

Abstract
Capping of mRNA occurs shortly after transcription initiation, preceding other mRNA processing events such as mRNA splicing and polyadenylation. To determine the mechanism of coupling between transcription and capping, we tested for a physical interaction between capping enzyme and the transcription machinery. Capping enzyme is not stably associated with basal transcription factors or the RNA polymerase II (Pol II) holoenzyme. However, capping enzyme can directly and specifically interact with the phosphorylated form of the RNA polymerase carboxy-terminal domain (CTD). This association occurs in the context of the transcription initiation complex and is blocked by the CTD–kinase inhibitor H8. Furthermore, conditional truncation mutants of the Pol II CTD are lethal when combined with a capping enzyme mutant. Our results provide in vitro and in vivo evidence that capping enzyme is recruited to the transcription complex via phosphorylation of the RNA polymerase CTD.
Keywords: mRNA capping enzyme, transcription initiation complex, RNA Pol II CTD
Transfer of information from DNA to protein is mediated by mRNA. In eukaryotes, mRNA is produced by RNA polymerase II (Pol II), but several modifications are required before the transcript can be used for protein synthesis. The first of these modifications is the addition of a reversed orientation 7-methyl-guanosine cap to the 5′ end of the RNA. Capping occurs soon after transcription initiation, around the time the transcript reaches a length of 25–30 nucleotides (Jove and Manley 1984; Rasmussen and Lis 1993)—before other modifications such as splicing of introns or addition of the poly(A) tail. The cap structure acts as a marker for Pol II-transcribed mRNA. The cap stimulates mRNA splicing in vitro (Konarska et al. 1984; Edery and Sonenberg 1985) and in vivo (Fresco and Buratowski 1996; Schwer and Shuman 1996) and is used for recognition of mRNA by the protein synthesis machinery (Sonenberg 1993).
The capping reaction occurs in three steps (for review, see Mizumoto and Kaziro 1987). The nascent transcript is synthesized with a 5′-triphosphate end, and the γ phosphate is removed in the first step. The second part of the reaction is transfer of the guanylyl group. Guanylyltransferase reacts with GTP to form a covalent enzyme–GMP intermediate. The GMP is then transferred onto the mRNA 5′-diphosphate end. In the final step, a methyl group is transferred from S-adenosylmethionine to the N7 position of the cap guanine. The triphosphatase and guanylyltransferase activities are physically associated, whereas the methyltransferase purifies separately. The yeast capping enzyme has two subunits: The 53-kD guanylyltransferase subunit is the product of the CEG1 gene (Itoh et al. 1987; Shibagaki et al. 1992), whereas the gene for the 80-kD triphosphatase subunit has not yet been cloned. In higher eukaryotes, both activities reside on a single protein (Mizumoto and Kaziro 1987). Cloning and characterization of the Caenorhabditis elegans capping enzyme reveals that it consists of a carboxy-terminal guanylyltransferase domain related to yeast Ceg1 and an amino-terminal triphosphatase domain that is related to the tyrosine phosphatase family (Takagi et al. 1997).
We sought to address the mechanism by which capping is restricted to Pol II-transcribed RNA. Because capping enzyme has no known RNA sequence requirements and can act upon RNA as short as dimers and trimers in vitro (for review, see Mizumoto and Kaziro 1987; T. Takagi, unpubl.), the most likely hypothesis is that the mRNA capping enzyme is functionally or physically associated with the RNA Pol II transcription complex. A series of experiments was performed to test this hypothesis. No stable interactions of capping enzyme were detected with basal transcription factors or the RNA Pol II holoenzyme. However, we found that the mRNA capping enzyme can be recruited to the transcription complex via an interaction with the phosphorylated form of the RNA Pol II carboxy-terminal domain (CTD). Because CTD phosphorylation occurs during or shortly after initiation of transcription (Cadena and Dahmus 1987; Laybourn and Dahmus 1990; Lu et al. 1991), our results suggest a simple but elegant mechanism for coupling mRNA capping with Pol II transcription initiation.
Results
Capping enzyme is not associated with the RNA Pol II holoenzyme or basal transcription factors
To test for an association of capping enzyme with the RNA Pol II transcription machinery, we performed coimmunoprecipitation assays with antibodies against basal transcription factors. Yeast whole cell extracts were precipitated with polyclonal antibodies against TATA-binding protein (TBP), TFIIB, TFIIE, or the TFG2 subunit of TFIIF. In no case was Ceg1 detected in the pellets (data not shown; also see below). Ceg1 also failed to coprecipitate with monoclonal antibody 8WG16 (Thompson et al. 1989), which recognizes the unphosphorylated CTD of RNA Pol II (data not shown; also see Fig. 3, below). Therefore, capping enzyme did not appear to be stably associated with free basal transcription factors. To test whether capping enzyme was associated with the RNA Pol II holoenzyme (for review, see Koleske and Young 1994), we tested a highly purified holoenzyme fraction (the gift of David Chao and Rick Young, MIT, Cambridge, MA) for the presence of Ceg1 by both quantitative immunoblotting and formation of the enzyme–GMP complex. Antibodies against SRB5 and TFIIB were used as positive controls, and recombinant proteins were used as quantitation standards. Only trace amounts of capping enzyme were detected (<5% the molar amount of SRB5 or TFIIB), indicating that capping enzyme is not a subunit of the holoenzyme (data not shown).
Figure 3.

