

Abstract
Though potent anti-HIV therapy has spectacularly reduced the morbidity and mortality of human immunodeficiency virus (HIV)-1 infection in the advanced countries, it continues to be associated with substantial toxicity, drug-drug interactions, difficulties in adherence, and abnormal cost. As a result, better effective, safe antiretroviral drugs and treatment strategies keep on to be pursued. In this process, CCR5 (chemokine receptor 5) inhibitors are a new class of antiretroviral drug used in the treatment of HIV. They are designed to prevent HIV infection of CD4 T-cells by blocking the CCR5. When the CCR5 receptor is unavailable, ‘R5-tropic’ HIV (the variant of the virus that is common in earlier HIV infection) cannot engage with a CD4 T-cell to infect the cell. In August 2007, the FDA approved the first chemokine (C-C motif) CCR5 inhibitor, maraviroc, for treatment-experienced patients infected with R5-using virus. Studies from different cohort in regions, affected by clad B HIV-1, demonstrate that 81-88% of HIV-1 variants in treatment naïve patients are CCR5 tropic and that virtually all the remaining variants are dual/mixed tropic i.e., are able to utilize both CCR5 and CXCR4 coreceptors. In treatment experienced patients, 49–78% of the variants are purely CCR5 tropic, 22–48% are dual/mixed tropic, and 2-5% exclusively utilize CXCR4. A 32 bp deletion in the CCR5 gene, which results in a frame shift and truncation of the normal CCR5 protein, was identified in a few persons who had remained uninfected after exposure to CCR5 tropic HIV-1 virus. This allele is common in white of European origin, with prevalence near to 10%, but is absent among East Asian, American Indian, Tamil Indian, and African ethnic groups. HIV-infected individuals, who are heterozygous for CCR5 delta 32, have slower rates of disease progression. The currently available data supports the continuation of the development of CCR5 antagonists in different settings related to HIV-1 infection. If safety issues do not emerge, these compounds could be positioned for use from very early stage of HIV infection to salvage strategies that would be an emerging therapeutic novel strategy for HIV/AIDS patients.
Keywords: aeR5-tropicae, CCR5 inhibitors, CXCR4 coreceptors
INTRODUCTION
CCR5 inhibitors are a new class of antiretroviral drug used in the treatment of human immunodeficiency virus (HIV). They are designed to prevent HIV infection of CD4 T-cells by blocking the CCR5 receptor. When the CCR5 receptor is unavailable, ‘R5-tropic’ HIV (the variant of the virus that is common in earlier HIV infection) cannot engage with a CD4 T-cell to infect the cell. Maraviroc, the first drug from this class to be marketed (as Selzentry in the US, Celsentri in Europe), was licensed in the summer of 2007.
Drugs in this class exploit the knowledge gained, when trying to understand why a small minority of people of northern European descent had a degree of naturally occurring immunity against HIV. It was discovered that a harmless genetic mutation meant that their CCR5 receptors were blocked (which some have speculated must have developed as a defense against the plague epidemics). Thus, denying HIV of its usual means of gaining a foothold.
Genetic testing For HIV/AIDS progression
HIV mirror™ is a revolutionary scientific test, based on the results of National Institute of Health-funded research studies. HIV mirror™ tests the DNA for CCR5 delta 32 and CCR2-64I genes, the two genes that are well-known in the scientific community that may slow down the process in which HIV becomes AIDS. Till recent past, the test for these genes was only available to participants in research studies; but now, these tests are done at many NBA labs.
CCR5 delta 32 and HIV
In the process of HIV infecting the CD4 cells, HIV requires a receptor a “doorway” to gain entry into the CD4 cells. It must bind receptors on the surface of CD4 cells or open the “locks” to gain entry. The first “lock” is opened by binding to the CD4 cell receptor on white blood cells. It then needs to open a second “lock” in order to open the “door” to CD4 cells and infect them; this second “lock” or receptor is called CCR5. Some people have been found to have mutations in the CCR5 gene that codes for this receptor. This mutated gene is called CCR5 delta 32. The CCR5 delta 32 mutation is missing 32 base pairs or pieces of genetic code. This mutation alters the structure of the receptor, so HIV cannot efficiently open the “lock” and enter the cell. Another mutation that has significant effect on HIV disease progression is found in the CCR2 gene.
In August 2007, the FDA approved the first chemokine (C-C motif) CCR5 inhibitor, maraviroc, for treatment-experienced patients infected with R5-using virus. In an effort to better educate physicians and patients regarding these new drugs, this article will review the available clinical data on CCR5 antagonists and discuss implications for clinical practice. Chemokine receptor antagonists are the first antiretrovirals to bind a cellular protein of the host and, as such, stimulate unique safety concerns. Based on the available clinical evidence, a preliminary framework to guide the rational use of these agents in HIV-infected patients is provided [Figure 1A].[1]
Figure 1.
