

Abstract
We have demonstrated previously that cyclo-oxygenase-2 (COX2), the rate-limiting enzyme in the biosynthesis of prostaglandins (PGs), is essential for blastocyst implantation and decidualization. However, the candidate PG(s) that participates in these processes and the mechanism of its action remain undefined. Using COX2-deficient mice and multiple approaches, we demonstrate herein that COX2-derived prostacyclin (PGI2) is the primary PG that is essential for implantation and decidualization. Several lines of evidence suggest that the effects of PGI2 are mediated by its activation of the nuclear hormone receptor PPARδ, demonstrating the first reported biologic function of this receptor signaling pathway.
Keywords: COX2, prostaglandins, PPARδ, mouse, implantation
Prostaglandins (PGs) participate in a variety of cellular functions including modulation of vascular tone, cell proliferation, and differentiation. These lipid mediators are generated via the cyclo-oxygenase (COX) pathway; COX is the rate-limiting enzyme for the conversion of arachidonic acid into prostaglandin H2 (PGH2), the common substrate for various PG synthases (Smith and DeWitt 1996). COX exists in two isoforms that are encoded by two separate genes. Gene targeting in mice has established distinct functions for these isoforms. Whereas COX1-deficient females are fertile with specific parturition defects, COX2-deficient females exhibit multiple reproductive failures (Dinchuk et al. 1995; Langenbach et al. 1995; Lim et al. 1997). For example, COX2 mutant females exhibit ovulation and fertilization defects, suggesting a role for ovarian COX2 in ovulation and oocyte maturation. Further, the expression of uterine COX2 in an implantation-specific manner in wild-type mice and defective implantation and decidualization in COX2-deficient mice establish that uterine COX2 is essential for these processes (Chakraborty et al. 1996; Lim et al. 1997). However, the candidate PG(s) derived from uterine COX2 and its mode of action in these events remain unanswered.
COX1 and COX2 are expressed in both the endoplasmic reticular (ER) membrane and the nuclear envelope (Spencer et al. 1998), suggesting that PGs are poised to function via two different classes of receptor systems. PGs synthesized in the ER can exit cells and function through G protein-coupled cell surface receptors that are linked to different cytoplasmic signaling pathways (Negishi et al. 1995). In the mouse, cell surface receptors for the prostaglandins PGE2, PGF2α, PGD2, prostacyclin (PGI2), and thromboxanes have been cloned as EP, FP, DP, IP, and TP, respectively (for review, see Negishi et al. 1995). Furthermore, PGE2 binds and activates a set of functionally distinct EP receptor subtypes, EP1, EP2, EP3, and EP4, which are classified on the basis of their responses to various PGE2 agonists or antagonists (Negishi et al. 1995). In contrast, PGs produced via nuclear COX can exert their effects directly on the nucleus by interacting with peroxisome proliferator-activated receptors (PPARs), which belong to nuclear hormone receptor superfamily (Mangelsdorf and Evans 1995). In the mouse, three members of the PPAR family have been cloned and identified as PPARα, PPARγ, and PPARδ(β) (Kliewer et al. 1994). These different isoforms exhibit different expression patterns and ligand dependency (Forman et al. 1995, 1997; Kliewer et al. 1994, 1995, 1997). For example, PPARα is highly expressed in the liver and is implicated in lipid homeostasis. It is activated by synthetic hypolipidemic drugs of the fibrate class, long-chain fatty acids, leukotriene B4 and PGI2 agonists, and regulates expression of several genes involved in lipid metabolism (for review, see Devchand et al. 1996; Schoonjans et al. 1996a,b; Forman et al. 1997; Kliewer et al. 1997; Motojima et al. 1998). PGI2 agonists carbaprostacyclin (cPGI) and iloprost can also directly interact with PPARδ (Forman et al. 1997; Kliewer et al. 1997). PPARγ is mainly expressed in white adipose tissue and is implicated in adipocyte differentiation. Ligands for PPARγ include the antidiabetic thiazolidinedione (TZD) drugs and a metabolite of PGJ2, 15-deoxy-Δ12,14-PGJ2 (Tontonoz et al. 1994; Forman et al. 1995, 1997; Kliewer et al. 1995, 1997). The activation of this receptor also terminally differentiates liposarcoma and breast carcinoma cells, suggesting a role in cell cycle regulation (Altoik et al. 1997; Tontonoz et al. 1997; Mueller et al. 1998). The identification of PPAR as a receptor for liver peroxisome proliferators stimulated extensive investigations regarding the role of these receptors in lipid metabolism and homeostasis (Reddy and Krishnakantha 1975; Isseman and Green 1990; Schoonjans et al. 1996a). However, emerging evidence suggests that PPARα and PPARγ mediate other biological functions in different cellular contexts (Jiang et al. 1998; Ricote et al. 1998; Staels et al. 1998; Tontonoz et al. 1998). In contrast, information regarding potential roles of PPARδ in any physiological and/or developmental processes is very limited, although this gene is expressed in various embryonic and adult tissues (Kliewer et al. 1994; Braissant et al. 1996; Braissant and Wahli 1998).
PPARs modulate transcription by binding to sequence-specific PPAR response elements (PPRE) in promoters of target genes, resulting in activation or suppression of transcription with respect to promoter context (Muerhoff et al. 1992; Tugwood et al. 1992). PPARs must heterodimerize with retinoid X receptors (RXRs) that functions as promiscuous partners for various nuclear receptors during transcriptional regulation (Kliewer et al. 1992). This heterodimer recruits transcriptional coactivators such as steroid receptor coactivator-1 (SRC-1) and CREB-binding protein (CBP) for transcriptional activation when exposed to PPAR-specific ligands (Direnzo et al. 1997; Zhou et al. 1998). In the presence of both PPAR- and RXR-specific ligands, this type of interaction confers synergistic activation of target genes (Mukherjee et al. 1997; Schulman et al. 1998).
Although our previous investigation demonstrated that uterine COX2 is essential for normal blastocyst implantation and decidualization (Lim et al. 1997), the specific PG(s) involved in these processes and its mechanism of action were not clearly understood. The present investigation establishes PGI2 as a key mediator in the process of implantation and decidualization. Moreover, it participates in these processes via the PPARδ and RXRα signaling pathway in the uterus.
Results
PGI2 is the most abundant PG at the implantation sites
In the rodent, the first sign for the attachment reaction is an increase in endometrial vascular permeability at the sites of blastocyst apposition. This coincides with the initial attachment reaction between the uterine luminal epithelium and blastocyst trophectoderm. In the mouse, these events occur at 2200–2300 hr on day 4 (day 1 = vaginal plug) of pregnancy (Dey 1996). Because of their vasoactive nature, PGs have been implicated in this process. To address which of the PGs mediates the process of implantation and decidualization, we first measured levels of various PGs at the implantation and interimplantation sites on day 5 of pregnancy in wild-type mice. Implantation and interimplantation sites were pooled from 3–10 wild-type mice and assayed for PGs by gas chromatography/mass spectrometry (GC/MS). As shown in Figure 1, the level of PGI2 was the highest followed by PGE2 and other prostanoids. In addition, the level of PGI2 was significantly higher at the implantation sites compared with the interimplantation sites. These PGs are primarily generated by uterine COX2, as uterine COX1 expression is very low or undetectable during this time (Chakraborty et al. 1996). Because PGI2 is a potent vasoactive agent (Moncada 1983) and participates in vascular permeability changes (Murohara et al. 1997; Wheeler-Jones et al. 1997), this lipid mediator may be associated with increased localized vascular permeability in the uterus during implantation.
Figure 1.

Levels of PGs in implantation and interimplantation sites on day 5 of pregnancy. Implantation (open bars) or interimplantation (patterned bars) sites were pooled from 3–10 wild-type mice. PGI2 was measured as 6-keto-PGF1α. Mean ± s.e.m.. The levels of PGI2 were higher in the implantation sites than those in the interimplantation sites as well as the PGE2 levels in the implantation sites (P < 0.05, paired t-test). These experiments were repeated several times as indicated within parentheses.
PGI synthase is expressed at the implantation sites
Because PGI2 is the most abundant PG at the implantation sites in wild-type mice, we examined cellular sites of PGI2 synthesis in the wild-type mouse uterus during early pregnancy. PGI2 is synthesized from PGH2 by PGI synthase (PGIS), a membrane-bound heme protein of the P450 family (Tone et al. 1997). Spatiotemporal expression of PGIS was examined by Northern blot, in situ hybridization, and immunocytochemistry (Smith et al. 1983; Geraci et al. 1997). A transcript of ∼2.0 kb, as reported previously (Tone et al. 1997), was detected in the pregnant uterus (data not shown). In situ hybridization revealed temporal and cell-specific expression in the peri-implantation uterus (Fig. 2). On days 1–4 (0900 hr), modest levels of autoradiographic signal were noted in the myometrium and subsets of stromal cells. In contrast, an abundant accumulation of PGIS mRNA was localized around the implanting blastocyst with distinct accumulation in the stromal vasculature on day 5. With the progression of implantation (days 6–8), strikingly condensed expression of PGIS was noted in the ectoplacental cone and trophoblast cells at the maternal–embryonic interface as well as in the embryo proper. In addition, cells lining the vascular sinusoids at the mesometrial pole also expressed this mRNA. These cells are implicated in angiogenesis and/or placentation, as they also express vascular endothelial growth factor (VEGF) and its receptors in a similar pattern (Chakraborty et al. 1995). Immunocytochemistry on day 5 implantation sites revealed differential subcellular expression of PGIS. As shown in Figure 2, e–f, PGIS immunoreactivity was noted both in the nucleus and cytoplasm of stromal cells surrounding the implanting embryo. This is consistent with the previous report of localization of PGIS in the nuclear and ER membranes of smooth muscle cells of various organs (Smith et al. 1983). It is to be noted that uterine COX2 protein on day 5 of pregnancy exhibits similar differential subcellular pattern (Lim et al. 1997), suggesting generation of PGI2 at the same subcellular sites.
Figure 2.
