

Abstract
NF-κB is a family of related, dimeric transcription factors that are readily activated in cells by signals associated with stress or pathogens. These factors are critical to host defense, as demonstrated previously with mice deficient in individual subunits of NF-κB. We have generated mice deficient in both the p50 and p52 subunits of NF-κB to reveal critical functions that may be shared by these two highly homologous proteins. We now demonstrate that unlike the respective single knockout mice, the p50/p52 double knockout mice fail to generate mature osteoclasts and B cells, apparently because of defects that track with these lineages in adoptive transfer experiments. Furthermore, these mice present markedly impaired thymic and splenic architectures and impaired macrophage functions. The blocks in osteoclast and B-cell maturation were unexpected. Lack of mature osteoclasts caused severe osteopetrosis, a family of diseases characterized by impaired osteoclastic bone resorption. These findings now establish critical roles for NF-κB in development and expand its repertoire of roles in the physiology of differentiated hematopoietic cells.
Keywords: NF-κB, knockout mice, osteopetrosis, B cell, development, osteoclasts
Mammalian NF-κB transcription factors are dimers composed of various combinations of the structurally related proteins p50 (NFKB1), p52 (NFKB2), p65 (RelA), c-Rel (Rel), and RelB. These proteins all share a so-called “Rel homology domain,” that encodes DNA-binding motifs, the dimerization interface, and a nuclear localization sequence. p65, c-Rel, and RelB, but not p50 and p52, also contain transcriptional activation domains unique to each of these proteins. p50 and p52 are generated by proteolytic processing from p105 and p100 precursors, respectively. NF-κB dimers are generally retained in the cytoplasm by association with the inhibitory IκB proteins. Upon appropriate cellular stimulation, most often by signals related to stress or pathogens, the inhibitors are proteolytically degraded and the NF-κB factors translocate to the nucleus. There they bind to κB DNA elements to induce transcription of a large number of genes, especially those concerned with immune or stress responses (for review, see Baeuerle and Henkel 1994; Siebenlist et al. 1994; Verma et al. 1995; Baldwin 1996).
Analyses of mice deficient in individual members of the NF-κB and IκB families of polypeptides have revealed essential roles for these factors in immune responses; in particular, they indicate a need for NF-κB in signal-induced responses of differentiated cells of the immune system. Each of these knockout mice presented with a distinct spectrum of defects (Beg et al. 1995a,b; Burkly et al. 1995; Kontgen et al. 1995; Sha et al. 1995; Weih et al. 1995, 1996, 1997; Gerondakis et al. 1996; Grigoriadis et al. 1996; Klement et al. 1996; Michaelson et al. 1996; Snapper et al. 1996a,b; DeKoning et al. 1997; Doi et al. 1997; Franzoso et al. 1997; Horwitz et al. 1997; Schwarz et al. 1997; Franzoso et al. 1998). Thus, individual mammalian NF-κB proteins have unique and essential functions in defense of the host against pathogens.
Little is known of the possible roles of NF-κB in mammalian development. Although the Drosophila NF-κB homolog Dorsal is responsible for development of all embryonic ventral structures (Belvin and Anderson 1996), a similar role has not been shown in mammals. Nevertheless, recent studies in mice are beginning to uncover other developmental roles for NF-κB. Mouse fetal liver cells deficient in both p50 and p65 have been postulated to lack an extracellular-acting factor necessary for development of early lymphocyte precursors, although neither cellular origin nor identity of this factor is known (Horwitz et al. 1997). Lack of p65 results in hepatocyte apoptosis in utero (Beg et al. 1995b), but it remains to be determined whether this reflects a developmental defect intrinsic to these cells. Finally, transgenic mice expressing a dominant-negative IκB-α protein in T cells are impaired, albeit only partially, in generation of mature, peripheral T cells, in particular CD8+ cells (Boothby et al. 1997).
