

Abstract
Specific inhibitors of histone deacetylase, such as trichostatin A (TSA) and trapoxin (TPX), are potent inducers of HIV-1 transcription in latently infected T-cell lines. Activation of the integrated HIV-1 promoter is accompanied by the loss or rearrangement of a positioned nucleosome (nuc-1) near the viral RNA start site. Here we show that TSA strongly induces HIV-1 transcription on chromatin in vitro, concomitant with an enhancer factor-assisted increase in the level of acetylated histone H4. TSA treatment, however, did not detectably alter enhancer factor binding or the positioning of nuc-1 on the majority of the chromatin templates indicating that protein acetylation and chromatin remodeling may be limiting steps that occur only on transcriptionally competent templates, or that remodeling of nuc-1 requires additional factors. To assess the number of active chromatin templates in vitro, transcription was limited to a single round with low levels of the detergent Sarkosyl. Remarkably, HIV-1 transcription on chromatin was found to arise from a small number of active templates that can each support nearly 100 rounds of transcription, and TSA increased the number of active templates in each round. In contrast, transcription on naked DNA was limited to only a few rounds and was not responsive to TSA. We conclude that HIV-1 enhancer complexes greatly facilitate transcription reinitiation on chromatin in vitro, and act at a limiting step to promote the acetylation of histones or other transcription factors required for HIV-1 enhancer activity.
Keywords: HIV-1 enhancer, histone acetylation, chromatin remodeling
Transcription activation of eukaryotic genes frequently leads to specific alterations in chromatin structure surrounding the induced promoter. In yeast, for example, induction of transcription at the PHO5 gene is accompanied by the loss of four translationally positioned nucleosomes that are situated over the TATA box and the proximal upstream region of the promoter (for review, see Svaren and Hörz 1997). Chromatin remodeling at the PHO5 promoter requires the binding of its natural transcriptional activator Pho4 and can occur when the transcription activation domain of Pho4 is replaced with a heterologous transactivation domain, or even with regions from the Gal11 or Srb2 proteins that direct their interaction with an RNA polymerase II (RNAP II) holoenzyme complex (Svaren et al. 1994; Gaudreau et al. 1997). These studies indicate that the assembly of a functional preinitiation complex by RNAP II may be a prerequisite for nucleosome disruption at inducible promoters. Because chromatin remodeling at PHO5 does not require an intact TATA box or actual initiation of transcription (Fascher et al. 1993), the assembly of a functional RNAP II preinitiation complex may even be sufficient to induce chromatin remodeling at the PHO5 gene in vivo (Gaudreau et al. 1997).
Transcription induction of the integrated human immunodeficiency virus-1 (HIV-1) provirus in mitogenically stimulated T cells and macrophages is also accompanied by changes in chromatin structure at the promoter (Verdin 1991; Verdin et al. 1993). The repressed HIV-1 promoter in unstimulated ACH2 T cells contains a positioned nucleosome located immediately upstream of the enhancer (nucleosome 0; −415 to −255), and another close to the viral RNA start site (nucleosome 1; +10 to +155). Transcription induction in activated T cells is accompanied by the specific disruption of the nucleosome at position 1 (nuc-1) and an extension of a DNase I hypersensitive site to include DNA sequences (from −5 to +130) that were protected previously by nuc-1 (Verdin et al. 1993; El Kharroubi and Verdin 1994; see Fig. 1A). Nuc-1 obscures the HIV-1 initiator as well as the binding sites for several transcription factors, including LBP-1 (−15 to +30; Yoon et al. 1994), AP-1, and NF-AT (+87 to +177; Van Lint et al. 1997). Because T-cell activation enhances the activity of LBP-1 (Volker et al. 1997) and AP-1 (Karin 1995), and induces nuclear translocation of NF-AT (Timmerman et al. 1996), these DNA-binding proteins could play a role in the disruption of nuc-1 in stimulated T cells. Moreover, transcription elongation promoted by the HIV-1 Tat protein could contribute to chromatin remodeling, although de novo initiation of HIV-1 transcription is known not to be required (Verdin et al. 1993). A DNase I hypersensitive site that lies downstream of nuc-1 (HS-IV; +201 to +265) becomes attenuated in activated T-cells, possibly because of nucleosome rearrangements that occur subsequent to the loss of nuc-1 (Van Lint et al. 1996a). Individually, the downstream AP-1, NF-AT and IRF-1 sites do not influence HIV-1 transcription, although these proteins may contribute collectively to HIV-1 promoter activity in infected cells (El Kharroubi and Martin 1996; Van Lint et al. 1997).
Figure 1.



Addition of the histone deacetylase inhibitor TSA to chromatin assembly extracts strongly induces HIV-1 enhancer activity in vitro. (A) Schematic diagram of the changes in chromatin structure that accompany TSA-mediated induction of HIV-1 transcription in latently infected T cell lines (Van Lint et al. 1996a). (B) Addition of TSA at early stages in chromatin assembly enhances HIV-1 transcription on chromatin templates in vitro. HIV-1 DNA-binding proteins were added together with pHIV-1/LUC DNA during nucleosome assembly at the following concentrations: NF-κB (p65 subunit; 200 nm), LEF-1 (50 nm), Ets-1 (100 nm), TFE-3 (20 nm), Sp1 (20 nm). These experiments used a partially purified Sp1 fraction that was obtained by wheat germ agglutinin chromatography of Jurkat nuclear extracts. α-Globin gene DNA was added to the HeLa transcription extract as nonchromatin DNA. Relative HIV-1 transcription levels from duplicate reactions were quantified by phosphorImager scanning, and the specific HIV-1 and α-globin RNA transcripts are indicated with arrowheads. (C) TSA does not affect transcription from naked pHIV-1/LUC DNA. Parallel transcription reactions were carried out in HeLa nuclear extracts with or without TSA, as indicated above the panels. Reactions either lacked HIV-1 enhancer-binding proteins (lanes marked −), or contained HIV-1 enhancer-binding proteins at identical levels to those listed in A (lanes marked +) or at levels sixfold higher (lanes marked ++). The TSA stocks were prepared as suspensions in 10% DMSO and equivalent levels of DMSO were added to the control reactions lacking TSA (although this had no effect on resulting transcription levels).
A central question raised by studies of HIV-1 and other highly inducible genes is whether the disruption of positioned nucleosomes is a necessary early step for transcription on nucleosomal DNA or an indirect effect of transcription initiation at active genes. The assembly of a positioned nucleosome over the TATA box or transcription start site has been shown to be sufficient to inhibit transcription in vitro (Lorch et al. 1992), and, in general, positioned nucleosomes are thought to repress transcription by blocking the ability of DNA-binding proteins or the general transcription machinery to bind to the promoter. It is clear that chromatin disruption alone is not sufficient to induce transcription, however, because mutant transcription factors that bind DNA but cannot activate transcription frequently retain the ability to alter chromatin structure (Pazin et al. 1994; Mymryk and Archer 1995). Consequently, it is difficult to distinguish whether the disruption of positioned nucleosomes near the TATA box or initiator is an important step in gene activation or an inevitable consequence of the formation of an active RNAP II initiation complex.
Strong support for the idea that localized nucleosome disruption is important for promoter activation comes from studies in which histones have been altered either by genetic manipulation (for review, see Grunstein 1997) or by chromatin modification (for reviews, see Rothe and Allis 1996; Sternglanz 1996; Wade et al. 1997). For example, genetic alteration of histones H3 and H4 in yeast is sufficient to induce PHO5 gene transcription, although the resulting RNA levels are significantly lower than those observed for the fully induced gene (Straka and Hörz 1991; Wechser et al. 1997). Moreover, acetylation of specific lysine residues within nucleosomal histones is closely linked to chromatin disruption and transcriptional activation for many genes (Rothe and Allis 1996; Sternglanz 1996; Wade et al. 1997). This correlation was strengthened significantly by the recent discovery that a number of transcriptional coactivators, including GCN5, TAFII250, and CREB-binding protein (CBP)/p300, possess intrinsic histone acetyltransferase (HAT) activity that is critical for their function in vivo (Bannister and Kouzarides 1996; Brownell et al. 1996; Mizzen et al. 1996; Ogryzko et al. 1996; Candau et al. 1997; Wang et al. 1997), and that these coactivators may also exist in the cell in complexes with other HATs (Yang et al. 1996; Chen et al. 1997). Similarly, transcriptional repressors, such as Mad/Max and unliganded nuclear receptors, have been shown to recruit the histone deacetylases HDAC1 and HDAC2 as part of larger transcriptional corepressor complexes (Ayer et al. 1997; Heinzel et al. 1997; Laherty et al. 1997; Nagy et al. 1997). Collectively, these studies provide strong evidence that both activation and silencing is mediated through the recruitment of enzymes that control protein acetylation. It has not been established that the nucleosome is the sole target for transcriptional control by acetylation, however, and other highly acetylated transcription factors and chromatin-associated proteins may also be critical targets for enhancer-bound acetyltransferases (Gu and Roeder 1997; for review, see Pazin and Kadonaga 1997).
