication checkpoint (Francesconi et al. 1997). Cdc2 kinase is affected by Chk1, which was recently shown to regulate Tyr15 phosphorylation of Cdc2 through Cdc25 and/or Wee1 (O’Connell et al. 1997; Rhind et al. 1997).
We showed that a fraction of Crb2 was transiently modified, probably phosphorylated, after UV irradiation. In sucrose gradient centrifugation, the modified Crb2 was slowly sedimenting, possibly released from the large complex structures. Similarly, the phosphorylated form of Chk1 kinase, which was reported to be activated by autophosphorylation when damaged (Walworth and Bernards 1996), was also present in the slowly sedimenting fractions. We speculate that damage leads to Crb2 modification, and the modified Crb2 can produce a signal to activate Chk1, possibly by direct interaction. This hypothesis is consistent with two-hybrid interaction, the suppression of Δcrb2 by the elevated gene dosage of chk1+ and the absence of Chk1 phosphorylation in Δcrb2 cells. The occurrence of Crb2 modification induced by damage in Δchk1 cells supports the upstream nature of Crb2 to Chk1.
Following UV irradiation, the products of the damage-sensing checkpoint genes (rad1+, rad3+, rad9+, rad17+, and rad26+; Al-Khodairy et al. 1994) may act in conjunction with Crb2 to generate the signal for the cell-cycle block. Functional relationship between Crb2 and these five checkpoint genes is unclear. The fact that Crb2 is not modified upon UV irradiation in these rad mutants and that plasmids carrying any one of these rad+ genes fail to suppress the UV phenotype of Δcrb2 cells suggests that Crb2 functions in the downstream of these Rad gene products. Crb2 may be activated by the Rad gene products. It is also possible, however, that Crb2 independently senses damaged DNAs and sends an essential signal for promoting cell-cycle arrest, separately from the Rad genes. A more complex relationship is likely, as pCRB2 can suppress the UV sensitivity of rad1-1, but not that of rad3, rad9, or rad17 mutants (F. Esashi and M. Yanagida, unpubl.). Crb2 may thus have a close functional link to Rad1. The S. cerevisiae Rad17, an S. pombe Rad1 homolog, has an exonuclease activity to produce a gap in DNA and plays an important role in the damage checkpoint (Lyddall and Weinert 1995; Nugent et al. 1996).
The predicted amino acid sequence of Crb2 is significantly similar to the budding yeast Rad9 (Weinert and Hartwell 1988; Hartwell and Weinert 1989) that acts in the processing of DNA damage for repair (Lydall and Weinert 1995), as a component in the sensor/transducer pathway of UV damage outside of S phase (Navas et al. 1996). However, the similarity between budding yeast Rad9 and fission yeast Crb2 is restricted to the carboxyl termini. Hence, RAD9 and crb2+ are unlikely to be true homologs. Elevated dosage of the budding yeast RAD9 or the fission yeast crb2+ gene introduced into the fission yeast Δcrb2 or the budding yeast rad9 mutant, respectively, did not suppress the UV sensitivity (S. Mochida and M. Yanagida, unpubl.).
Mammalian proteins involved in oncogenesis, 53BP1 (Iwabuchi et al. 1994) and BRCA1 (Koonin et al. 1996), contain regions similar to the carboxyl termini of Crb2. 53BP1 binds to a tumor suppressor protein p53 by the two-hybrid method, and the carboxyl terminus of 53BP1 is the site for binding to p53 (Iwabuchi et al. 1994). Mutations in BRCA1 lead to familial breast and ovarian cancers. The common carboxyl terminal motifs present in Crb2, Rad9, 53BP1, and BRCA1 (and other related proteins, Koonin et al. 1996) may be the sites for interaction with proteins implicated in the maintenance of the genome in response to damages. It is of considerable interest to determine whether any protein bound to the carboxyl terminus of Crb2 exists in fission yeast, and whether such protein(s) is a transcription factor sharing a property with p53.