Because mammalian NF-κB complexes may have redundant activities, mice lacking individual family members may not reveal the full range of critical functions executed by this transcription factor family. To eliminate possible redundancies and to better understand the biologic roles of NF-κB factors in vivo, including possible developmental roles, we have generated mice deficient in both p50 and p52. These two subunits are highly homologous and are usually coexpressed, and they are the most frequent partners of the transactivating family members p65, c-Rel, and RelB (for review, see Baeuerle and Henkel 1994; Siebenlist et al. 1994; Verma et al. 1995; Baldwin 1996). Consistent with the elimination of a majority of partly redundant NFκB complexes, the double knockout mice exhibited defects not seen with either the p50 or the p52 single knockout mice (Sha et al. 1995; Franzoso et al. 1998). Double knockout mice are impaired in the development of osteoclasts and B cells, and the defects appear to lie within the respective cell lineages. Mutant mice also displayed multiple defects in differentiated cell functions not seen in the single knockout mice. The inability to generate mature osteoclasts in double knockout mice resulted in severe osteopetrosis. A critical involvement of NF-κB in osteoclast development and, therefore, bone remodeling was unexpected, and a necessary role in development of mature B cells had not been shown previously.
Results
Severe osteopetrosis in p50 and p52 double knockout mice
Double knockout mice were derived from p50 and p52 single knockouts by interbreeding first generation mice heterozygous for each locus (Sha et al. 1995; Franzoso et al. 1998; see Materials and Methods). In the progeny, double knockout mice were identified with the expected frequency of ∼1 in 16. However, they had retarded growth, which became apparent within 1 week of birth. Typically, the pups succumbed shortly after weaning when they were 3–4 weeks old, presumably because of failure of their incisor teeth to erupt (although other defects probably contributed; see below). Failure of tooth eruption is characteristic of osteopetrosis, a disorder of bone remodeling caused by impaired osteoclast formation or function (Popoff and Marks 1995). These cells normally function to resorb bone. Mice in which any three of the four alleles for p50 and p52 were mutant also had retarded growth and reduced survival but generally lived up to a few months and their teeth erupted normally.
Further indication of osteopetrosis was obtained by whole-body anteroposterior radiographs of 2- to 4-week-old double knockout mice and their littermate controls. The mutant mice had shortened long bones that were radio-opaque and lacked proper bone marrow cavities, (Fig. 1A, left panels). Furthermore, lateral radiographs of mutant animals showed that the incisors had failed to erupt into the oral cavity (Fig. 1A, right panels). Histologic analysis of longitudinal sections cut through fore- and hindlimbs of the double knockout mice confirmed severe osteopetrosis, the marrow cavities being filled with unremodeled osteocartilaginous matrix (Fig. 1B). Staining of bone sections for tartrate-resistant acid phosphatase (TRAP)—an enzyme that is highly expressed in osteoclasts (Minkin et al. 1982)—showed abundant, strongly TRAP+ cells in the metaphyseal bone of wild-type mice. No TRAP+ osteoclasts were seen in most sections cut from the bones of double knockout mice (Fig. 1C; occasionally, one or two osteoclasts were seen in sections of osteopetrotic limbs, but the intensity of the TRAP staining in these few cells was very weak). Multinucleated mature osteoclasts (TRAP+), as well as their mononuclear, TRAP+ precursors, were missing in mutant but not wild-type mice, suggesting that p50 or p52 is required for TRAP+ osteoclast precursor formation. Furthermore, we saw no evidence of apoptosis of osteoclasts or of their mononuclear precursors to account for the absence of osteoclasts. Thus, in the absence of these transcription factors, osteoclast development was halted at a stage prior to expression of TRAP, resulting in severe osteopetrosis.
Figure 1.