Capping enzyme is recruited to the transcription complex by phosphorylation of the Pol II CTD. (A) Capping enzyme does not associate with unphosphorylated transcription complexes. Transcription complexes were assembled by incubating hexokinase-treated yeast whole cell extract with beads containing the CYC1 promoter (lane 2) or a control fragment without a promoter (lane 1). The immobilized templates were washed extensively and tested for the presence of various transcription factors by SDS-PAGE and immunoblotting. Rpb1 is the largest subunit of Pol II, Tfb1 is a subunit of TFIIH, Tfa1 is a subunit of TFIIE, Sua7 is TFIIB, and TBP is a TBP subunit of TFIID. Before gel electrophoresis, the beads were incubated with [α-32P]GTP and the capping enzyme–GMP intermediate (Ceg1–*pG) was detected by autoradiography. (B) Capping enzyme associates with transcription complexes containing a phosphorylated Pol II CTD. Immobilized transcription complexes were assembled in hexokinase-treated extracts. (Lane 1) A reaction with a promoter-less DNA template; (lanes 2–4) reaction with the CYC1 promoter fragment. (Lanes 1,3,4) The complexes were incubated with [γ-32P]ATP to allow CTD phosphorylation, although the reaction in lane 4 also contained the CTD kinase inhibitor H8. All four reactions received creatine phosphate (CP) and creatine phosphate kinase (CPK) to counteract the hexokinase. The immobilized templates were precipitated and washed, and the pellet was tested for the presence of the Pol II largest subunit (Rpb1), TFIIH (Tfb1), and TFIID (TBP) by immunoblotting. In addition, capping enzyme was labeled by adding [α-32P]GTP to form the capping enzyme–GMP complex (Ceg1–*pG). Phosphorylation of the Pol II CTD (Rpb1–*Pi) and the capping enzyme–GMP intermediate (Ceg1–*pG) was monitored by autoradiography of the blot (bottom panel).
Capping enzyme associates with the phosphorylated CTD of RNA Pol II
The possibility of an association between capping enzyme and Pol II was examined further because the monoclonal antibody against the Pol II CTD is known to dissociate CTD-bound proteins (Kim et al. 1994) and may have also disrupted interactions with capping enzyme. Also, the mAb 8WG16 only recognizes the unphosphorylated CTD (Thompson et al. 1989), and we wanted to examine both the phosphorylated and unphosphorylated forms of Pol II. Partially purified capping enzyme and Pol II were incubated together and the mixture precipitated with anti-Ceg1 antibody. In the absence of phosphorylation, no association between capping enzyme and Pol II was detected (data not shown; see Fig. 3, below).
RNA polymerase was labeled with [γ-32P]ATP and TFIIH. The TFIIH kinase phosphorylates the CTD of RNA Pol II (Feaver et al. 1991; Lu et al. 1992; Serizawa et al. 1993). When mixed with capping enzyme, phosphorylated RNA Pol II could be efficiently coimmunoprecipitated by anti-Ceg1 antibodies but not preimmune serum (Fig. 1, lanes 1,2). To determine whether this interaction was mediated by the CTD of Pol II, binding was competed with either glutathione S-transferase (GST, lane 3), or a GST–CTD fusion protein that was either untreated (GST–CTD, lane 4) or phosphorylated with TFIIH [GST–CTD(P), lane 5]. Only the phosphorylated CTD fusion protein competed for binding, suggesting that interaction of the capping enzyme with RNA Pol II is mediated by the phosphorylated CTD.
Figure 1.
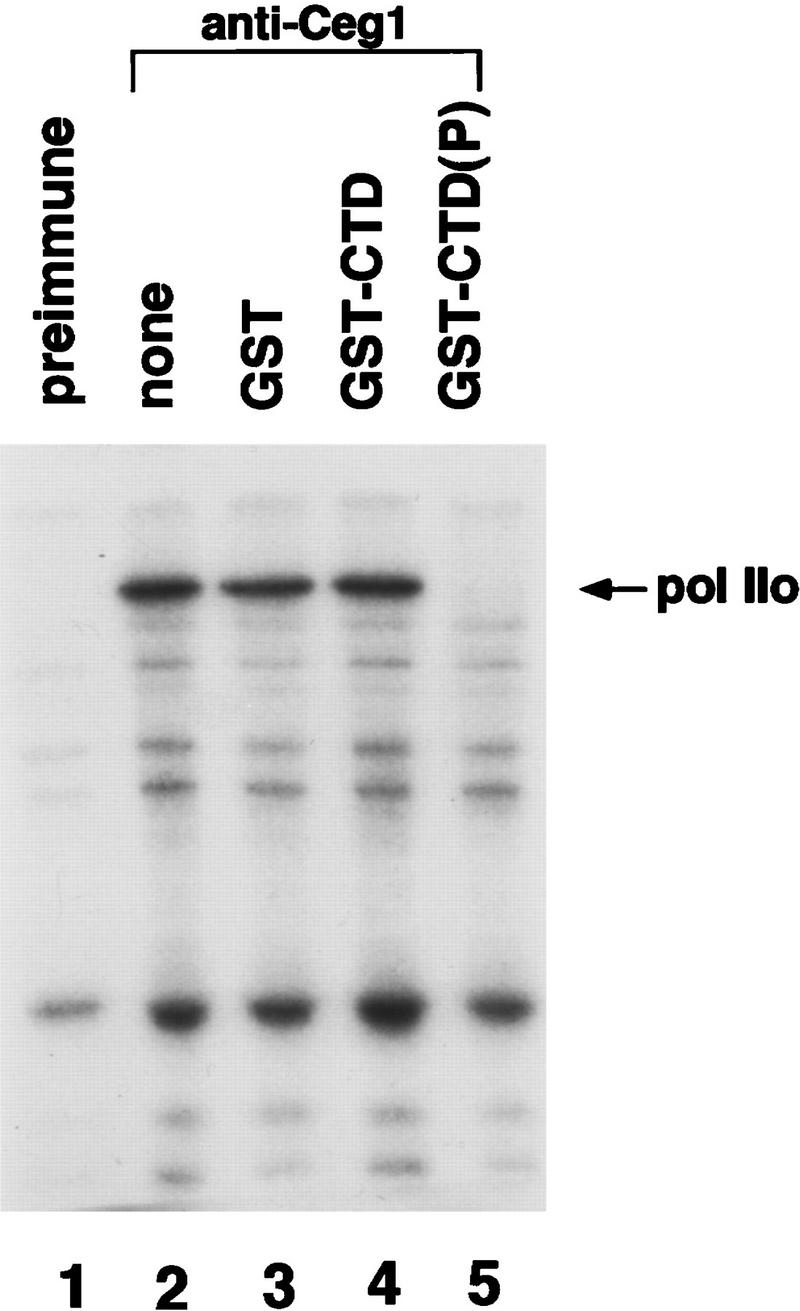
Capping enzyme binds to phosphorylated RNA Pol II. RNA Pol II was phosphorylated with TFIIH and [γ-32P]ATP. Capping enzyme was added and the mixture was precipitated with preimmune serum (lane 1) or anti-Ceg1 antibodies (lanes 2–5). Precipitated proteins were separated by SDS-PAGE and the labeled largest subunit of polymerase (Pol IIo) was visualized by autoradiography. As competitors for binding to the capping enzyme, parallel reactions contained GST (lane 3), GST–CTD (lane 4), or phosphorylated GST–CTD (lane 5).