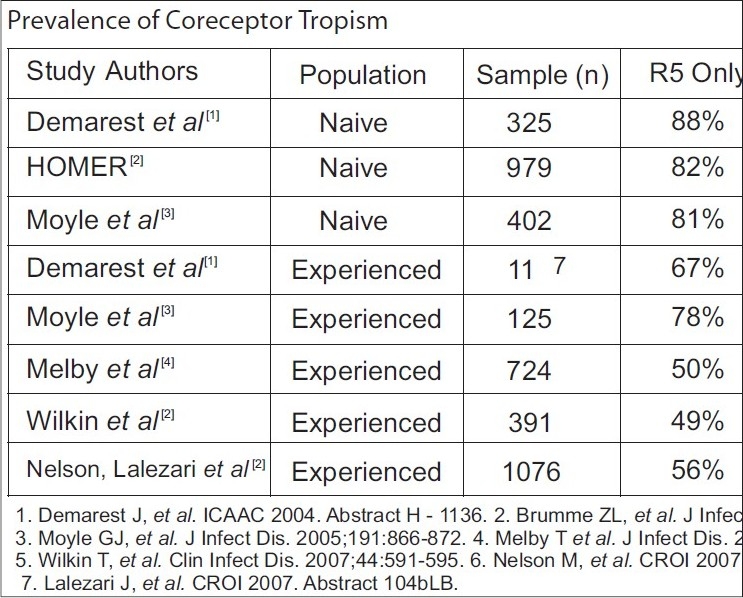
Prevalence of coreceptor tropism in naïve and experienced populations
Recently, an analysis of existing data sets from the AIDS Clinical Trials Group 5211 (ACTG 5211) and the T-20 Versus Optimized Regimen Only (TORO) trials, using the Trofile™ tropism assay, interestingly demonstrates that patients with only X4 virus have CD4 counts similar to patients with only R5 virus and have lower baseline HIV RNA levels than either R5 or dual-mixed viral populations.[5,6] That is, the more rapid disease progression seen with the X4 virus may, in fact, be due to infections with dual-tropic or mixed R5X4 viruses, intimating that the R5 component may have been missed with prior assays geared to detect the syncytium-inducing phenotype alone. As such, infection with pure X4 virus would be less of a concern than infection with dual-tropic or mixed viral populations.
Determining coreceptor usage in clinical samples
Since CCR5 antagonists have anti-HIV activity only against R5-using viruses, all patients being considered for CCR5 antagonist therapy will need a blood test to determine their virus’ coreceptor usage. The drug label for MARAVIROC states that it should be used in the treatment-experienced patients with documented R5 virus (only). This test is required because the use of an R5 antagonist as part of a triple regimen in patients with pure X4 virus exposes the patient to suboptimal therapy with all the concomitant associated risks of virologic failure. Further, the use of an R5 antagonist in patients with R5/X4 dual/mixed-tropic virus may select for the emergence of X4 virus. Clearly, knowing the patient's viral coreceptor usage, or tropism, is critical to the appropriate use of these drugs in day-to-day clinical practice.
In the US, the Trofile™ HIV coreceptor tropism assay (Monogram Biosciences) is commercially available to determine viral tropism and should have a turnaround time of between two and three weeks. Before discussing the two types of tests, one point deserves clarification: viral tropism technically refers to the types of cells (for example, T-cells, macrophages) that a virus can infect, while coreceptor usage defines a virus as either R5 or X4.[7] However, since “tropism” is so often used interchangeably in the literature with coreceptor usage, we will follow the updated common usage of tropism and will refer to viruses as either R5 or X4 tropic.
The Trofile™ HIV Co- Receptor Tropism Assay
The Trofile™ assay, developed and marketed by Monogram Biosciences, determines tropism by isolating and cloning full-length, patient-derived viral env from patient blood samples into a proprietary vector system.[8] The cost of this test is approximately $1,960. As with resistance testing, viral loads above 1,000 copies per mL are required to perform this test with maximum accuracy. Two separate vectors are used: one contains the cloned patient envelope genes and the other the remainder of the HIV genome. The HIV genomic vector contains an intentional self-inactivating deletion in the long terminal repeat (LTR) region that effectively prevents more than one round of viral replication. Thus, the Trofile™ assay is an example of a single-cycle assay [Figure 1].
Test results of this assay supplied to the clinician will indicate whether a virus is R5-tropic, X4-tropic, or dual/mixed. The term dual/mixed refers to the fact that the Trofile™ assay cannot distinguish between the presence of one virus that uses either receptor for viral entry (dual-tropic) or mixed viral populations in the same patient sample that uses either CCR5 or CXCR4. For clinical purposes, this distinction is not as important as knowing whether the sample contains any X4-using virus or not.
The tropism recombinant test (PHENOSCRIPT™ ASSAY)
The Phenoscript™ assay (Euro fins VIRalliance, Inc.) is another recombinant phenotypic assay that will be commercially available for the determination of viral coreceptor usage, although it has not been cross-validated with the Monogram Trofile™ assay. Virus is isolated from patient blood and only a portion of the HIV envelope gene is amplified. With Phenoscript™, infection produces color (rather than light) that can be measured and quantified. Again, these test results will report either R5-tropism, X4-tropism, or dual/mixed-tropism. Although, this assay is in development, it has not been used in any of the clinical trials of CCR5 antagonists, so the interpretation of results will be more challenging.