During the preparation of this paper, Wilson et al. (1997) reported that one of the fission yeast methylmethane sulfonate (MMS)-sensitive alleles, rhp9+, was required for the DNA damage checkpoint but not the replication checkpoint. Comparison of the sequences indicated that Rhp9 was identical to Crb2. Though their conclusion apparently differed from ours, the experimental results on rhp9 null were not inconsistent with ours, because only the replication checkpoint induced by HU was investigated for rhp9 null. Crb2/Rhp9 may respond to a variety of DNA toxins.
XRCC1, a human repair protein, the amino acid sequence of which partly resembles Cut5, plays a scaffold role in interactions with polymerase β and ligase III (Kubota et al. 1996). Cut5 might also have a scaffold structure, which makes complex regulations possible through protein–protein interactions. The amino-terminal T45, which appears to be necessary for interacting with Crb2, is the temperature sensitive mutation site. Because the mutants were also defective in replication, the amino terminus might also interact with an unidentified protein(s) essential for replication. The central-to-carboxy-terminal region is probably the site for interaction with Chk1 and Crb3. The amino-terminal and the central-to-carboxy-terminal regions contain multiple BRCT motifs (Saka et al. 1994; Bork et al. 1997). As both Cut5 and Crb2 contain BRCT motif, pursuing their protein functions will shed light on the actual role of BRCT motif in maintaining the genome.
It is not a simple matter to explain the reason cut5 mutants are sensitive to UV because the UV- and 4NQO-induced checkpoint is maintained in cut5 mutant at both 26°C and 36°C (Saka et al. 1994a,b; Saka et al., unpubl.). We speculate that the interaction between Cut5 and Crb2 is needed for promoting efficient repair of the damaged DNA. In cut5/rad4 mutants, where the interaction of Cut5 with Crb2 might be lost even at 26°C, repair replication may become inefficient, leading to UV hypersensitivity. However, Crb2, which is responsible for generating a damage signal for cell-cycle arrest is present. The reason for the HU sensitivity in Δcrb2 is also unclear because the HU-induced checkpoint is retained in Δcrb2. We propose that only Cut5 is required for HU-induced checkpoint and that the Cut5–Crb2 interaction is needed for an unidentified function to facilitate replication under an inhibited nucleotide supply. Under the HU-induced checkpoint arrest, Cut5 may send an arrest signal through a protein other than Crb2 or directly to Chk1. Two-hybrid interaction favors the latter case. To explain the result that both Crb2 and Cut5 are necessary for the cell-cycle arrest by defects in DNA polymerases or ligase, we propose that neither Crb2 nor Cut5 alone can detect the replication defect caused by defects of polymerases or ligase.
It remains to be determined whether the stable complex between Crb2 and Cut5 is actually present in the nucleus in vivo but difficult to detect in S. pombe extracts, or if the stable complex never exists in vivo and the interaction is only transient such as is true of a substrate–enzyme relationship. In sucrose gradient centrifugation, fractions of Crb2 and Cut5 were sedimented as large heterogeneous materials that might contain both Crb2 and Cut5. A small fraction of Chk1 was also in the large complex form. The properties of these large complexes are unknown, and whether Cut5, Crb2, and Chk1 exist in the same large structure remains to be determined. Alternatively, slowly sedimenting forms of Crb2, Cut5, and Chk1 were obtained after UV irradiation, and these smaller forms possibly phosphorylated may interact each other in a transient style.
Materials and Methods
Strains and media
Haploid wild-type S. pombe h− 972 and h+ 975 (Gutz et al. 1974) and their derivative mutant strains were used. cut5 mutants (cut5-T401, cut5−580, rad4-116; Duck et al. 1976; Saka and Yanagida 1993; Samejima et al. 1993), rad mutants (rad1-1, rad3::ura4+, rad9::ura4+, rad17::ura4+, rad26::ura4+, and rad27::ura4+ mutants; Al-Khodairy et al. 1994; gifts of Dr. A.M. Carr), cdc mutants (cdc6-121, cdc17-K42, cdc20-M10, cdc22-C1, Nurse et al. 1976; Gordon and Fantes 1986) and swi7-H4 (Singh and Klar 1993) mutants were described previously. Rich YPD (2% glucose, 2% polypeptone, and 1% yeast extract) and minimal EMM2 (Mitchison 1970) were employed.