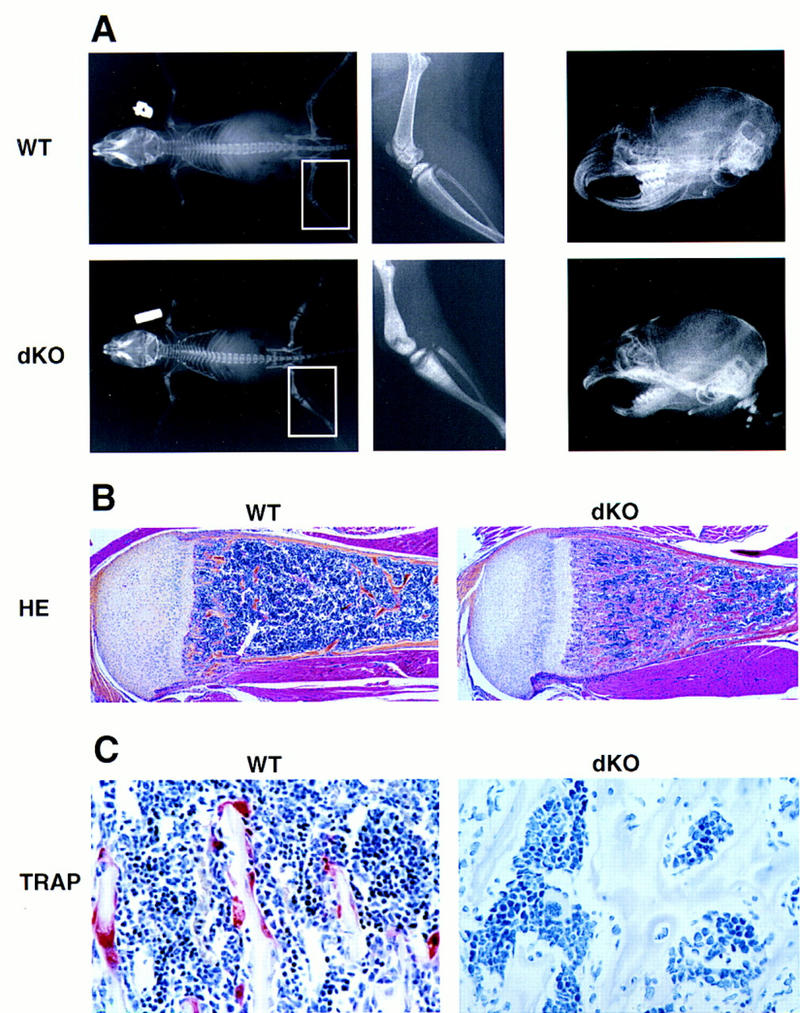
Severe osteopetrosis in p50 and p52 double knockout mice. (A) Radio-opaque long bone shafts and failure of incisor teeth to erupt in double knockout mice. Shown are whole body anterior–posterior (left panels) and skull lateral (right panels) radiographs of a 4-week-old double knockout mouse (dKO, bottom panels) and a wild-type littermate control (WT, top panels). (Middle panels) Enlargement of the boxed areas in the left panels to document the absence of a normal marrow cavity in the femur of a mutant mouse. (B) Normal marrow cavity of an 8-day-old control heterozygote mouse (WT) (p50(+/−), p52(+/−), which was indistinguishable from a true wild-type mouse) and marrow cavity from an 8-day-old double knockout (dKO) mouse, which is largely filled with unremodeled osteocartilaginous matrix. Shown are hematoxylin and eosin (HE) (B) and TRAP (C) staining of longitudinal sections through proximal tibiae. (C) Absence of TRAP+ cells in double knockout mice. A different pair of 8-day-old mutant mouse (dKO) and control littermate (WT) was used in C [WT control in C was p50(+/+), p52(+/−)], which was indistinguishable from a true wild-type animal). Osteopetrosis and lack of osteoclasts were confirmed histologically in six double knockout mice, aged 3–4 weeks. Mean values (±s.d.) are for cancellous bone volumes in sections of humeri from five dKO mice 42 ± 7% vs. 3.4 ± 1% in seven p50(+/+), p52(+/−) mice. Original magnifications were 4× (HE) and 40× (TRAP).