Analysis: Advantages and challenges in tropism assays
Considerable technical differences between the two tests preclude a direct comparison of the tropism results obtained. A recent article that attempted to do that comparison, found an 85% concordance between these two tests in 74 clinical isolates tested; although, the absence of a gold-standard test made it impossible to determine which test, in fact, delivers the “correct” answer. One important limitation of both tests is the inability to reliably detect minority tropism variants that comprise less than 10% of the total viral population. Monogram has published data reporting limits of detection down to 5%, but no information is provided on the actual amount of virus used to generate those results. In other words, the detection of a tropism variant comprising 5% viral population in a patient with a viral load of 5,000 copies per mL is quite different, and considerably more difficult, than being able to detect 5% of a population in a patient with a viral load of 500,000 copies per mL.
Aplaviroc
Aplaviroc, a GlaxoSmithKline compound, demonstrated antiviral activity with minimal toxicities during short-term monotherapy studies.[9] Aplaviroc in various dosing regimens led to a 1.0 to 1.6 log10 reduction in viral loads during 10 days of treatment.[10] In phase 2b trials, however, four out of roughly 300 patients developed severe hepatotoxicity, that on liver biopsy, was found to be consistent with drug-induced hepatitis. This finding led to the cessation of the trial.[11] Similarly, one out of 26 patients participating in a phase 3 trial of aplaviroc demonstrated elevation of alanine aminotransferase to 24 times normal levels. No deaths occurred, and the hepatitis resolved with aplaviroc discontinuation. This trial was also stopped, and clinical development of aplaviroc was terminated.
Maraviroc
Maraviroc is a CCR5 antagonist with potent in vitro and in vivo anti-HIV-1 activity, recently approved by the FDA for treatment-experienced patients with R5-tropic virus. In a 10-day monotherapy trial, conducted in HIV-1-infected subjects with R5 virus, administration of maraviroc at doses up to 600 mg daily, resulted in a reduction of plasma HIV-1 of at least 1.6 log10.[12] However, a shift in viral tropism was noted in two subjects. One subject experienced transient emergence of a mixed tropic population on 11th day of therapy, which was not detectable on day 40. The other one developed a dual- or mixed-tropic virus by 11th day, which remained detectable through day 433 of follow-up. Additional studies suggested that these viruses emerged from a pretreatment reservoir of X4 viruses that was not detected on initial tropism testing[13] [Figure 2].
Figure 2.
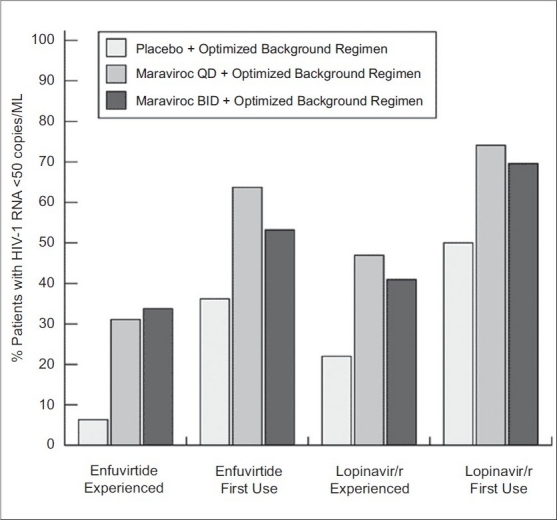
Efficacy of maraviroc with ART study
The maraviroc versus efavirenz regimens as initial therapy (MERIT) study evaluated the activity of maraviroc versus efavirenz, both in combination with zidovudine/lamivudine (ZDV/3TC), in treatment-naive subjects with HIV-1 infection.[14] Inferior efficacy compared with a standard efavirenz-based regimen led to discontinuation of the 300 mg, once-daily arm in the treatment-naive trial, but the 300 mg, twice-daily arm continued. Over 48 weeks, twice-daily maraviroc plus ZDV/3TC failed to demonstrate noninferiority to efavirenz plus ZDV/3TC, in the primary endpoint of HIV-1 RNA below 50 copies per mL. One subject receiving maraviroc at a dose of 300 mg daily, in combination with ZDV/3TC, developed severe hepatotoxicity that necessitated liver transplantation. The subject was infected with hepatitis C virus (HCV), without detectable HCV RNA and had recently initiated other potentially hepatotoxic medications, including isoniazid and trimethoprim/sulfamethoxazole. This patient had elevated transaminases prior to receiving the first dose of maraviroc. The causal role of maraviroc in this patient's liver failure is therefore unclear. Similar cases of severe hepatotoxicity have not been observed in ongoing or completed maraviroc studies.