Determination of UV sensitivity
The procedures for examining UV sensitivity were described previously (Saka and Yanagida 1993; Saka et al. 1994). Briefly, S. pombe cells diluted were plated and incubated for 30 min, followed by UV irradiation (0–300 J/m2; Stratalinker, Stratagene) and culturing for several days at 26°C (the permissive temperature for temperature sensitive mutants). The percent septation index was obtained by Calicoflour staining after glutaraldehyde fixation. To study the checkpoint response, exponentially growing S. pombe cells were irradiated by 100 J/m2.
FACScan analysis, Southern transfer, and sequencing
The procedures for FACScan were described previously (Costello et al. 1986; Saka and Yanagida 1993). Southern hybridization was performed by the procedures described (Saka et al. 1994a). The dye-terminator method was employed for nucleotide sequencing by use of the ABI 373A Sequencing System.
Cell extracts and immunoblotting
Cells were collected and suspended in ice chilled phosphate buffer-saline at pH 7.5 containing 10 mm NaN3 and 50 mm NaF, rapidly frozen by liquid nitrogen and kept at −20°C. Cells were then thawed and suspended at a concentration of 1 × 109/ml in HB buffer (25 mm Tris–HCl at pH 7.5 containing 15 mm MgCl2, 15 mm EGTA, 0.1% NP-40, 1 mm DTT, 60 mm β-glycerophosphate, 15 mm p-nitrophenylphosphate, 0.5 mm Na3V04, 0.1 mm NaF, 1 mm PMSF). Cells were disrupted by glass beads and centrifuged at 4°C at 14,000 rpm for 20 min. Supernatants were used for immunoblotting. As secondary antibodies, HRP-labeled protein A (200-fold dilution, Bio-Rad) or HRP-labeled sheep anti-mouse antibody (Amersham, 500-fold dilution) was employed.
Two-hybrid screening
The S. pombe cDNA library adapted to the two-hybrid system in yeast was purchased from Clontech, Inc. (XL4000AA), and the instruction procedures for screening were followed by use of the HF7c strain. Yeast cells were grown in the SD synthetic medium supplemented with the dropout solution at 30°C. DNAs of different domains of the cut5+ gene were obtained by amplification by the PCR method (the R1R2 region) or by isolating the restriction fragments (the central acidic domain, R3, R4, and the carboxy-terminal region). These DNAs were ligated in frame with pGBT9 containing the GAL4 DNA-binding domain. All of these plasmids were verified by nucleotide sequencing. The filter assay for β-galactosidase activity was done in accordance with the company’s instructions. Strong signals were obtained from colonies by incubating them on the filter for 30 min or up to 2 hr. Liquid assay for the β-galactosidase activity was also done following the company’s instructions by use of the SFY526 strain.
Preparation of GST fusion proteins and in vitro binding assay
NdeI–BamHI fragment of cut5+ containing the R1R2 region was blunt-end ligated at the XhoI site of plasmid pGEX-KG (Guan and Dixon 1991), whereas BamHI–SalI fragment containing the R3 region was inserted at the same site of pGEX-KG. Resulting plasmids pYS583 and pR308, respectively, were used for producing fusion proteins that were dissolved in the presence of 8 m urea, followed by successive dialysis in PBS containing reduced concentrations of urea and 10 mm DTT. Resulting GST proteins in the absence of urea were incubated with glutathione beads. In vitro translation of Crb2 and Crb3 proteins was performed with the Promega TNT Coupled reticulocyte lysate system and [35S]methionine (ICN). Luciferase, Crb2, and Crb3 proteins were in vitro translated and mixed with the beads bound to GST–R1R2 or GST–R3, and incubated at 4°C for 1 hr. Beads were washed three times by 10 volumes of 20 mm Tris-HCl at pH 7.5 containing 1 mm EDTA, 10% glycerol, 1 mm DTT, 0.1 mm PMSF, 1 μg/ml of leupeptin, 0.5 μg/ml of aprotinin, followed by SDS-PAGE and autoradiography by use of the Amersham Amplify system.
