

Abstract
Malignant small round cell tumors are characterised by small, round, relatively undifferentiated cells. They generally include Ewing's sarcoma, peripheral neuroectodermal tumor, rhabdomyosarcoma, synovial sarcoma, non-Hodgkin's lymphoma, retinoblastoma, neuroblastoma, hepatoblastoma, and nephroblastoma or Wilms’ tumor. Other differential diagnoses of small round cell tumors include small cell osteogenic sarcoma, undifferentiated hepatoblastoma, granulocytic sarcoma, and intraabdominal desmoplastic small round cell tumor. Differential diagnosis of small round cell tumors is particularly difficult due to their undifferentiated or primitive character. Tumors that show good differentiation are generally easy to diagnose, but when a tumor is poorly differentiated, identification of the diagnostic, morphological features is difficult and therefore, no definitive diagnosis may be possible. As seen in several study reports, fine needle aspiration cytology (FNAC) has become an important modality of diagnosis for these tumors. The technique yields adequate numbers of dissociated, viable cells, making it ideally suitable for ancillary techniques. Typically, a multimodal approach is employed and the principal ancillary techniques that have been found to be useful in classification are immunohistochemistry and immunophenotyping by flow cytometry, reverse transcriptase polymerase chain reaction (RT-PCR), fluorescence in situ hybridization (FISH), and electron microscopy. However, the recent characterization of chromosomal breakpoints and the corresponding genes involved in malignant small round cell tumors means that it is possible to use molecular genetic approaches for detection.
Keywords: Fine needle aspiration cytology, malignant small round cell tumors, ancillary techniques
Introduction
Malignant small round cell tumors (MSRCT) is a term used for tumors composed of malignant round cells that are slightly larger or double the size of red blood cells in air-dried smears.[1] This group of neoplasms is characterized by small, round, relatively undifferentiated cells. They generally include Ewing's sarcoma (EWS), peripheral neuroectodermal tumor, rhabdomyosarcoma, synovial sarcoma, non-Hodgkin's lymphoma, retinoblastoma, neuroblastoma, hepatoblastoma, and nephroblastoma.[2] Other differential diagnoses of small round cell tumors include small cell osteogenic sarcoma, undifferentiated hepatoblastoma, granulocytic sarcoma, and intraabdominal desmoplastic small round cell tumor. Differential diagnosis of small round cell tumors is particularly difficult due to their undifferentiated or primitive character. Tumors that show good differentiation are generally easy to diagnose, but identification of the diagnostic, morphological features is difficult when a tumor is poorly differentiated, therefore, no definitive diagnosis may be possible.[3] As seen in several study reports, fine needle aspiration cytology (FNAC) has become an important modality of diagnosis for these tumors.[4,5] It yields adequate numbers of dissociated, viable cells making it ideally suitable for ancillary techniques.
Cytomorphological Features of MSRCT
Fine needle aspiration cytology is a successful diagnostic tool when used by a skilled cytopathologist for documenting primary and recurrent MSRCTs in pediatric patients.[6] During the past decade, a number of articles highlighting the role of FNAC in the diagnosis of MSRCT have been published.[6–8] MSRCTs that show good differentiation, are amenable to cytological diagnosis. However, exact categorization is not possible at the light microscopic level in the case of poorly differentiated tumors. This poses a considerable challenge to the diagnostic skill of the cytopathologist.[3,7] In fact, a definite diagnosis based on clinical and cytomorphological criteria was possible in 57% of the cases in one study,[9] and in 68% of the cases in another study.[8]
Cytologically, these tumors are composed of a nearly uniform population of round to oval cells with scanty, basophilic cytoplasm; mixed populations of small and large cells are present in some tumors like neuroblastomas.[8] These are also called small round blue cell tumors as the cells are blue, in the sense that they have large hyperchromatic nuclei and a thin rim of cytoplasm. In general, these neoplasms consist of cells having diameters up to approximately three times that of a small mature lymphocyte and typically possess only a single hyperchromatic nucleus having finely granular, evenly distributed chromatin. Nucleoli can be unapparent, small, or quite prominent. The cytoplasm is generally scanty, resulting in a very high nuclear to cytoplasmic ratio.
The salient features of the individual tumors will be briefly reviewed here under the broad category of MSRCT.
Ewing's Sarcoma / Primitive Neuroectodermal Tumor
This neoplasm predominates in the second decade of life and the pelvis and femur are the most commonly affected sites.[10] Primitive neuroectodermal tumor (PNET) is a small round cell malignancy of presumedly primitive, neuroectodermal tissue or pluripotential, migratory neural crest cells arising from the soft tissue or bone, predominantly in older children and adults. The term, “PNET” includes malignant small round cell tumors of the thoracopulmonary region (Askin's tumor), extraskeletal Ewing's sarcoma, peripheral neuroblastoma, and peripheral neuroepithelioma.[11] In view of the shared cytogenetic abnormality, the term, “EWS/PNET” is currently favored for this tumor family. It has been commented that while Ewing's sarcoma of the bone tends to be undifferentiated, PNET shows variable degrees of neuroectodermal differentiation.[12] The cell of origin of this tumor is uncertain. Originally, it was thought that EWS/PNET might arise from the neuroectoderm, but recent data have suggested that this tumor is more likely to originate from primitive stem cells and that the degree of malignancy appears to depend upon the stage of stem cell arrest during differentiation.[13]
In FNAC, the tumor cells tend to be uniform and are usually arranged in relatively small, tight clusters. The nuclei may be round or irregular and usually lack nucleoli. These small blue cells tend to have a high nucleocytoplasmic ratio [Figure 1a]. Two population of cells have been described: large chief cells and smaller dark cells.[14] The cytoplasm is pale blue and contains variable numbers of punched-out vacuoles which correspond to glycogen deposits. In the majority of cases, abundant cytoplasmic glycogen can be demonstrated by periodic acid Schiff staining.[2] But the presence of large amounts of intracellular glycogen is not a specific finding as while up to 35% of all Ewing's sarcoma cases do not contain detectable glycogen, many other childhood tumors do contain detectable glycogen.[15] Variable numbers of pseudorosettes may be seen;[6,11] fibrillary matrix and Homer Wright rosettes are seen at times and mitotic figures are rarely detected. Atypical variants of EWS may reveal large cells with abundant cytoplasm, large irregular nuclei, vesicular chromatin, and prominent eosinophilic nucleoli.[2,8] Several studies have confirmed that CD99 or MIC2, which shows membrane positivity [Figure 1b], was helpful in distinguishing the EWS/PNET group from other MSRCTs.[16–20] The FLI1 protein, the gene product ofFLI1 , t (11:22), is positive in 85% of all EWS / PNET cases.[21]
Figure 1.