In MOTIVATE 1 (US and Canada), of the 601 patients enrolled, 585 were treated. Roughly 90% of patients were male and 80% were white. The median entry CD4 counts were 150 to 168 cells per mm3 with viral loads of 4.84 to 4.86 log10 copies per mL. A total of 40% patients received a regimen containing enfuvirtide, while approximately 70% subjects had two or fewer active drugs in their OBR arm.
In MOTIVATE 2 (US, Europe, and Australia), 464 of 475 enrolled subjects received treatment with CD4 counts of 174 to 182 cells per mm3 and mean viral loads between 4.84 and 4.89 log10 copies per mL. Mean change in HIV-1 viral load at 24 weeks was -0.93 log reduction for placebo, and -1.95 and -1.97 for OBR + maraviroc once and twice daily, respectively. Viral load reductions below 50 copies per mL, were seen in 45.6% of subjects receiving maraviroc QD compared to 40.9% receiving maraviroc BID, and 20.9% receiving placebo. Also, increase in Mean CD4 counts were: 64 cells per mm3 in placebo, 112 cells per mm3 in OBR + maraviroc QD, and 102 cells per mm3 in OBR + maraviroc BID. In contrast to MOTIVATE 1, no increase in liver enzyme elevations was observed in subjects taking maraviroc over those taking placebo [Figures 3–5].
Figure 3.
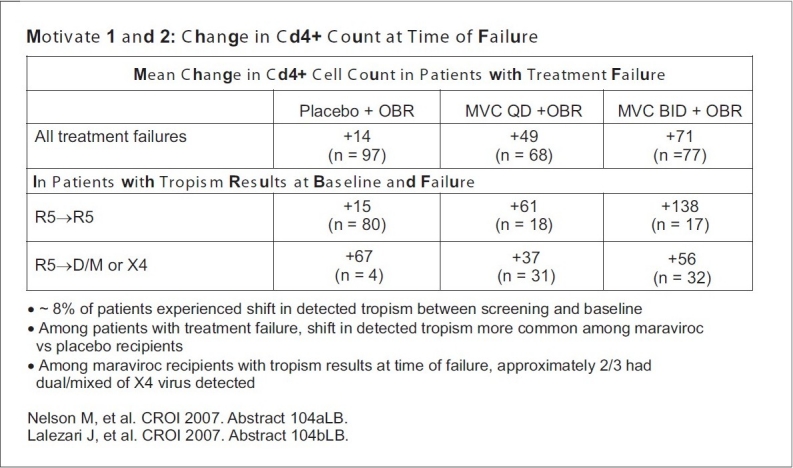
Changes in CD4 counts in treatment failure cases
Figure 5.
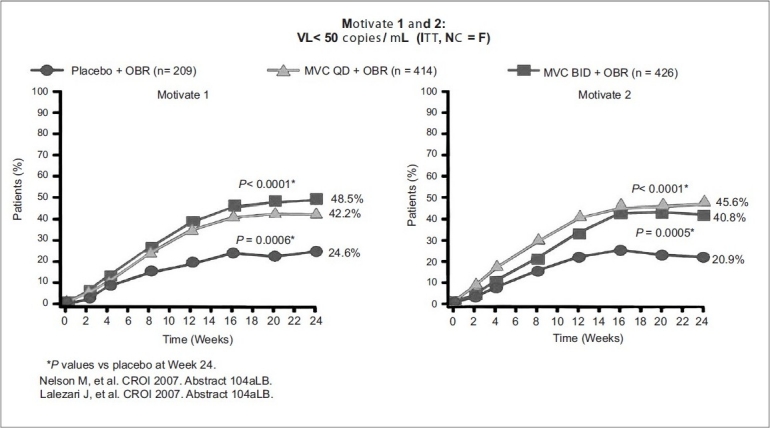
Efficacy of maraviroc OD and BID dosage plus OBR Vs placebo study VL <50 copies /ml
Figure 4.
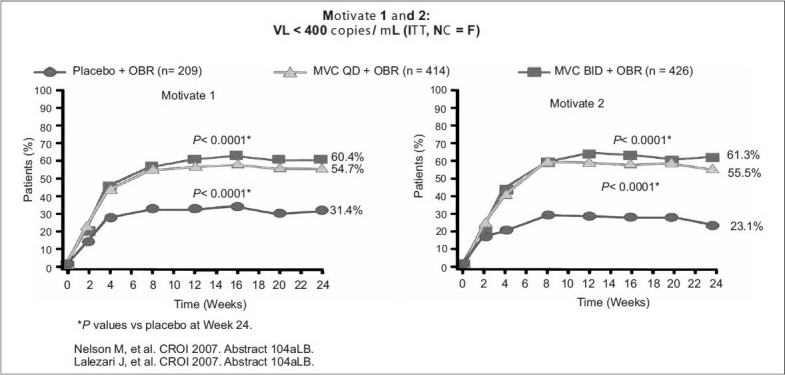
Efficacy of maraviroc OD and BID dosage plus OBR Vs placebo study VL <400 copies /ml
Treatment outcome In Motivate 1 and 2 studies
Inclusion of maraviroc and at least one other active antiretroviral, either enfuvirtide or lopinavir/ritonavir depicted here, led to a greater percentage of subjects achieving viral loads < 50 copies/mL. Note that only twice-daily maraviroc, is FDA approved for clinical use; adapted with permission from van der Ryst E, et al. Efficacy of maraviroc in combination with at least one other potent new antiretroviral drug: 24-week combined analysis of the MOTIVATE 1 and 2 studies.