Sucrose gradient centrifugation and phosphatase treatment
The procedure of sucrose gradient centrifugation was followed to that reported previously (Yamashita et al. 1996; Yamada et al. 1997). Extracts prepared from each 4 × 108 cells nonirradiated or irradiated with UV (100 J/m2) and cultured 1 hr at 26°C were run at 40,000 rpm for 12 hr at 4°C with a SW50.1 rotor. BSA (4.5S) and thyroglobulin (16.5–19S) were used as the markers for the S values. The procedure of Walworth and Bernards (1996) was followed for treatment of Crb2 and Chk1 in cell extracts by λ protein phosphatase (New England Biolab).
GFP tagging and immunofluorescence microscopy
The jellyfish GFP gene was introduced to the amino-terminal site of the crb2+ gene by creating a new BamHI site at the amino-terminus. A mild inducible promoter REP41 (Maundrell 1990) was placed upstream of the fused GFP–Crb2 gene by use of pGFT41 (Heim et al. 1995). Resulting plasmid was introduced into Δcrb2 strain and fluorescence was observed in the presence (repressed) or the absence (induced) of thiamine (Nabeshima et al. 1995). Cells cultured in the presence or the absence of thiamine carrying pGFT–Crb2 were observed by blue light without fixation.
Gene disruption
The procedure of gene disruption was as reported previously (Rothstein 1983).
Acknowledgments
Y.S and F.E. contributed equally to the present work. We are greatly indebted to Drs. N. Walworth, A. Carr, and S. Francesconi for strains, plasmids, and helpful comments. This work was supported by grants of the Specially Promoted Research from the Ministry of Education, Science, and Culture of Japan and the Core Research for Evolutional Science and Technology research fund of the Japan Science and Technology Corporation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL yanagida@kozo.biophys.kyoto-u.ac.jp; FAX 81 75 753 4208.
References
- Al-Khodairy F, Carr AM. DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. EMBO J. 1992;11:1343–1350. doi: 10.1002/j.1460-2075.1992.tb05179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khodairy F, Fotou E, Sheldrick KS, Griffiths DJ, Lehmann AR, Carr AM. Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol Biol Cell. 1994;5:147–160. doi: 10.1091/mbc.5.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki H, Leem SH, Phongdara A, Sugino A. Dpb11, which interacts with DNA polymerase II epsilon in Saccharomyces cerevisiae, has a dual role in S-phase progression at a cell cycle checkpoint. Proc Natl Acad Sci. 1995;92:11791–11795. doi: 10.1073/pnas.92.25.11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley N, Holtzman D, Flaggs G, Keegan K, DeMaggio A, Ford J, Hoekstra M, Carr A. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- Bork P, Hoffmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. A superfamily of conserved domains in DNA damage responsive cell cycle checkpoint proteins. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- Callaebaut I, Mornon JP. From BRCA1 to RAP1: A widespread BRCT module closely associated with DNA repair. FEBS Lett. 1997;400:25–30. doi: 10.1016/s0014-5793(96)01312-9. [DOI] [PubMed] [Google Scholar]
- Costello G, Rodgers L, Beach D. Fission yeast enters the stationary phase G0 state from either mitotic G1 or G2. Curr Genet. 1986;11:119–125. [Google Scholar]
- Duck P, Nasim A, James AP. Temperature-sensitive mutant of Schizosaccharomyces pombe exhibiting enhanced radiation sensitivity. J Bact. 1976;128:536–539. doi: 10.1128/jb.128.2.536-539.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: Preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Enoch T, Carr T, Nurse P. Fission yeast genes involved in coupling mitosis to completion of DNA replication. Genes & Dev. 1992;6:2035–2046. doi: 10.1101/gad.6.11.2035. [DOI] [PubMed] [Google Scholar]