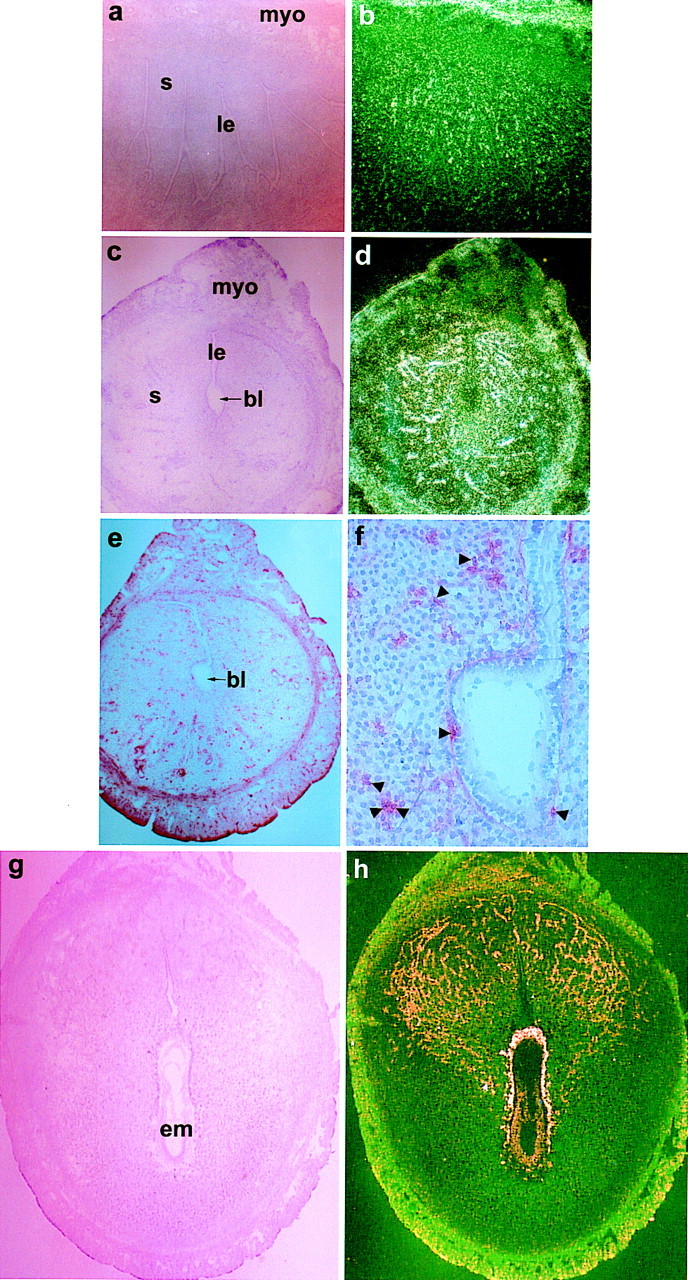
Expression of PGIS in the peri-implantation mouse uterus. In situ hybridization of PGIS mRNA at 0900 hr on days 4 (a,b), 5 (c,d), and 8 (g,h) of pregnancy using a 35S-labeled cRNA probe are shown in bright-field (a,c,g) and dark-field (b,d,h) photomicrographs at 40× (a–d) or at 20× (g,h). Immunolocalization of PGIS in day 5 implantation site is shown at 40× (e) and 100× (f). (le) Luminal epithelium; (s) stroma; (myo) myometrium; (bl) blastocyst; (em) embryo. (Arrowheads) Perinuclear localization of PGIs.
Nuclear PGI2 receptor PPARδ and its obligate partners RXRs are expressed at implantation sites
The above results suggest that PGI2 is generated by the COX2/PGIS system in stromal cells at the site of implantation. To understand the possible mechanism by which COX2-derived PGI2 exerts its effects during implantation, we examined the spatiotemporal expression of known cell surface and nuclear receptors for PGI2. A G protein-coupled cell surface receptor that activates cAMP and inositol phosphate production has been identified for PGI2 as IP (Namba et al. 1994). However, this gene is apparently not critical in reproduction, as both female and male homozygous null mutant mice for IP are fertile (Murata et al. 1997). We also observed that IP mRNA levels are very low or undetectable in the uterus before and after implantation (Fig. 3) suggesting that this receptor is not crucial for PGI2 action during implantation. Thus, we examined the expression of known nuclear receptors that are activated by PGI2 agonists, that is, PPARα and PPARδ, as well as PPARγ, a receptor for 15-deoxy-Δ12,14-PGJ2 (Forman et al. 1995, 1997). In situ hybridization demonstrated that PPARα expression is undetectable or very low in the uterus during the peri-implantation period as is PPARγ (Fig. 3). In contrast, uterine PPARδ expression during this period has a distinctive pattern. For example, this gene is undetectable in the preimplantation uterus (days 1–4, 0900 hr) but is markedly induced in the stroma surrounding the implanting blastocyst on the night of day 4 (2200–2300 hr) when the attachment reaction occurs (Fig. 4A). Furthermore, this expression becomes localized around the embryo on the morning of day 5 and is abundantly expressed in the decidua during the postimplantation period (days 6–8) (Fig. 4A). The prominent induction of PPARδin the absence of either IP or PPARα expression strongly suggests that PPARδ mediates PGI2 effects in implantation and decidualization. Normal fertility in PPARα-deficient female mice also supports this notion (Lee et al. 1995). Furthermore, this observation suggests a novel role for PGI2 as a nuclear PG ligand that regulates gene expression during these processes. Although our observation demonstrates for the first time a unique pattern of developmentally regulated expression pattern of PPARδ, a ubiquitous nature of PPARδ expression has been previously reported (Braissant et al. 1996; Braissant and Wahli 1998).
Figure 3.
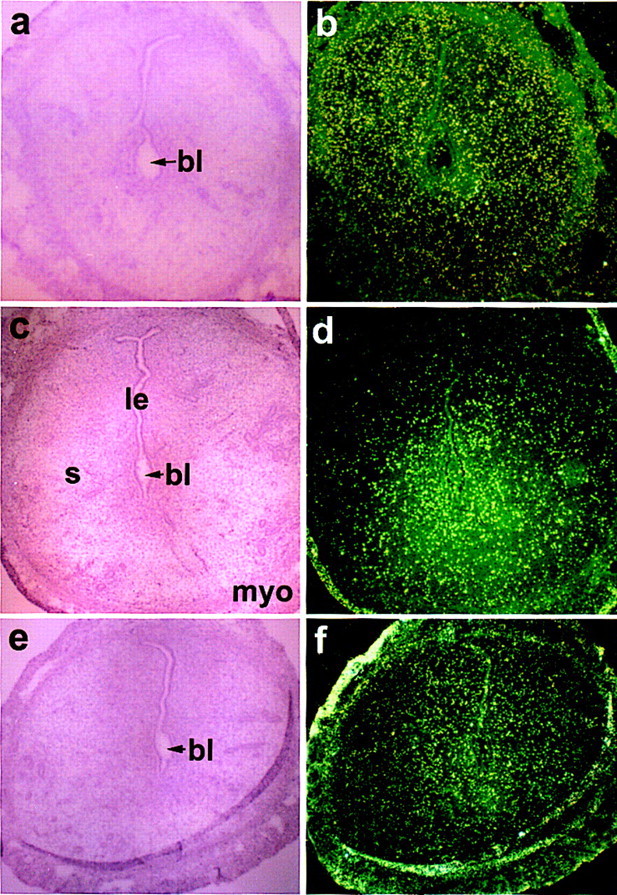
In situ hybridization of prostacyclin receptor (IP) (a,b), PPARα (c,d), and PPARγ (e,f) mRNAs in the day 5 pregnant mouse uterus. Bright-field (a,c,e) and dark-field (b,d,f) photomicrographs are shown at 40×. (le) Luminal epithelium; (s) stroma; (myo) myometrium; (bl) blastocyst. Note absence of autoradiographic signals in sections of implantation sites.
Figure 4.
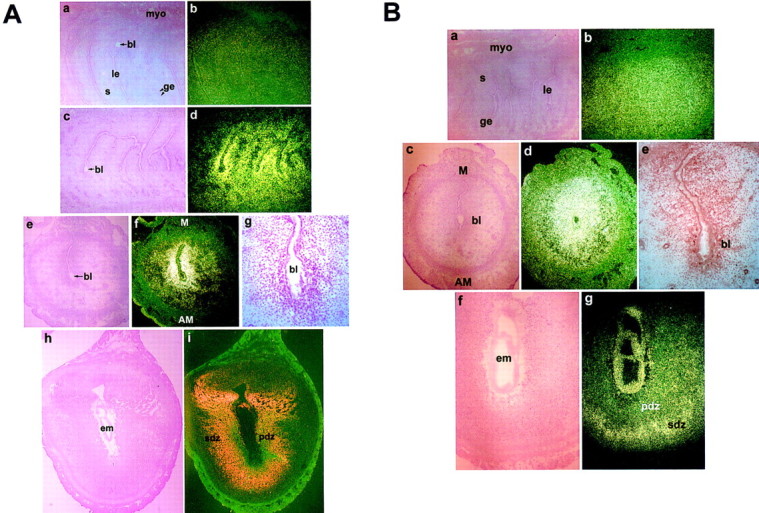
Expression of PPARδ and RXRα in the peri-implantation mouse uterus. (A) In situ hybridization of PPARδ mRNA at 0900 hr (a,b) and 2300 hr (c,d) on day 4 and at 0900 hr on day 5 (e,f) and day 8 (h,i) of pregnancy are shown in bright-field (a,c,e,h) and dark-field (b,d,f,i) photomicrographs at 40× (a–f) or at 20× (h,i). (g) Immunolocalization of PPARδ in day 5 implantation site is shown at 100×. (B) In situ hybridization of RXRα mRNA at 0900 hr on days 4 (a,b), 5 (c,d), and 8 (f,g) of pregnancy are shown in bright-field (a,c,f) and dark-field (b,d,g) photomicrographs at 40×. (e) Immunolocalization of RXRα in day 5 implantation site is shown at 100×. (le) Luminal epithelium; (ge) glandular epithelium; (s) stroma; (myo) myometrium; (bl) blastocyst; (em) embryo; (pdz) primary decidual zone; (sdz) secondary decidual zone; (AM) antimesometrial pole; (M) mesometrial pole.