Vicriviroc
Vicriviroc, formerly known as SCH-D, is 2- to 40-fold more potent in vitro than the first-generation compound, Schering C (SCH-C). Similar to maraviroc, this molecule blocks signaling by the C-C chemokines at nanomolar concentrations. Testing in healthy volunteers or HIV-1-infected subjects has not, to date, revealed QTc prolongation or central nervous system (CNS) adverse effects. Vicriviroc is metabolized by CYP3A4 and, thus, can be boosted with ritonavir. Data from a 14-day monotherapy trial demonstrated a reduction of plasma HIV-1 RNA by approximately 1.0 to 1.5 log10 copies per mL [Figure 6].
Figure 6.
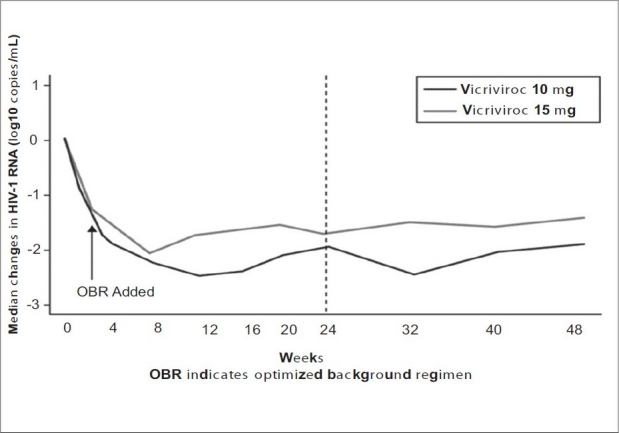
Vicriviroc 48 week study
Vicriviroc was recently evaluated in a phase 2b trial in antiretroviral-experienced patients by the ACTG 5211. That study compared the safety and efficacy of three different vicriviroc dosages in combination with an OBR to OBR alone.[15] The primary endpoint was change in plasma HIV-1 RNA at day 14, with a secondary endpoint of the safety, tolerability, and HIV-1 RNA changes at 24 weeks. At 14 days and 24 weeks, mean changes in HIV-1 RNA (log10 copies/mL) were greater in the vicriviroc groups: –0.87 and –1.51 (5 mg), –1.15 and –1.86 (10 mg), and –0.92 and –1.68 (15 mg), than in the placebo group, +0.06 and –0.29 (P < 0.01). Grade 3/4 adverse events were similar across groups. The 48-week results were recently reported and demonstrated a sustained virologic response in subjects receiving Vicriviroc.[16]
48 -week data from ACTG 5211
The benefits in viral load reductions previously reported after 24 weeks of follow-up were maintained at 48 weeks in both the vicriviroc 10 mg and 15 mg, once-daily arms of this trial. Virologic failure was, however, associated with a change in coreceptor usage in 35% of vicriviroc-treated subjects. Interim results raised concern, when five cases of malignancy occurred in subjects receiving vicriviroc: one case of recurrent Hodgkin's disease; one case of nonHodgkin's disease in a subject with previously treated Hodgkin′s; two de novo cases of lymphoma (one Hodgkin's and one nonHodgkin′s); and one case of gastric adenocarcinoma. No lymphomas have been reported to date in the control arm.
Because of the small sample size (N = 118) and the participants’ advanced stage of HIV disease, the significance of these results is unclear, and a causal link with vicriviroc has yet to be established. No link to increased Epstein-Barr virus (EBV) viral replication was seen in subjects receiving vicriviroc as part of this study.[17] Vicriviroc has not previously been shown to be carcinogenic or mutagenic, and preclinical toxicology studies in animals showed no increased malignancy risk. To date, severe hepatitis has not been observed in clinical trials of vicriviroc.