In the previous experiment, the capping enzyme bound to phosphorylated Pol II in the context of the entire polymerase and in the presence of TFIIH. To determine whether the phosphorylated CTD was sufficient for capping enzyme binding, the GST–CTD protein was phosphorylated by TFIIH and ATP and then repurified on glutathione–agarose. In parallel, unphosphorylated GST–CTD beads and GST beads were prepared. Upon mixing with capping enzyme, only the phosphorylated GST–CTD beads bound capping enzyme (Fig. 2, lanes 4–6), indicating that capping enzyme binds directly to the phosphorylated CTD. The interaction requires the presence of both capping enzyme subunits, as the recombinant guanylyltransferase subunit alone was not precipitated with the phosphorylated GST–CTD beads (lane 3). Because the gene for the triphosphatase has not yet been cloned, we cannot conclude whether CTD directly contacts the triphosphatase or some other protein tightly associated with the capping enzyme.
Figure 2.

Capping enzyme binds directly to the phosphorylated CTD. Recombinant guanylyltransferase (his7–CEG1) or purified capping enzyme was incubated with glutathione–agarose beads carrying GST, GST–CTD, or phosphorylated GST–CTD protein. The beads were pelleted and washed extensively. Capping enzyme in the pellet and supernatant (one-tenth of the total) was detected by formation of the enzyme–GMP complex followed by SDS-PAGE and autoradiography.
Capping enzyme is recruited to the RNA Pol II transcription complex by CTD phosphorylation
We next tested whether the capping enzyme could associate with transcription complexes assembled at a promoter using an immobilized template assay (adapted from a protocol provided by J. Reese, University of Massachusetts, Amherst). Template DNA carrying the CYC1 promoter upstream of a G-less cassette was linked to magnetic beads, and transcription complexes were assembled in whole cell extracts that had been treated with hexokinase to deplete endogenous nucleotide triphosphates (Fig. 3). As a negative control for background binding to the beads, parallel binding reactions were carried out with beads linked to a DNA fragment carrying the G-less cassette but lacking promoter sequences. The beads were washed and tested for the presence of capping enzyme by formation of the Ceg1–GMP complex and for the presence of several transcription factors by immunoblotting. Binding of basal transcription factors was dependent on the presence of a functional promoter (Fig. 3A). RNA Pol II showed some binding to the TATA-less beads (presumably because of nonspecific interactions with the DNA), but levels were significantly increased by the presence of the promoter. In contrast to the transcription factors, levels of Ceg1 were very low for beads with or without a promoter (Fig. 3A, bottom). Therefore, it appears that capping enzyme does not associate with a fully assembled transcription complex in the absence of CTD phosphorylation.
To determine the effect of CTD phosphorylation, transcription complexes were assembled in NTP-depleted extracts. The transcription complex was then phosphorylated on the Pol II CTD by the addition of [γ-32P]ATP, which is a substrate for the TFIIH-associated CTD kinase. All of the reactions also received creatine phosphate and creatine phosphate kinase to counteract the hexokinase. The immobilized templates were then washed, precipitated, and assayed for transcription factors and capping enzyme. In the presence of ATP, much higher levels of capping enzyme were coprecipitated with the transcription complex beads (Fig. 3B, cf. lanes 2 and 3). This association correlated with the presence of the phosphorylated CTD (visualized by autoradiography). A small amount association of capping enzyme in the absence of added ATP was observed (lane 2), presumably because of regeneration of endogenous ATP by the creatine phosphate kinase (cf. lane 2 with A). ATP did not produce binding of capping enzyme to control beads carrying a promoter-less DNA fragment, demonstrating that association of capping enzyme was dependent on transcription complex formation (lane 1). To further demonstrate the correlation between CTD phosphorylation and capping enzyme association, the CTD kinase inhibitor H8 was added to a reaction identical to that in lane 3. H8 effectively inhibited both CTD phosphorylation and association of capping enzyme.
Genetic interaction between capping enzyme and the CTD
To establish an in vivo interaction between the capping enzyme and Pol II CTD, we tested whether a yeast strain carrying mutations in both the CTD and capping enzyme would exhibit phenotypes not associated with either single mutation. A temperature-sensitive allele of the capping enzyme guanylyltransferase gene CEG1 (Fresco and Buratowski 1996) was crossed into a yeast strain carrying a deletion of the chromosomal copy of the gene encoding the largest subunit of Pol II (RPB1). A wild-type copy of RPB1 on a URA3-marked plasmid supports viability (Nonet and Young 1989). The wild-type polymerase CTD has 27 repeats of the heptapeptide sequence PTSPSYS. Using the technique of plasmid shuffling (Guthrie and Fink 1991), various RPB1 alleles were introduced into the strain and loss of the RPB1/URA3 plasmid was selected using 5-fluoro-orotic acid (5-FOA). Introduction of a wild-type RPB1/LEU2 plasmid allows loss of the URA3 plasmid and growth on 5-FOA-containing media (Fig. 4). This complementation occurs in both a CEG1 and ceg1-250 background. In a CEG1+ background, the partial CTD truncation alleles rpb1Δ101, rpb1Δ103, and rpb1Δ104 (112/7, 105/7, and 113/7 repeats, respectively; Nonet and Young 1989) also support growth on 5-FOA, although these alleles exhibit slightly slower growth and are sensitive to high or low temperature (Nonet and Young 1989). When these same alleles were tested in a ceg1-250 background at 26°C or 30°C (permissive temperature for the individual capping enzyme or CTD mutants), they were unable to support growth on 5-FOA. Therefore, the capping enzyme mutation confers dependence on the full-length Pol II CTD. This phenotype was specific for mutations in the CTD, as three other temperature-sensitive alleles of RPB1 with mutations outside the CTD (rpb1-15, rpb1-18, and rpb1-19; see Scafe et al. 1990) were able to support viability in the ceg1-250 background (data not shown). The “synthetic lethal” phenotype of the capping enzyme/CTD double mutant demonstrates a genetic link between mRNA capping and the CTD of RNA Pol II.
Figure 4.