HIV-1 transcription is also likely to be regulated by enhancer-recruited HAT complexes, because treatment of latently infected T-cell lines with specific histone deacetylase inhibitors such as trichostatin A (TSA) has been shown to induce viral transcription and chromatin remodeling events that are the same as those observed in activated T cells (Van Lint et al. 1996a). Although little is known about the specific HAT and deacetylase complexes that are targeted to the HIV-1 enhancer in resting and activated T cells, the p65 subunit of NF-κB may regulate protein acetylation in stimulated T cells through its ability to interact with CBP/p300 (Gerritsen et al. 1997; Parker et al. 1997). Because TSA can induce viral transcription in resting T cells that lack NF-κB, other HIV-1 enhancer-binding proteins must be able to recruit acetyltransferase complexes as well.
To better understand the mechanisms that regulate HIV-1 transcription, we have used a cell-free transcription system with nucleosome-assembled DNA templates (Kamakaka et al. 1993; Bulger and Kadonaga 1994) that supports HIV-1 enhancer activity in vitro (Sheridan et al. 1995; Pazin et al. 1996). We have shown previously in this system that binding of Sp1 and NF-κB induces DNase I hypersensitivity at the HIV-1 promoter and positions nuc-1 in its appropriate location at the promoter (Pazin et al. 1996). Recent studies from other groups have demonstrated that Sp1 and NF-κB can access their binding sites on nucleosomal DNA in an ATP-dependent manner in vitro to organize chromatin structure and form DNase I hypersensitive sites (Steger and Workman 1997; Widlak et al. 1997). In this report, we extend our studies with the cell-free chromatin transcription system to examine HIV-1 enhancer activity and remodeling of nuc-1 under conditions that promote histone acetylation. The results presented here indicate that protein acetylation is a critical step underlying HIV-1 enhancer activity in vitro.
Results
Histone deacetylase inhibitors strongly induce HIV-1 enhancer activity on chromatin templates in vitro
Previously, we have shown that HIV-1 enhancer activity can be modeled in a cell-free transcription system programmed with nucleosome-assembled DNA templates and purified HIV-1 enhancer-binding proteins (Sheridan et al. 1995; Pazin et al. 1996). To assess whether nucleosome acetylation plays an important role in HIV-1 enhancer activity in vitro, we tested two different chemical inhibitors of histone deacetylase, TSA and trapoxin (TPX) (Yoshida et al. 1995), which have been shown previously to be potent transcriptional inducers of latent integrated HIV-1 proviruses in vivo (Van Lint et al. 1996a). HIV-1 enhancer complexes were assembled in vitro with purified NF-κB (p65), LEF-1, ETS-1, TFE-3, and Sp1, and TSA was added at different times during nucleosome assembly, as indicated in the reaction time-line shown in Figure 1B. HIV-1 RNA was measured by primer extension after transcription of the chromatin templates with HeLa nuclear extract. Interestingly, addition of TSA to the Drosophila S-190 extract before nucleosome assembly enhanced HIV-1 transcription by as much as 50-fold in vitro, most notably in reactions that were supplemented with purified HIV-1 enhancer-binding proteins (Fig. 1B). TSA also potentiated HIV-1 enhancer activity, albeit to a lesser extent, when added at later times to the chromatin assembly reaction. In contrast, TSA had no effect on transcription from naked α-globin DNA that was mixed with the HIV-1 chromatin templates during transcription (Fig. 1B), nor did it affect basal HIV-1 promoter activity on naked DNA in the HeLa transcription extract, even when the extract was supplemented with Sp1 and purified HIV-1 enhancer-binding factors (Fig. 1C). Different levels of TSA ranging from 0.1 to 5 μm were found to be equally effective at inducing HIV-1 transcription in vitro (data not shown), and thus TSA acts in these reactions at levels comparable to those used to induce viral transcription in vivo (Van Lint et al. 1996a). We conclude from this experiment that HIV-1 enhancer activity is strongly regulated in vitro by the enzymes that control histone acetylation.
To determine whether transcription induction by TSA correlates with an increase in histone acetylation, and more generally whether changes in histone acetylation accompany enhancer activation even in the absence of TSA, aliquots of the nucleosome assembly reactions were analyzed in parallel for transcriptional activity and for acetylated histone H4 content before transcription. As shown in Figure 2A, addition of TSA at late times in the assembly reaction (t = 4.5 hr) potentiated HIV-1 transcription only in the presence of enhancer-binding proteins. In contrast, addition of TSA at the beginning of the nucleosome assembly (t = 0 hr) enhanced transcription both in the presence and in the absence of enhancer factors, although the magnitude of the induction was significantly higher when the enhancer-binding proteins were present. These results suggest that TSA treatment modifies the chromatin template even in the absence of exogenous enhancer-binding proteins to an extent that permits a low level of transcription by factors present in the HeLa nuclear extract.
Figure 2.
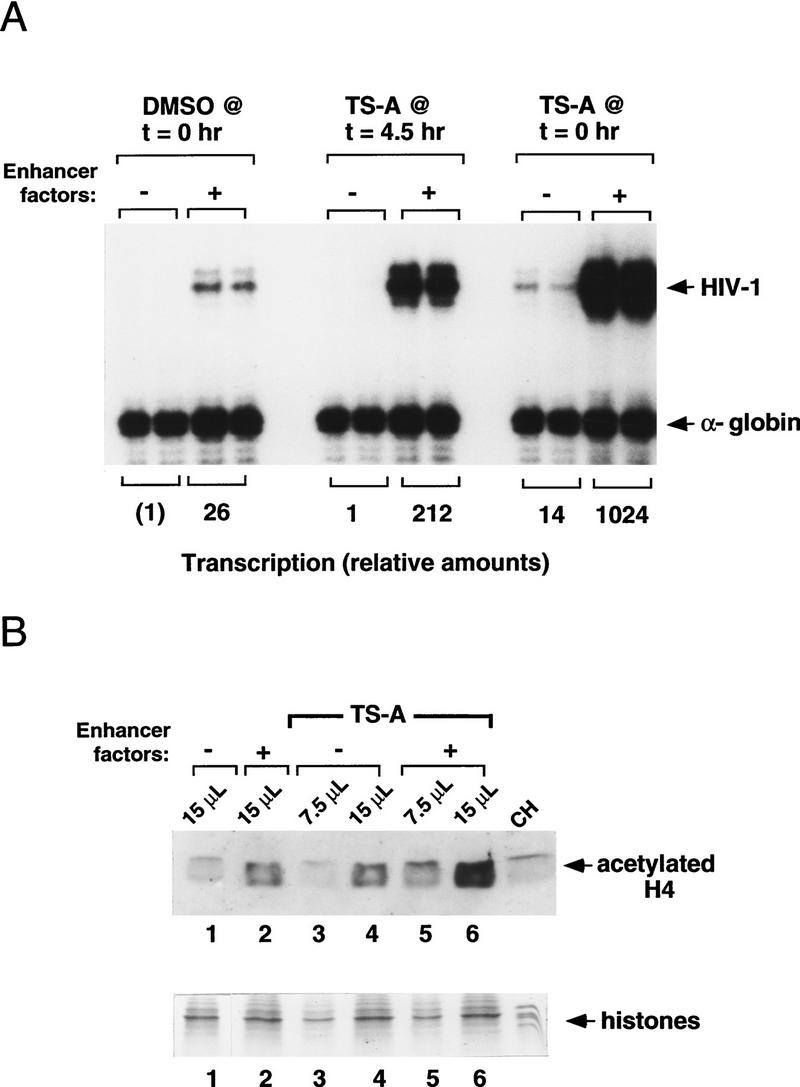
Activation of the HIV-1 enhancer in vitro is accompanied by increased levels of acetylated histone H4. (A) HIV-1 transcription reactions prepared in the presence and absence of TSA. HIV-1 enhancer-binding proteins were added at the concentrations indicated in the legend to Figure 1A, except that the concentration of Ets-1 was increased to 150 nm. (B) Western blot analysis of the level of acetylated histone H4 from the same chromatin assembly reactions shown in A, in which DMSO or TSA was added at the beginning of nucleosome assembly (t = 0 hr). Immunoblotting was carried out using antiserum specific for the acetylated form of histone H4 (Lin et al. 1989). Reaction volumes loaded on the SDS-polyacrylamide gel (18% polyacrylamide) are indicated above each lane. Total histone levels were assessed by Coomassie blue staining of aliquots from the same reactions that were analyzed by Western blot.
The level of acetylated histones in the nucleosome assembly reaction before transcription was assessed by immunoblot experiments with an antiserum specific for the acetylated form of histone H4 (Fig. 2B, top). Control aliquots of the same chromatin reactions were run in parallel on SDS-polyacrylamide gels and visualized with Coomassie blue stain to confirm that the samples tested contained comparable amounts of histones (Fig. 2B, bottom). Interestingly, the level of acetylated histone H4 increased commensurately with HIV-1 enhancer activity in these reactions. Thus, acetylated histone H4 levels were consistently higher in reactions that contained exogenous enhancer-binding proteins, regardless of whether the reactions contained TSA (Fig. 2B, top, cf. lane 1 with lane 2), indicating that the HIV-1 enhancer complexes are capable of recruiting acetyltransferases from the Drosophila chromatin assembly extract. In addition, acetylated histone levels were higher in reactions that had been supplemented with TSA (Fig. 2B, cf. lane 2 with lane 6), either with or without exogenous enhancer factors (Fig. 2B, cf. lane 1 with lane 4). Histone acetylation was the highest in reactions that contained both enhancer-binding proteins and TSA, which also supported the highest levels of transcription. Although the changes in histone acetylation observed here are relatively modest, a clear correlation was observed between the overall level of histone acetylation and transcriptional activity, indicating that HIV-1 enhancer activity is accompanied by increases in HAT activity in vitro.