HIV resistance to CCR5 inhibitors
The high error rate of HIV-1 reverse transcriptase and rapid turnover of the viral population facilitate the emergence of drug-resistant mutants under conditions of partial drug efficacy. In the case of CCR5 inhibitors, the drug target is a host cell protein that will not undergo mutation in response to CCR5 antagonist therapy. However, viral adaptation to CCR5 inhibitors could involve changes in the viral envelope protein that alters dependence on CCR5. Greatest theoretical concern is the potential selection of CXCR4-tropic virus in response to CCR5 blockade. Whether emergence of X4 viruses in this setting would accelerate disease progression, is the subject of much speculation. True resistance appears to be mediated by changes in HIV-1 gp120 that allows binding to the complex of the CCR5 receptor and bound drug.[18,19]
Maraviroc-resistant HIV-1 variants have been generated by serial passage in vitro.[20] For one virus isolate, two mutations in the V3 loop, T316A and V323I, were associated with maraviroc resistance; a third V3 loop mutation, A319S, was not consistently observed. Standard drug susceptibility testing showed a decrease in the percent maximal inhibition achievable with maraviroc, without an appreciable shift in the IC50. This pattern has been attributed to the noncompetitive nature of maraviroc inhibition of gp120 binding to CCR5, and it suggests that resistant viruses have developed the capacity to bind the CCR5 receptor-drug complex. Maraviroc-resistant recombinants retained sensitivity to both aplaviroc and enfuvirtide. Aplaviroc-resistant viruses also have been isolated after serial in vitro passage. All viruses retained CCR5 tropism. However, the cells used for passage had either no or low levels of CXCR4, possibly limiting the opportunity for CXCR4 variants to emerge. In contrast to maraviroc resistance, aplaviroc resistance was characterized by a rightward shift in the IC50 and no change or plateau in the percent maximal inhibition.[21] Mutations that conferred decreased sensitivity to aplaviroc did not cluster in the V3 loop and were found in the C1-5, V1, and V3 regions of gp120 and within gp41, as well. In addition, the phenotypic effects of these mutations depended on the env context in which they were expressed. That is, mutations that emerge in a given env backbone do not confer resistance when engineered into env from a different viral strain.
Clonal analysis of V3 loop sequences obtained from subjects experiencing virologic failure while taking vicriviroc, showed evidence of genetic selection of the V3 loop sequences, although no common pattern of mutations could be identified across samples from different subjects.[22] As in the case of maraviroc resistance, the V3 loop changes that emerged with vicriviroc treatment were associated with a decrease in the percent maximal inhibition achievable. The contribution to vicriviroc susceptibility of other envelope regions outside the V3 loop still remains to be determined.
Resistance to CCR5 antagonists emerges as viruses, which acquire the ability to use the inhibitor-bound form of CCR5 for viral entry. Decreases in the percent maximal suppression seen with the emergence of CCR5 antagonist resistance is a function of the allosteric, noncompetitive nature of small-molecule CCR5 antagonism and reflects the ability of the virus to use either inhibitor-bound CCR5 or unbound CCR5 for entry. As the virus becomes more efficient at using antagonis bound CCR5 for entry, the height of the plateau decreases. Eventually, plateau height will decrease below 50%, making it impossible to calculate an IC50 for the resistant virus.
CCR5 blockade
Although G-protein-coupled receptors have been the targets of pharmacologic inhibition in the past, chemokine inhibitors are the first antiretroviral drugs that target host proteins. The apparent absence of significant immunologic deficits among individuals with naturally occurring mutations (i.e., CCR5-Δ32 homozygotes) that result in a lack of functional CCR5, provides some reassurance that pharmacologic blockade of CCR5 will be relatively benign.[23] Presumably, redundancy in the chemokine network allows other chemokine receptors to subsume the function of CCR5. However, pharmacologic blockade of a receptor in mature individuals may have different consequences than congenital absence of the receptor. Thus, the long-term safety of CCR5 blockade remains to be proven.
The murine studies have been corroborated by studies in humans showing that CCR5Δ32 heterozygotes have a six-fold increased risk for severe morbidity from West Nile virus infection and a five-fold increased risk of mortality.[24] The immunologic importance of functional CCR5 is underscored by several studies demonstrating improved outcomes in patients with immunologically mediated conditions such as rheumatoid arthritis and organ transplantation, among others, in CCR5Δ32 homozygotes.[25,26]
Clinical uses of CCR5 antogonists
For at least the next one to two years, and perhaps into the foreseeable future, maraviroc is the only FDA-approved CCR5 inhibitor available for clinical use. As such, a discussion of its most appropriate use in three different segments of the patient population is warranted.
Antiretroviral NAÏVE patients
Evidence of increased rates of treatment failures has been reported for vicriviroc and maraviroc. Given the impressive potency of currently recommended first-line regimens, and the theoretical concerns of adverse events (i.e., infection, malignancy with vicriviroc), the use of a CCR5 antagonist in antiretroviral-naive patients is currently not recommended.
Antiretroviral-experienced patients
Most of the clinical data with maraviroc have been derived from its use in this patient population. As such, the available phase-3 data supports the use of maraviroc for this patient subgroup. All patients being considered for maraviroc therapy should first be screened by a tropism assay. Results demonstrating the presence of dual-mixed or X4 virus should preclude the use of maraviroc. Since achieving an undetectable viral load is the goal of therapy in all patients, maraviroc will best be used when it can be combined with at least two and preferably three, other active drugs. The availability of newer-generation protease, nonnucleoside reverse transcriptase, and integrase inhibitors makes this a more realistic goal in patients for perhaps the first time in years, if ever. A recent comparison of once- and twice-daily maraviroc in the MOTIVATE studies suggested that twice-daily dosing was more effective, and the drug labeling recommends maraviroc be dosed twice daily. Because of drug interactions, the dose of maraviroc must be adjusted for concomitant medications as follows: 300 mg twice daily (nucleoside reverse transcriptase inhibitor [NRTI], enfuvirtide, tipranavir/ritonavir, nevirapine); 150 mg twice daily (all other ritonavir-boosted PIs); and 600 mg twice daily (efavirenz, rifampin) [Figure 7].