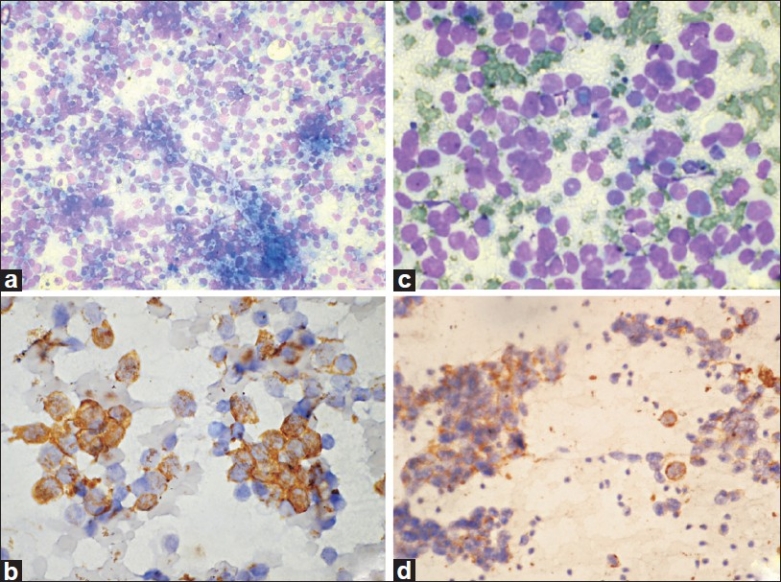
Ewing's sarcoma / PNET. (a) Cellular smears with dispersed monomorphic cells in a vacuolated tigroid background (MGG, ×100); (b) Cells show CD99/MIC2 membrane positivity (IHC, ×400). Rhabdomyosarcoma. (c) Undifferentiated tumor cells with scanty to moderate cytoplasm (MGG, ×400); (d) Smears show desmin positivity (IHC, ×200)
Neuroblastoma
Neuroblastoma (NB) is the third most common, solid, malignant tumor of infancy and childhood. Ninety percent of these tumors occur in patients less than ten years of age; there is a slight male preponderance. The tumor arises from the neuroblasts, the undifferentiated precursor cells of the sympathetic nervous system. About 70% of the neuroblastomas occur in the retroperitoneum and the majority of these involve the adrenal gland. Synchronous or metachronous bilaterality is extremely unusual.[22] The tumor cells may be disposed singly or arranged in small clusters in FNAC. The cells are small and undifferentiated with a higher nuclear to cytoplasmic ratio [Figure 2a]. Small clusters of cells may be separated by pale blue to light purple fibrillar matrix.[2] These pseudorosettes may be seen in as low as 18%[23] to as high as 72% of all cases.[24] Neuroblastomas are characteristically positive for neuron-specific enolase (NSE) [Figure 2b], which is an isoenzyme of the glycolytic enzyme, enolase, which has been shown to be highly specific for neurons and neuroendocrine cells.[25] Classical neuroblastomas are neither immunoreactive for vimentin [Figure 2c] nor for HBA71.[26] Other markers evaluated in neuroblastoma include S-100, chromogranin, and synaptophysin, which are not usually helpful in distinguishing undifferentiated and poorly differentiated tumors.[27] Recently, a new antibody, the anti-GD2 antibody, has been described that is directed against a ganglioside and may be applied on tissues.[28] This is a sensitive and reproducible marker and is used to detect and quantify minimal residual disease in neuroblastoma.
Figure 2.
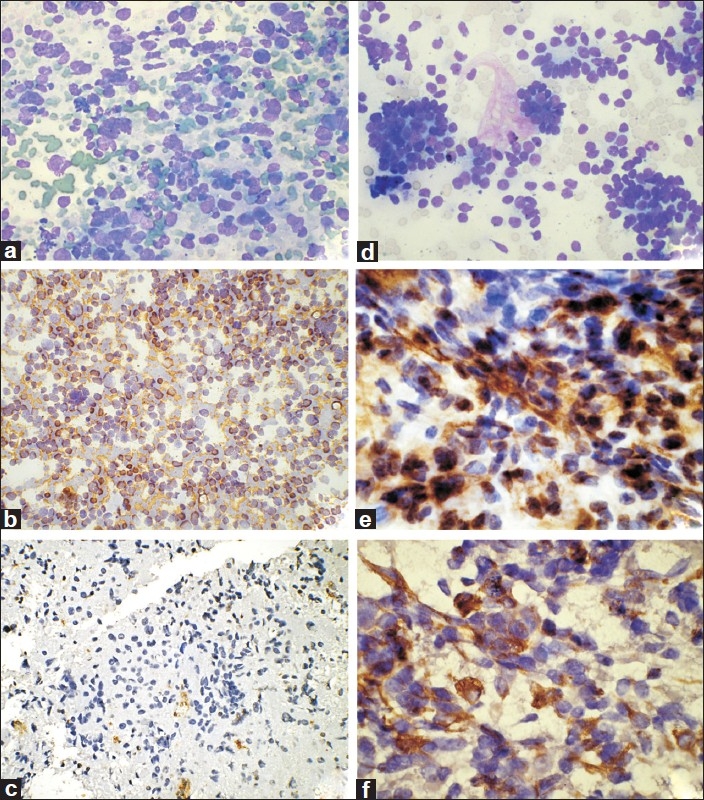
Neuroblastoma. (a) Cellular smears with dispersed undifferentiated cells (MGG, ×200); (b) Tumor cells are positive for NSE (IHC, 100); (c) Tumor cells are negative for vimentin (IHC, ×100). Wilms’ tumor. (d) Smears showing undifferentiated blastemal cells with focal tubule formation (MGG, ×200); (e) Tumor cells are positive for vimentin (IHC, ×400); (f) Tumor cells are positive for cytokeratin (IHC, ×400)
Chromogranin A (CgA), the main component of chromaffin granules, is considered to be a specific marker of neuroendocrine differentiation.[29] Reverse transcriptase PCR (RT-PCR) is another sensitive and specific test for neuroblastoma as it can help to detect chromogranin A transcript, PGP 9.5,[30] β1, 4-N acetylgalactosaminyl transferase or GD2 synthase, neurofilament, and synaptophysin, MAGE and GAGE[31] , and the most useful target, tyrosine hydroxylase (TH) mRNA. Gilbert et al.[32] analysed 84 pediatric malignancies including 55 neuroblastomas for the expression of tyrosine hydroxylase and DOPA decarboxylase by using the RT-PCR technique. Out of 55 neuroblastoma cases, 54 expressed clearly detectable levels of both genes. In another study, TH mRNA was found to be expressed in all NB cell lines (100%) and in 23/25 (92%) clinical NB tumor samples.[33] Thus, the expression of TH and DOPA decarboxylase may prove to be a valuable aid in the differential diagnosis of neuroblastoma from other small round cell tumors of childhood.