Different subregions of the HIV-1 enhancer are equally responsive to inhibitors of histone deacetylase
The integrated HIV-1 promoter is very strongly induced by TSA in vivo relative to most cellular genes (Van Lint et al. 1996b); however, the reasons for this are not well understood, and in particular it is not clear which enhancer elements mediate this response. NF-κB may normally play a role in recruiting protein acetyltransferase complexes to the promoter through specific interactions between the transactivation domain of p65 and the CBP/p300 coactivator complex (Gerritsen et al. 1997; Parker et al. 1997). NF-κB is clearly not the sole target for acetyltransferases on the HIV-1 enhancer, however, because latent HIV-1 proviruses in resting T-cell lines are strongly induced by TSA even in the absence of NF-κB (Van Lint et al. 1996a). To determine which factors are capable of responding to TSA in vitro, HIV-1 enhancer complexes were assembled with different combinations of NF-κB (p65), LEF-1, ETS-1, and TFE-3 and analyzed by in vitro transcription reactions in the presence or absence of TSA. For these studies, TSA was added late in the assembly reaction to ensure that basal transcription would not be affected, and Sp1 was included to enhance the overall level of transcription. High levels of transcription were observed with chromatin templates containing either NF-κB or a combination of LEF-1, ETS-1, and TFE-3, and both of these enhancer constructs were induced by TSA (Fig. 3). In contrast, TSA did not strongly induce transcription from chromatin templates that contained only recombinant Sp1 or TFE-3 (Fig. 3B). The irreversible histone deacetylase inhibitor TPX was also able to activate HIV-1 transcription in these studies (Fig. 3A). Induction of HIV-1 transcription by TSA did not require Sp1 (see below), although partially purified fractions of Sp1 derived by wheat germ affinity chromatography of Jurkat (T cell) nuclear extracts were capable of responding to TSA when it was present throughout the assembly reaction (Fig. 4). These data indicate that different HIV-1 enhancer complexes are activated by TSA in vitro, presumably reflecting their ability to act independently to recruit protein acetyltransferase complexes to the HIV-1 promoter.
Figure 3.

The two functional subregions of the HIV-1 enhancer can be induced independently by TSA in vitro. (A) NF-κB (p65 homodimer) enhancer complexes are activated by histone deacetylase inhibitors. In vitro transcription experiments were carried out with p65 (200 nm) and Sp1 (20 nm), in the presence or absence of TSA and TPX, as indicated above each lane. (B) Induction by TSA of enhancer complexes containing LEF-1 (100 nm), Ets-1 (150 nm), TFE-3 (20 nm), and Sp1 (20 nm), or equivalent levels of recombinant Sp1 or TFE-3 in the absence of any other proteins, as indicated above each lane.
Figure 4.
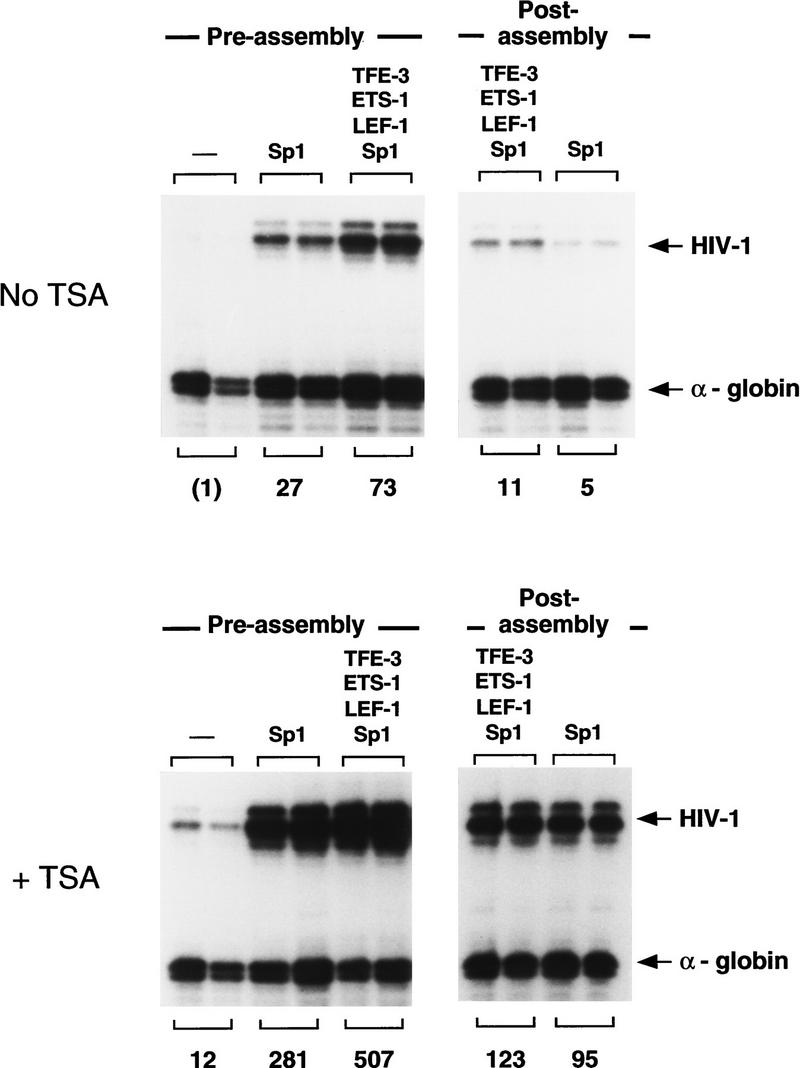
TSA activates HIV-1 transcription when the HIV-1 DNA-binding proteins are added simultaneously with the DNA at an early (preassembly) step, or when the enhancer factors are added at a late (postassembly, at t = 4.5 hr) step. Reactions were carried out in the absence (top) or presence (bottom) of TSA. These experiments used a partially purified Sp1 fraction that was obtained by wheat germ agglutinin chromatography of Jurkat nuclear extracts, which was tested alone or together with recombinant LEF-1 (100 nm), Ets-1 (150 nm), and TFE-3 (20 nm). The relative HIV-1 transcription levels are listed below each lane.
In the experiments described above, TSA was added simultaneously with the DNA and purified HIV-1 enhancer-binding proteins prior to nucleosome assembly. We also asked whether TSA would induce transcription when added together with the enhancer-binding factors at a later step, after nucleosome assembly. As shown in Figure 4, nucleosome assembly markedly reduced the ability of the enhancer-binding proteins to activate HIV-1 transcription in vitro, presumably by inhibiting the ability of these factors to access their binding sites in DNA. Nevertheless, enhancer-dependent transcription from preassembled chromatin templates was strongly induced (10- to 20-fold) by TSA under these conditions, and we conclude that TSA potentiates HIV-1 enhancer activity regardless of whether the enhancer factors are added before or after nucleosome assembly.
Transcription induction by TSA is not accompanied by detectable changes in enhancer factor binding or chromatin structure
To determine whether TSA had any effect on chromatin structure under the conditions that were used in these experiments, we examined the nucleosomal array and DNase I hypersensitive sites on the same chromatin templates that had been analyzed for transcription. Micrococcal nuclease (MNase) digestion experiments (Fig. 5A) revealed that TSA had no detectable effect on nucleosome assembly or the regularity of nucleosome spacing, either for bulk chromatin (as determined using a probe located ∼1 kb upstream of the enhancer), or for chromatin in the vicinity of the promoter (detected using a probe complementary to the TATA box). Because core histone acetylation can facilitate the binding of transcription factors to nucleosome-assembled DNA (for review, see Wolffe and Pruss 1996; Wade et al. 1997), and because protein acetylation can markedly alter the DNA-binding activity of transcription factors (Gu and Roeder 1997), we also examined whether TSA had any effect on the binding of the enhancer factors to the chromatin templates. Under conditions that support strong induction of transcription by TSA, no changes in enhancer factor binding were evident by DNase I footprint analysis of the chromatin templates (Fig. 5B; data for LEF-1, ETS-1, and TFE-3 not shown), indicating that TSA did not induce transcription by altering the affinity of any of the factors for HIV-1 DNA.
Figure 5.


TSA treatment does not detectably alter HIV-1 enhancer factor binding or chromatin structure before transcription. (A) Analysis of nucleosome assembly by micrococcal nuclease digestion of the chromatin templates. Reactions shown in each panel were incubated with micrococcal nuclease for 3, 8, 16, and 25 min at 37°C. Blots were hybridized to a control plasmid DNA probe (located ∼1 kb upstream of the enhancer) to assess the regularity of the nucleosomal array in bulk chromatin, and with an HIV-1 primer that anneals to the TATA box to analyze the disruption in the nucleosomal array in the promoter region. DNA-binding factors present in this experiment were: NF-κB (p50; 30 nm) and Sp1 (20 nm). (B) DNase I footprint analysis of the binding of NF-κB (p50; 200 nm) and Sp1 to pHIV-1/LUC chromatin templates in the presence or absence of TSA. (C) (DNase I) This panel displays the DNase I hypersensitive sites that were generated with binding of different factors to pLTR/LUC chromatin templates, as indicated above each lane. The panel marked MNase shows the result of a micrococcal nuclease indirect end-labeling experiment to assess the positioning of nuc-1 on pLTR/LUC chromatin templates in the presence and absence of TSA. Enhancer factors were used at the following concentrations: NF-κB (p50; 60 nm), Sp1 (20 nm), IRF-1 (45 nm), and LBP-1 (150 nm). Sp1 was purified from HeLa nuclear extracts by WGA chromatography. Solid boxes are used to indicate DNase I hypersensitive sites, and the location of nuc-1 is indicated with a circle.