As mentioned earlier, PPARs heterodimerize with their obligate partner receptors, the RXRs, to bind to a PPRE and function as transcriptional regulators (Muerhoff et al. 1992; Tugwood et al. 1992). Thus, we examined the spatiotemporal expression of RXR members (α, β, and γ) in the uterus during implantation to identify the potential RXR member(s) available as a putative heterodimerization partner for PPARδ in this process. As shown in Figure 4B, RXRα is highly expressed in endometrial cells on all days of pregnancy examined (days 1–8, data shown for days 4, 5, and 8). Abundant expression of RXRα in the decidua was reported previously (Mangelsdorf et al. 1992). RXRγ was undetectable by in situ hybridization during this period (data not shown). Although RXRβ is also expressed at high levels in the peri-implantation uterus (data not shown), we focused on RXRα for further experiments. The RXRα null mutant mice are embryonic lethal (Kastner et al. 1994) and its importance as a critical member of the RXR family was established using compound knockout mice in which it was shown that one copy of RXRα is sufficient for the viability of mice in the absence of other RXR members (Krezel et al. 1996). It should be noted that all three RXR members are capable of binding to PPRE in vitro with any given PPAR (Kliewer et al. 1994).
COX2 and PPARδ are coordinately expressed during decidualization
Previously, we showed that the application of a deciduogenic stimulus in the pseudopregnant uterus on day 4 induces rapid and transient expression of COX2 in the epithelial cells by 2 hr (Lim et al. 1997). This observation suggested that COX2 induction is important for the initiation of decidualization (Lim et al. 1997). Because COX2 induction during the attachment reaction in normal pregnancy occurs in the epithelial and stromal cells at the site of blastocyst apposition, we examined whether similar induction of COX2 occurs in the stroma during experimentally induced decidualization. As shown in Figure 5, the application of a deciduogenic stimulus did markedly induce COX2 expression in the uterine stroma of the infused horn in a focal fashion by 24 hr, similar to the pattern observed during normal implantation (Chakraborty et al. 1996). Furthermore, PPARδ was coordinately, but broadly, induced in the stroma of the infused horn in a pattern coincident with COX2 induction (Fig. 5). Both COX2 and PPARδ expressions were very low or undetectable in the noninfused horn at this time, suggesting that these genes are associated with the onset of decidualization.
Figure 5.
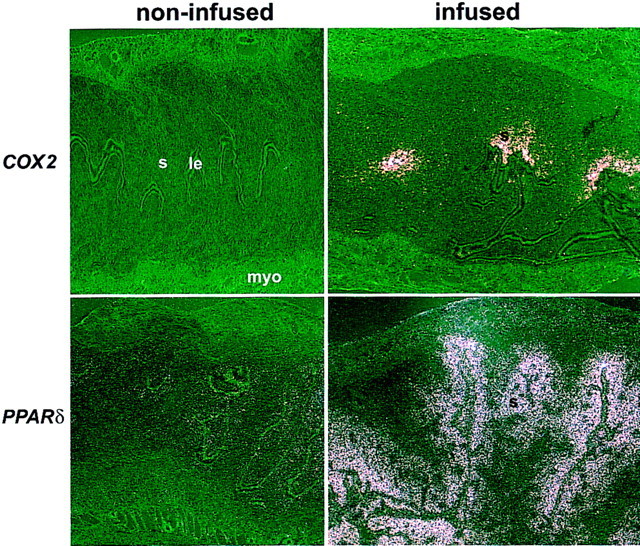
In situ hybridization of COX2 and PPARδ mRNAs in day 5 pseudopregnant wild-type uteri with or without intraluminal oil infusion. Uteri were collected 24 hr after the oil infusion. (le) Luminal epithelium; (s) stroma; (myo) myometrium. Dark-field photomicrographs are shown at 40×.
cPGI amplifies heterodimerization of PPARδ and RXRα in decidual cell nuclei
RXRs function as promiscuous partner receptors for various nuclear hormone receptors including PPARs, vitamin D receptor (VDR), thyroid hormone receptor (TR), and retinoic acid receptors (RARs). Experiments in vitro suggest that PPAR/RXR heterodimerization and subsequent DNA binding are independent of ligand when both receptors are in excess (Mangelsdorf and Evans 1995). However, it has also been demonstrated that if either of the receptors is limiting, then the ligands in fact promote receptor dimerization (Forman et al. 1997). This study specifically showed that cPGI induces heterodimerization of PPARδ/PPARα and RXR. Further, earlier studies have established that ecdysone (Yao et al. 1993), vitamin D (Cheskis and Freedman 1997), and 9-cis-retinoic acid (9-cis-RA) (Zhang et al. 1992) all can promote dimerization of their specific receptors. Thus, under normal physiological conditions, where the amount of receptor could be limiting, a ligand can promote both receptor dimerization and cofactor recruitment. To examine if a ligand for PPARδ, cPGI, can augment or stabilize heterodimer formation between PPARδ and RXRα in decidual cells, we monitored heterodimerization of PPARδ and RXRα in intact nuclei isolated from day 8 decidua by coimmunoprecipitation. As shown in Figure 6A, when PPARδ immunoprecipitates were subjected to Western blotting with both PPARδ and RXRα antibodies, a more prominent RXRα immunoreactive product was noted in the presence of cPGI; the relative amount of RXRα to PPARδ was approximately twofold. This demonstrates that cPGI is able to induce or stabilize the heterodimerization of these two nuclear receptors within the decidual cell nucleus. This is the first reported observation that PPARδ–RXRα dimerization can be enhanced by cPGI in a physiological context. The results reinforce our hypothesis that cPGI-mediated restoration of implantation and decidualization in COX2−/− mice is through this receptor system.
Figure 6.
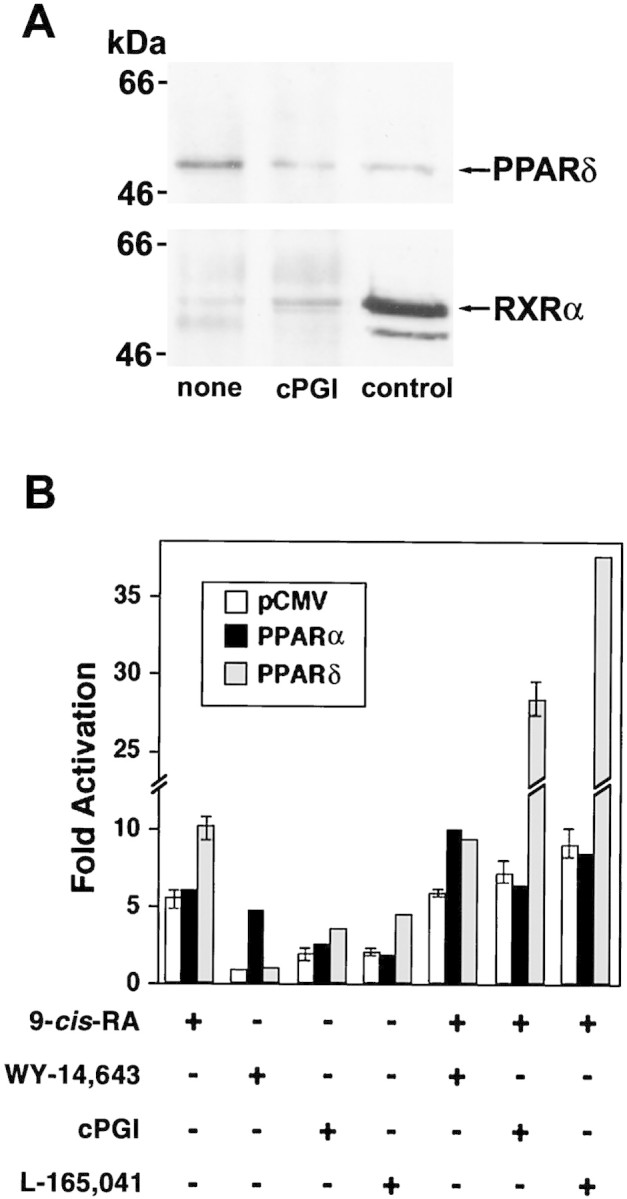
(A) Heterodimerization of PPARδ and RXRα by cPGI in the intact nuclei isolated from decidua on day 8 of pregnancy. Intact nuclei were incubated with (5 μm) or without cPGI for 30 min at 25°C followed by a brief sonication to disrupt the nuclear membranes. Supernatants after centrifugation were immunoprecipitated with goat polyclonal anti-mouse PPARδ antibody. Western blotting was performed in two separate blots with rabbit polyclonal anti-mouse PPARδ and anti-mouse RXRα antibodies as indicated. Nuclear extracts (control) were also run on the gels along with the samples for Western blotting to monitor the sizes of the immunoprecipitated products. Similar results were obtained from three separate sets of experiment. (B) Potent activation of PPARδ-mediated transcription by cPGI or L-165,041 in combination with 9-cis-RA in AN3CA cells. AN3CA cells were transiently transfected with PPRE3–tk–luciferase, pRL-TKRL, and either an empty expression vector (pCMV) (open bars) or CMV-driven expression vectors for either PPARα (solid bars) or PPARδ (shaded bars). Transfected cells were treated with the indicated ligand for 24 hr at the following concentrations: 5 μm 9-cis-RA, 10 μm WY-14,643, 1 μm cPGI, and 1 μm L-165,041. Cells were harvested and dual-luciferase assays were performed as described in Materials and Methods. Data are presented as fold activation relative to vehicle-treated (0.1% Me2SO) cells and represent the mean from three independent transfections. Mean ± s.e.m..
cPGI induces transcription of a PPRE-containing reporter gene in the presence of PPARδ in an endometrial cell line
To determine whether cPGI serves as an activating ligand for PPARδ in uterine cells, we performed transient transfection assays in a human uterine carcinoma AN3CA cell line (Dawe et al. 1964). This endometrial cell line, which expresses RXRα but not PPARδ as assessed by immunoblotting (data not shown), was transiently transfected with the PPRE3–tk–luciferase reporter (which contains three tandem repeats of the PPAR response element from the acyl CoA oxidase gene promoter) and either an empty expression vector or expression vectors for either PPARα or PPARδ. As shown in Figure 6B, although addition of cPGI alone modestly induced PPARδ activity, the combination of cPGI with the RXR ligand 9-cis-RA led to a profound synergistic induction of PPARδ activity. We also employed a novel synthetic PPARδ agonist, L-165,041, (an investigational product of Merck Pharmaceutical Company) which has been shown to exhibit >100-fold selectivity for PPARδ versus PPARγ or PPARα (Berger et al. 1999). Similar results were observed in AN3CA cells when L-165,041 was used (Fig. 6B). Although cPGI was reported as an activating ligand for PPARα (Forman et al. 1997), it was unable to induce PPARα-mediated transcription to the levels observed in PPARδ-mediated transcription. However, the PPARα-specific ligand WY-14,643 activated PPARα-mediated transcription by fivefold. Further, the inability of L-165,041 to activate PPARα-mediated transcription confirms its specificity toward PPARδ (Fig. 6B).