The RT-PCR technique is so sensitive that it can detect even small numbers of neuroblastoma cells in peripheral blood and bone marrow and is hence useful in providing strong supporting evidence in the determination of treatment.[34] Tyrosine hydroxylase RT-PCR was reported to be able to detect a single neuroblastoma cell in 105 normal bone marrow mononuclear cells (MNCs),[35] whereas PGP9.5 RT-PCR was sufficiently sensitive for one tumor cell to be found in 107 control peripheral MNCs.[30]
Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is the most commonly found soft tissue sarcoma in children wherein the cancer cells are thought to arise from skeletal muscle progenitors. These tumors are currently classified into embryonal rhabdomyosarcoma (ERMS), alveolar rhabdomyosarcoma (ARMS), and pleomorphic rhabdomyosarcoma (PRMS) subtypes. ERMS typically occurs in young children, whereas ARMS usually occurs in older children and young adults, and PRMS occurs in older adults.[36] Whilst the younger age group is generally associated with better survival, the particularly excellent outcomes of children less than one year of age observed in the case of other round cell tumors is a feature not shared by rhabdomyosarcoma.[10]
FNAC smears from RMS tumors reveal cells that vary considerably in size and shape, but usually contain moderate to abundant amounts of cytoplasm that stain deep blue and contain occasional, small cytoplasmic vacuoles [Figure 1c].[3,37,38] The presence of strap or tadpole cells is deemed to be strongly associated with rhabdomyosarcoma.[5,7] The finding of cells with more abundant cytoplasm, eccentrically located nuclei, and bi/multinucleated tumor cells in a background of mucosubstance helps to distinguish RMS from other small round cell tumors.[39,40]
It is difficult to subtype RMS into embryonal and alveolar subtypes based on morphology alone because of a wide spectrum of overlapping morphology within the same histological type.[41] However, alveolar RMS shows predominantly dissociated cells or chance formations, whereas the embryonal type shows large tissue fragments with abundant eosinophilic material and various numbers of dissociated cells. The relative proportion of poorly to better and well-differentiated rhabdomyoblasts varies in both types and in all patterns.[41] In a previous study, the embryonal type was found to be composed of mainly early rhabdomyoblasts.[38] Large, tadpole or ribbon-shaped tumor cells were also observed in embryonal RMS. In contrast, most tumor cells were small and lymphocyte-like in alveolar RMS, with finely granular chromatin. The use of desmin on cytological material has been reported as being useful in differentiating between different small round cell tumors of childhood, and is a specific marker for RMS [Figure 1d].[39,41] Desmin positivity is seen not only in large rhabdomyoblasts, but also in more numerous, smaller, less well differentiated tumor cells because it is expressed during the early differentiation of skeletal and smooth muscle cells. Immunohistochemical reagents such as antibodies recognizing the muscle-specific intermediate filament-desmin, muscle-specific actin, and myoglobin, are valuable in differentiating them but may lack sensitivity or specificity in some cases.[42] Myf-4/myogenin belonging to the family of MyoD genes, regulates the differentiation of pluripotent, primitive mesodermal cells in the skeletal muscle. A very useful monoclonal antibody against MyoD1 detects myogenic regulatory proteins and was introduced for use in pathology in 1990.[43] In a study conducted by Wang et al.[44] on paraffin sections, myogenin and MyoDl nuclear expression was noted in 91% of RMS cases, whereas neither was detected in any of the neuroblastomas or Ewing's sarcomas/ PNETs. In a large series of 956 RMS cases reported by Morotti et al.,[45] the two markers, MyoD1 and myogenin, were positive in 97% of the cases but in none of the 96 non-RMS tumors tested in the series. Both ARMS and ERMS stain positively for myogenin, although the number of cells staining in the latter subtype is clearly fewer. Thus, MyoD1 and myogenin are highly sensitive and specific markers for RMS as compared to desmin, and can differentiate alveolar RMS from the embryonal type.
Wilms’ Tumor
Wilms’ tumor (WT) or nephroblastoma is a malignant neoplasm of the kidney, which morphologically resembles embryonal renal tissue. It is a common pediatric malignant tumor and is one of the seven, most common, malignant tumors in childhood. More than 90% of cases are children under the age of ten years. This tumor is believed to arise from the cells of the nephrogenic blastema. Wilms’ tumor comprises of a variety of histological patterns including blastematous or epithelial-predominant, mixed blastematous, and epithelial and sarcomatous types.[46] The first cytomorphological description of Wilms’ tumor was from our Institute by Dey et al.[47] who described 15 cases of renal, extrarenal, and metastatic Wilms’ tumor; this was followed by several other reports.[48,49] Smears from typical, triphasic Wilms’ tumor reveal blastemal cells with evidence of epithelial and mesenchymal differentiation. The blastemal cells which are found in 90% of the cases, have scanty, deep blue cytoplasm with ill-defined borders and round to oval nuclei having fine, regular, evenly distributed chromatin [Figure 2d].[47] The epithelial component shows cells forming tubules or solid strands or cords, and the cells exhibit a paler blue cytoplasm.[47] Stromal elements are usually composed of spindle-shaped cells. Occasional, large, pleomorphic cells with abundant cytoplasm may be seen.[47] Anaplastic Wilms’ tumor can also be recognized cytologically: the cells are two to three times larger than the blastemal cells, displaying marked pleomorphism and atypia; atypical and multipolar mitosis are also seen in smears. Other recently described variants include mesenchyme-predominant Wilms’ tumor, which shows clusters of spindle cells associated with the matrix material, with or without evidence of rhabdomyoblastic differentiation.[50] These uncommon variants pose a challenge to the cytopathologist and have to be distinguished from mesoblastic nephroma and the spindle cell variant of clear cell sarcoma of the kidney. On the other hand, blastema-predominant Wilms’ tumor should be distinguished mainly from neuroblastoma with which it shares clinical and cytological overlaps.
The WT1 gene, identified as a tumor suppressor gene located at 11p13, is involved in the development of Wilms’ tumor.[51] Brahmi et al.[39] reported positivity for cytokeratin [Figure 2f], NSE, epithelial membrane antigen, and vimentin [Figure 2e] in Wilms’ tumor. However, both the studies concluded that immunocytochemistry (ICC) is useful in the diagnosis of Wilms’ tumor. RT PCR cannot be used in Wilms’ tumor cases as there is no translocation.
Synovial Sarcoma
Synovial sarcoma (SS) was originally described by Simon in 1865, although the first use of the term is ascribed to Fisher.[52] Typically, these tumors are spindle cell sarcomas but some variants of synovial sarcoma such as the small cell (or round cell variant) may be considered under the category of MSRCT.
Synovial sarcoma (SS) is a highly malignant tumor that occurs mainly in adolescents and young adults between 15 and 30 years of age, and is usually seen in the extremities in the vicinity of joints, most commonly the knee and lower thigh region. The peak incidence is in the 3rd decade and males are affected more often than females. Synovial sarcoma is the fourth most common soft tissue sarcoma following malignant fibrous histiocytoma, liposarcoma, and rhabdomyosarcoma. These tumors account for 5–10% of soft tissue sarcomas. There are four recognized subtypes of synovial sarcoma: biphasic tumors, monophasic fibrous tumors, monophasic epithelial tumors, and poorly differentiated tumors.[53,54] The three types of poorly differentiated SS are a large cell epithelioid variant, a small cell variant, and a high-grade spindle cell variant.
The cytomorphology of conventional synovial sarcoma has been recently described by Akerman et al.[55] The yield was reported to be rich with dispersed cells, stripped nuclei, and tissue fragments; a branching network of vessels was also observed. Tumor cells were small to medium in size, with rounded, ovoid, or fusiform bland nuclei with inconspicuous nucleoli. Small glandular or acinar-like structures were seen in some biphasic variant cases. Cellular pleomorphism was seen in the pleomorphic variant. Ewing et al.[56] have elaborated on the cytological features of the monophasic variant of synovial sarcoma wherein tissue fragments and single cells containing scant granular cytoplasm, medium-sized nuclei, and coarse chromatin were seen. A monotonous spindle pattern with comma-shaped or oval and spindled or round nuclei were also seen.
The cytology of the small cell variant of synovial sarcoma shows numerous, small round cells with high nucleocytoplasmic ratio. Its cytomorphological features could overlap with those of other malignant small round cell tumors.[57] Cytomorphology of the rare variants of synovial sarcoma such as the poorly differentiated, small cell, and monophasic variants has also been reported recently, wherein the importance of ancillary techniques such as immunocytochemistry and molecular analysis have been emphasised.[58] ICC was not found to be a very helpful ancillary technique; only two out of ten cases in which ICC was applied were diagnostic and eight cases were negative for both cytokeratin and the epithelial membrane antigen (EMA).