Genetic interaction between capping enzyme and the Pol II CTD. Plasmid shuffling was used to introduce the wild-type (RPB1) or CTD truncation alleles (rpb1Δ101, rpb1Δ103, rpb1Δ104) of the RNA polymerase large subunit into a wild-type (strain N418, CEG1+) or mutant (strain YSB516, ceg1-250) capping enzyme background. Transformants were selected using the LEU2 marker on the plasmids, and the ability of the introduced alleles to support viability was tested by growth on 5-FOA, which selects against the original RPB1/URA3 plasmid. The plates were photographed after 2 days of incubation at 30°C.
Discussion
How are mRNA-specific modifications such as capping, splicing, and polyadenylation targeted to transcripts produced by RNA Pol II? Several mechanisms appear to contribute to the specificity. For mRNA splicing and polyadenylation, targeting is in part accomplished by having the splicing and polyadenylation machinery recognize specific RNA sequences in the transcript. Spliceosomes can be assembled in vitro on a synthetic RNA as long as the transcript contains the appropriate splice junctions. Similarly, polyadenylation can be uncoupled from RNA Pol II transcription in vitro as long as the synthetic RNA contains the appropriate sequences.
In addition to the recognition of specific RNA sequences, both in vitro (Konarska et al. 1984; Edery and Sonenberg 1985) and in vivo (Fresco and Buratowski 1996; Schwer and Shuman 1996), experiments suggest that the cap structure itself acts as a marker to help the splicing machinery recognize RNA produced by RNA Pol II. This recognition is mediated by the cap binding complex (CBC), which is associated with the splicing machinery (Izaurralde et al. 1994; Colot et al. 1996). The cap is apparently not required for normal polyadenylation in yeast (Fresco and Buratowski 1996), but some in vitro experiments suggest that the cap and/or CBC may stimulate polyadenylation in higher eukaryotes (Cooke and Alwine 1996; Flaherty et al. 1997).
Unlike the splicing and polyadenylation enzymes, purified capping enzyme can use many RNAs as a substrate, including di- and trinucleotides (for review, see Mizumoto and Kaziro 1987; T. Takagi, unpubl.). Therefore, the targeting of the cap to RNA Pol II transcripts cannot be mediated by a specific RNA sequence. Rather, our results indicate that the capping enzyme is physically linked to the RNA Pol II transcription initiation complex. Binding of the capping enzyme is dependent on phosphorylation of the CTD, an event closely linked to the initiation of transcription (Cadena and Dahmus 1987; Laybourn and Dahmus 1990; Lu et al. 1991; Serizawa et al. 1993). However, as our experiments were done in the absence of transcription, the association of the capping enzyme apparently does not require the presence of the RNA transcript. In support of the idea that recruitment of capping enzyme to the transcription complex is necessary for proper gene expression, we observe synthetic lethality between mutations in capping enzyme and the Pol II CTD. Furthermore, McCracken et al. (1997b) have shown that mRNA is inefficiently capped in vivo when it is synthesized by a polymerase lacking the CTD (D. Bentley, pers. comm.).
A similar mechanism has been proposed recently to help couple transcription by RNA Pol II with splicing and polyadenylation (Greenleaf 1993; McCracken et al. 1997a; Steinmetz 1997). A deletion of the RNA Pol II CTD in vivo results in accumulation of unspliced pre-mRNA and loss of proper 3′-end processing (McCracken et al. 1997a). Furthermore, overexpression of a CTD fusion protein interferes with mRNA splicing in vivo (Du and Warren 1997), and antibodies against the CTD inhibit in vitro splicing (Yuryev et al. 1996). Microscopy experiments also reveal a colocalization of splicing factors with sites of transcription by phosphorylated polymerase (Weeks et al. 1993; Neugebauer and Roth 1997).
These effects may be attributable to a direct interaction between the CTD and RNA processing factors. Splicing factors and phosphorylated Pol II can be coimmunoprecipitated from extracts (Mortillaro et al. 1996; Vincent et al. 1996; Kim et al. 1997), and polyadenylation factors can be copurified with an RNA Pol II-containing complex (Dantonel et al. 1997; McCracken et al. 1997a). Furthermore, in vitro associations with the CTD have been demonstrated for some polyadenylation factors (McCracken et al. 1997a) and some serine/arginine-rich putative splicing factors (Yuryev et al. 1996). However, these CTD interactions differ from that with capping enzyme, as they are not dependent on CTD phosphorylation.
With the finding that capping enzyme is recruited to the transcription complex by phosphorylation of the CTD, earlier results from CTD deletion or interference experiments (Yuryev et al. 1996; Du and Warren 1997; McCracken et al. 1997a) should be interpreted carefully. As the CTD mediates association of capping enzyme (this paper), and the cap is in turn required for efficient splicing (Konarska et al. 1984; Edery and Sonenberg 1985; Fresco and Buratowski 1996; Schwer and Shuman 1996), it becomes necessary to consider whether the effects of CTD mutations on splicing are, at least in part, mediated indirectly by the capping process. Loss of capping enzyme in yeast does not appear to affect polyadenylation (Fresco and Buratowski 1996), but there may be a connection between the cap and polyadenylation in mammalian cells (Cooke and Alwine 1996; Flaherty et al. 1997).
Whether by a direct or indirect mechanism, it is clear that the Pol II CTD acts as a platform for coupling RNA processing to transcription. Before initiation, the CTD is associated with holoenzyme proteins involved in initiation of transcription (for review, see Koleske and Young 1994). Concurrent with initiation and escape into elongation, the CTD becomes phosphorylated. Presumably the initiation factors are removed at this point and the capping enzyme and other RNA processing enzymes can associate with the polymerase in a manner that allows them to modify the transcript while it is being synthesized.