We also examined whether treatment of the chromatin assembly extract with TSA influenced DNase I hypersensitive site formation and positioning of nuc-1 at the HIV-1 promoter. Previously, we reported that binding of NF-κB (p65) and Sp1 generates strong DNase I hypersensitive sites in the region corresponding to HS-II and HS-III in vivo, but does not generate DNase I hypersensitivity in the HS-IV region that is located downstream of nuc-1 (Pazin et al. 1996). To determine which factor might generate HS-IV, we tested the abilities of recombinant AP-1 (c-jun), NF-ATc, or IRF-1, which bind to the HIV-1 promoter DNA in the vicinity of HS-IV (Van Lint et al. 1997), to generate hypersensitive sites in vitro. Binding of IRF-1, but not any of the other proteins, induced strong DNase I hypersensitivity in the region corresponding to HS-IV (Fig. 5C; other data not shown). None of these factors, including IRF-1, significantly activated HIV-1 transcription on chromatin, either when tested alone or in the presence of the upstream enhancer-binding proteins (data not shown). Importantly, TSA had no effect on the pattern or intensity of DNase hypersensitive sites generated by IRF-1 or the enhancer factors. To examine whether TSA altered nuc-1 in particular, chromatin templates prepared with IRF-1 and the upstream enhancer-binding factors were treated with MNase and analyzed by an indirect end-labeling technique, as described in Materials and Methods. The positioned nuc-1 was readily detected under these conditions (Fig. 5C, right panel) and was unaffected in the presence of TSA. Identical results were obtained when nuc-1 was positioned by the binding of Sp1 and NF-κB to the pHIV/LUC promoter, and we did not detect disruption of nuc-1 after transcription in the HeLa nuclear extract (data not shown). Because the number of transcriptionally active templates in cell-free transcription systems is generally quite low, we cannot exclude the possibility that nuc-1 has been disrupted exclusively on a small fraction of chromatin templates that are capable of supporting transcription in vitro (see below).
Binding of the cellular factor LBP-1 to the HIV-1 promoter blocks the formation of nuc-1
The promoter-proximal boundary of nuc-1 overlaps with the DNA-binding site for a cellular DNA-binding protein called LBP-1 (also known as CP2 or LSF), which binds most strongly to HIV-1 sequences between −15 and +33 of the HIV-1 promoter (Lim et al. 1992; Yoon et al. 1994). T-cell activation induces phosphorylation of LBP-1 and enhances its DNA-binding activity (Volker et al. 1997); consequently, LBP-1 is most likely to bind to the HIV-1 LTR in activated T cells, either before or after the disruption of nuc-1. As a result, we asked whether nuc-1 is capable of assembling on templates that are bound by LBP-1 before nucleosome assembly. MNase digestion experiments indicate that binding of LBP-1 prevented formation of nuc-1 in vitro (Fig. 5C), and this was confirmed by restriction enzyme susceptibility experiments (see below). DNase I footprint experiments revealed that the binding of LBP-1 to the chromatin was unaffected by TSA, regardless of whether LBP-1 was added before or after nucleosome assembly (data not shown).
Analysis of nuc-1 remodeling by restriction enzyme digestion of bulk and transcriptionally active chromatin
Remodeling of nuc-1 was examined further using sensitive restriction enzyme accessibility experiments that rely on the relative inability of enzymes to cleave nucleosomal DNA as compared to naked DNA. Chromatin templates prepared without HIV-1 enhancer-binding proteins were found to be resistant to cleavage by restriction enzymes that cut either at the promoter (PvuII), or within the nuc-1 region (AflII; HindIII). In contrast, nucleosomal DNA templates prepared with exogenous enhancer-binding proteins were much more susceptible to cleavage at the TATA box (−20) by PvuII, although they remained resistant to digestion by AflII (+63) and HindIII (+76; Fig. 6, top panel). Enhanced cleavage by PvuII is consistent with our previous observation that the enhancer factors disrupt the nucleosomal array specifically at the promoter (Pazin et al. 1996). Addition of LBP-1 to the assembly reaction greatly facilitated cleavage of nuc-1 sequences by AflII and HindIII, indicating that nuc-1 has been disrupted or rearranged on these templates. Binding of LBP-1 to the DNA before nucleosome assembly resulted in only a very modest (10-fold) activation or derepression of HIV-1 transcription, which was significantly below the RNA levels observed with enhancer-bound templates (data not shown), indicating that the loss of nuc-1 alone does not lead to a high level of transcription. TSA had no effect on restriction enzyme digestion of nuc-1 sequences, indicating that nuc-1 remains intact on most of the templates under these conditions.
Figure 6.


Analysis of HIV-1 chromatin structure by restriction enzyme digestion. (Top) Restriction enzyme accessibility studies carried out with HIV-1 chromatin templates. pLTR/LUC DNA was incubated with a mixture of enhancer factors at the following concentrations: NF-κB (30 nm), LEF-1 (40 nm), TFE-3 (7.5 nm), Sp1 (WGA; 20 nm), IRF-1 (45 nm), or LBP-1 (150 nm) before transcription. The chromatin templates were digested with 15 units of the various restriction enzymes indicated above each lane, and the DNA was analyzed by indirect end-labeling. The PvuII and AflII restriction enzyme digestion products were detected using the SphI primer, whereas the HindIII digests were detected using the XmnI primer. (Bottom) In vitro transcription experiments carried out with chromatin templates that were either uncut or incubated with AflII or HindIII (10 units per reaction). The location of the restriction enzyme cleavage sites relative to a downstream primer in the luciferase gene is indicated in the diagram at the bottom of the figure. Enhancer factors were present at the concentrations listed above, except that LBP-1 and IRF-1 were omitted. No TSA was added to these transcription reactions.
From these findings, we conclude that if nuc-1 is disrupted before transcription, it does not occur on the majority of the chromatin templates. Consequently, the disruption of nuc-1 may be a limiting step that occurs only on those templates that are actively transcribed, or, alternatively, transcription might occur independently of chromatin remodeling under the conditions we have examined here. To assess whether nuc-1 is intact on the transcriptionally active templates, we tested whether transcription was affected by digestion of the chromatin templates with enzymes that cleave nuc-1 DNA sequences. For these experiments, the chromatin templates were cut with AflII and HindIII prior to transcription in the HeLa nuclear extract, and transcription was assessed using a primer that anneals downstream of the restriction enzyme cleavage site. As shown in Figure 6 (bottom), cleavage with AflII and HindIII significantly reduced the number of HIV-1 transcripts, indicating that nuc-1 is disrupted on the majority of the actively transcribed templates. In these and repeated experiments, however, a significant proportion of the active chromatin templates (10%–15%) were resistant to digestion by AflII and HindIII, whereas comparable levels of these enzymes completely eliminated transcription from naked HIV-1 DNA templates. These data suggest that chromatin remodeling of nuc-1 has occurred on most, but not all, of the actively transcribed templates, even though it was not detected from structural studies of the bulk chromatin.
Assembled HIV-1 enhancer complexes support many rounds of transcription in vitro
To assess the number of transcriptionally active HIV-1 enhancer templates in the chromatin transcription system, reactions were carried out in the presence of low concentrations of the detergent Sarkosyl, which effectively prevents reinitiation and limits transcription to a single round (Hawley and Roeder 1985, 1987). RNA polymerase II preinitiation complexes were formed on the chromatin templates, and initiation was allowed to proceed for a short time before Sarkosyl was added to prevent transcription reinitiation. Remarkably, the addition of low levels of Sarkosyl (0.1%) to the reactions reduced dramatically transcription from the chromatin templates, indicating that the number of active HIV-1 enhancer templates in a single round of transcription is extremely low (Fig. 7A). PhosphorImager scanning of the gel indicates that each of the HIV-1 chromatin templates supports an average of ∼100 rounds of transcription in vitro. Although the number of active templates in the first round was increased severalfold in the presence of TSA, the total number of transcriptionally competent templates in these reactions remained quite low. Thus, if chromatin remodeling occurs only on the small fraction of transcriptionally active templates, we would not have detected it in the structural studies shown in Figures 5 and 6. In contrast, comparable levels of Sarkosyl reduced HIV-1 promoter activity six- to eightfold on naked DNA (Fig. 7B), indicating that a larger fraction of the naked HIV-1 DNA templates is transcribed actively and that each active nonchromatin template supports only a few (six to eight) rounds of transcription. Induction of transcription by TSA on chromatin was found to be two- to threefold lower in the presence of Sarkosyl than in its absence (data not shown), indicating that TSA increases the number of active chromatin templates in each round. We conclude from these experiments that the HIV-1 enhancer-binding proteins greatly facilitate transcription reinitiation on chromatin, and activate transcription through a mechanism that is regulated by protein acetylation.
Figure 7.