The translocation, t(X: 18) (p11:q11), was first identified in 1989 by Limon et al. and was further confirmed in more than 90% of synovial sarcomas, including biphasic and monophasic tumors.[59] Cloning of the translocation, t(X;18) (p11.2;q11.2), from human synovial sarcoma revealed a fusion between theSYTgene located on chromosome 18 and theSSXgene located on the X chromosome, resulting in the formation of chimeric proteins localization.[60] The Xp11 breakpoint actually involves two closely related genes, SSX1 and SSX2, located in the vicinity of the ornithine aminotransferase-like (OATL) pseudogenes 1 and 2 respectively.[61] Although representing only a limited number of cases, the study by Janz et al.[62] supports the hypothesis that the biphasic type of SS has a breakpoint within the ornithine aminotransferase OATL1 cluster, whereas the majority of the monophasic type of SS shows a breakpoint mapping to the OATL2 cluster. The t(X; 18) results in the formation of a chimeric protein that probably deregulates transcription and hence, expression of specific target genes.[63] The predicted protein sequence encoded by the SYT-SSX chimeric transcript consisted of 396 amino-terminal amino acids of SYT fused to 78 carboxyl terminal amino acids of either SSX1 or SSX2.[64,65] SYT contains a predicted glutamine-proline-glycine-rich region suggestive of a transcriptional activation domain, which replaces a region of KRAB (Kruppel-associated box) homology within the 5’ portion of SSX genes[64,65] A translocation t(X;18) (p11.2;q11.2) is detectable in more than 90% of the cases: 75% with SYT-SSX1 and 25% with SYT-SSX2 transcripts.[59,63]
The diagnosis of SS in unusual locations has been confirmed by RT-PCR in tumors of the lung,[66] pleura,[67] prostate,[68] and peritoneum.[69]
A number of studies have suggested that i) patients with SYT-SSX-containing synovial sarcoma have worse overall and five-year metastasis-free survival[63,70] and ii) SYT-SSX1 has a higher proliferative activity.[63,70] Ladayni et al.[71] also demonstrated that tumors with the SYT-SSX2 transcript were confined to monophasic tumors and had better prognosis than tumors with the SYT-SSX1 transcript. At the same time, there were some studies which could not find such correlation.[54,72] There is an overall reduction in survival in cases of SS with SYT-SSX1 transcripts, possibly due to the higher cellular proliferation rates seen in these tumors; the five-year metastasis-free interval was significantly higher for SS cases with SYT- SSX2 transcripts.[70] A series of 243 patients were evaluated by Ladanyi et al.[71] and the SYT-SSX2 fusion type was confirmed as a significant positive prognostic factor for overall survival through an association with a lower prevalence of metastatic disease at the time of diagnosis.
Desmoplastic Small Round Cell Tumor
Desmoplastic small round cell tumor (DSRCT) is a rare neoplasm that was first described by Gerald and Rosai in 1989. It is an extremely rare, high-grade tumor that most often causes diffuse involvement of the abdominal cavity and visceral organs. It differs from other childhood tumors because of its clinical features, morphology, and its immunohistochemistry staining pattern.[2] It is a highly malignant mesenchymal neoplasm growing along serosal surfaces, mostly involving the abdominal cavity of young males, and is associated with poor prognosis. Involvement of the pleura,[73] scrotum,[74] and ovary[75] has also been documented. Clinically, complete excision is often impossible in DSRCT because of the presence of multiple implants in the peritoneum. The prognosis of DSRCT is extremely poor and the average survival is less than two years.[76] Gerald et al.[76] followed up on 17 DSRCT patients: 15 patients died of tumor progression with an average survival of about 22 months. In the vast majority of cases, the disease follows a course toward lethal progression. DSRCT is also characterised by a distinctive clinical profile.[77] Males are affected more commonly than females in a ratio of approximately 4:1. Patients are affected during adolescence or early adulthood; the median patient age is approximately 21 years .[77] The cytomorphology of DSRCT has been described in some recent reports which consist of small numbers of cases.[78–80]
FNAC smears are characterized by high cellularity-undifferentiated, round cells arranged in sheets and clusters. Nuclei are round to oval, some of them with inconspicuous nucleoli and display nuclear molding. The cells have high nuclear/cytoplasmic ratios with granular chromatin. The cytoplasm is scant to moderate, pale blue, and occasionally vacuolated. Pseudorosettes are also seen frequently. Stromal fragments which stain metachromatically have also been identified in all cases.[78,79]
The application of ICC in DSRCT has been described in an occasional report wherein destained smears showed positivity for cytokeratin, EMA, desmin, and WT1 in two cases.[79] Finally, it may be said that the cytological diagnosis of DSRCT is aided by multiple factors such as a typical clinical presentation, the characteristic cytomorphological presentations, and immunocytochemistry and/or demonstration of the cytogenetic abnormality. Although cytology coupled with the demonstration of a polyphenotypic nature by ICC may not be considered as a gold standard, it can serve as a mode of preoperative diagnosis in select cases where a biopsy may be difficult to obtain.[79]
In 1992, Sawyer et al. identified a novel reciprocal translocation of chromosomes 11 and 22, t (11;22)(p13;q12), in desmoplastic small round cell tumors that was later confirmed by several other investigators.[81] Detection of this chimeric message by the RT-PCR method is a very powerful and specific aid in the differential diagnosis of small round cell tumors. Alava et al.[82] detected EWS-WT1 chimeric transcripts in 11/12 DSRCTs by RT-PCR by using primers for the EWS gene exon 7 (EWS 22.3) and the WT1 gene exon 10 (WT1 10.1). Ten out of the 11 positive cases showed in-frame fusions of EWS exon 7 to WT1 exon 8, and one case showed a fusion of EWS exon 8 to WT1 exon 8. Several other case reports have also reported the successful application of this technique for the diagnosis of DSCRT.[83]
Flowcytometric Immunophenotyping
Flowcytometry offers the advantage of automation, speed, and a higher degree of objectivity. It is also possible to detect a small population of monoclonal cells in a background of reactive cells by the use of multiple markers with dual labeling.[85] On the other hand, the main advantage of ICC over flowcytometric immunophenotyping (FCI) is the requirement of a small number of malignant cells and the preservation of cellular morphology. Immunocytochemical methods are also aesthetically pleasing to the morphologist who can visualize and correlate the immunological reaction with malignant cells, staining pattern, and intensity of staining, and easily assess background. Moreover, if neoplastic cells are fragile, they may be destroyed during FCI analysis but successfully assessed in cytospins. However, it is not possible to evaluate double antigen expression with ICC and scoring is semiquantitative. FNAC yields adequate numbers of dissociated, viable cells, making it ideally suited for FCI.[86] The main disadvantage of FCI is the lack of awareness of cytomorphology. A close cooperation between cytopathologists and the FCI laboratory is required for that reason.[87]
FCI has been used extensively for the immunophenotypic evaluation of bone marrow specimens in acute and chronic leukemia and lymphoma.[88] The minimum required number of cells needed for FCI varies in different studies, however, it is generally accepted to average 1 × 105.[88] FCI in conjunction with FNAC is a valuable method for diagnosing and subclassifying NHL and can eliminate the need for a more invasive surgical biopsy in many cases.[85,88–90] Several recent studies have shown the reliability of FNAC for the diagnosis of NHL and its subclassification on the basis of the WHO classification.[91,92] In contrast to NHL, Hodgkin's lymphoma is less readily diagnosed by FCI and more accurately by ICC.[93]
Brahmi et al.[86] included 24 cases of MSRCT which were analyzed by FCI. Semiquantitation of various MSRCT antigens was performed and the sensitivity of FCI was compared to ICC. FCI analysis of cell suspensions prepared from FNAC material was helpful to quantify both cell membrane and intracellular antigens along with the measurement of DNA content to estimate ploidy and proliferative capacity of tumors. Additional positivity for antibodies was observed with the help of FCI in cases of Ewing's sarcoma, neuroblatoma, and PNET. This additional positivity of FCI was due to the increased sensitivity of this technique.[86] A recent study by Leon et al.[94] evaluated the role of FCI in adult SRCTs. The study included four patients, three with EWS/PNET and one with neuroblastoma. FCI done on FNAC and the bone marrow aspirate demonstrated an abnormal population of cells that were positive for CD99 in the former and showed a phenotype of CD45- and CD16/CD56+ in the latter.