Materials and methods
RNA Pol II purification
Dry yeast (Fleischmann’s Yeast, Oakland, CA) was used for purification of Saccharomyces cerevisiae RNA Pol II. Cells were disrupted, extracted, and clarified by ultracentrifugation. The clear supernatant was chromatographed over columns of Bio-Rex 70 (Bio-Rad), DEAE–Sephacel (Pharmacia), and hydroxyapatite (Macro-Prep Ceramic Type I, Bio-Rad) as described (Sayre et al. 1992). The hydroxyapatite fractions containing RNA Pol II were applied to a Mono Q (HR 5/5) column (Pharmacia), which was pre-equilibrated in buffer A [20 mm HEPES–KOH at pH 7.6, 1 mm EDTA, 1 mm DTT, 20% (vol/vol) glycerol, and protease inhibitors mixture]. The column was washed and eluted with a 15-ml gradient of 0.5–1.2 m potassium acetate in buffer A. The fractions containing RNA polymerase (eluting at ∼1 m potassium acetate) were pooled and dialyzed against the storage buffer [20 mm HEPES–KOH at pH 7.6, 0.1 m potassium acetate, 1 mm EDTA, 1 mm DTT, 50% (vol/vol) glycerol]. Pol II was monitored by immunoblotting with mAb 8WG16 (Thompson et al. 1989).
TFIIH purification
YSB471 (MATα, ura3-52, leu2-3,112, his3Δ200, tfb1Δ::LEU2) strain harboring plasmid YCp50/TFB1-6HIS (Svejstrup et al. 1994; a gift from R.D. Kornberg, Stanford University, CA) produces TFIIH with a histidine tag on the Tfb1 subunit. This strain was grown in YPD medium (2% yeast extract, 1% Bacto-Peptone, 2% glucose). Extract preparation and column chromatography over Bio-Rex 70, DEAE–Sephacel, and hydroxyapatite were performed as described above for RNA Pol II preparation. Hydroxyapatite column fractions that contained (His)6-tagged TFIIH were pooled, dialyzed against buffer A, and loaded onto a Mono S (HR 5/5) column (Pharmacia), which was developed with a 30-ml gradient of 0–0.5 m KOAc in buffer A. The TFIIH fractions were pooled and dialyzed overnight against buffer B [20 mm Tris-acetate at pH 7.3, 5 mm β-mercaptoethanol, 250 mm KOAc, protease inhibitors, 0.01% NP-40, 20% (vol/vol) glycerol]. The dialysate was mixed with 2 ml of Ni2+–NTA–agarose (Qiagen) and then centrifuged to collect the matrix. The matrix was washed sequentially with 15 ml of buffer B with 5, 10, 20, and 30 mm imidazole. Then TFIIH was eluted with 15 ml of buffer B containing 100 mm imidazole. The active fractions were pooled and dialyzed against the storage buffer. Proteins were monitored by either CTD kinase assay or immunoblot assay using anti-Tfb1 antibody (a gift from Drs. W.J. Feaver and R.D. Kornberg).
Capping enzyme purification
S. cerevisiae capping enzyme was partially purified as follows. Crude extracts (100,000g supernatant fraction) were prepared as described (Sayre et al. 1992) except for a lysis buffer, which was 20 mm Tris-acetate (pH 7.9), 1 mm EDTA, 10 mm MgSO4, 1 mm DTT, 20% (vol/vol) glycerol, 1 mm PMSF, 1 μg/ml of pepstatin A, and 0.3 m ammonium sulfate (7% saturation). Lysate was precipitated with ammonium sulfate to 50% saturation. The pellet was resuspended in and dialyzed against buffer A containing 100 mm KOAc and then loaded onto a Bio-Rex 70 column. Capping enzyme activity was eluted with 300 mm KOAc in buffer A, concentrated with 80% ammonium sulfate, and resuspended in and dialyzed against buffer C [20 mm HEPES–KOH (pH 7.9), 1 mm EDTA, 20% (vol/vol) glycerol, 1 mm DTT, 1 mm PMSF, 1 μg/ml of pepstatin A] containing 50 mm KOAc. The sample was then loaded onto a column of DEAE–Sephacel. After washing the column with buffer C containing 150 mm KOAc, bound protein was eluted with buffer C containing 450 mm KOAc. The fraction was concentrated with ammonium sulfate and chromatographed on a heparin–Sepharose CL-6B (Pharmacia) column with a linear gradient of KOAc (50–400 mm) in buffer A. Activity was assayed by formation of enzyme–GMP complex as described (Fresco and Buratowski 1994). The presence of two bands in the enzyme–GMP assay indicates that some proteolysis (which does not appear to affect activity) of the Ceg1 subunit has occurred as described previously (Itoh et al. 1987). Approximately 200-fold purification was obtained with a recovery of 20% starting from the crude extracts.
Recombinant protein purification
Recombinant GAL4–VP16 fusion protein was produced as follows: Escherichia coli strain BL21(DE3) was transformed with plasmid pRJRGAL4–VP16 (a gift from M. Ptashne, Harvard University, Cambridge, MA), which expresses polyhistidine-tagged amino-terminal 95 amino acids of GAL4 protein fused with VP16. Strains were grown at 30°C to an OD600 of 0.5 and induced by the addition of 0.05 mm IPTG. Cells were further cultured for 3 hr at 30°C and harvested. Lysates were prepared by sonication in buffer D [20 mm HEPES–KOH (pH 7.5), 10 μm ZnCl2, 5% (vol/vol) glycerol, 300 mm KCl, 0.1% NP-40, 1 mm PMSF], and soluble extracts were incubated in batch with Ni2+–NTA–agarose resin overnight on a rotator at 4°C. The resin was washed extensively with buffer E [20 mm HEPES–KOH (pH 7.5), 10 μm ZnCl2, 5% (vol/vol) glycerol, 30 mm KCl, 1 mm PMSF] containing 15 mm imidazole. Bound proteins were eluted with 600 mm imidazole in buffer E. Fractions were collected and loaded onto a Mono Q (HR 5/5) column, which was developed with a linear gradient of 0.1–1.0 m NaCl in buffer A. Proteins were monitored by SDS-PAGE and Coomassie blue staining.
GST–CTD and GST were produced in E. coli and were kindly supplied by N.H. Kuldell (Harvard Medical School, Boston, MA). Polyhistidine-tagged guanylyltransferase subunit of S. cerevisiae (his7–Ceg1) was expressed and purified as described (Fresco and Buratowski 1994).