Figure 7.
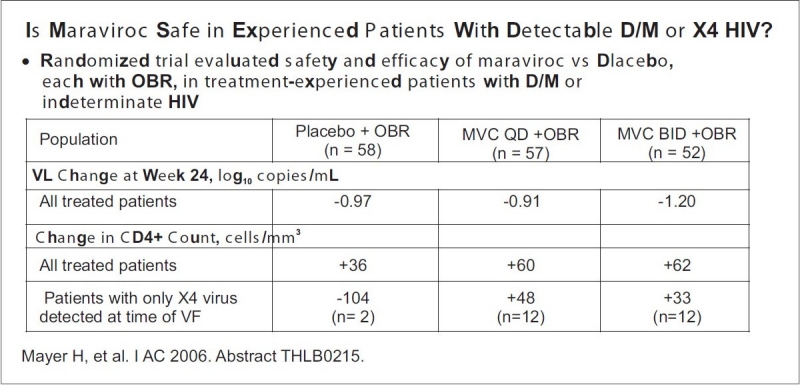
Safety and efficacy of maraviroc Vs placebo study
SUMMARY
The CCR5 antagonists are a welcome addition to the therapeutic armamentarium available for antiretroviral-experienced patients. Currently, their use in antiretroviral-naive patients should be restricted to enrollment in ongoing or planned clinical trials. The CCR5 antagonist maraviroc is FDA-approved for treatment-experienced patients with R5 virus (only), and no patient should receive maraviroc without first undergoing a tropism assay. Although, the cost of this additional test is substantial, the possibility of combining maraviroc with at least two other active drugs to achieve undetectable viral loads may justify the added expense. Ongoing clinical monitoring will provide important follow-up data to address the theoretical safety concerns surrounding CCR5 inhibition. The approval of drugs from several new drug classes onto the market in 2007 through 2008 will herald a major step forward in the treatment of antiretroviral-experienced patients with multidrug-resistant HIV-1.
Footnotes
Delivered the prestigious Institute of Venereology silver jubilee oration during IASSTD & AIDS {ASTICON 2008} at kodaikanal on 4th Oct 2008.
Source of Support: Nil,
Conflict of Interest: None declared.
REFERENCES
- 1.Kwong PD, Wyatt R, Sattentau QJ, Sodroski J, Hendrickson WA. Oligomeric modeling and electrostatic analysis of the gp120 envelope glycoprotein of human immunodeficiency virus. J Virol. 2000;74:1961–72. doi: 10.1128/jvi.74.4.1961-1972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilkin TJ, Su Z, Kuritzkes DR, Hughes M, Flexner C, Gross R, et al. HIV type 1 chemokine coreceptor use among antiretroviral-experienced patients screened for a clinical trial of a CCR5 inhibitor: AIDS Clinical Trial Group A5211. Clin Infect Dis. 2007;44:591–5. doi: 10.1086/511035. [DOI] [PubMed] [Google Scholar]
- 3.Goodenow MM, Collman RG. HIV-1 coreceptor preference is distinct from target cell tropism: A dual-parameter nomenclature to define viral phenotypes. J Leukoc Biol. 2006;80:965–72. doi: 10.1189/jlb.0306148. [DOI] [PubMed] [Google Scholar]
- 4.Whitcomb JM, Huang W, Fransen S, Limoli K, Toma J, Wrin T, et al. Development and characterization of a novel single-cycle recombinant-virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Antimicrob Agents Chemother. 2007;51:566–75. doi: 10.1128/AAC.00853-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trouplin V, Salvatori F, Cappello F, Obry V, Brelot A, Heveker N, et al. Determination of coreceptor usage of human immunodeficiency virus type 1 from patient plasma samples by using a recombinant phenotypic assay. J Virol. 2001;75:251–9. doi: 10.1128/JVI.75.1.251-259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lalezari J, Thompson M, Kumar P, Piliero P, Davey R, Patterson K, et al. Antiviral activity and safety of 873140, a novel CCR5 antagonist, during short-term monotherapy in HIV-infected adults. AIDS. 2005;19:1443–8. doi: 10.1097/01.aids.0000183633.06580.8a. [DOI] [PubMed] [Google Scholar]
- 7.Nichols W, Steel HM, Bonny T, Min S, Curtis L, Kabeya K, Clumeck N. Targeting HIV Entry—1st International Workshop. Bethesda, Md: 2005. Hepatotoxicity observed in clinical trials of aplaviroc (APL, 873140) [abstract] pp. 2–3. [Google Scholar]
- 8.Fätkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11:1170–2. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 9.Westby M, Lewis M, Whitcomb J, Youle M, Pozniak AL, James IT, et al. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J Virol. 2006;80:4909–20. doi: 10.1128/JVI.80.10.4909-4920.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saag M, Ive P, Heera J, et al. Program and abstracts of the 4th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention. Sydney, Australia: 2007. Jul 22-25, A multicenter, randomized, double-blind, comparative trial of a novel CCR5 antagonist, maraviroc versus efavirenz, both in combination with Combivir (zidovudine ZDV]/lamivudine [3TC]), for the treatment of antiretroviral naive subjects infected with R5 HIV-1: Week results of the MERIT study [abstract] Abstract WESS104. [Google Scholar]