Impaired development of osteoclasts tracks with this cell lineage in adoptive transfers
Previous studies of naturally occurring or genetically engineered forms of osteopetrosis indicate that failure of osteoclast formation may result from defects associated with either accessory (osteoblast/stromal) cells or with cells of the osteoclast lineage (Yoshida et al. 1990; Soriano et al. 1991; Takahashi et al. 1991; Wang et al. 1992; Grigoriades et al. 1994; Tondravi et al. 1997). To address this issue we performed in vitro coculture experiments in which the generation of functional osteoclasts from splenic precursors is dependent on supportive stromal cells/osteoblasts. Primary osteoblasts, derived from calvariae of newborn mice were cocultured with splenocytes on thin slices of dentine, the bone-like matrix of teeth. Functional osteoclasts were formed from wild-type precursors present in spleen, regardless of the source of osteoblasts, wild-type or mutant, as determined by TRAP positivity and formation of resorption pits (Fig. 2A). However, functional, mature osteoclasts were not formed with splenocytes of double knockout mice as the source of osteoclast precursors (Fig. 2A). These findings suggest that the primary defect tracks with cells of the osteoclast lineage, rather than with cells of stromal origin, such as osteoblasts.
Figure 2.
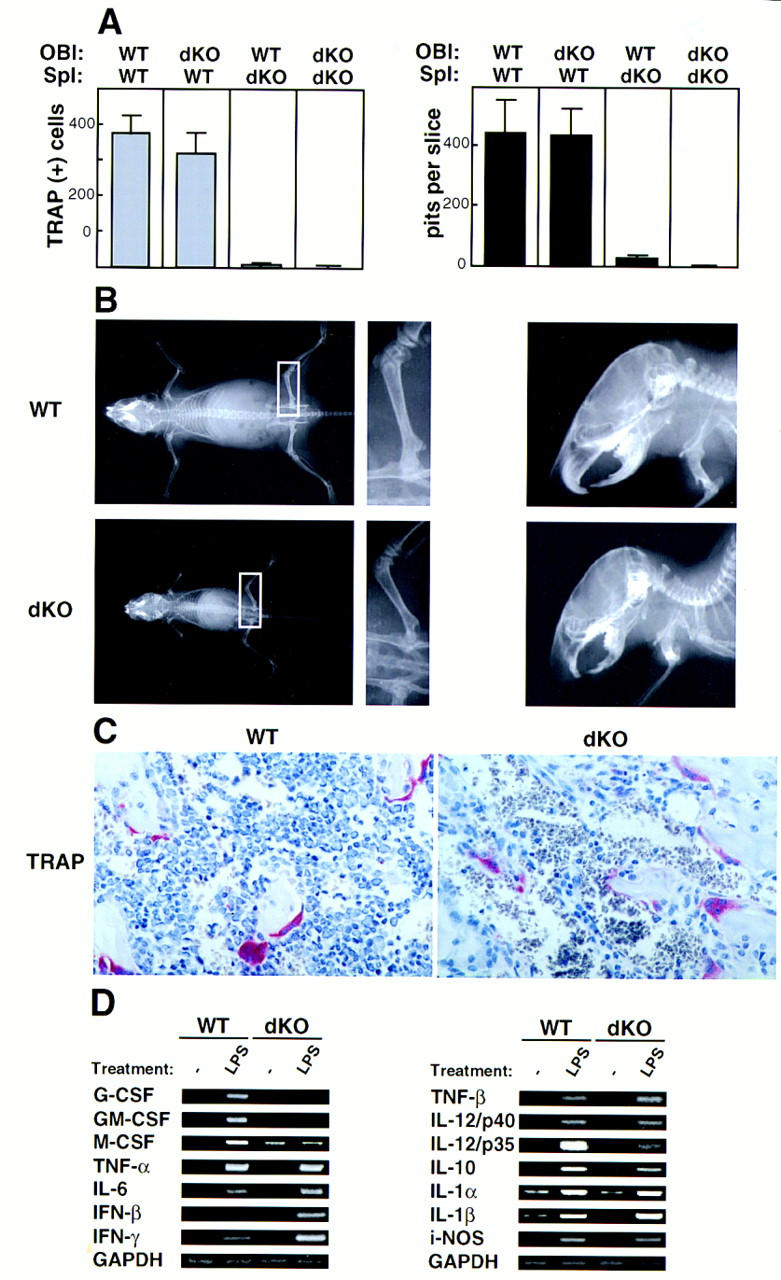
The osteopetrotic phenotype is due to a defect intrinsic to the osteoclast cell lineage. (A) Impaired development of double knockout osteoclasts in vitro. Primary osteoblasts (OBl) isolated from the calvariae of newborn wild-type (WT) or double knockout (dKO) mice were cocultured with spleen cells (Spl) isolated from either wild-type or mutant mice for 20 days on dentine slices in the presence of 10−8 m 1,25 dihydroxyvitamin D3. TRAP+ osteoclasts (left) and resorption pits (right) were counted after the culture period. Means and standard errors were calculated from six dentine slices per group. Data shown are representive of three separate experiments. (B) Osteopetrosis is cured by wild-type fetal liver transfer. Formation of radiologically clear marrow cavities within the shafts of long bones (left and middle panels) and normal tooth eruption (right panels) in adoptively transferred double knockout (dKO, bottom panels) and littermate control (WT, p50(+/−), p52(+/−), top panels) mice. Four-day-old double knockout mice and littermate controls were lethally irradiated and injected intraperitoneally (i.p.) with wild-type fetal liver cells (day 14). Whole body (left and middle panels) and skull lateral (right panels) radiographs were taken 4.5 weeks after injection. Middle panels represent an enlargement of the inset in the left panels. (C) Presence of numerous