Analysis of single and multiple rounds of transcription on chromatin or nonchromatin HIV-1 DNA templates. (A) Transcription on chromatin templates in the presence and absence of TSA. Where indicated above the lanes, transcription was limited to a single round by the addition of Sarkosyl (0.1%) shortly after initiation of transcription. Assembly reactions either lacked (−) or contained (+) the following mixture of recombinant HIV-1 enhancer-binding proteins: NF-κB (p50/p65; 25 nm), LEF-1 (40 nm), and TFE-3 (7.5 nm). (B) Transcription of naked pHIV-1/LUC DNA templates in the presence of different levels of Sarkosyl, as indicated above each lane. Relative transcription levels determined by phosphorImager scanning are listed below each lane.
Discussion
The results presented in this study provide strong biochemical evidence that the acetylation of histones or other, as yet undefined, regulatory transcription factors plays a critical role in the regulation of HIV-1 enhancer activity in vitro. First, we show that a specific inhibitor of histone deacetylase (TSA) induces HIV-1 transcription on nucleosomal DNA as much as 50-fold in vitro, and that HIV-1 transcription in the chromatin transcription system is invariably accompanied by an enhancer-dependent increase in the level of acetylated histones. Second, histone acetylation was also correlated with increased HIV-1 transcription even in the absence of TSA, indicating that TSA potentiates enhancer function without altering the normal mechanism of enhancer action. De novo acetylation of histone H4 did not require that the assembly extract be supplemented with exogenous acetyl coenzyme A (CoA) substrate, indicating that the standard conditions we used previously to study enhancer function (e.g., Sheridan et al. 1995; Pazin et al. 1996; Mayall et al. 1997) can support significant levels of protein acetylation. Thus, our results provide general support for the proposal that the recruitment of coactivator complexes that contain associated HAT activities is a critical step in enhancer function (for reviews, see Rothe and Allis 1996; Sternglanz 1996; Grunstein 1997; Wade et al. 1997).
Although HIV-1 transcription was strongly induced by TSA in vitro, we detected no specific remodeling of the nucleosome located near the viral RNA start site (nuc-1) by indirect end-labeling or restriction enzyme accessibility experiments carried out with the chromatin templates (Figs. 5 and 6). However, nuc-1 DNA sequences were accessible to restriction enzyme cutting on most of the actively transcribed HIV-1 chromatin templates (Fig. 6). Taken together, these findings raise the possibility that acetylation and chromatin remodeling are limiting steps that occur only on the small percentage of the templates that are competent for transcription in vitro (Fig. 7), and any changes in chromatin structure that might occur only on active templates would therefore be difficult to detect by structural studies of the bulk chromatin. In contrast, DNase I footprint experiments indicate that HIV-1 enhancer complexes are assembled efficiently on the majority of the chromatin templates, and therefore the interaction of DNA-binding proteins with the enhancer is not a limiting step under the conditions that were used here. Because evidence obtained from nuclease susceptibility studies is indirect, and because nuclease digestion of the chromatin templates did not eliminate all HIV-1 transcription, it is possible that nuc-1 remains intact on some of the active templates. Therefore the specific loss or rearrangement of nuc-1 might also require particular DNA-binding proteins (e.g., LBP-1, AP-1) or actual transcription elongation through nuc-1, which is stimulated by the HIV-1 Tat protein. Thus, further studies will be needed to establish whether nucleosome remodeling can ever be dissociated from transcription initiation in vitro. From the available evidence, we conclude that protein acetylation plays a major role in the regulation of HIV-1 enhancer activity in vitro, and that protein acetylation and nucleosome remodeling may constitute limiting steps that restrict the number of functional enhancer templates in vitro. The target for acetylation could be either the nucleosome (i.e., nuc-1), or a critical transcription factor that regulates HIV-1 enhancer activity on chromatin.
A model for the regulation of HIV-1 enhancer activity by HAT complexes
These findings have several implications for models of the regulation of HIV-1 enhancer activity by HAT complexes, as outlined in Figure 8. The observation that histone acetylation levels increased when HIV-1 enhancer-binding proteins were added to the assembly reaction (Fig. 2) suggests that these factors can recruit HAT complexes from the Drosophila chromatin assembly extract, which is known to contain functional homologs to the human CBP/p300 and TAFII250 proteins. The CBP/p300 protein has been implicated specifically in the regulation of HIV-1 enhancer activity by virtue of its ability to interact with the transactivation domain of the p65 subunit of NF-κB (Gerritsen et al. 1997; Parker et al. 1997). Because HIV-1 enhancer complexes that lack NF-κB are also responsive to TSA, both in vitro (Fig. 3) and in vivo (Van Lint et al. 1996a), we postulate that one or more of the other enhancer-binding proteins (e.g., LEF-1, ETS-1, or TFE-3) can also recruit functional HAT coactivators, such as CBP/p300 or TAFII250. As suggested in Figure 8, it is possible that several HIV-1 enhancer-binding proteins interact with different regions of CBP/p300 (or with different components of a CBP/p300 coactivator complex) to stabilize its association with the enhancer. In this regard, the ability of LEF-1 to bend DNA strongly (Giese et al. 1992) might play an important role in configuring the structure of the enhancer to allow for optimal interactions between the various enhancer-binding proteins and the CBP HAT complex. Transcriptional activation by LEF-1 has been shown to be highly sensitive to the location of its binding site relative to those of other enhancer-binding proteins (Carlsson et al. 1993; Giese and Grosschedl 1993), and it is interesting to note that the distance between the LEF-1 and NF-κB-binding sites on the HIV-1 enhancer is nearly identical to that between LEF-1 and CREB (another CBP/p300-responsive transcription factor) on the human T-cell receptor α (TCRα) enhancer. Therefore, the HIV-1 and TCRα enhancers may be structured in a similar manner that facilitates the recruitment of the CBP/p300 HAT complex in T cells.
Figure 8.
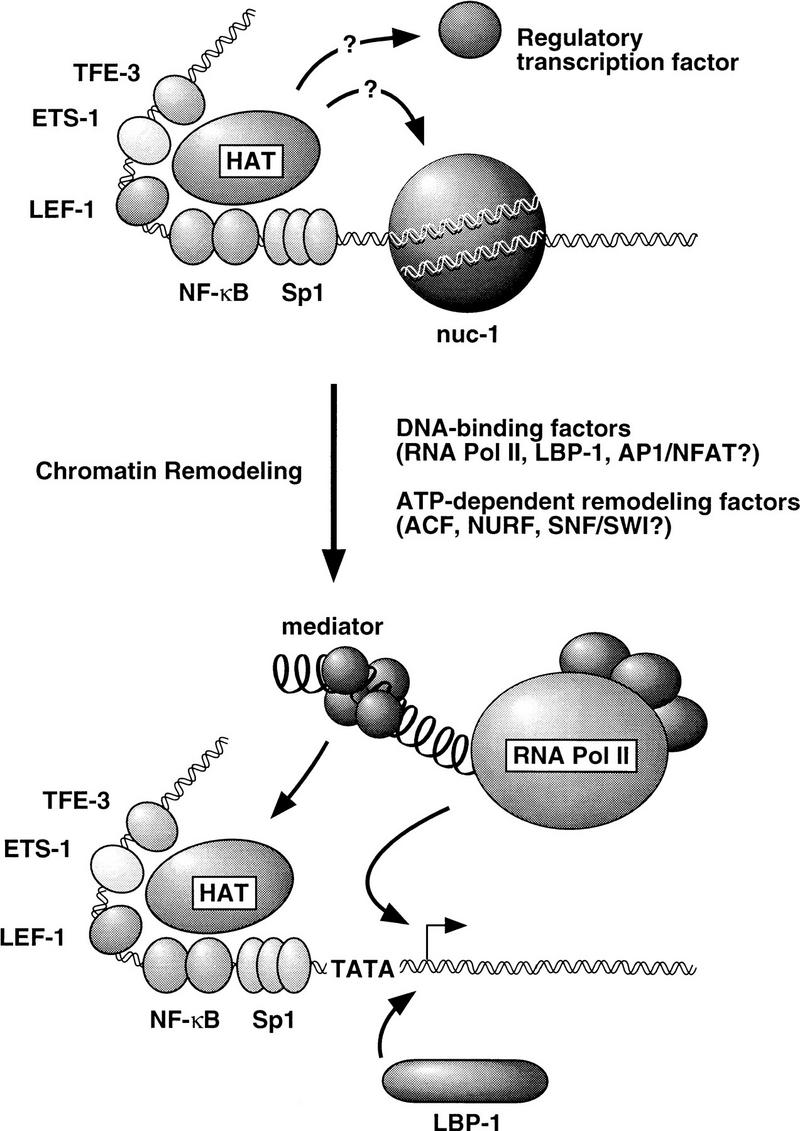
A model for the role of histone acetylation and chromatin remodeling in regulating HIV-1 enhancer activity. Possible roles for DNA-binding proteins and chromatin remodeling activities in the disruption of nuc-1 and establishing an open chromatin structure within the initiator region of the HIV-1 promoter are discussed in the text.