Protein interaction experiments
Recombinant GST–CTD fusion protein was phosphorylated with TFIIH and ATP as described (Kim et al. 1994). GST–CTD (500 ng, either prephosphorylated or nonphosphorylated) or GST was immobilized on a 20-μl bed volume of glutathione–agarose beads (Sigma). The beads were incubated for 1 hr at room temperature with his7–Ceg1 or purified native capping enzyme in a final volume of 200 μl in buffer A containing 100 mm potassium acetate, 0.03% NP-40, and 600 μg/ml of BSA. The beads were washed five times with buffer A plus 100 mm potassium acetate and 0.03% NP-40 (1 ml each time) and resuspended in 20 μl of an enzyme–GMP complex assay buffer (20 mm HEPES–KOH at pH 7.5, 5 mm magnesium acetate, 5 mm DTT, 5% glycerol). Then 3 μm [α-32P]GTP (30,000–60,000 cpm/pmole; NEN/DuPont) was added, and the reaction mixture was incubated for 20 min at 30°C. The labeled guanylyltransferase–GMP complex was resolved by SDS-PAGE and visualized by autoradiography.
Immunoprecipitation with anti-Ceg1 antibody (Fresco and Buratowski 1996) was performed as follows. Pol II (450 ng) was phosphorylated with TFIIH and [γ-32P]ATP and then incubated for 1 hr at room temperature with capping enzyme in buffer A containing 100 mm potassium acetate, 0.03% NP-40, and 600 μg/ml of BSA for 1 hr. The mixture was precipitated with either preimmune serum or anti-Ceg1 serum bound to protein A–Sepharose CL-4B beads (Sigma). For the competition assay, 500 ng of each competitor protein, GST, GST–CTD, or phosphorylated GST–CTD was added to the binding reaction mixture. To prevent further phosphorylation of GST–CTD, kinase inhibitor H8 (Sigma) was added to 2 mm final concentration in all reactions. The precipitate was washed five times (1 ml each time) and resuspended in loading buffer for SDS-PAGE. The labeled Rpb1 protein was resolved by SDS-PAGE and visualized by autoradiography.
Immobilized template assay
The CYC1 promoter-containing plasmid pGAL4CG− was digested with HindIII and the single-stranded overhang filled in using Klenow fragment of DNA polymerase I (New England Biolabs). The DNA was then digested with AflIII and end-labeled with biotin-14–dATP (GIBCO/BRL). As a promoter-less control, SmaI–AflIII fragment of p(C2AT)19 was biotinylated by end-filling with Klenow at the AflIII site. The two fragments contain identical 400-nucleotide G-less cassettes, and the pGAL4CG− additionally contains ∼200 nucleotides carrying one GAL4-binding site and the CYC1 basal promoter region. The biotinylated DNA fragments from pGAL4CG− or p(C2AT)19 were separated from vector DNA by agarose gel electrophoresis, and electroeluted from gel slices. The purified DNA fragments were bound to Streptavidin-linked magnetic beads (Dynabeads M280 Streptavidin, Dynal, Inc.) in buffer F (2 m NaCl, 10 mm Tris-HCl at pH 7.4, 0.01% NP-40). The DNA bound beads were washed several times in buffer F and subsequently in TE buffer (10 mm Tris-HCl at pH 7.9, 1 mm EDTA). Finally, the beads were preincubated with 50 μg/ml of BSA for 30 min and washed twice with transcription reaction buffer (25 mm HEPES–KOH at pH 7.6, 10 mm magnesium acetate, 5 mm EGTA, 2.5 mm DTT, 100 mm potassium acetate, 10% glycerol).
Whole cell extracts were prepared from YSB143 (MATa, ura3-52, leu2-3,112, his3Δ200, sua7Δ::LEU2, [pRS313-SUA7]) as described in Woontner et al. (1991). Transcription initiation complexes were assembled on immobilized template DNA by incubation of 120 μg of whole cell extract and 250 ng of GAL4–VP16 with 3 μl of beads in 60 μl of transcription reaction buffer. Two units of hexokinase and 2 mm glucose were added to the mixture during the assembly reaction to deplete endogenous NTPs. After 40 min, 5 μCi of [γ-32P]ATP, phosphocreatine, and creatine kinase (Sigma) were added to label the CTD of the RNA Pol II largest subunit. To inhibit phosphorylation of the CTD, kinase inhibitor H8 (Sigma) was added to a final concentration of 2 mm, where indicated. After 30 min of labeling, 1 μm unlabeled dATP was added to complete phosphorylation of the CTD and incubation continued for another 30 min. The transcription complexes were magnetically purified and thoroughly washed with simple transcription buffer containing 0.003% NP-40. Beads were resuspended in the enzyme–GMP complex assay buffer with 3 μm [α-32P]GTP, and incubated at 30°C to label the Ceg1 protein. The reaction was stopped after 30 min by addition of loading buffer for SDS-PAGE and electrophoresed on a 4%–20% gradient polyacrylamide gel. The proteins were analyzed by immunoblotting with polyclonal antibodies against TFIIB (anti-Sua7), TFIIE (anti-Tfa1), TFIID, (anti-TBP), TFIIH (anti-Tfb1, provided by W.J. Feaver and R.D. Kornberg), and mAb 8WG16 against the unphosphorylated Pol II CTD (Thompson et al. 1989). Autoradiography was used to detect phosphorylated Pol II and capping enzyme.
Yeast strains
(PY469) MATa, ura3, leu2, trp1, his3, ade2, ade3, can1, CEG1, RPB1; (N418) MATa, ura3, leu2, TRP1, his3, CEG1, rpb1Δ187::HIS3, [pRP112]; (YSB516) MATα, ura3, leu2, trp1, his3, ceg1-250, rpb1Δ187::HIS3, [pRP112]; (YSB517) MATα, ura3, leu2, trp1, his3, ade2, ade3, can1, ceg1-250, RPB1.
The mating type of PY469 (gift of D. Pellman, Dana-Farber Cancer Institute, Boston, MA) was switched and the resulting strain was transformed with the BamHI-digested integration construct pRS306–ceg1-250, and Ura+ transformants were selected. This resulted in a strain with wild-type and mutant CEG1 alleles flanking URA3. A single Ura+ colony was grown in YEPD medium to allow loss of the URA3 marker by recombination, and a Ura− ts− colony was identified and designated YSB517. The temperature-sensitive phenotype was complemented upon transformation with pRS315–CEG1 but not pRS315. N418 (Nonet and Young 1989) was mated with YSB517, the diploid was sporulated, and tetrads were dissected. YSB516 was identified as a Ura+ His+ ts− spore; the temperature-sensitive phenotype could be complemented upon transformation with pRS315–CEG1 but not pRS315.