In view of the advantages of multicolor flow cytometric analysis which provides an opportunity to analyse a smaller number of cells, FCI holds a lot of promise in the evaluation of MSRCT.[95]
Conclusions
Ancillary diagnostic techniques of immunocytochemistry, flow cytometric immunophenotyping, and reverse-transcriptase polymerase chain reaction are extremely useful in cases when tumors are undifferentiated and distinction is not possible on morphological basis alone. These studies are extremely effective in establishing a definite diagnosis. This would enable the institution of appropriate therapeutic protocols, including neo-adjuvant chemotherapy in advanced malignancy. Most of the studies reported to date, have applied these techniques on surgically resected material or on biopsies that involve an invasive technique. However, FNAC is a minimally invasive technique yielding a smaller amount of material and requiring that the applicability of these newer techniques be proven.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Kocjan G. Diagnostic dilemas in FNAC cytology: small round cell tumors. In: Schroder G, editor. Fine needle aspiration cytology diagnostic principles and dilemmas. Berlin: Springer-Verlag; 2006. pp. 133–4. [Google Scholar]
- 2.Akhtar M, Iqbal MA, Mourad W, Ali MA. Fine-needle aspiration biopsy diagnosis of small round cell tumors of childhood: a comprehensive approach. Diagn Cytopathol. 1999;21:81–91. doi: 10.1002/(sici)1097-0339(199908)21:2<81::aid-dc2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 3.Akhtar M, Ali MA, Sabbah R, Bakry M, Nash JE. Fine-needle aspiration biopsy diagnosis of round cell malignant tumors of childhood: A combined light and electron microscopic approach. Cancer. 1985;55:1805–17. doi: 10.1002/1097-0142(19850415)55:8<1805::aid-cncr2820550828>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 4.McGahey BE, Moriarty AT, Nelson WA, Hull MT. Fine-needle aspiration biopsy of small round blue cell tumors of childhood. Cancer. 1992;69:1067–73. doi: 10.1002/1097-0142(19920215)69:4<1067::aid-cncr2820690439>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 5.Layfield LJ, Liu K, Dodge RK. Logistic regression analysis of small round cell neoplasms: a cytologic study. Diagn Cytopathol. 1999;20:271–7. doi: 10.1002/(sici)1097-0339(199905)20:5<271::aid-dc5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 6.Cohen MC, Pollano D, Tomarchio SA, Drut R. Cytologic characteristics of peripheral neuroectodermal tumors in fine needle aspiration smear: A retrospective study of three pediatric cases. Diagn Cytopathol. 1997;16:513–7. doi: 10.1002/(sici)1097-0339(199706)16:6<513::aid-dc8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 7.Das DK, Bhambhani S, Chachra KL, Murthy NS, Tripathi RP. Small round cell tumors of the abdomen and thorax: Role of fine needle aspiration cytologic feature in the diagnosis and differential diagnosis. Acta Cytol. 1997;41:1035–47. doi: 10.1159/000332785. [DOI] [PubMed] [Google Scholar]
- 8.Layfield LJ, Glasgow B, Ostrzega N, Reynolds CP. Fine-needle aspiration cytology and the diagnosis of neoplasms in the pediatric age group. Diagn Cytopathol. 1991;7:451–61. doi: 10.1002/dc.2840070504. [DOI] [PubMed] [Google Scholar]
- 9.Pilotti S, Rilke F, Alasio L, Garbagnati F. The role of fine needle aspiration in the assessment of renal masses. Acta Cytol. 1988;32:1–10. [PubMed] [Google Scholar]
- 10.Variend S. Small cell tumors in childhood: A review. J Pathol. 1985;145:25. doi: 10.1002/path.1711450102. [DOI] [PubMed] [Google Scholar]
- 11.Silverman JF, Berns LA, Holbrook CT, Neil JSA, Joshi W. Fine needle aspiration cytology of primitive neuroectodermal tumors: A report of these cases. Acta Cytol. 1992;36:541–50. [PubMed] [Google Scholar]
- 12.Roessner A, Jürgens H. Round cell tumors of bone. Pathol Res Pract. 1993;189:111–36. [PubMed] [Google Scholar]
- 13.Horowitz ME, Delaney TF, Malawer MM, Woo SY, Hicks MJ. Principles and practice of pediatric oncology. Philadelphia: Lippincott; 1997. Ewing's sarcoma of bone and soft tissue and peripheral primitive neuroectodermal tumor; pp. 831–63. [Google Scholar]
- 14.Sahu K, Pai RR, Khadilkar UN. Fine needle aspiration cytology of the Ewing's sarcoma family of tumors. Acta Cytol. 2000;44:332–6. doi: 10.1159/000328474. [DOI] [PubMed] [Google Scholar]
- 15.Park YK, Chi SG, Park HR, Yang MH, Unni KK. Detection of t(11; 22)(q24; q12) translocation of Ewing's sarcoma in paraffin embedded tissue by nested reverse transcription-polymerase chain reaction. J Korean Med Sci. 1998;13:395–9. doi: 10.3346/jkms.1998.13.4.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for Ewing's sarcoma and peripheral primitive neuroectodermal tumors: Evidence for a common histogenesis of Ewing's sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67:1886–93. doi: 10.1002/1097-0142(19910401)67:7<1886::aid-cncr2820670712>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 17.Fellinger EJ, Garin-Chesa P, Triche TJ, Huvos AG, Rettig WJ. Immunohistochemical analysis of Ewing's sarcoma cell surface antigen p30/32 MIC2. Am J Pathol. 1991;139:317–25. [PMC free article] [PubMed] [Google Scholar]
- 18.Dehner LP. Primitive neuroectodermal tumor and Ewing's sarcoma. Am J Surg Pathol. 1993;17:1–13. doi: 10.1097/00000478-199301000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Halliday BE, Slagel DD, Elsheikh TE, Silverman JF. Diagnostic utility of mic-2 immunocytomchemical staining in the differential diagnosis of small blue cell tumors. Diagn Cytopathol. 1998;19:410–6. doi: 10.1002/(sici)1097-0339(199812)19:6<410::aid-dc2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 20.Collins BT, Cramer HM, Frain BE, Davis MM. Fine needle aspiration biopsy of metastatic Ewing's sarcoma with MIC2 (CD99) immunocytomchemistry. Diagn Cytopathol. 1998;19:382–4. doi: 10.1002/(sici)1097-0339(199811)19:5<382::aid-dc15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 21.Llombart-Bosch A, Navarro S. Immunohistochemical detection of EWS and FLI1 proteins in Ewing's sarcoma and primitive neuroectodermal tumors: Comparative analysis with CD99 (MIC-2) expression. Appl Immunohistochem Mol Morphol. 2001;9:255–60. doi: 10.1097/00129039-200109000-00010. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki H, Honzumi M, Funada M, Tomiyama H. Metachronous bilateral adrenal neuroblastoma. Cancer. 1985;56:1490–2. doi: 10.1002/1097-0142(19850915)56:6<1490::aid-cncr2820560645>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 23.Akhtar M, Ali MA, Sabbah RS, Bakry M, Sackey K, Nash EJ. Aspiration cytology of neuroblastoma.Light and electron microscopic correlations. Cancer. 1986;57:797–803. doi: 10.1002/1097-0142(19860215)57:4<797::aid-cncr2820570419>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 24.Silverman JF, Dabbs DJ, Ganick DJ, Holbrook CT, Geisinger KR. Fine needle aspiration cytology of neuroblastoma, including peripheral neuroectodermal tumor, with immunocytochemical and ultrastructural confirmation. Acta Cytol. 1988;32:367–76. [PubMed] [Google Scholar]