Although histone deacetylase inhibitors, such as TSA, strongly induced HIV-1 transcription in resting T cells, only ∼2% of the active cellular genes were similarly induced by TSA, as determined by differential display analysis (Van Lint et al. 1996b). The ability of the HIV-1 enhancer to respond so markedly to TSA might reflect the architectural organization of the repressed HIV-1 promoter, including the specific location of nuc-1 relative to the RNA start site, as well as the nature of the histone deacetylase complexes that repress HIV-1 transcription in resting T cells. Disruption of nuc-1 in activated T cells could be initiated by the binding of proteins such as LBP-1 and AP-1 to the promoter, or alternatively, it might be accomplished more simply through the recruitment and assembly of a functional RNAP II preinitiation complex, as has been proposed for the PHO5 gene promoter (Gaudreau et al. 1997). Nucleosome remodeling is an ATP-dependent process (Pazin et al. 1994, 1997) that is carried out by multisubunit chromatin remodeling complexes, such as ACT (Ito et al. 1997), NURF (Tsukiyama et al. 1995), and SWI–SNF (Peterson and Tamkun 1995). Therefore, the remodeling of nucleosome structure at the HIV-1 promoter is likely to involve the coordinated assembly of an RNAP II preinitiation complex and recruitment of chromatin reconfiguring enzymes, and it will be interesting to learn in which order these events occur and what signals attract the chromatin remodeling activities to the HIV-1 promoter.
The observation that prebinding of LBP-1 to DNA is sufficient to block the formation of nuc-1 (Fig. 6), raises the possibility that LBP-1 might be needed to establish or maintain an open chromatin structure at the HIV-1 promoter in vivo. Remodeling of nuc-1 in activated T cells exposes the high-affinity LBP-1 sites (−15 to +30), and the DNA-binding activity of LBP-1 is also enhanced in activated T cells (Volker et al. 1997). Thus, LBP-1 might play a positive role in HIV-1 transcription by binding to the HIV-1 promoter after disruption of nuc-1 to ensure that the RNA start site and Tat-responsive transactivation response (TAR) region are not obscured by a repressive nucleosome for subsequent rounds of transcription. The LBP-1-binding site on the HIV-2 promoter spans an extensive (150 bp) region of the HIV-2 TAR region (Jones et al. 1988), which suggests that LBP-1 may have a conserved role in viral transcription. A positive role for LBP-1 in the regulation of HIV-1 chromatin structure contrasts with its repressive effect on HIV-1 transcription on naked DNA, which is observed only at very high concentrations of LBP-1 and results from its binding to low affinity sites that surround the HIV-1 TATA box (Kato et al. 1991). In contrast, the interaction of LBP-1 with its high affinity sites does not repress transcription, and might instead serve to maintain an open promoter configuration.
The disruption of nuc-1 by prebinding of LBP-1 to DNA caused only a very modest derepression of HIV-1 transcription, which was not augmented by TSA, indicating that the loss of nuc-1 is not sufficient to activate transcription to the high levels that are observed with an enhancer. It has not been established whether LBP-1 contains a functional transactivation domain, and our results suggest that it is incapable of attracting HAT complexes to the promoter. Thus, the failure of LBP-1 to induce transcription strongly could reflect its inability to sustain the high levels of transcription reinitiation, which are observed with enhancer factors that are capable of recruiting the general transcription machinery to the HIV-1 promoter. The conclusion that disruption of nucleosome structure is not sufficient to activate transcription is in general agreement with results of genetic studies that have analyzed the consequence of specific histone mutations on gene transcription in yeast (for review, see Grunstein 1997). Thus, for example, PHO5 transcription is only modestly induced in yeast strains that contain altered histones, and genes that are already highly expressed appear not to be further induced in such strains (Straka and Hörz 1991; Wechser et al. 1997). Thus, it appears that the simple disruption of repressive nucleosomes, such as nuc-1, does not usually result in high levels of transcription in the absence of a functional enhancer.
Enhancer complexes assembled on chromatin are stable to multiple rounds of transcription reinitiation
An interesting observation made in this study is that HIV-1 enhancer-assembled complexes support extremely high levels of transcription reinitiation on chromatin templates relative to naked DNA (Fig. 7). Whereas transcription complexes assembled on naked DNA are stable to only a few rounds of transcription, enhancer complexes on chromatin were found to support ∼100 rounds of transcription in vitro. Thus, transcription on naked DNA is dramatically different from transcription in a chromatin environment. Previous studies have documented other important effects of chromatin on transcription in vitro, including the ability of enhancer-assembled chromatin templates to support long-range activation from distal upstream or downstream enhancers and to discriminate between bona fide and cryptic transactivation domains (for review, see Paranjape et al. 1994). In general, enhancers do not act to increase the rate of transcription initiation from a promoter, but, rather, increase the probability that a promoter will be active (Walters et al. 1996). Therefore, the differences we observe between transcription on chromatin and naked DNA templates most likely reflect the influence of a chromatin environment, rather than any intrinsic difference between an enhancer and a promoter. Most important, these data suggest that once an enhancer complex has successfully recruited a coactivator complex and remodeled chromatin structure, it becomes highly stable to multiple rounds of transcription initiation.
In summary, we have shown that HIV-1 enhancer activity is dramatically up-regulated under conditions that support histone acetylation in vitro. The available evidence indicates that enhancer complexes recruit protein acetyltransferases with different substrate specificities and potentially different target proteins, and the studies presented here support the idea that protein acetylation regulates a critical step in enhancer activity. To fully understand the mechanism underlying the induction of HIV-1 transcription in stimulated T cells, it will be important to determine the events leading to the selective reconfiguration of nuc-1 and to identify the histone deacetylase and acetyltransferase complexes that control HIV-1 enhancer activity. In addition, it will be very important to determine which regulatory proteins are the relevant targets for acetylation. The results presented in this paper indicate that further experiments with the cell-free chromatin transcription system should be useful in resolving some of these interesting questions.
Materials and methods
DNA
pHIV-1/LUC contains HIV-1 (ARV-2) sequences from −340 to +80 (relative to the RNA start site) in a luciferase vector (Sheridan et al. 1995). The plasmid pLTR/LUC has been described previously (Van Lint et al. 1997), and contains pLAI sequences from −453 to +340. The HIV-1 primer (GCTTTATTGAGGCTTAAGCAGTGGG; +56/+79) was used to detect HIV-1 transcripts. The +56/+79 and −267/−233 primers (GGAGAGAACAACAGCTTGTTACACCCTATGAGCC) were used for DNase I footprint analysis of pHIV-1/LUC templates, whereas the +56/+79 or +340/+310 primers (GATCGCTAGACTCTCTCCTTCTAGCCTCCGCTAG) were used to assess factor binding to pLTR/LUC in DNase I footprint experiments. Nucleosomal ladder disruption studies carried out with pHIV-1/LUC DNA were detected with a promoter-specific primer (−47/−15; GGCGTCCCTCAGATGCTGCATATAAGCAGCTGCTTTTTGC) and a control (distal upstream region) primer (NdeI; GCACTCTCAGTACAATCTGCTCTGATGCCGC). DNase I hypersensitive site studies and indirect end-labeling analyses of pHIV-1/LUC templates were detected using either the NdeI or the SphI primers (CGCAGGCAGTTCTATGCGGAAGGGCCACACCC), whereas structural studies with pLTR/LUC were carried out using the SphI and XmnI primers (TCGGGGCGAAAACTCTCAAGGATCTTACCGC), as indicated in the individual figure legends.
Purification of HIV-1 enhancer-binding proteins
Full-length human NF-κB (p50), LEF-1, Ets-1, and TFE-3 proteins were expressed in bacteria and purified as described previously (Sheridan et al. 1995; Pazin et al. 1996). Homodimer preparations of the p65 subunit of NF-κB were derived from recombinant vaccinia virus-infected HeLa cells (Pazin et al. 1996). The experiment shown in Figure 7 was carried out using a chimeric p50/p65 NF-κB protein that contains the high-affinity DNA-binding region of murine p50 (amino acids 1–485) extended at the carboxyl terminus with the transcriptional activation of the murine p65 (amino acids 426–549). The composite cDNA was cloned into pET28α(+) (Novagen) for bacterial expression as an amino-terminal (His)6-tagged protein (pET28p50/65). The full-length human IRF-1 protein was expressed in bacteria as a carboxy-terminal (His)6 fusion protein (pET28IRF-1). This expression plasmid was constructed by inserting an AflIII/HindIII PCR fragment from pUCIRF-1 (Maruyama et al. 1989) into the NcoI/HindIII site of pET28α(+) (Novagen). Both p50/65 and IRF-1 were purified as described previously for other His-tagged proteins (Mayall et al. 1997). Briefly, plasmids were transferred to Escherichia coli strain BL21(DE3) (Novagen) and protein induction initiated with 1 mm IPTG for 2 hr. Bacterial pellets were resuspended in lysis buffer [50 mm TRIS (pH 7.9), 0.1 m KCl, 1% Triton X-100, 12.5 mm MgCl2, 5 mm β-mercaptoethanol, 0.1 mm PMSF, and 2 mg/ml of benzamidine] and disrupted by sonication. The soluble proteins were removed by spinning at 100,000g for 30 min and the pellet was resuspended in lysis buffer containing 8 m urea. After repeated centrifugation the supernatant was supplemented with 10 mm imidazole and the salt was raised to 500 mm KCl before incubating with Ni–NTA agarose resin (Qiagen) for 1 hr with constant agitation. After extensive washing, the proteins were eluted with 200 mm imidazole and peak fractions were pooled and dialyzed stepwise to 25 mm HEPES (pH 8), 12.5 mm MgCl2, 0.4 m KCl, 10% glycerol, 2 mm DTT, and 0.1 mm PMSF before freezing. Murine CP2/LBP-1 (Lim et al. 1992) was produced as a glutathione S-transferase fusion and purified as described by Ron and Dressler (1992) using glutathione–Sepharose 4B affinity resin (Pharmacia). Sp1 was isolated from Jurkat T cells using wheat germ agglutinin (WGA) affinity chromatography as described by Sheridan et al. (1995) or was added as recombinant Sp1 (Promega), as indicated in the legends to Figures 1, 2, 3, 4, 5, 6 (no Sp1 was used in Fig. 7). DNA-binding activity and protein concentrations were established by gel mobility-shift assay and SDS-PAGE. HeLa nuclear extracts (10–20 mg/ml) and purified recombinant proteins (0.3–1 mg/ml) were stored at −100°C before use.