PGI2and PPARδ agonists restore implantation in COX2−/− mice
Synchronized development of the embryo to the blastocyst stage and differentiation of the uterus to the receptive state are essential for implantation (Paria et al. 1993; Dey 1996). In the mouse, day 4 of pregnancy is the day of uterine receptivity for implantation and is achieved by coordinated effects of ovarian progesterone (P4) and estrogen (Huet-Hudson et al. 1989; Dey 1996). On this day, the luminal epithelium becomes differentiated for its interaction with the blastocyst trophectoderm for the attachment reaction. This interaction is readily followed by stromal cell proliferation and differentiation into decidual cells (decidualization) restricted to the implantation sites.
Because PGI2 is the most abundant PG at the implantation sites (Fig. 1) and because carbaprostacyclin (cPGI) can function as a ligand for PPARδ that is expressed in the uterus at the sites of implantation (Figs. 4A and 6B), we sought to improve defective implantation in COX2−/− mice by exogenous administration of cPGI. In these experiments, day 4 wild-type blastocysts were transferred into wild-type or COX2−/− recipient uteri on day 4 of pseudopregnancy and implantation sites were recorded on day 6 by the blue dye method (Paria et al. 1993; Lim et al. 1997). Mice were treated with vehicle, cPGI, cicaprost, or PGE2. Because COX2−/− mice also have defective ovulation (Lim et al. 1997), it was surmised that they have reduced P4 levels. Thus, they were treated with P4 (2 mg/mouse) from days 3–5. As shown in Table 1, 55/112 (∼49%) blastocysts had implanted in all wild-type recipients (n = 9). In contrast, only 7/139 (∼5%) blastocysts had implanted in 3 of 10 COX2−/− recipients. Administration of cPGI considerably improved the implantation rate; 32/98 (∼33%) blastocysts had implanted in all COX2−/− recipients (n = 6). However, administration of cicaprost (a PGI2 agonist) that can activate the IP receptor, but not PPARδ (Forman et al. 1995), failed to induce implantation in all COX2−/− recipients (n = 4). PGE2 alone showed a meager improvement in implantation; only 16/98 (∼16%) blastocysts had implanted in 4 of 6 COX2−/− recipients. Although a small number of blastocysts implanted in vehicle-treated COX2−/− recipients, they all exhibited reduced endometrial vascular permeability on day 6 as determined by the blue dye method. Our previous observation shows that the attachment reaction does not occur in COX2−/− recipients when examined on day 5 in similar blastocyst transfer experiments (Lim et al. 1997). Collectively, these results suggest that not only the implantation process is defective in COX2−/− mice, but also it was delayed in those which showed only a few implantation sites.
Table 1.
Implantation of wild-type blastocysts transferred into pseudopregnant wild-type or COX2−/− mice treated with various agonists
|
Genotype
|
Treatment
|
Day
of sacrifice
|
No. of blastocysts transferred
|
No.
of recipients
|
No. of mice with IS (%)
|
No. of
mice without IS
|
No. of IS
(%)
|
|---|---|---|---|---|---|---|---|
| (+/+) | vehicle | 6 | 112 | 9 | 9 (100) | 0 | 55 (49.1) |
| (+/+) | 8 | 199 | 12 | 12 (100) | 0 | 83 (41.7) | |
| (−/−) | vehicle | 6 | 139 | 10 | 3 (30) | 7a | 7 (5) |
| (−/−) | 8 | 120 | 10 | 3 (30) | 7a | 27 (22.5) | |
| (−/−) | cPGI | 6 | 98 | 6 | 6 (100) | 0 | 32 (32.7) |
| (−/−) | 8 | 99 | 8 | 8 (100) | 0 | 44 (44.4) | |
| (−/−) | cicaprost | 6 | 64 | 4 | 0 (0) | 4a | 0 (0) |
| (−/−) | L-165,041 | 8 | 107 | 7 | 7 (100) | 0 | 43 (40.2) |
| (−/−) | PGE2 | 6 | 98 | 6 | 4 (66.7) | 2 | 16 (16.3) |
| (−/−) | 8 | 57 | 5 | 3 (60) | 2a | 10 (17.5) | |
| (−/−) | L-165,041 + 9-cis-RA | 6 | 95 | 8 | 8 (100) | 0 | 59 (62.1) |
| (−/−) | cPGI + PGE2 | 8 | 124 | 10 | 9 (90) | 1 | 60 (48.4) |
| (−/−) | L-165,041 + PGE2 | 8 | 80 | 6 | 5 (83.3) | 1 | 36 (45) |
Day 4 wild-type blastocysts were transferred into day 4 pseudopregnant uteri of wild-type or COX2−/− recipients. Mice were injected (ip) with the vehicle, cPGI, cicaprost, L-165,041, PGE2, L-165,041 plus 9-cis-RA, cPGI plus PGE2, or L-165,041 plus PGE2 at 1700 hr on the day of blastocyst transfers and each day thereafter until sacrificed. Recipients were examined for implantation sites (IS) on days 6 or 8 by the blue dye method. The doses of various agonists are described in Materials and Methods. Uteri from mice without IS were flushed with saline to recover unimplanted blastocysts. All COX2−/− recipients received progesterone (2 mg/mouse) from day 3 until sacrificed.
A number of delayed blastocysts was recovered from uterine flushings
Because a small number of wild-type blastocysts implant in COX2−/− recipients albeit with delayed response, we speculated that embryonic and decidual growth are retarded in these mice. Thus, we examined gross morphology and histology of implantation sites on day 8 of pregnancy after the blastocyst transfer. Mice were treated with vehicle, cPGI, or PGE2 alone or in combination. As shown in Table 1, 83/199 (∼42%) blastocysts had implanted in all of the wild-type recipients on day 8, whereas only 27/120 (∼22%) blastocysts had implanted in 3 of 10 COX2−/− recipients. P4 was supplemented in COX2−/− recipients from days 3–7 of pseudopregnancy to compensate for any P4 deficiency resulting from reduced ovulation in these mice. In contrast, cPGI administration restored implantation in these mice to the level observed in wild-type recipients; 44/99 (∼44%) blastocysts implanted in the uteri of all COX2−/− recipients (n = 8). Administration of PGE2 alone was not effective in this response; only 10/57 (∼17%) blastocysts implanted in 3 of 5 COX2−/− recipients. Moreover, PGE2 did not further increase the cPGI-induced implantation rate; 60/124 (∼48%) blastocysts implanted in 9 of 10 COX2−/− recipient uteri treated with cPGI and PGE2. These results suggest that PGI2 is a key player in the process of implantation in the mouse.
To further confirm that activation of PPARδ indeed participates in this process, we examined the effects of a PPARδ-selective agonist, L-165,041, on implantation in COX2−/− mice using embryo transfer protocol as described above. All of the COX2−/− recipients receiving L-165,041 showed implantation; 43/107 (∼40%) blastocysts had implanted in 7 of 7 recipients (Table 1). Administration of PGE2 with L-165,041 did not further improve implantation in COX2−/− recipients; 36/80 (45%) blastocysts had implanted in 5 of 6 recipients. In contrast, administration of L-165,041 with 9-cis-RA, a RXR agonist, greatly improved implantation rate in COX2−/− recipients, 59/95 (∼62%) blastocysts had implanted in 8 of 8 COX2−/− recipients. These results suggest that PPARδ agonists are effective in improving implantation in COX2−/− mice and this effect is further potentiated by RXR agonists. It is interesting to note that a combined treatment with PPARδ and RXR agonists induces implantation in COX2−/− recipients more efficiently than in wild-type recipients after blastocyst transfers. PPARδ and RXRα are expressed in the COX2−/− uterine stroma on day 5 (data not shown), but the expression is not concentrated at the sites of blastocysts not undergoing implantation as opposed to highly localized stromal expression surrounding blastocysts undergoing implantation in wild-type mice. This observation suggests that these receptors are present in the COX2−/− uterus in the presence of blastocysts to respond to exogenous PPARδ ligands for implantation. Further, a modest COX1 expression was only restricted to the luminal epithelium regardless of the location of unimplanted blastocysts (data not shown). Thus, COX2, which is localized in the stroma surrounding the blastocyst during implantation in wild-type mice, could not be compensated by COX1 in COX2−/− mice for implantation.