- 25.Schmechel D, Marangos PJ, Brightman M. Neurone-specific enolase is a molecular marker for peripheral and central neuroendocrine cells. Nature. 1978;276:834–6. doi: 10.1038/276834a0. [DOI] [PubMed] [Google Scholar]
- 26.Osborn M, Dirk T, Käser H, Weber K, Altmannsberger M. Immunohistochemical localization of neurofilaments and neuron-specific enolase in 29 cases of neuroblastoma. Am J Pathol. 1986;122:433–42. [PMC free article] [PubMed] [Google Scholar]
- 27.Carter RL, McCarthy KP. Divergent differentiation in round-cell tumors of soft tissues: An interim appraisal. Histopathology. 1993;23:93–7. doi: 10.1111/j.1365-2559.1993.tb01191.x. [DOI] [PubMed] [Google Scholar]
- 28.Swerts K, Ambros PF, Brouzes C, Navarro JM, Gross N, Rampling D, et al. Standardization of the immunocytochemical detection of neuroblastoma cells in bone marrow. J Histochem Cytochem. 2005;53:1433–40. doi: 10.1369/jhc.5C6661.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winkler H, Fischer-Colbrie R. The chromogranins A and B: The first 25 years and future perspectives. Neuroscience. 1992;49:497–528. doi: 10.1016/0306-4522(92)90222-N. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattano LA, Jr, Moss TJ, Emerson SG. Sensitive detection of rare circulating neuroblastoma cells by the reverse transcriptase-polymerase chain reaction. Cancer Res. 1992;52:4701–5. [PubMed] [Google Scholar]
- 31.Cheung IY, Barber D, Cheung NK. Detection of microscopic neuroblastoma in marrow by histology, immunocytology and reverse transcription-PCR of multiple molecular markers. Clin Cancer Res. 1998;4:2801–5. [PubMed] [Google Scholar]
- 32.Gilbert J, Haber M, Bordow SB, Marshall GM, Norris MD. Use of tumor-specific gene expression for the differential diagnosis of neuroblastoma from other pediatric small round cell malignancies. Am J Pathol. 1999;155:17–21. doi: 10.1016/S0002-9440(10)65093-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito R, Asami S, Kagawa S, Motohashi S, Shichino H, Chin M, et al. Usefulness of tyrosine hydroxylase mRNA for diagnosis and detection of minimal residual disease in neuroblastoma. Biol Pharm Bull. 2004;27:315–8. doi: 10.1248/bpb.27.315. [DOI] [PubMed] [Google Scholar]
- 34.Miyajima Y, Horibe K, Fukuda M, Matsumoto K, Numata S, Mori H, et al. Sequential detection of tumor cells in the peripheral blood and bone marrow of patients with stage IV neuroblastoma by the reverse transcription-polymerase chain reaction for tyrosine hydroxylase mRNA. Cancer. 1996;77:1214–9. doi: 10.1002/(sici)1097-0142(19960315)77:6<1214::aid-cncr31>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 35.Naito H, Kuzumaki N, Uchino J, Kobayash R, Shikano T, Ishikawa Y, et al. Detection of tyrosine hydroxylase mRNA and minimal neuroblastoma cells by the reverse transcription polymerase chain reaction. Eur J Cancer. 1991;27:762–5. doi: 10.1016/0277-5379(91)90184-f. [DOI] [PubMed] [Google Scholar]
- 36.Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults: Report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002;26:1175–83. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 37.Rajwanshi A, Rao KL, Marwaha RK, Nijhawan VS, Gupta SK. Role of fine-needle aspiration cytology in childhood malignancies. Diagn Cytopathol. 1989;5:378–82. doi: 10.1002/dc.2840050407. [DOI] [PubMed] [Google Scholar]
- 38.Akhtar M, Ali MA, Bakry M, Hug M, Sackey K. Fine-needle aspiration biopsy diagnosis of rhabdomyosarcoma: Cytologic, histologic, and ultrastructural correlations. Diagn Cytopathol. 1992;8:465–74. doi: 10.1002/dc.2840080507. [DOI] [PubMed] [Google Scholar]
- 39.Brahmi U, Rajwanshi A, Joshi K, Ganguly NK, Vohra H, Gupta SK, et al. Role of immunocytochemistry and DNA flowcytometry in the fine-needle aspiration diagnosis of malignant small round-cell tumors. Diagn Cytopathol. 2001;24:233–9. doi: 10.1002/dc.1050. [DOI] [PubMed] [Google Scholar]
- 40.Das DK. Fine-needle aspiration (FNA) cytology diagnosis of small round cell tumors: value and limitations. Indian J Pathol Microbiol. 2004;47:309–18. [PubMed] [Google Scholar]
- 41.Pohar-Marinsek Z, Bracko M. Rhabdomyosarcoma. Cytomorphology, subtyping and differential diagnostic dilemmas. Acta Cytol. 2000;44:524–32. doi: 10.1159/000328524. [DOI] [PubMed] [Google Scholar]
- 42.Truong LD, Rangdaeng S, Cagle P, Ro JY, Hawkins H, Font RL. The diagnostic utility of desmin: A study of 584 cases and review of the literature. Am J Clin Pathol. 1990;93:305–14. doi: 10.1093/ajcp/93.3.305. [DOI] [PubMed] [Google Scholar]
- 43.Dias P, Parham DM, Shapiro DN, Webber BL, Houghton PJ. Myogenic regulatory protein (MyoD1) expression in childhood solid tumors: Diagnostic utility in rhabdomyosarcoma. Am J Pathol. 1990;137:1283–91. [PMC free article] [PubMed] [Google Scholar]
- 44.Wang NP, Marx J, McNutt MA, Rutledge JC, Gown AM. Expression of myogenic regulatory proteins (myogenin and MyoD1) in small blue round cell tumors of childhood. Am J Pathol. 1995;147:1799–810. [PMC free article] [PubMed] [Google Scholar]
- 45.Morotti RA, Nicol KK, Parham DM, Teot LA, Moore J, Hayes J, et al. An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children's Oncology Group experience. Am J Surg Pathol. 2006;30:962–8. doi: 10.1097/00000478-200608000-00005. [DOI] [PubMed] [Google Scholar]
- 46.Berkley C, Stanley MW, Wolpert J, Rainwater LM, Smith C. Adult Wilms’ tumor: Intraoperative cytology and ancillary studies performed in a case as an adjunct to the histologic diagnosis. Acta Cytol. 1990;34:79–83. [PubMed] [Google Scholar]