Chromatin assembly reactions
HIV-1 chromatin templates were assembled in vitro with Drosophila S-190 extract, core histones, histone H1, and an ATP-regenerating system prepared as described previously (Kamakaka et al. 1993; Bulger and Kadonaga 1994). Typically, the factors were allowed to bind their sites on DNA for 10–20 min on ice before the addition of the S-190 assembly extract and purified histone components as previously described (Sheridan et al. 1995; Pazin et al. 1996). TSA (5 μm final concentration; Sigma) and TPX (1 μm final concentration) were added together with acetyl CoA (3 μm final concentration) either before (t = 0 hr), during, or after (t = 4.5 hr) the start of assembly of the chromatin templates. The assembly reactions were incubated for a total of 5 hr before aliquots of the chromatin were removed for transcription and structural studies. Although all of the chromatin transcription reactions shown in this paper were supplemented with exogenous acetyl CoA substrate, subsequently we determined that this was unnecessary, indicating that the assembly extracts contain sufficient levels of endogenous substrate to support histone acetylation without supplementation.
For analysis of acetylated histone H4 levels by Western blot, aliquots (>0.5 μl or 15 μl each) of individual chromatin assembly reactions were resolved on an 18% polyacrylamide denaturing gel (SDS-PAGE). Total histone levels were visualized by Coomassie blue staining and immunoblots were carried out using anti-acetylated histone H4 antibody (Lin et al. 1989; 1:500 dilution) using the enhanced chemiluminescence detection system (Amersham Life Science).
In vitro transcription reactions
HIV-1 transcription from chromatin and nonchromatin templates was analyzed by primer extension as described previously (Sheridan et al. 1995; Pazin et al. 1996). Briefly, 100 ng of plasmid DNA or chromatin (taken as a 20-μl aliquot of the chromatin reconstitution reaction) together with 25 ng of α-globin template, was preincubated in a 100-μl reaction with HeLa transcription extract and buffer, followed by addition of rNTPs. For the Sarkosyl inhibition experiments, transcription reactions were incubated with different levels of Sarkosyl a short time (90 sec) after transcription was initiated by the addition of dNTPs. HIV-1 and control α-globin RNAs were detected by primer extension and quantified using a phosphorImaging scanner (Molecular Dynamics).
Chromatin structure analyses
Chromatin structure was analyzed by indirect end-labeling of micrococcal nuclease-digested chromatin and DNaseI hypersensitive site formation as described previously (Pazin et al. 1996). For DNase I hypersensitive site mapping, the aliquots of the same DNase I samples that were analyzed by primer extension footprinting were instead digested with NdeI (−1062) and SphI (+756) (for pHIV-1/LUC DNA), or SphI (+756) and XmnI (−1096) (for pLTR/LUC DNA). The digests were analyzed by agarose gel electrophoresis, transferred to nitrocellulose, and sequentially hybridized with a template-specific primer as described in the legend to Figure 5. For indirect end-labeling experiments, MNase-treated chromatin was digested with NdeI and SphI (for pHIV-1/LUC DNA), or XmnI and SphI (for pLTR/LUC DNA), resolved by agarose gel electrophoresis, and the nitrocellulose blot was probed sequentially with primers specific for pHIV-1/LUC or pLTR/LUC DNA, as indicated in the legend to Figure 5. In all cases, chromatin structure was analyzed before transcription on aliquots of the same templates that were used in the in vitro transcription reactions. Restriction enzyme accessibility experiments were carried out by incubation of 40 μl of unpurified chromatin with individual restriction enzymes (at levels indicated in the legend to Fig. 6) for 15 min at 37°C. The DNA was isolated and digested with either SphI and NdeI (for pHIV-1/LUC DNA) or SphI and XmnI (for pLTR/LUC DNA) before Southern blot transfer to nitrocellulose. Blots were probed subsequently with template-specific primers as defined in the legend to Figure 6.
Acknowledgments
We thank James T. Kadonaga and Beverly M. Emerson for their helpful suggestions and advice throughout the course of this study, Carine Van Lint for providing the plasmid pLTR/LUC before its publication, C. David Allis for providing antibodies to acetylated histone H4, Timothy Hoey for supplying the IRF-1 cDNA plasmid, Adrian Clausell for purified NF-AT protein, and Amy Andrews for her excellent technical assistance on this project. This research was supported by grants to K.A.J. from the National Institutes of Health (GM 38166), The G. Harold and Leila Y. Mathers Foundation, and the California Universitywide State Task Force on AIDS.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jones@salk.edu; FAX (619) 535-8194.
References
- Ayer DE, Lawrence QA, Eisenman RN. Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell. 1995;89:341–347. doi: 10.1016/0092-8674(95)90355-0. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmonson DG, Roth SY, Allis CD. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- Bulger M, Kadonaga JT. Biochemical reconstitution of chromatin with physiological nucleosome spacing. Methods Mol Genet. 1994;5:241–262. [Google Scholar]
- Candau R, Zhou JX, Allis CD, Berger SL. Histone acetyltransferase activity and interaction with ADA2 are critical for GCN5 function in vivo. EMBO J. 1997;16:555–565. doi: 10.1093/emboj/16.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson P, Waterman ML, Jones KA. The hLEF/TCF-1α HMG protein contains a context-dependent activation domain that induces the TCRα enhancer in T cells. Genes & Dev. 1993;7:2418–2430. doi: 10.1101/gad.7.12a.2418. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin R, Schlitz R, Chakravarti D, Nash A, Nagy L, Privalsky M, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- El Kharroubi A, Verdin E. Protein–DNA interactions within DNase I-hypersensitive sites located downstream of the HIV-1 promoter. J Biol Chem. 1994;269:19916–19924. [PubMed] [Google Scholar]
- El Kharroubi A, Martin MA. cis-acting sequences located downstream of the human immunodeficiency virus type 1 promoter affect its chromatin structure and transcriptional activity. Mol Cell Biol. 1996;16:2958–2966. doi: 10.1128/mcb.16.6.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fascher K-D, Schmitz J, Hörz W. Structural and functional requirements for the chromatin transition at the PHO5 promoter in Saccharomyces cerevisiae upon PHO5 activation. J Mol Biol. 1993;231:658–667. doi: 10.1006/jmbi.1993.1317. [DOI] [PubMed] [Google Scholar]
- Gaudreau L, Schmid A, Blaschke D, Ptashne M, Hörz W. RNA polymerase II holoenzyme recruitment is sufficient to remodel chromatin at the yeast PHO5 promoter. Cell. 1997;89:55–62. doi: 10.1016/s0092-8674(00)80182-8. [DOI] [PubMed] [Google Scholar]
- Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese K, Grosschedl R. LEF-1 contains an activation domain that stimulates transcription only in a specific context of factor-binding sites. EMBO J. 1993;12:4667–4676. doi: 10.1002/j.1460-2075.1993.tb06155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese K, Cox J, Grosschedl R. The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell. 1992;69:185–196. doi: 10.1016/0092-8674(92)90129-z. [DOI] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 c-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Hawley DK, Roeder RG. Separation and partial characterization of three functional steps in transcription initiation by human RNA polymerase II. J Biol Chem. 1985;260:8163–8172. [PubMed] [Google Scholar]
- ————— Functional steps in initiation and reinitiation from the major late promoter in a HeLa nuclear extract. J Biol Chem. 1987;262:3452–3461. [PubMed] [Google Scholar]
- Heinzel T, Lavinsky R, Mullen T-M, Soderstrom M, Laherty C, Torchia W-M, Glass C, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- Ito T, Bulger M, Pazin M, Kobayashi R, Kadonaga JT. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell. 1997;90:145–155. doi: 10.1016/s0092-8674(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Jones KA, Luciw PA, Duchange N. Structural arrangements of transcription control domains within the 5′-untranslated leader regions of the HIV-1 and HIV-2 promoters. Genes & Dev. 1988;2:1101–1114. doi: 10.1101/gad.2.9.1101. [DOI] [PubMed] [Google Scholar]
- Kamakaka RT, Bulger M, Kadonaga JT. Potentiation of RNA polymerase II transcription by Gal4-VP16 during but not after DNA replication and chromatin assembly. Genes & Dev. 1993;7:1779–1795. doi: 10.1101/gad.7.9.1779. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Kato H, Horikoshi M, Roeder RG. Repression of HIV-1 transcription by a cellular protein. Science. 1991;251:1476–1479. doi: 10.1126/science.2006421. [DOI] [PubMed] [Google Scholar]