Plasmids were introduced into yeast using modified lithium acetate transformation protocol (Guthrie and Fink 1991). Media preparation, plasmid shuffling, and all other yeast manipulations were performed by standard methods as described (Guthrie and Fink 1991). 5-FOA was purchased from PCR Incorporated.
Plasmids
(pRP112) CEN/ARS, URA3, RPB1 (27 PTSPSYS heptapeptide repeats); (pRP114) CEN/ARS, LEU2, RPB1 (27 heptapeptide repeats); (pRP1-101) CEN/ARS, LEU2, rpb1Δ101 (112/7 heptapeptide repeats); (pRP1-103) CEN/ARS, LEU2, rpb1Δ103 (105/7 heptapeptide repeats); (pRP1-104) CEN/ARS, LEU2, rpb1Δ104 (113/7 heptapeptide repeats); (pRS306–ceg1-250) URA3, ceg1-250; (pRS315–CEG1) CEN/ARS, LEU2, CEG1.
pRP plasmids (Nonet and Young 1989) and pRS315–CEG1 (Fresco and Buratowski 1996) have been described previously. pRS306–ceg1-250 was created by cloning a 1.7-kb SpeI–SalI fragment from pRS315–ceg1-250 bearing the ceg1-250 allele (Fresco and Buratowski 1996) into SpeI–SalI-digested pRS306. Plasmid cloning and bacterial transformation were performed using standard methods.
Acknowledgments
We thank Dave Chao and Rick Young for Pol II holoenzyme fractions, anti-SRB5 antibody, and RPB1 strains and plasmids; Joe Reese for help with the immobilized template protocol; John Feaver and Roger Kornberg for TFB1 plasmids and antibodies; Nancy Thompson and Dick Burgess for the hybridoma producing 8WG16; Dave Pellman for strain PY469; Mark Ptashne and Jeff Parvin for the Gal4–VP16 plasmid; and Natalie Kuldell for GST and GST–CTD proteins. We are grateful to Greg Gilmartin and Sean Flaherty for sharing results before publication. The helpful advice and discussions with members of the Buratowski laboratory are deeply appreciated. This work was supported by a research grant from the Council for Tobacco Research (USA). C.R.M. is a recipient of a Department of Defense predoctoral fellowship. S.B. is supported by a Pew Scholar Award and a Junior Faculty Research Award from the American Cancer Society.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL steveb@warren.med.harvard.edu; FAX (617) 738-0516.
References
- Cadena DL, Dahmus ME. Messenger RNA synthesis in mammalian cells is catalyzed by the phosphorylated form of RNA polymerase II. J Biol Chem. 1987;262:12468–12474. [PubMed] [Google Scholar]
- Colot HV, Stutz F, Rosbash M. The yeast splicing factor Mud13 is a commitment complex component and corresponds to CBP20, the small subunit of the nuclear cap-binding complex. Genes & Dev. 1996;10:1699–1708. doi: 10.1101/gad.10.13.1699. [DOI] [PubMed] [Google Scholar]
- Cooke C, Alwine JC. The cap and the 3′ splice site similarly affect polyadenylation efficiency. Mol Cell Biol. 1996;16:2579–2584. doi: 10.1128/mcb.16.6.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantonel JC, Murthy KGK, Manley JL, Tora L. Transcription factor TFIID recruits factor CPSF for formation of 3′ end of mRNA. Nature. 1997;389:399–402. doi: 10.1038/38763. [DOI] [PubMed] [Google Scholar]
- Du L, Warren SL. A functional interaction between the carboxy-terminal domain of RNA polymerase II and pre-mRNA splicing. J Cell Biol. 1997;136:5–18. doi: 10.1083/jcb.136.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edery I, Sonenberg N. Cap-dependent RNA splicing in a HeLa nuclear extract. Proc Natl Acad Sci. 1985;82:7590–7594. doi: 10.1073/pnas.82.22.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feaver WJ, Gileadi O, Li Y, Kornberg RD. CTD kinase associated with yeast RNA polymerase II initiation factor b. Cell. 1991;67:1223–1230. doi: 10.1016/0092-8674(91)90298-d. [DOI] [PubMed] [Google Scholar]
- Flaherty, S.M., P. Fortes, E. Izaurralde, I.W. Mattaj, and G.M. Gilmartin. 1997. Participation of the nuclear cap binding complex in pre-mRNA 3′ processing. Proc. Natl. Acad. Sci. (in press). [DOI] [PMC free article] [PubMed]
- Fresco LD, Buratowski S. Active site of the mRNA capping enzyme guanylyltransferase from Saccharomyces cerevisiae: Similarity to the nucleotidyl attachment motif of DNA and RNA ligases. Proc Natl Acad Sci. 1994;91:6624–6628. doi: 10.1073/pnas.91.14.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Conditional mutants in the yeast mRNA capping enzyme show that the cap enhances, but is not required for, mRNA splicing. RNA. 1996;2:584–596. [PMC free article] [PubMed] [Google Scholar]
- Greenleaf A. Positive patches and negative noodles: Linking RNA processing to transcription? Trends Biochem Sci. 1993;18:117–119. doi: 10.1016/0968-0004(93)90016-g. [DOI] [PubMed] [Google Scholar]
- Guthrie, C. and G.R. Fink. 1991. Guide to yeast genetics and molecular biology. Methods Enzymol. 194: [PubMed]
- Itoh N, Yamada H, Kaziro Y, Mizumoto K. Messenger RNA guanylyltransferase from Saccharomyces cerevisiae: Large scale purification, subunit functions, and subcellular localization. J Biol Chem. 1987;262:1989–1995. [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–668. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]
- Jove R, Manley JL. In vitro transcription from the adenovirus 2 Major Late Promoter utilizing templates truncated at promoter-proximal sites. J Biol Chem. 1984;259:8513–8521. [PubMed] [Google Scholar]
- Kim E, Du L, Bregman DB, Warren SL. Splicing factors associate with hyperphosphorylated RNA polymerase II in the absence of pre-mRNA. J Cell Biol. 1997;136:19–28. doi: 10.1083/jcb.136.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Bjorklund S, Li Y, Sayre MH, Kornberg RD. A multiprotein mediator of transcriptional activation and its interaction with the carboxy-terminal repeat domain of RNA polymerase II. Cell. 1994;77:599–608. doi: 10.1016/0092-8674(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Koleske AJ, Young RA. An RNA polymerase II holoenzyme responsive to activators. Nature. 1994;368:466–469. doi: 10.1038/368466a0. [DOI] [PubMed] [Google Scholar]