- 47.Dey P, Radhika S, Rajwanshi A, Rao KL, Khajuria A, Nijhawan R, et al. Aspiration cytology of Wilms’ tumor. Acta Cytol. 1993;37:477–82. [PubMed] [Google Scholar]
- 48.Hazarika D, Narasimhamurthy KN, Rao CR, Gopinath KS. Fine needle aspiration cytology of Wilms’ tumor: A study of 17 cases. Acta Cytol. 1994;38:355–60. [PubMed] [Google Scholar]
- 49.Ravindra S, Kini U. Cytomorphology and morphometry of small round-cell tumors in the region of the kidney. Diagn Cytopathol. 2005;32:211–6. doi: 10.1002/dc.20225. [DOI] [PubMed] [Google Scholar]
- 50.Radhika S, Bakshi A, Rajwanshi A, Nijhawan R, Das A, Kakkar N, et al. Cytopathology of uncommon malignant renal neoplasms in the pediatric age group. Diagn Cytopathol. 2005;32:281–6. doi: 10.1002/dc.20242. [DOI] [PubMed] [Google Scholar]
- 51.Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–20. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 52.Fisher C. Synovial sarcoma: Ultrastructural and immunohistochemical features of epithelial differentiation in monophasic and biphasic tumors. Hum Pathol. 1986;17:996–1008. doi: 10.1016/s0046-8177(86)80083-1. [DOI] [PubMed] [Google Scholar]
- 53.Ladanyi M. The emerging molecular genetics of sarcoma translocations. Diagn Mol Pathol. 1995;4:162–73. doi: 10.1097/00019606-199509000-00003. [DOI] [PubMed] [Google Scholar]
- 54.Tsuji S, Hisaoka M, Morimitsu Y, Hashimoto H, Shimajiri S, Komiya S, et al. Detection of SYT-SSX fusion transcripts in synovial sarcoma by reverse transcription-polymerase chain reaction using archival paraffin-embedded tissues. Am J Pathol. 1998;153:1807–12. doi: 10.1016/S0002-9440(10)65695-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akerman M, Ryd W, Skytting B. Scandinavian Sarcoma Group. Fine-needle aspiration of synovial sarcoma: Criteria for diagnosis: Retrospective reexamination of 37 cases, including ancillary diagnostics: A Scandinavian Sarcoma Group study. Diagn Cytopathol. 2003;28:232–8. doi: 10.1002/dc.10265. [DOI] [PubMed] [Google Scholar]
- 56.Ewing CA, Zakowski MF, Lin O. Monophasic synovial sarcoma: A cytologic spectrum. Diagn Cytopathol. 2004;30:19–23. doi: 10.1002/dc.10413. [DOI] [PubMed] [Google Scholar]
- 57.Silverman JF, Landreneau RJ, Sturgis CD, Raabm SS, Fox KR, Jasnosz KM, et al. Small-cell variant of synovial sarcoma. Diagn Cytopathol. 2000;23:118–23. doi: 10.1002/1097-0339(200008)23:2<118::aid-dc11>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 58.Akerman M, Domanski HA. The complex cytological features of synovial sarcoma in fine needle aspirates: An analysis of four illustrative cases. Cytopathology. 2007;18:234–40. doi: 10.1111/j.1365-2303.2007.00458.x. [DOI] [PubMed] [Google Scholar]
- 59.Sreekantaiah C, Ladanyi M, Rodriguez E, Chaganti RSK. Chromosomal aberrations in soft tissue tumors: Relevance to diagnosis, classification and molecular mechanisms. Am J Pathol. 1994;144:1121–34. [PMC free article] [PubMed] [Google Scholar]
- 60.dos Santos NR, de Bruijn DR, Balemans M, Janssen B, Gärtner F, Lopes JM, et al. Nuclear localization of SYT, SSX and the synovial sarcoma-associated SYT-SSX fusion proteins. Hum Mol Genet. 1997;6:1549–58. doi: 10.1093/hmg/6.9.1549. [DOI] [PubMed] [Google Scholar]
- 61.de Leeuw, Balemans M, OIde Weghuis D, Geurts van Kessel A. Identification of two alternative fusion genes, SYT-SSX1 and SYTSSX2, in t(X; 18)(p11.2; q11.2)-positive synovial sarcomas. Hum Mol Genet. 1995;6:1097–9. doi: 10.1093/hmg/4.6.1097. [DOI] [PubMed] [Google Scholar]
- 62.Janz M, De Leeuw B, Weghuis DO, Werner M, Nolte M, Geurts Van Kessel A, et al. Interphase cytogenetic analysis of distinct X chromosomal translocation breakpoints in synovial sarcoma. J Pathol. 1995;175:391–6. doi: 10.1002/path.1711750405. [DOI] [PubMed] [Google Scholar]
- 63.Kawai A, Woodruff J, Healey JH, Brennan MF, Antonescu CR, Ladanyi M. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med. 1998;338:153–60. doi: 10.1056/NEJM199801153380303. [DOI] [PubMed] [Google Scholar]
- 64.Crew AJ, Clark J, Fisher C, Gill S, Grimer R, Chand A. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel associated box in human synovial sarcoma. EMBO J. 1995;14:2333–40. doi: 10.1002/j.1460-2075.1995.tb07228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark J, Rocques PJ, Braun T, Bober E, Arnold HH, Fisher C, et al. Expression of members of the myf gene family in human rhabdomyosarcomas. Br J Cancer. 1991;64:1039–42. doi: 10.1038/bjc.1991.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hisaoka M, Hashimoto H, Iwamasa T, Ishikawa K, Aoki T. Primary synovial sarcoma of the lung: report of two cases confirmed by molecular detection of SYT-SSX fusion gene transcripts. Histopathology. 1999;34:205–10. doi: 10.1046/j.1365-2559.1999.00630.x. [DOI] [PubMed] [Google Scholar]
- 67.Aubrey MC, Bridge JA, Wickert R, Tazelaar HD. Primary monophasic synovial saromas of the pleura.Five cases confirmed by the presence of SYT-SSX fusion transcript. Am J Surg Pathol. 2001;25:776–81. doi: 10.1097/00000478-200106000-00009. [DOI] [PubMed] [Google Scholar]
- 68.Fritsch M, Epstein JI, Perlman EJ, Watts JC, Argani P. Molecularly confirmed primary prostatic synovial sarcoma. Hum Pathol. 2000;31:246–50. doi: 10.1016/s0046-8177(00)80228-2. [DOI] [PubMed] [Google Scholar]
- 69.Kashima T, Matsushita H, Kuroda M, Takeuchi H, Udagawa H, Ishida T, et al. Biphasic synovial sarcoma of the peritoneal cavity with t(X; 18) demonstrated by reverse transcriptase polymerase chain reaction. Pathol Int. 1997;47:637–41. doi: 10.1111/j.1440-1827.1997.tb04555.x. [DOI] [PubMed] [Google Scholar]
- 70.Inagaki H, Nagasaka T, Otsuka T, Sugiura E, Nakashima N, Eimoto T. Association of SYT-SSX fusion types with proliferative activity and prognosis in synovial sarcoma. Mod Pathol. 2000;13:482–8. doi: 10.1038/modpathol.3880083. [DOI] [PubMed] [Google Scholar]
- 71.Ladanyi M, Antonescu CR, Leung DH, Woodruff JM, Kawai A, Healey JH, et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi institutional retrospective study of 243 patients. Cancer Res. 2002;62:135–40. [PubMed] [Google Scholar]
- 72.Hill DA, O’Sullivan MJ, Zhu X, Vollmer RT, Humphrey PA, Dehner LP, et al. Practical application of molecular genetic testing as an aid to the surgical pathologic diagnosis of sarcomas: a prospective study. Am J Surg Pathol. 2002;26:965–77. doi: 10.1097/00000478-200208000-00001. [DOI] [PubMed] [Google Scholar]