- Fenech M, Carr AM, Murray J, Watts FZ, Lehmann AR. Cloning and characterization of the rad4 gene of Schizosaccharomyces pombe; a gene showing short regions of sequence similarity to the human XRCC1 gene. Nucleic Acids Res. 1991;19:6737–6741. doi: 10.1093/nar/19.24.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Sarabia M-J, McInerny C, Harris P, Gordon C, Fantes P. The cell cycle genes cdc22+ and suc22+ of the fission yeast Schizosaccharomyces pombe encode the large and small subunits of ribonucleotide reductase. Mol & Gen Genet. 1993;238:241–251. doi: 10.1007/BF00279553. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Ford JC, Al-Khodairy F, Fotou E, Sheldrick KS, Griffiths DJ, Carr AM. 14-3-3 protein homologs required for the DNA damage checkpoint in fission yeast. Science. 1994;265:533–535. doi: 10.1126/science.8036497. [DOI] [PubMed] [Google Scholar]
- Francesconi S, De Recondo AM, Baldacchi G. DNA polymerase delta is required for the replication feedback control of cell cycle progression in Schizosaccharomyces pombe. Mol Gen Genet. 1995;246:561–569. doi: 10.1007/BF00298962. [DOI] [PubMed] [Google Scholar]
- Francesconi S, Grenon M, Bouvier D, Baldacci G. p56chk1 protein kinase is required for the DNA replication checkpoint at 37°C in fission yeast. EMBO J. 1997;16:1332–1341. doi: 10.1093/emboj/16.6.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CB, Fantes P. The cdc22 gene of Schizosaccharomyces pombe encodes a cell cycle regulated transcript. EMBO J. 1986;5:2981–2985. doi: 10.1002/j.1460-2075.1986.tb04595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths DJ, Barbet NC, McCready S, Lehmann AR, Carr AM. Fission yeast rad17: A homologue of budding yeast RAD24 that shares regions of sequence similarity with DNA polymerase accessory proteins. EMBO J. 1995;14:5812–5823. doi: 10.1002/j.1460-2075.1995.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan K, Dixon JE. Eucaryotic proteins expressed in Escherichia coli: An improved thrombin cleavage and purification procedure of fusion proteins with glutathion S-transferase. Analytical Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- Gutz H, Heslot H, Leupold U, Lopreno N. Schizosaccharomyces pombe. In: King RC, editor. Handbook of genetics 1. New York, NY: Plenum Press; 1974. pp. 395–446. [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoint: Controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Hayles J, Fisher D, Woollard A, Nurse P. Temporal order of S phase and mitosis in fission yeast is determined by the state of the p34cdc2-mitotic B cyclin complex. Cell. 1994;78:813–822. doi: 10.1016/s0092-8674(94)90542-8. [DOI] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Hirano T, Funahashi S, Uemura T, Yanagida M. Isolation and characterization of Schizosaccharomyces pombe cut mutants that block nuclear division but not cytokinesis. EMBO J. 1986;5:2973–2979. doi: 10.1002/j.1460-2075.1986.tb04594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci. 1994;91:6098–6102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TJ, Martin S, Forsburg SL, Stephen RJ, Ausso A, Nurse P. The fission yeast cdc18+ gene product couples S phase to start and mitosis. Cell. 1993;74:371–382. doi: 10.1016/0092-8674(93)90427-r. [DOI] [PubMed] [Google Scholar]
- Kitazuno, A. and T. Matsumoto. 1997. Isogaba maware, Quality control of gene DNA by checkpoints. BioEssays (in press). [DOI] [PubMed]
- Koonin EV, Altschul SF, Bork P. Functional motifs. Nature Genet. 1996;13:266–268. doi: 10.1038/ng0796-266. [DOI] [PubMed] [Google Scholar]