We evaluated the quality of embryos and decidua under these treatments. As shown in Figure 7A, the few implantation sites that occurred in COX2−/− recipients treated with P4 in the absence of cPGI showed severely retarded embryonic and decidual growth. All of the six implantation sites analyzed contained remnants of degenerating embryonic structures. Although administration of cPGI improved embryonic and decidual growth in COX2−/− recipients (n = 6), their growth was not comparable to that of the wild-type implantation site. PGE2 treatment alone was also not effective in correcting these abnormalities (n = 4). Interestingly, coadministration of cPGI with PGE2 markedly improved embryonic and decidual growth in COX2−/− mice. Analysis of implantation sites (n = 5) from this group showed proper development of vascular sinusoids (the presumptive site of placentation) at the mesometrial pole and the size of the decidua was comparable to that of the wild-type decidua. Further, unlike embryonic structures in vehicle- or cPGI-treated implantation sites, embryonic cells were compact in this treatment group. The decidual and embryonic growth of implantation sites induced by the PPARδ agonist L-165,041 or PGE2 plus L-165,041 in COX2−/− recipients were superior to those of the vehicle-treated group (data not shown). The inability of exogenously administered PGs or agonists to fully restore normal decidual and embryonic growth could be due to inappropriate selection of time and doses, rapid metabolism, and suboptimal delivery of these agents to the target cells. Nonetheless, these results suggest that PGI2 is the primary mediator of implantation, whereas PGE2 functions as a supplementary factor for the embryonic and decidual growth in the presence of PGI2. The retarded embryonic development could be due to improper development of the decidua and/or reduced angiogenesis as described below. This is consistent with the evidence that underdeveloped decidua are incapable of supporting normal embryonic development and placentation as demonstrated in IL-11 receptor α null mutant mice (Bilinski et al. 1998).
Figure 7.

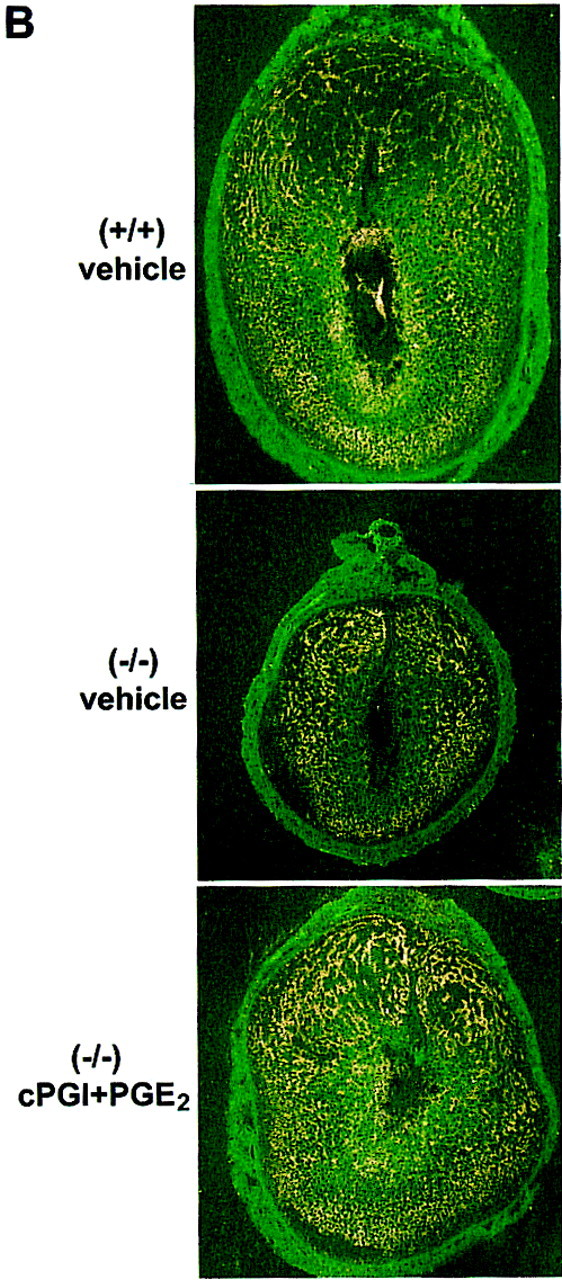
Examination of the implantation sites from wild-type and COX2−/− recipients on day 8. (A) Histological sections of implantation sites. Paraffin-embedded sections were stained with hematoxylin and eosin. Implantation sites from COX2−/− recipients treated with vehicle (a,b), cPGI (c,d), PGE2 (e,f), cPGI + PGE2 (g,h), and from wild-type recipients (i,j) are shown at 20× (a,c,e,g,i) and 100× (b,d,f,h,j). (epc) Ectoplacental cone; (xec) extraembryonic ectoderm; (xen) extraembryonic endoderm; (c) chorion; (em) embryonic mesoderm; (ec) embryonic ectoderm. (B) In situ hybridization of Flk1 mRNA in implantation sites of wild-type and COX2−/− recipients treated with vehicle or cPGI + PGE2 on day 8. Dark-field photomicrographs are shown at 40×.
Because PGI2 actively participates in vascular events, we speculated that the retarded embryonic and decidual growth in COX2−/− mice could be due to aberrant angiogenesis. Thus, we examined expression of the VEGF receptor gene Flk1 in day 8 implantation sites of wild-type and COX2−/− recipients. Flk1 was aberrantly expressed in COX2−/− mice. As shown in Figure 7B, the expression of this mRNA was markedly reduced in the deciduum immediately surrounding the embryo and within the degenerating embryo, suggesting impairment of angiogenesis. In contrast, coadministration of cPGI and PGE2 partially restored Flk1 expression in these cell types. Because Flk1 is a marker for angiogenesis (Chakraborty et al. 1995), these results suggest that these PGs could be involved in angiogenesis that is important for initiation and progression of implantation. Others have reported a role for COX in angiogenesis (Tsujii et al. 1998).
PGI2 agonist restores decidualization in COX2−/− mice
To ascertain the role of PGs in decidualization in the absence of embryonic influence, we employed an experimentally induced decidualization model. COX2+/− and COX2−/− mice received an intraluminal oil infusion on the morning of day 4 of pseudopregnancy and were killed on day 8 to record the decidual response (Lim et al. 1997). Again, P4 was supplemented in COX2−/− mice from days 3–7 of pseudopregnancy. Although vehicle-treated COX2+/− mice exhibited ∼14-fold increases in uterine weights, only 1 of 10 (10%) COX2−/− mice showed little response (∼2-fold) (Fig. 8). In contrast, 10 of 15 (70%) COX2−/− mice showed more than fivefold induction in decidualization when treated with cPGI. However, PGE2 was not as effective as cPGI; 3 of 10 (30%) COX2−/− mice showed approximately threefold induction (Fig. 8). These results establish that PGI2 is not only important for the initial stages of implantation, but also for subsequent decidualization.
Figure 8.

Restoration of decidualization in COX2−/− mice by a PGI2 agonist. Pseudopregnant wild-type or COX2−/− mice were given an intraluminal oil infusion (25 μl) at 1000 hr on day 4 to induce decidualization. cPGI and/or PGE2 (4 μg each/mouse) was injected at 1700 hr on day 4 followed by an injection at 0900 hr on days 5–7. On day 8, mice were killed and uterine weights were recorded. Fold increases denote comparison of weights between infused and noninfused uterine horns.
Discussion
COX2-derived PGs are involved in a variety of physiological and pathological processes (DuBois et al. 1998). Our previous investigation evaluating COX2-deficient mice has established the essential role of COX2 in female reproductive processes including ovulation, fertilization, implantation, and decidualization (Lim et al. 1997). However, the candidate PG(s) or its mechanism of action in these processes has remained ill-defined. A recent observation provides evidence that COX2-derived PGE2 participates in ovulation and fertilization, but not in implantation, in the mouse via the EP2 receptor subtype (Kennedy et al. 1999). In contrast, our present results demonstrate that COX2-derived PGI2 participates in implantation and decidualization via the nuclear receptor PPARδ in the uterus. These two observations illustrate an intriguing distinction as to how specific PGs elicit unique physiological function through utilization of differential signaling pathways. Further, our present observation constitutes the first evidence for specific physiological functions of PPARδ. Among the PPARs, PPARα and PPARγ exhibit various biological functions including lipid homeostasis, cell cycle regulation, and modulation of immune function (Schoonjans et al. 1996a; Jiang et al. 1998; Mueller et al. 1998; Ricote et al. 1998; Staels et al. 1998; Tontonoz et al. 1998). However, in spite of its widely distributed expression in adult and embryonic tissues (Kliewer et al. 1994; Braissant et al. 1996; Braissant and Wahli 1998), evidence for the role of PPARδ in any normal or abnormal physiological process has yet to be reported. Our investigation demonstrates that this receptor manifests a unique function as a mediator of nuclear PGI2 signaling during implantation. Our present study demonstrates that exogenous administration of a PGI2 agonist cPGI significantly improves implantation rate and incidence of decidual response in COX2−/− mice comparable to those in wild-type mice. However, histological examination of implantation sites from COX2−/− recipients revealed that embryonic growth and decidual development are not fully complemented. This could be explained by difficulties in delivering the agonist at appropriate times to the target cells in vivo. Nonetheless, these results provide strong evidence that PGI2 is the primary PG mediator of implantation. The role of PGE2 in these uterine events is still uncertain, as it was not effective in improving the implantation rate in COX2−/− mice when administered alone or in combination with cPGI. However, PGE2 was able to improve embryonic and decidual growth in COX2−/− recipients when coadministered with cPGI, suggesting an ancillary role in implantation. Further investigation is required to address the role of PGE2 in implantation and decidualization. It should be noted that PGE2 receptor subtypes exhibit uterine cell type-specific expression at the time of implantation (Lim and Dey 1997; Yang et al. 1997). The role of EP1, EP2, and EP3 subtypes appears to be insignificant as female mice lacking these genes do not exhibit implantation defects (Ushikubi et al. 1998; Kennedy et al. 1999). However, the role of EP4 in implantation has not yet been established (Nguyen et al. 1997). Extensive neovascularization that occurs at the onset of implantation is accompanied with increased expression of VEGF and its receptors (Chakraborty et al. 1995). Aberrant expression of Flk1 in the implantation sites of COX2−/− recipients on day 8 (Fig. 7B) reflects poor angiogenesis. Whether this aberrant angiogenesis is the cause or the consequence of defective implantation in COX2−/− recipients is not yet known.