- 11.Mayer H. Targeting HIV Entry—1st International Workshop. Bethesda, Md: 2005. Dec 2-3, Maraviroc: A case of severe hepatotoxicity [abstract] Abstract LB. [Google Scholar]
- 12.Gulick RM, Su Z, Flexner C, et al. Phase II study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-infected, treatment-experienced patients: ACTG 5211. J Infect Dis. 2007 doi: 10.1086/518797. In press. [DOI] [PubMed] [Google Scholar]
- 13.Gulick RM. Program and abstracts of the 4th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention. Sydney, Australia: 2007. Jul 22-25, ACTG 5211: Phase II study of the safety and efficacy of vicriviroc (VCV) in HIV+ treatment-experienced subjects: 48-week results [abstract] Abstract TUAB102. [Google Scholar]
- 14.Cohen C, DeJesus E, Mills A, et al. Program and abstracts of the 4th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention. Sydney, Australia: 2007. Jul 22-25, Potent antiretroviral activity of the once-daily CCR5 antagonist INCB009471 over 14 days of monotherapy [abstract] Abstract TUAB106. [Google Scholar]
- 15.Pugach P, Marozsan AJ, Ketas TJ, Landes EL, Moore JP, Kuhmann SE. HIV-1 clones resistant to a small molecule CCR5 inhibitor use the inhibitor-bound form of CCR5 for entry. Virology. 2007;361:212–28. doi: 10.1016/j.virol.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westby M, Smith-Burchnell C, Mori J, Lewis M, Mosley M, Stockdale M, et al. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol. 2007;81:2359–71. doi: 10.1128/JVI.02006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosley M, Mori J, Smith-Burchnell C, et al. Targeting HIV Entry—1st International Workshop. Bethesda, Md: 2005. Dec 2-3, Resistance to the CCR5 antagonist maraviroc is associated with V3 loop-specific mutations and dose response curves characterized by a reduction in maximal inhibition (plateau) [abstract] Abstract 11. [Google Scholar]
- 18.LaBranche C, Kitrinos K, Howell R, McDanal C, Harris S, Jeffrey J, Demarest J. Targeting HIV Entry—1st International Workshop. Bethesda, Md: 2005. Jul 22-25, Clonal analysis of in vitro-derived viruses with reduced susceptibility to aplaviroc (873140, APL) shows wide ranges in IC50 and sequence changes [abstract] Abstract 9. [Google Scholar]
- 19.Landovitz R, Faetkenhauer G, Hoffmann C, et al. XV International HIV Drug Resistance Workshop. Sitges, Spain: 2006. Characterization of susceptibility profiles for the CCR5 antagonist vicriviroc in treatment-naive HIV-infected subjects. [Google Scholar]
- 20.de Silva E, Stumpf MP. HIV and the CCR5-Delta32 resistance allele. FEMS Microbiol Lett. 2004;241:1–12. doi: 10.1016/j.femsle.2004.09.040. [DOI] [PubMed] [Google Scholar]
- 21.Huffnagle GB, McNeil LK, McDonald RA, Murphy JW, Toews GB, Maeda N, et al. Cutting edge: Role of C-C chemokine receptor 5 in organ-specific and innate immunity to Cryptococcus neoformans. J Immunol. 1999;163:4642–6. [PubMed] [Google Scholar]
- 22.Prahalad S. Negative association between the chemokine receptor CCR5-Delta32 polymorphism and rheumatoid arthritis: A meta-analysis. Genes Immun. 2006;7:264–8. doi: 10.1038/sj.gene.6364298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischereder M, Luckow B, Hocher B, Wuthrich RP, Rothenpieler U, Schneeberger H, et al. CC chemokine receptor 5 and renal-transplant survival. Lancet. 2001;357:1758–61. doi: 10.1016/s0140-6736(00)04898-4. [DOI] [PubMed] [Google Scholar]
- 24.Gulick RM, van der Ryst E, Lampiris H, et al. 4th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention. Sydney, Australia: 2007. Efficacy and safety of once-daily (QD) compared with twice-daily (BID) maraviroc plus optimized background therapy (OBT) in treatment-experienced patients infected with CCR5-tropic-HIV-1: 24-week combined analysis of the MOTIVATE 1 and 2 studies [abstract] Abstract WEPEB116LB. [Google Scholar]