- Konarska MM, Padgett RA, Sharp PA. Recognition of cap structure in splicing in vitro of mRNA precursors. Cell. 1984;38:731–736. doi: 10.1016/0092-8674(84)90268-x. [DOI] [PubMed] [Google Scholar]
- Laybourn PJ, Dahmus ME. Phosphorylation of RNA polymerase IIA occurs subsequent to interaction with the promoter and before the initiation of transcription. J Biol Chem. 1990;265:13165–13173. [PubMed] [Google Scholar]
- Lu H, Flores O, Weinmann R, Reinberg D. The nonphosphorylated form of RNA polymerase II preferentially associates with the preinitiation complex. Proc Natl Acad Sci. 1991;88:10004–10008. doi: 10.1073/pnas.88.22.10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Zawel L, Fisher L, Egly J-M, Reinberg D. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature. 1992;358:641–645. doi: 10.1038/358641a0. [DOI] [PubMed] [Google Scholar]
- McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson SD, Wickens M, Bentley DL. The carboxy-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997a;385:357–361. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- McCracken, S., N. Fong, E. Rosonina, K. Yankulov, G. Brothers, D. Siderovski, A. Hessel, S. Foster, Amgen EST Program, S. Shuman, and D.L. Bentley. 1997b. 5′ Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Mizumoto K, Kaziro Y. Messenger RNA capping enzymes from eukaryotic cells. Prog Nucleic Acid Res. 1987;34:1–28. doi: 10.1016/s0079-6603(08)60491-2. [DOI] [PubMed] [Google Scholar]
- Mortillaro MJ, Blencowe BJ, Wei X, Nakayasu H, Du L, Warren SL, Sharp PA, Berezney R. A hyperphosphorylated form of the large subunit of RNA polymerase II is associated with splicing complexes and the nuclear matrix. Proc Natl Acad Sci. 1996;93:8253–8257. doi: 10.1073/pnas.93.16.8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer KM, Roth MB. Distribution of pre-mRNA splicing factors at sites of RNA polymerase II transcription. Genes & Dev. 1997;11:1148–1159. doi: 10.1101/gad.11.9.1148. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Young RA. Intragenic and extragenic suppressors of mutations in the heptapeptide repeat domain of Saccharomyces cerevisiae RNA polymerase II. Genetics. 1989;123:715–724. doi: 10.1093/genetics/123.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen EB, Lis JT. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci. 1993;90:7923–7927. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre MH, Tschochner H, Kornberg RD. Reconstitution of transcription with five purified initiation factors and RNA polymerase II from Saccharomyces cerevisiae. J Biol Chem. 1992;267:23376–23382. [PubMed] [Google Scholar]
- Scafe C, Martin C, Nonet M, Podos S, Okamura S, Young RA. Conditional mutants accur predominantly in highly conserved residues of RNA polymerase II subunits. Mol Cell Biol. 1990;10:1270–1275. doi: 10.1128/mcb.10.3.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Shuman S. Conditional inactivation of mRNA capping enzyme affects yeast pre-mRNA splicing in vivo. RNA. 1996;2:574–583. [PMC free article] [PubMed] [Google Scholar]
- Serizawa H, Conaway JW, Conaway RC. Phosphorylation of C-terminal domain of RNA polymerase II is not required in basal transcription. Nature. 1993;363:371–374. doi: 10.1038/363371a0. [DOI] [PubMed] [Google Scholar]
- Shibagaki Y, Itoh N, Yamada H, Nagata S, Mizumoto K. mRNA capping enzyme: Isolation and characterization of the gene encoding mRNA guanylyltransferase subunit from Saccharomyces cerevisiae. J Biol Chem. 1992;267:9521–9528. [PubMed] [Google Scholar]
- Sonenberg N. Remarks on the mechanism of ribosome binding to eukaryotic mRNAs. Gene Expression. 1993;3:317–323. [PMC free article] [PubMed] [Google Scholar]
- Steinmetz EJ. Pre-mRNA processing and the CTD of RNA polymerase II: The tail that wags the dog? Cell. 1997;89:491–494. doi: 10.1016/s0092-8674(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Svejstrup JQ, Feaver WJ, LaPointe J, Kornberg RD. RNA polymerase transcription factor IIH holoenzyme from yeast. J Biol Chem. 1994;269:28044–28048. [PubMed] [Google Scholar]
- Takagi T, Moore CR, Diehn F, Buratowski S. An RNA 5′-triphosphatase related to the protein tyrosine phosphatases. Cell. 1997;89:867–873. doi: 10.1016/s0092-8674(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Thompson NE, Steinberg TH, Aronson DB, Burgess RR. Inhibition of in vivo and in vitro transcription by monoclonal antibodies prepared against wheat germ RNA polymerase II that react with the heptapeptide repeat of eukaryotic RNA polymerase II. J Biol Chem. 1989;264:11511–11520. [PubMed] [Google Scholar]
- Vincent M, Lauriault P, Dubois MF, Lavoie S, Bensaude O, Chabot B. The nuclear matrix protein p255 is a highly phosphorylated form of RNA polymerase II largest subunit which associates with spliceosomes. Nucleic Acids Res. 1996;24:4649–4652. doi: 10.1093/nar/24.23.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks JR, Hardin SE, Shen J, Lee JM, Greenleaf AL. Locus-specific variation in phosphorylation state of RNA polymerase II in vivo: Correlations with gene activity and transcript processing. Genes & Dev. 1993;7:2329–2344. doi: 10.1101/gad.7.12a.2329. [DOI] [PubMed] [Google Scholar]
- Woontner M, Wade PA, Bonner J, Jaehning JA. Transcriptional activation in an improved whole-cell extract from Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:4555–4560. doi: 10.1128/mcb.11.9.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuryev A, Patturajan M, Litingtung Y, Joshi RV, Gentile C, Gebara M, Corden JL. The C-terminal domain of the largest subunit of RNA polymerase II interacts with a novel set of serine/arginine-rich proteins. Proc Natl Acad Sci. 1996;93:6975–6980. doi: 10.1073/pnas.93.14.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]