- Kostrub C, Al-Khodairy F, Ghazizadeh H, Carr A, Enoch T. Molecular analysis of hus1+, a fission yeast gene required for S-M and DNA damage checkpoints. Mol Gen Genet. 1997;254:389–399. doi: 10.1007/pl00008606. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Nash RA, Klugland A, Schär P, Barnes DB, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase β and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- Larimer FW, Perry JR, Hardigree AA. The REV1 gene of Saccharomyces cerevisiae: Isolation, sequence, and functional analysis. J Bacteriol. 1989;171:230–237. doi: 10.1128/jb.171.1.230-237.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman HB, Hopkins KM, Nass M, Demetrick D, Davey S. A human homologue of the Schizosaccharomyces pombe rad9+ checkpoint gene. Proc Natl Acad Sci. 1996;93:13890–13895. doi: 10.1073/pnas.93.24.13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydall D, Weinert T. Yeast checkpoint genes in DNA damage processing: Implications for repair and arrest. Genes & Dev. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- Maundrell K. nmt1 of fission yeast. J Biol Chem. 1990;265:10857–10864. [PubMed] [Google Scholar]
- Miki T, Smith CL, Long JE, Eva A, Fleming TP. Oncogene ect2 is related to regulators of small GTP-binding protein. Nature. 1993;362:462–465. doi: 10.1038/362462a0. [DOI] [PubMed] [Google Scholar]
- Mitchison JM. Physiological and cytological methods for Schizosaccharomyces pombe. Methods Cell Physiol. 1970;4:131–165. [Google Scholar]
- Mizukami T, Chang WI, Gargavitsev I, Kaplan N, Lombardi D, Matsumoto T, Niwa O, Kounousu A, Yanagida M, Marr TG, Beach D. A 13 kb resolution cosmid map of the 14 Mb fission yeast genome by nonrandom sequence-tagged site mapping. Cell. 1993;73:121–132. doi: 10.1016/0092-8674(93)90165-m. [DOI] [PubMed] [Google Scholar]
- Moreno S, Nurse P. Regulation of progression through the G1 phase of the cell cycle by the rum1+ gene. Nature. 1994;367:236–242. doi: 10.1038/367236a0. [DOI] [PubMed] [Google Scholar]
- Murakami H, Okayama H. A kinase from fission yeast responsible for blocking mitosis in S-phase. Nature. 1995;374:817–819. doi: 10.1038/374817a0. [DOI] [PubMed] [Google Scholar]
- Nabeshima K, Kurooka H, Takeuchi M, Kinoshita K, Nakaseko Y, Yanagida M. p93dis1 required for sister chromatid separation is a novel microtubule and spindle pole body associating protein phosphorylated at the Cdc2 target sites. Genes & Dev. 1995;9:1572–1585. doi: 10.1101/gad.9.13.1572. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. Viewpoint: Putting the cell cycle in order. Science. 1996;274:1643–1645. doi: 10.1126/science.274.5293.1643. [DOI] [PubMed] [Google Scholar]
- Navas TA, Sanchez Y, Elledge SJ. RAD9 and DNA polymerase ε form parallel sensory branches for transducing the DNA damage checkpoint signal in Saccharomyces cerevisiae. Genes & Dev. 1996;10:2632–2643. doi: 10.1101/gad.10.20.2632. [DOI] [PubMed] [Google Scholar]
- Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Nurse P. p65cdc18 plays a major role controlling the initiation of DNA replication in fission yeast. Cell. 1995;83:397–405. doi: 10.1016/0092-8674(95)90117-5. [DOI] [PubMed] [Google Scholar]
- Nugent CI, Hughes TR, Lue NF, Lundblad V. Cdc13p: A single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science. 1996;274:249–251. doi: 10.1126/science.274.5285.249. [DOI] [PubMed] [Google Scholar]
- Nurse P, Thuriaux P, Nasmyth K. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol Gen Genet. 1976;146:167–178. doi: 10.1007/BF00268085. [DOI] [PubMed] [Google Scholar]
- Paulovich AG, Hartwell LH. A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell. 1995;82:841–847. doi: 10.1016/0092-8674(95)90481-6. [DOI] [PubMed] [Google Scholar]