- Laherty CD, Yan W-M, Sun J-M, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate Mad transcriptional repression. Cell. 1997;89:349–356. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- Lim LC, Swendeman SL, Sheffery M. Molecular cloning of the α-globin transcription factor CP2. Mol Cell Biol. 1992;12:828–835. doi: 10.1128/mcb.12.2.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Leone JW, Cook RG, Allis CD. Antibodies specific to acetylated histones document the existence of deposition- and transcription-related histone acetylation in Tetrahymena. J Cell Biol. 1989;108:1577–1588. doi: 10.1083/jcb.108.5.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorch Y, LaPointe JW, Kornberg RD. Initiation on chromatin templates in a yeast RNA polymerase II transcription system. Genes & Dev. 1992;6:2282–2287. doi: 10.1101/gad.6.12a.2282. [DOI] [PubMed] [Google Scholar]
- Maruyama M, Fujita T, Taniguchi T. Sequence of a cDNA coding for human IRF-1. Nucleic Acids Res. 1989;17:3292. doi: 10.1093/nar/17.8.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayall TP, Sheridan PL, Montminy MR, Jones KA. Distinct roles for P-CREB and LEF-1 in TCRα enhancer assembly and activation on chromatin templates in vitro. Genes & Dev. 1997;11:887–899. doi: 10.1101/gad.11.7.887. [DOI] [PubMed] [Google Scholar]
- Mizzen CA, Yang X-J, Kokubo T, Brownell JE, Bannister AJ, Owen-Hughes T, Workman J, Wang L, Berger SL, Kouzarides T, Nakatani Y, Allis CD. The TAFII250 subunit of TFIID has histone acetyltransferase activity. Cell. 1996;87:1261–1270. doi: 10.1016/s0092-8674(00)81821-8. [DOI] [PubMed] [Google Scholar]
- Mymryk J, Archer T. Dissection of progesterone receptor-mediated chromatin remodeling and transcriptional activation in vivo. Genes & Dev. 1995;9:1366–1376. doi: 10.1101/gad.9.11.1366. [DOI] [PubMed] [Google Scholar]
- Nagy L, Kao H-Y, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A and histone deacetylase. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Paranjape SM, Kamakaka RT, Kadonaga JT. Role of chromatin structure in the regulation of transcription by RNA polymerase II. Annu Rev Biochem. 1994;63:265–297. doi: 10.1146/annurev.bi.63.070194.001405. [DOI] [PubMed] [Google Scholar]
- Parker SF, Felzien LK, Perkins ND, Imperiale MJ, Nabel GJ. Distinct domains of adenovirus E1A interact with specific cellular factors to differentially modulate human immunodeficiency virus transcription. J Virol. 1997;71:2004–2012. doi: 10.1128/jvi.71.3.2004-2012.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazin MJ, Kadonaga JT. What’s up and down with histone deacetylation and transcription? Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- Pazin MJ, Kamakaka RT, Kadonaga JT. ATP-dependent nucleosome reconfiguration and transcriptional activation from preassembled chromatin templates. Science. 1994;266:2007–2011. doi: 10.1126/science.7801129. [DOI] [PubMed] [Google Scholar]
- Pazin MJ, Sheridan PL, Cannon K, Cao Z, Keck JG, Kadonaga JT, Jones KA. NF-κB medicated chromatin reconfiguration and transcriptional activation of the HIV-1 enhancer in vitro. Genes & Dev. 1996;10:37–49. doi: 10.1101/gad.10.1.37. [DOI] [PubMed] [Google Scholar]
- Pazin M, Bhargava P, Geiduschek E, Kadonaga J. Nucleosome mobility and the maintenance of nucleosome positioning. Science. 1997;276:809–812. doi: 10.1126/science.276.5313.809. [DOI] [PubMed] [Google Scholar]
- Peterson C, Tamkun J. The SWI-SNF complex: A chromatin remodeling machine? Trends Biochem Sci. 1995;20:143–146. doi: 10.1016/s0968-0004(00)88990-2. [DOI] [PubMed] [Google Scholar]
- Ron D, Dressler H. pGSTag—A versatile bacterial expression plasmid for enzymatic labeling of recombinant proteins. Biotechnology. 1992;13:866–869. [PubMed] [Google Scholar]
- Rothe SY, Allis CD. Histone acetylation and chromatin assembly: A single escort, multiple dances. Cell. 1996;87:5–8. doi: 10.1016/s0092-8674(00)81316-1. [DOI] [PubMed] [Google Scholar]
- Sheridan PL, Sheline CT, Cannon K, Voz ML, Pazin MJ, Kadonaga JT, Jones KA. Activation of the HIV-1 enhancer by the LEF-1 HMG protein on nucleosome-assembled DNA in vitro. Genes & Dev. 1995;9:2090–2104. doi: 10.1101/gad.9.17.2090. [DOI] [PubMed] [Google Scholar]
- Steger DJ, Workman JL. Stable co-occupancy of transcription factors and histones at the HIV-1 enhancer. EMBO J. 1997;16:2463–2472. doi: 10.1093/emboj/16.9.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternglanz R. Histone acetylation: A gateway to transcriptional activation. Trends Biochem Sci. 1996;21:357–358. [PubMed] [Google Scholar]
- Straka C, Hörz W. A functional role for nucleosomes in the repression of a yeast promoter. EMBO J. 1991;10:361–368. doi: 10.1002/j.1460-2075.1991.tb07957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svaren J, Hörz W. Transcription factors vs nucleosomes: Regulation of the PHO5 promoter in yeast. Trends Biochem Sci. 1997;22:93–97. doi: 10.1016/s0968-0004(97)01001-3. [DOI] [PubMed] [Google Scholar]
- Svaren J, Schmitz J, Horz W. The transactivation domain of Pho4 is required for nucleosome disruption at the PHO5 promoter. EMBO J. 1994;13:4856–4862. doi: 10.1002/j.1460-2075.1994.tb06812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature. 1996;383:837–840. doi: 10.1038/383837a0. [DOI] [PubMed] [Google Scholar]
- Tsukiyama T, Becker P, Wu C. ISWI, a member of the SWI2/SNF2 ATPase family, encodes the 140kDa subunit of the nucleosome remodeling factor. Cell. 1995;83:1021–1026. doi: 10.1016/0092-8674(95)90217-1. [DOI] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996a;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996b;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Amella CA, Emiliani S, John M, Jie T, Verdin E. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J Virol. 1997;71:6113–6127. doi: 10.1128/jvi.71.8.6113-6127.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of intergrated human immunodeficiency virus type 1. J Virol. 1991;65:6790–6799. doi: 10.1128/jvi.65.12.6790-6799.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Para Jr P, Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12:3249–3259. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volker JL, Rameh LE, Zhu Q, DeCaprio J, Hansen U. Mitogenic stimulation of resting T cells causes rapid phosphorylation of the transcription factor LSF and increased DNA-binding activity. Genes & Dev. 1997;11:1435–1446. doi: 10.1101/gad.11.11.1435. [DOI] [PubMed] [Google Scholar]
- Wade DA, Pruss D, Wolffe AP. Histone acetylation: Chromatin in action. Trends Biochem Sci. 1997;22:128–132. doi: 10.1016/s0968-0004(97)01016-5. [DOI] [PubMed] [Google Scholar]
- Walters M, Magis W, Fiering S, Eidemiller J, Scalzo D, Groudine M, Martin D. Transcriptional enhancers act in cis to suppress position-effect variegation. Genes & Dev. 1996;10:185–195. doi: 10.1101/gad.10.2.185. [DOI] [PubMed] [Google Scholar]
- Wang L, Mizzen C, Ying C, Candau R, Barlev N, Brownell J, Allis CD, Berger SL. Histone acetyltransferase activity is conserved between yeast and human GCN5 and is required for complementation of growth and transcriptional activation. Mol Cell Biol. 1997;17:519–527. doi: 10.1128/mcb.17.1.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechser MA, Kladde MP, Alfieri JA, Peterson CL. Effects of Sin-versions of histone H4 on yeast chromatin structure and function. EMBO J. 1997;16:2086–2095. doi: 10.1093/emboj/16.8.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlak P, Gaynor RB, Garrard WT. In vitro chromatin assembly of the HIV-1 promoter. ATP-dependent polar positioning of nucleosomes by Sp1 and NFkappaB. J Biol Chem. 1997;272:17654–17661. doi: 10.1074/jbc.272.28.17654. [DOI] [PubMed] [Google Scholar]
- Wolffe AP, Pruss D. Targeting chromatin disruption: Transcription regulators that acetylate histones. Cell. 1996;84:817–819. doi: 10.1016/s0092-8674(00)81059-4. [DOI] [PubMed] [Google Scholar]
- Yang X-J, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- Yoon JB, Li G, Roeder RG. Characterization of a family of related cellular transcription factors which can modulate human immunodeficiency virus type 1 transription in vitro. Mol Cell Biol. 1994;14:1776–1785. doi: 10.1128/mcb.14.3.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Horinouoshi S, Beppu T. Trichostatin A and trapoxin: Novel chemical probes for the role of histone acetylation in chromatin structure and function. BioEssays. 1995;17:423–430. doi: 10.1002/bies.950170510. [DOI] [PubMed] [Google Scholar]