- 73.Parkash V, Gerald WL, Parma A, Miettinen M, Rosai J. Desmoplastic small round cell tumor of the pleura. Am J Surg Pathol. 1995;19:659–65. doi: 10.1097/00000478-199506000-00006. [DOI] [PubMed] [Google Scholar]
- 74.Roganovich J, Bisogno G, Cecchetto G, D’Amore ES, Carli M. Paratesticular desmoplastic small round cell tumor: case report and review of the literature. J Surg Oncol. 1999;71:269–72. doi: 10.1002/(sici)1096-9098(199908)71:4<269::aid-jso13>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 75.Zaloudek C, Miller TR, Stern JL. Desmoplastic small cell tumor of the ovary: a unique polyphenotypic tumor with an unfavorable prognosis. Int J Gynecol Pathol. 1995;14:260–5. doi: 10.1097/00004347-199507000-00011. [DOI] [PubMed] [Google Scholar]
- 76.Gerald WL, Miller HK, Battifora H, Miettinen M, Silva EG, Rosai J. Intraabdominal desmoplastic small round cell tumor.Report of 19 cases of a distinctive type of high grade polyphenotypic malignancy affecting young individuals. Am J Surg Pathol. 1991;15:499–513. [PubMed] [Google Scholar]
- 77.Kretschmar CS, Colbach C, Bhan I, Crombleholme TM. Desmoplastic small cell tumor: A report of three cases and a review of the literature. J Pediatr Hematol Oncol. 1996;18:293–8. doi: 10.1097/00043426-199608000-00012. [DOI] [PubMed] [Google Scholar]
- 78.Crapanzano JP, Cardillo M, Lin O, Zakowski MF. Cytology of desmoplastic small round cell tumor. Cancer. 2002;96:21–31. doi: 10.1002/cncr.10316. [DOI] [PubMed] [Google Scholar]
- 79.Dave B, Shet T, Chinoy R. Desmoplastic round cell tumor of childhood: can cytology with immunocytochemistry serve as an alternative for tissue diagnosis? Diagn Cytopathol. 2005;32:330–5. doi: 10.1002/dc.20244. [DOI] [PubMed] [Google Scholar]
- 80.Presley AE, Kong CS, Rowe DM, Atkins KA. Cytology of desmoplastic small round-cell tumor: comparison of pre- and post-chemotherapy fine-needle aspiration biopsies. Cancer. 2007;111:41–6. doi: 10.1002/cncr.22421. [DOI] [PubMed] [Google Scholar]
- 81.Gerald WL, Ladanyi M, de Alava E, Rosai J. Desmoplastic small round-cell tumor: a recently recognized tumor type associated with a unique gene fusion. Adv Anatomic Pathol. 1995;2:341–5. [Google Scholar]
- 82.de Alava E, Ladanyi M, Rosai J, Gerald WL. Detection of chimeric transcripts in desmoplastic small round cell tumor and related developmental tumors by reverse transcriptase polymerase chain reaction.A specific diagnostic assay. Am J Pathol. 1995;147:1584–91. [PMC free article] [PubMed] [Google Scholar]
- 83.Argatoff LH, Connell JX, Mathers JA, Gilks CB, Sorensen PH. Detection of the EWS/WT1 gene fusion by reverse transcriptase-polymerase chain reaction in the diagnosis of intra-abdominal desmoplastic small round cell tumor. Am J Surg Pathol. 1996;20:406–12. doi: 10.1097/00000478-199604000-00002. [DOI] [PubMed] [Google Scholar]
- 84.Chiu LL, Koay ES, Chan NH, Salto-Tellez M. Sequence confirmation of the EWS-WT1 fusion gene transcript in the peritoneal effusion of a patient with desmoplastic small round cell tumor. Diagn Cytopathol. 2003;29:341–3. doi: 10.1002/dc.10397. [DOI] [PubMed] [Google Scholar]
- 85.Simsir A, Fetsch P, Stetler-Stevenson M, Abati A. Immunophenotypic analysis of non-Hodgkin's lymphomas in cytologic specimens: a correlative study of immunocytochemical and flow cytometric techniques. Diagn Cytopathol. 1999;20:278–84. doi: 10.1002/(sici)1097-0339(199905)20:5<278::aid-dc6>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 86.Brahmi U, Rajwanshi A, Joshi K, Dey P, Vohra H, Ganguly NK, et al. Flow cytometric immunophenotyping and comparison with immunocytochemistry in small round cell tumors. Anal Quant Cytol Histol. 2001;23:405–12. [PubMed] [Google Scholar]
- 87.Laane E, Tani E, Bjorklund, Elmberger G, Everaus H, Skoog L, et al. Flowcytometric immunophenotyping including Bcl-2 detection on fine needle aspirates in the diagnosis of reactive lymphadenopathy and non Hodgkin's lymphoma. Cytometry B Clin Cytom. 2005;64:34–42. doi: 10.1002/cyto.b.20043. [DOI] [PubMed] [Google Scholar]
- 88.Dunphy CH. Combining morphology and flow cytometric immunophenotyping to evaluate bone marrow specimens for B-cell malignant neoplasms. Am J Clin Pathol. 1998;109:625–30. doi: 10.1093/ajcp/109.5.625. [DOI] [PubMed] [Google Scholar]
- 89.Saleh H, Masood S. Value of ancillary studies in fine-needle aspiration biopsy. Diagn Cytopathol. 1995;13:310–5. doi: 10.1002/dc.2840130407. [DOI] [PubMed] [Google Scholar]
- 90.Dunphy CH, Dunphy FR, Visconti JL. Flowcytometric immunophenotyping of bone marrow core biopsies: report of 8 patients with previously undiagnosed hematologic malignancy and failed bone marrow aspiration. Arch Pathol Lab Med. 1999;123:206–12. doi: 10.5858/1999-123-0206-FCIOBM. [DOI] [PubMed] [Google Scholar]
- 91.Siebert JD, Weeks LM, List LW, Kugler JW, Knost JA, Fishkin PA, et al. Utility of flow cytometry immunophenotyping for the diagnosis and classification of lymphoma in community hospital clinical needle aspiration/biopsies. Arch Pathol Lab Med. 2000;124:1792–9. doi: 10.5858/2000-124-1792-UOFCIF. [DOI] [PubMed] [Google Scholar]
- 92.Dey P, Amir T, Al Jassar A, Al Shemmari S, Jogai S, Bhat M G, et al. Combined applications of fine needle aspiration cytology and flowcytometric immunophenotyping for diagnosis and classification of non Hodgkins lymphoma. Cytojournal. 2006;3:24. doi: 10.1186/1742-6413-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morse EE, Yamase HT, Greenberg BR, Sporn J, Harshaw SA, Kiraly TR, et al. The role of flow cytometry in the diagnosis of lymphoma: a critical analysis. Ann Clin Lab Sci. 1994;24:6–11. [PubMed] [Google Scholar]
- 94.Leon ME, Hou JS, Galindo LM, Garcia FU. Fine-needle aspiration of adult small round-cell tumors studied with flow cytometry. Diagn Cytopathol. 2004;31:147–54. doi: 10.1002/dc.20074. [DOI] [PubMed] [Google Scholar]
- 95.Sigstad E, Dong HP, Davidson B, Berner A, Tierens A, Risberg B. The role of flow cytometric immunophenotyping in improving the diagnostic accuracy in referred fine-needle aspiration specimens. Diagn Cytopathol. 2004;31:159–63. doi: 10.1002/dc.20108. [DOI] [PubMed] [Google Scholar]