- O’Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997;16:545–554. doi: 10.1093/emboj/16.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Furnari B, Russell P. Cdc2 tyrosine phosphorylation is required for the DNA damage checkpoint in fission yeast. Genes & Dev. 1997;11:504–511. doi: 10.1101/gad.11.4.504. [DOI] [PubMed] [Google Scholar]
- Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Rowley R, Subramani S, Young PG. Checkpoint controls in Schizosaccharomyces pombe: rad1. EMBO J. 1992;11:1335–1342. doi: 10.1002/j.1460-2075.1992.tb05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka Y, Yanagida M. Fission yeast cut5+ gene required for the onset of S-phase and the restraint of M-phase is identical to the radiation-damage repair gene rad4+ Cell. 1993;74:383–393. doi: 10.1016/0092-8674(93)90428-s. [DOI] [PubMed] [Google Scholar]
- Saka Y, Fantes P, Sutani T, McInerny C, Creanor J, Yanagida M. Fission yeast cut5 links nuclear chromatin and M phase regulator in the replication checkpoint control. EMBO J. 1994a;13:5319–5329. doi: 10.1002/j.1460-2075.1994.tb06866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka, Y., P. Fantes, and M. Yanagida. 1994b. Coupling of DNA replication and mitosis by fission yeast rad4/cut5. J. Cell Sci. (suppl.) 18: 57–61. [DOI] [PubMed]
- Samejima I, Matsumoto T, Nakaseko Y, Beach D, Yanagida M. Identification of seven new cut genes involved in Schizosaccharomyces pombe mitosis. J Cell Sci. 1993;105:135–143. doi: 10.1242/jcs.105.1.135. [DOI] [PubMed] [Google Scholar]
- Sheldrick KS, Carr AM. Feedback controls and G2 checkpoints: Fission yeast as a model system. BioEssays. 1993;15:775–782. doi: 10.1002/bies.950151202. [DOI] [PubMed] [Google Scholar]
- Siede W, Friedberg AS, Friedberg EC. RAD9-dependent G1 arrest defines a second checkpoint for damaged DNA in the cell cycle of Saccharomyces cerevisiae. Proc Natl Acad Sci. 1993;90:7985–7989. doi: 10.1073/pnas.90.17.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Klar AJ. DNA polymerase-α is essential for mating-type switching in fission yeast. Nature. 1993;361:271–273. doi: 10.1038/361271a0. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Brookman KW, Jones JJ, Allen SA, Carrano AV. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strands repair and sister chromatid exchange. Mol Cell Biol. 1990;10:6160–6171. doi: 10.1128/mcb.10.12.6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NC, Bernards R. rad-Dependent response of the chk1-encoded protein kinase at the damage checkpoint. Science. 1996;271:353–356. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- Walworth N, Davey S, Beach D. Fission yeast chk1 protein kinase links rad checkpoint pathway to cdc2. Nature. 1993;363:368–371. doi: 10.1038/363368a0. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- Wilson J, Wilson S, Warr N, Watts FZ. Isolation and characterization of the Schizosaccharomyces pombe rhp9 gene: A gene required for the DNA damage checkpoint but not the replication checkpoint. Nucleic Acids Res. 1997;25:2138–2145. doi: 10.1093/nar/25.11.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Kumada K, Yanagida M. Distinct subunit functions and cell cycle regulated phosphorylation of 20S APC/Cyclosome required for anaphase in fission yeast. J Cell Sci. 1997;110:1793–1804. doi: 10.1242/jcs.110.15.1793. [DOI] [PubMed] [Google Scholar]
- Yamashita YM, Nakaseko Y, Samejima I, Kazuki K, Yamada H, Michaelson D, Yanagida M. 20S cyclosome complex formation and proteolytic activity inhibited by the cAMP/PKA pathway. Nature. 1996;384:276–279. doi: 10.1038/384276a0. [DOI] [PubMed] [Google Scholar]