Coordinate expression of COX2, PGIS, PPARδ, and RXRα in stromal cells surrounding the implanting blastocysts suggests a possible signaling cascade that is operative during implantation. Similar subcellular localization of COX2 and PGIS in stromal cell nuclei provides evidence that PGI2 is synthesized and used directly at the sites of its synthesis where PPARδ and RXRα are coexpressed during implantation. Our observation is consistent with linear increases in COX2 expression that are correlated with PGI2 production in endothelial cells (Jones et al. 1993). Further, exclusive localization of only PPARδ, but not other known PGI2 receptors, at the onset of the attachment reaction and during the progression of decidualization suggests a strong correlation between this receptor and the implantation process. This is further supported by our observation of failure of cicaprost to induce implantation in COX2−/− recipients.
RXRs are universal partners for a variety of nuclear hormone receptors. With respect to PPAR/RXR heterodimerization, ligands for both PPAR and RXR can contribute to synergistic transcriptional activation (Mangelsdorf and Evans 1995), whereas other receptors, including TR and RAR, act as repressive partners. Recent investigation suggests that the relative amount of RAR (repressive partner) and PPAR (permissive partner) within a cell system determines the functionality of RXR (Direnzo et al. 1997). It is possible that enhanced or stabilized dimerization of PPAR/RXR in decidual cell nuclei further enhances receptor responsiveness to a PPARδ ligand, which would then lead to increased recruitment of transcriptional coactivators such as SRC-1. The role of SRC-1 in uterine biology has recently been documented in SRC-1-deficient mice that exhibit a reduced decidual response (Xu et al. 1998).
Our results demonstrate that either cPGI or the selective PPARδ ligand L-165,041 rescues implantation failure in COX2−/− mice. Similarly, in a functional assay of transcriptional activity, both of these ligands with no structural homology had approximately equivalent activities in inducing PPARδ-mediated transcription that was profoundly upregulated further by 9-cis-RA. This upregulated transcription by combined addition of PPARδ and RXR ligands corroborates with heightened implantation rates in COX2−/− mice induced by L-165,041 and 9-cis-RA. Taken together, these two results present an agreeable correlation between the physiological phenotype (in vivo implantation experiments) and receptor activity (transcriptional assays), reinforcing our contention that PGI2 signals through PPARδ during blastocyst implantation. The fact that transient transfections were performed in an endometrial cell line provides further evidence that this signaling pathway is functional in uterine cells during implantation. Failure of PPARα activation by cPGI in AN3CA cells is surprising, as cPGI has been reported to be an activator of both PPARα and PPARδ (Forman et al. 1997). PPARα is expressed in these cells because treatment of transfected cells with the PPARα selective ligand WY-14,643 leads to an approximately fivefold induction not seen with empty vector or PPARδ-transfected cells. It is possible that cPGI induces the recruitment of a different set of transcriptional cofactors depending on whether it is bound to PPARα or PPARδ. Another possibility is the existence of an endometrial cell-specific cofactor that mediates the high level of induction seen with 9-cis-RA and either cPGI or L-165,041, as similar levels of induction were not observed in CV-1 cells, a green monkey kidney cell line (data not shown). The remarkable synergy observed with coaddition of either cPGI or L-165,041 with 9-cis-RA is consistent with a previous report demonstrating intermolecular synergy between PPAR and RXR (Schulman et al. 1998). It is possible that binding of a RXR ligand confers conformational change within PPAR, leading to efficient ligand binding by this receptor.
It is possible that cPGI and L-165,04 share a common unidentified receptor pathway independent of PPARδ and this receptor pathway influences implantation. However, heightened induction of implantation in COX2−/− mice by combined administration of L-165,041 and 9-cis-RA and the failure of cicaprost to induce such effect strongly suggest that neither the IP receptor nor an unidentified pathway is involved in inducing this effect. Furthermore, if the effect is not occurring through PPAR/RXR complex, we have to speculate that L-165,041, cPGI, and 9-cis-RA all share binding activity to a receptor pathway independent of PPAR/RXR.
Recent evidence demonstrates that COX2 is expressed either in the uterus, blastocyst, or both during implantation in a variety of species including sheep, mink, skunk, baboon, and pig (Charpigny et al. 1997; Song et al. 1998; Das et al. 1999; Kim et al. 1999; D.L. Davis, S.K. Das, and S.K. Dey, unpubl.). These results suggest a conserved function of COX2 in implantation. Therefore, future studies should be directed in addressing whether a similar PGI2-mediated signaling cascade is operative during implantation in these species. Finally, ligands for PPARα and PPARγ are being developed for clinical purposes because of their implications in pathological conditions, such as tumorigenesis, hypertriglyceridemia, and diabetes. Given the role of PPARδ in implantation, activity of these drugs under development toward PPARδ should also be carefully monitored to avoid possible complications in pregnancy.
Materials and methods
Material
cPGI and PGE2 were purchased from Cayman Chemical (Ann Arbor, MI); 9-cis-RA was purchased from Biomol (Plymouth Meeting, PA); and WY-14,643 was obtained from Chemsyn Science Laboratories (Lenexa, KS). L-165,041 was synthesized by Dr. R. Marquis (Merck Research Laboratories) and cicaprost was kindly provided by Dr. B. Forman (Beckman Research Institute, Duarte, CA). Mouse cDNAs for PPARs and the PPREx3–tk–luciferase construct were kindly provided by Dr. R. Evans (Salk Institute, La Jolla, CA). Dr. J. Reddy (Northwestern University, Evanston, IL) provided the PPARα cDNA, which was subsequently subcloned into the pCDNA3 expression vector and used in transfection assays. Mouse RXR cDNAs and rat PGI synthase clone were provided by Dr. P. Chambon (Institute de Genetique et de Biologie Moleculaire et Cellulaire, Illkirch Cedex, France) and Dr. M. Geraci (University of Colorado, Boulder), respectively. Anti-peptide polyclonal antibodies for mouse PPARδ (goat) and RXRα (rabbit) were purchased from Santa Cruz Biotechnology. Rabbit polyclonal antibody for mouse PPARδ was custom-raised by Research Genetics. Mouse monoclonal anti-PGIS was a generous gift from Dr. D. DeWitt (Michigan State University, Lansing).
Mice
The disruption of the COX2 gene was performed by introducing PGK–neo cassette in place of a 1.8-kb EcoRV genomic fragment housing exon 1 and surrounding sequences (Dinchuk et al. 1995). PCR analysis of tail genomic DNA and blood urea nitrogen test (Dinchuk et al. 1995) were used for genotyping. All of the mice used in the present investigation were housed in the animal care facility at the University of Kansas Medical Center according to National Institutes of Health (NIH) and institutional guidelines on the care and use of laboratory animals.
Measurement of PGs
PGs were quantitated utilizing gas chromatography/negative ion chemical ionization mass spectrometric assays as described previously (DuBois et al. 1994).
In situ hybridization
In situ hybridization was performed as described previously (Das et al. 1994). Sense or antisense 35S-labeled cRNA probes were generated using appropriate polymerases from mouse-specific cDNAs to COX1, COX2, PPARα, PPARγ, PPARδ, IP, RXRα, RXRβ, RXRγ, and Flk1 for in situ hybridization. For PGIS, rat-specific cDNA was used (Geraci et al. 1997). The probes had specific activities at 2 × 109 disintegrations per minute (dpm)/μg. Sections hybridized with the sense probes did not exhibit any positive autoradiographic signals and served as negative controls.
Immunohistochemistry
Immunolocalization of PGIS, RXRα, and PPARδ was performed in acetone-fixed frozen sections using a Zymed-Histostain-SP kit (Zymed) as described previously (Chakraborty et al. 1996). Incubation of sections with preneutralized antibodies with specific antigens served as negative controls.
Nuclei preparation
Decidual tissues from day 8 pregnant mouse uteri were homogenized in TMK buffer (0.1% Triton X-100, 10 mm Tris, 5 mm MgCl2, 25 mm KCl, 5 mm DTT, 20 μg/ml PMSF, 10 μg/ml aprotinin, leupeptin at pH 8.0) containing 0.32 m sucrose with glass–Teflon homogenizer. Intact nuclei were prepared with ascending concentration of sucrose in TMK buffer. They were washed in buffer A (10% glycerol, 20 mm Tris, 4 mm MgCl2, 5 mm DTT, 20 μg/ml PMSF at pH 8.0) and suspended in buffer B (40% glycerol, 50 mm Tris, 5 mm MgCl2, 0.1 mm EDTA at pH 8.0). Isolated intact nuclei were confirmed by microscopic observation.
Coimmunoprecipitation and Western blotting
To examine cPGI-induced heterodimerization of PPARδ and RXRα, same amount of intact nuclei were incubated with (5 μm) or without cPGI followed by a brief sonication to disrupt the nuclear envelope. The mixtures were then subjected to immunoprecipitation with goat polyclonal anti-mouse PPARδ antibody (1 μg). Antibody–antigen complexes were pulled down with Protein A/G–agarose (Santa Cruz) and washed with HNTG buffer (20 mm HEPES, 150 mm NaCl, 0.1× Triton X-100, 10% glycerol at pH 7.5). The samples were boiled in 1× SDS sample buffer and supernatants were run on SDS-PAGE gels (7.5%) in duplicate. After transferring onto nitrocellulose membranes, the membranes were subjected to Western blotting with PPARδ- and RXRα-specific antibodies. The relative amount of RXRα/PPARδ was quantitated by PhosphorImager analysis.
Cell culture, transfection, and luciferase assays
AN3CA uterine carcinoma cell line (Dawe et al. 1964) was obtained from Dr. M. Fukuda (The Burnham Institute, La Jolla, CA). Cells were grown in Dulbecco’s modified Eagle medium (GIBCO-BRL) supplemented with 10% FBS (Hyclone), l-glutamine (2 mm), penicillin (100 U/ml), and streptomycin (100 μg/ml) in a 5% CO2 atmosphere. Cells (5.0 × 105) were transfected with a mixture containing 20 μg/ml lipofectamine (GIBCO-BRL), 0.66 μg/ml PPRE3–tk–luciferase, 0.66 μg/ml pRL-TK, 0.66 μg/ml of either pCDNA3, pCDNA-PPARα, or pCMX-PPARδ in Opti-MEM (GIBCO-BRL) for 5 hr. All transfections were normalized to a total of 2.0 μg/ml with pCDNA3. The transfection mixture was replaced with complete media containing either the vehicle (0.1% Me2SO) or the indicated ligands. After 24 hr, cells were harvested in 1× luciferase lysis buffer. Relative light units from firefly luciferase activity were determined using a luminometer (MONO LIGHT 2010) and normalized to the relative light units from renilla luciferase using the Dual Luciferase kit (Promega).
Blastocyst transfer and implantation
Pseudopregnancy in wild-type or COX2−/− mice was induced by mating with wild-type vasectomized males. Day 4 wild-type blastocysts were transferred into the uteri of day 4 pseudopregnant recipients (Paria et al. 1993; Lim et al. 1997). On day 6 or 8, the number of implantation sites were recorded after intravenous injections of Chicago Blue B dye solution in saline (Paria et al. 1993). For reversal experiments, cPGI, cicaprost, or PGE2 were prepared in 10% ethanol/90% saline. The first injection (4 μg/mouse, i.p.) was given at 1700 hr on the day of blastocyst transfer followed by single injections each day until mice were sacrificed. L-165,041 was prepared in sesame oil (20 μg/mouse) and injected with or without PGE2 (20 μg/mouse) following the same injection schedule as described above. L-165,041 dissolved in 0.5% (wt/vol) methylcellulose and 0.1% (vol/vol) polysorbate 80 in water was administered (30 mg/kg body weight) by oral gavage when coadministered with 9-cis-RA (10 μg/mouse, i.p.) following the same schedule. COX2−/− recipients were supplemented with exogenous P4 (2 mg/mouse) from day 3 until sacrificed as indicated in Table 1 to eliminate any deficiency of this steroid attributable to ovulation defects.
Reversal of decidualization
To induce decidualization, pseudopregnant COX2+/− or COX2−/− mice received intraluminal oil infusion (25 μl sesame oil) on day 4 at 1000 hr and were killed on day 8. Uterine weights of the infused and noninfused (control) horns were recorded and the fold induction in uterine weights was used as an index of decidualization (Lim et al. 1997). For reversal experiments, PGs (4 μg/200 μl) were given following the same protocol as described above. COX2−/− mice were supplemented with P4 (2 mg/mouse) from days 3–7 of pseudopregnancy.
Histological examination of implantation sites
Implantation sites were isolated from embryo-transferred wild-type or COX2−/− mice on day 8 and were fixed in 10% formalin or Karnovsky’s fixative. Paraffin-embedded (6 μm) sections were stained with hematoxylin and eosin.
Acknowledgments
We thank Dr. Sanjoy Das for his constant advice with molecular biology techniques. We also thank J. Guo, Y. Zhu, and X. Zhao for their help with DNA subcloning, animal breeding, genotyping, and immunocytochemistry. We also appreciate advice and suggestions from Dr. Barry Forman. This work was supported by National Institutes of Health (NIH) grants HD 12304 and HD 29968, as part of the National Cooperative Program on Markers of Uterine Receptivity for Blastocyst Implantation (S.K.D.) and DK 48831 and CA 77839 (J.D.M). H.L. was a Kansas Health Foundation predoctoral fellow. Center grant in Reproductive Biology (HD 33994) and Mental Retardation (HD 02528) provided access to various core facilities.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sdey@kumc.edu; FAX (913) 588-5677.
References
- Altiok S, Xu M, Spiegelman BM. PPARγ induces cell cycle withdrawal: Inhibition of E2F/DP DNA-binding activity via down-regulation of PP2A. Genes & Dev. 1997;11:1987–1998. doi: 10.1101/gad.11.15.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J, Leibowitz MD, Doebber TW, Elbrecht A, Zhang B, Zhou G, Biswas C, Cullinan CA, Hayes NS, Li Y, Tanen M, Ventre J, Wu MS, Berger GD, Mosley R, Marquis R, Santini C, Sahoo SP, Tolman RL, Smith RG, Moller DE. Novel PPARγ and PPARδ ligands produce distinct biological effects. J Biol Chem. 1999;274:6718–6725. doi: 10.1074/jbc.274.10.6718. [DOI] [PubMed] [Google Scholar]
- Bilinski P, Roopentian D, Gossler A. Maternal IL-11Rα function is required for normal decidua and fetoplacental development in mice. Genes & Dev. 1998;12:2234–2243. doi: 10.1101/gad.12.14.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braissant O, Wahli W. Differential expression of peroxisome proliferator-activated receptors-α, -β, and -γ during rat embryonic development. Endocrinology. 1998;139:2748–2754. doi: 10.1210/endo.139.6.6049. [DOI] [PubMed] [Google Scholar]
- Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- Chakraborty I, Das SK, Dey SK. Differential expression of vascular endothelial growth factor and its receptor mRNAs in the mouse uterus around the time of implantation. J Endocrinol. 1995;147:339–352. doi: 10.1677/joe.0.1470339. [DOI] [PubMed] [Google Scholar]
- Chakraborty I, Das SK, Wang J, Dey SK. Developmental expression of the cyclo-oxygenase-1 and cyclo-oxygenase-2 genes in the peri-implantation mouse uterus and their differential regulation by the blastocyst and ovarian steroids. J Mol Endocrinol. 1996;16:107–122. doi: 10.1677/jme.0.0160107. [DOI] [PubMed] [Google Scholar]
- Charpigny G, Reinaud P, Tamby JP, Creminon C, Guillomot M. Cyclooxygenase-2 unlike cyclooxygenase-1 is highly expressed in ovine embryos during the implantation period. Biol Reprod. 1997;57:1032–1040. doi: 10.1095/biolreprod57.5.1032. [DOI] [PubMed] [Google Scholar]
- Cheskis B, Freedman LP. Ligand modulates the conversion of DNA-bound vitamin D3 receptor (VDR) homodimers into VDR-retinoic X receptor heterodimers. Mol Cell Biol. 1997;14:3329–3338. doi: 10.1128/mcb.14.5.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Wang X-N, Paria BC, Damm D, Abraham JA, Klagsbrun M, Andrews GK, Dey SK. Heparin-binding EGF-like growth factor gene is induced in the mouse uterus temporally by the blastocyst solely at the site of its apposition: A possible ligand for interaction with blastocyst EGF-receptor in implantation. Development. 1994;120:1071–1083. doi: 10.1242/dev.120.5.1071. [DOI] [PubMed] [Google Scholar]
- Das SK, Wang J, Dey SK, Mead RA. Spatiotemporal expression of cyclooxygenase-1 and cyclooxygenase-2 during delayed implantation and periimplantation period in the western spotted skunk. Biol Reprod. 1999;60:893–899. doi: 10.1095/biolreprod60.4.893. [DOI] [PubMed] [Google Scholar]
- Dawe CJ, Banfield WG, Morgan WD, Slatick MS, Curth HO. Growth in continuous culture, and in Hamsters, of cells from a neoplasm associated with Acanthosis Nigricans. J Nat Cancer Inst. 1964;33:441–456. [PubMed] [Google Scholar]
- Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARα-leukotriene B4pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- Dey SK. Implantation. In: Adashi EY, Rock JA, Rosenwaks Z, editors. Reproductive endocrinology, surgery, and technology. Philadelphia, PA: Lippincott-Raven; 1996. pp. 421–434. [Google Scholar]
- Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM, Gorry SA, Trzaskos JM. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- Direnzo J, Soderstrom M, Kurokawa R, Ogliastro M, Ricote M, Ingrey S, Horlein A, Rosenfeld MG, Glass CK. Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Mol Cell Biol. 1997;17:2166–2176. doi: 10.1128/mcb.17.4.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBois RN, Awad J, Morrow J, Roberts LJ, Bishop PR. Regulation of eicosanoid production and mitogenesis in rat intestinal epithelial cells by transforming growth factor-α and phorbol ester. J Clin Invest. 1994;93:493–498. doi: 10.1172/JCI116998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-deoxy-Δ12,14-prostaglandin J2is a ligand for the adipocyte determination factor PPARγ. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc Natl Acad Sci. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraci M, Shepherd D, Moore M, Vernon J, Allard J, Shannon J, Voelkel NF. Cloning and regulation of gene expression in the lung. Chest. 1997;111:129S. doi: 10.1378/chest.111.6_supplement.129s. [DOI] [PubMed] [Google Scholar]
- Huet-Hudson YM, Andrews GK, Dey SK. Cell type-specific localization of c-Myc protein in the mouse uterus: Modulation by steroid hormones and analysis of the periimplantation period. Endocrinology. 1989;125:1638–1690. doi: 10.1210/endo-125-3-1683. [DOI] [PubMed] [Google Scholar]
- Isseman I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Jones DA, Carlton DP, McIntyre TM, Zimmerman GA, Prescott SM. Molecular cloning of human prostaglandin endoperoxide synthase type II and demonstration of expression in response to cytokines. J Biol Chem. 1993;268:9049–9054. [PubMed] [Google Scholar]
- Kastner P, Grondona JM, Mark M, Gansmuller A, LeMeur M, Decimo D, Vonesch JL, Dolle P, Chambon P. Genetic analysis of RXR alpha developmental function: Convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell. 1994;78:987–1003. doi: 10.1016/0092-8674(94)90274-7. [DOI] [PubMed] [Google Scholar]
- Kennedy CRJ, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, Magnuson MA, Oates JA, Breyer MD, Breyer RM. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2receptor. Nat Med. 1999;5:217–220. doi: 10.1038/5583. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Wang J, Bambra C, Das SK, Dey SK, Fazleabas AT. Expression of cyclooxygenase-1 and -2 in the baboon endometrium during the menstrual cycle and pregnancy. Endocrinology. 1999;140:2672–2678. doi: 10.1210/endo.140.6.6716. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Umenoso K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cisretinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358:771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umenoso K, Evans RM. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2metabolite binds peroxisome proliferator activated receptor-γ and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisley GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, Lehmann JM. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors α and γ. Proc Natl Acad Sci. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krezel W, Dupe V, Mark M, Dierich A, Kastner P, Chambon P. RXRγ null mice are apparently normal and compound RXRα+/-/RXRβ-/-/RXRγ-/- mutant mice are viable. Proc Natl Acad Sci. 1996;93:9010–9014. doi: 10.1073/pnas.93.17.9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:438–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the α isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Dey SK. Prostaglandin E2 receptor subtype EP2gene expression in the mouse uterus coincides with differentiation of the luminal epithelium for implantation. Endocrinology. 1997;138:4599–4606. doi: 10.1210/endo.138.11.5528. [DOI] [PubMed] [Google Scholar]
- Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, Dey SK. Multiple female reproductive failures in cyclooxygenase-2 deficient mice. Cell. 1997;91:197–208. doi: 10.1016/s0092-8674(00)80402-x. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Kakizuka A, Evans RM. Characterization of three RXR genes that mediate the action of 9-cis-retinoic acid. Genes & Dev. 1992;6:329–344. doi: 10.1101/gad.6.3.329. [DOI] [PubMed] [Google Scholar]
- Moncada S. Biology and therapeutic potential of prostacyclin. Stroke. 1983;14:157–168. doi: 10.1161/01.str.14.2.157. [DOI] [PubMed] [Google Scholar]
- Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor α and γ activators in a tissue- and inducer-specific manner. J Biol Chem. 1998;273:16710–16714. doi: 10.1074/jbc.273.27.16710. [DOI] [PubMed] [Google Scholar]
- Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C, Singer S, Spiegelman BM. Terminal differentiation of human breast cancer through PPARγ. Mol Cell. 1998;1:465–470. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- Muerhoff AS, Griffin KJ, Johnson EF. The peroxisomal proliferator-activated receptor mediates the induction of CYP4A6, a cytochrome P450 fatty acid θ-hydroxylase, by clofibric acid. J Biol Chem. 1992;267:19051–19053. [PubMed] [Google Scholar]
- Mukherjee R, Davies PJ, Crombie DL, Bischoff ED, Cesario RM, Jow L, Hamann LG, Boehm MF, Mondon CE, Nadzan AM, Paterniti JR, Heyman RA. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature. 1997;386:407–410. doi: 10.1038/386407a0. [DOI] [PubMed] [Google Scholar]
- Murata T, Ushikubi F, Matsuoka T, Hirata M, Yamasaki A, Sugimoto Y, Ichikawa A, Aze Y, Tanaka T, Yoshida N, Ueno A, Oh-ishi S, Narumiya S. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–682. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- Murohara T, Horowitz J, Silver M, Tsurumi Y, Chen D, Sullivan A, Isner J. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998;97:99–107. doi: 10.1161/01.cir.97.1.99. [DOI] [PubMed] [Google Scholar]
- Namba T, Oida H, Sugimoto Y, Kakizuka A, Negishi M, Ichikawa A, Narumiya S. cDNA cloning of a mouse prostacyclin receptor. J Biol Chem. 1994;269:9986–9992. [PubMed] [Google Scholar]
- Negishi M, Sugimoto Y, Ichikawa A. Molecular mechanisms of diverse actions of prostanoid receptors. Biochim Biophys Acta. 1995;1259:109–120. doi: 10.1016/0005-2760(95)00146-4. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Camenisch T, Snouwaert JN, Hicks E, Coffman TM, Anderson PAW, Malouf NN, Koller BH. The prostaglandin receptor EP4triggers remodelling of the cardiovascular system at birth. Nature. 1997;390:78–81. doi: 10.1038/36342. [DOI] [PubMed] [Google Scholar]
- Paria BC, Huet-Hudson YM, Dey SK. Blastocyst’s state of activity determines the ‘window’ of implantation in the mouse receptive uterus. Proc Natl Acad Sci. 1993;90:10159–10162. doi: 10.1073/pnas.90.21.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy JK, Krishnakantha TP. Hepatic perxisome proliferation: Induction by two novel compounds structurally unrelated to clofibrate. Science. 1975;200:787–789. doi: 10.1126/science.1198095. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Wilson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391:79–81. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta. 1996a;1302:93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Peinado-Onsurbe J, Lefebvre A, Heyman RA, Briggs M, Deeb S, Staels B, Auwerx J. PPARα and PPARγ activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996b;15:5336–5348. [PMC free article] [PubMed] [Google Scholar]
- Schulman IG, Shao G, Heyman RA. Transactivation by retinoid X receptor-peroxisome proliferator-activated receptor γ (PPARγ) heterodimers: Intermolecular synergy requires only the PPARγ hormone-dependent activation function. Mol Cell Biol. 1998;18:3483–3494. doi: 10.1128/mcb.18.6.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL. Prostaglandin endoperoxide H synthase-1 and -2. Adv Immunol. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- Smith WL, Dewitt DL, Allen ML. Bimodal distribution of the prostaglandin I2synthase antigen in smooth muscle cells. J Biol Chem. 1983;258:5922–5926. [PubMed] [Google Scholar]
- Song JH, Sirois J, Houde A, Murphy BD. Cloning, developmental expression and immunohistochemistry of cyclooxygenase 2 (COX2) in the endometrium during embryo implantation and gestation in the mink. Endocrinology. 1998;139:3629–3636. doi: 10.1210/endo.139.8.6142. [DOI] [PubMed] [Google Scholar]
- Spencer AG, Woods JW, Arakawa T, Singer II, Smith WL. Subcellular localization of prostaglandin endoperoxide synthase-1 and -2 by immunoelectron microscopy. J Biol Chem. 1998;273:9886–9893. doi: 10.1074/jbc.273.16.9886. [DOI] [PubMed] [Google Scholar]
- Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, Najib J, Maclouf J, Tedgui A. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature. 1998;393:790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- Tone Y, Inoue H, Hara S, Yokoyma C, Hatae T, Oida H, Narumiya S, Shigemoto R, Yukawa S, Tanabe T. The regional distribution and cellular localization of mRNA encoding rat prostacyclin synthase. Eur J Cell Biol. 1997;72:268–277. [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPARγ2: Tissue-specific regulator of an adipocyte enhancer. Genes & Dev. 1994;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CDM, Brun RP, Mueller E, Altiok S, Oppenheim H, Evans RM, Spiegelman BM. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor γ and the retinoid X receptor. Proc Natl Acad Sci. 1997;94:237–241. doi: 10.1073/pnas.94.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARγ promotes monocyte-macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- Tugwood JD, Isseman I, Anderson RG, Bundell K, McPheat W, Green S. The mouse peroxisome proliferator activated receptors recognizes a response element in the 5′-flanking sequence of rat acyl CoA oxidase gene. EMBO J. 1992;11:433–439. doi: 10.1002/j.1460-2075.1992.tb05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushikubi F, Segi E, Sugimoto Y, Murata T, Matsuoka T, Kobayashi T, Hizaki H, Tuboi K, Katsuyama M, Ichikawa A, Tanaka T, Yoshida N, Narumiya S. Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature. 1998;395:281–284. doi: 10.1038/26233. [DOI] [PubMed] [Google Scholar]
- Wang ZM, Das SK, Wang J, Sugimoto Y, Ichikawa A, Dey SK. Potential sites of prostaglandin actions in the periimplantation mouse uterus: Differential expression and regulation of prostaglandin receptor genes. Biol Reprod. 1997;56:368–379. doi: 10.1095/biolreprod56.2.368. [DOI] [PubMed] [Google Scholar]
- Wheeler-Jones C, Abu-Ghazaleh R, Cospedal R, Houliston RA, Martin J, Zachary I. Vascular endothelial growth factor stimulates prostacyclin production and activation of cytosolic phospholipase A2 in endothelial cells via p42/p44 mitogen-activated protein kinase. FEBS Letts. 1997;420:28–32. doi: 10.1016/s0014-5793(97)01481-6. [DOI] [PubMed] [Google Scholar]
- Xu J, Qiu Y, DeMayo FJ, Tsai SY, Tsai M-J, O’Malley BW. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science. 1998;279:1922–1925. doi: 10.1126/science.279.5358.1922. [DOI] [PubMed] [Google Scholar]
- Yang ZM, Das SK, Wang J, Sugimoto Y, Ichikawa A, Dey SK. Potential sites of prostaglandin actions in the periimplantation mouse uterus: Differential expression and regulation of prostaglandin receptor genes. Biol Reprod. 1997;56:368–379. doi: 10.1095/biolreprod56.2.368. [DOI] [PubMed] [Google Scholar]
- Yao TP, Forman BM, Jiang Z, Cherbas L, Chen FO, McKeown M, Cherbas P, Evans RM. Functional ecdysone receptor is the product of EcR and Ultraspiracle genes. Nature. 1993;366:476–479. doi: 10.1038/366476a0. [DOI] [PubMed] [Google Scholar]
- Zhang XK, Lehmann J, Hoffmann B, Dawson MI, Cameron J, Graupner G, Tran P, Pfahl MM. Homodimer formation of retinoid X receptor induced by 9-cis-retinoic acid. Nature. 1992;358:587–591. doi: 10.1038/358587a0. [DOI] [PubMed] [Google Scholar]
- Zhou G, Cummings R, Li Y, Mitra S, Wilkinson HA, Elbrecht A, Hermes JD, Schaeffer JM, Smith RG, Moller DE. Nuclear receptors have distinct affinities for coactivators: Characterization by fluorescence resonance energy transfer. Mol Endocrinol. 1998;12:1594–1604. doi: 10.1210/mend.12.10.0176. [DOI] [PubMed] [Google Scholar]