TRAP+ osteoclasts in proximal tibia sections of adoptively transferred double knockout mice. The control littermate that was adoptively transferred was a p50(+/+), p52(+/−) mouse. Mice were analyzed 3.5 weeks after fetal lever cell transfer, when their cancellous bone volumes were 7.8% and 4.7% in two rescued double knockout mice and 5.5% and 6.2% in two rescued wild-type mice. (D) Altered gene expression in double knockout mice. RT–PCR was performed on RNA extracted from thioglycollate-elicited macrophages obtained from two pairs of 3-week-old littermates (WT, p50(+/+), p52(+/−), and dKO). Cells were isolated from the peritoneal cavity, allowed to adhere to plastic, and then treated with LPS for 2 hr or left untreated, as indicated. PCR primers and animal genotypes are as indicated. Identical results were obtained in two independent experiments, and data from a representative pair of mice are shown. Data shown in the left and right panels are from two independent RT reactions (GAPDH controls at the bottom of each set).
If defects reside in the osteoclast lineage, then the osteopetrotic phenotype should be rescued by adoptive transfers into newborn double knockout animals of cells from wild-type fetal livers, which contain osteoclast and other hematopoietic precursors but not stromal precursors (Lowe et al. 1993). When wild-type fetal liver cells were injected into lethally irradiated 3- to 6-day-old double knockout mice, tooth eruption was observed in these mice 5 weeks after birth, a finding consistent with rescue of the osteoclast defect (Fig. 2B, right panels). Rescue of the osteopetrosis could also be demonstrated both radiologically and histologically by the appearance of normal bone marrow cavities and cortices of normal thickness (Fig. 2B, left panels). Numerous TRAP+ osteoclasts and mononuclear cells were now observed along the bone surfaces (Fig. 2C). Rescue of the osteopetrosis was also observed in mutant mice given a transfer of wild-type bone marrow cells (data not shown). These results support the conclusion that a defect tracking with the osteoclast lineage is responsible for the developmental block in osteoclast formation in p50 and p52 double knockout mice.
Impaired macrophage functions
To address whether development in p50- and p52-deficient mice might be arrested at a precursor common to both macrophages and osteoclasts (Yoshida et al. 1990; Takahashi et al. 1991; Tondravi et al. 1997), we stained liver and spleen sections for the macrophage marker F4/80 (Leenen et al. 1994). As we observed F4/80+ cells in both of these organs of double knockout mice, the block to osteoclast formation could not have occurred at an early precursor stage common to both cell types (for spleen sections, see Fig. 4B, below).
Figure 4.
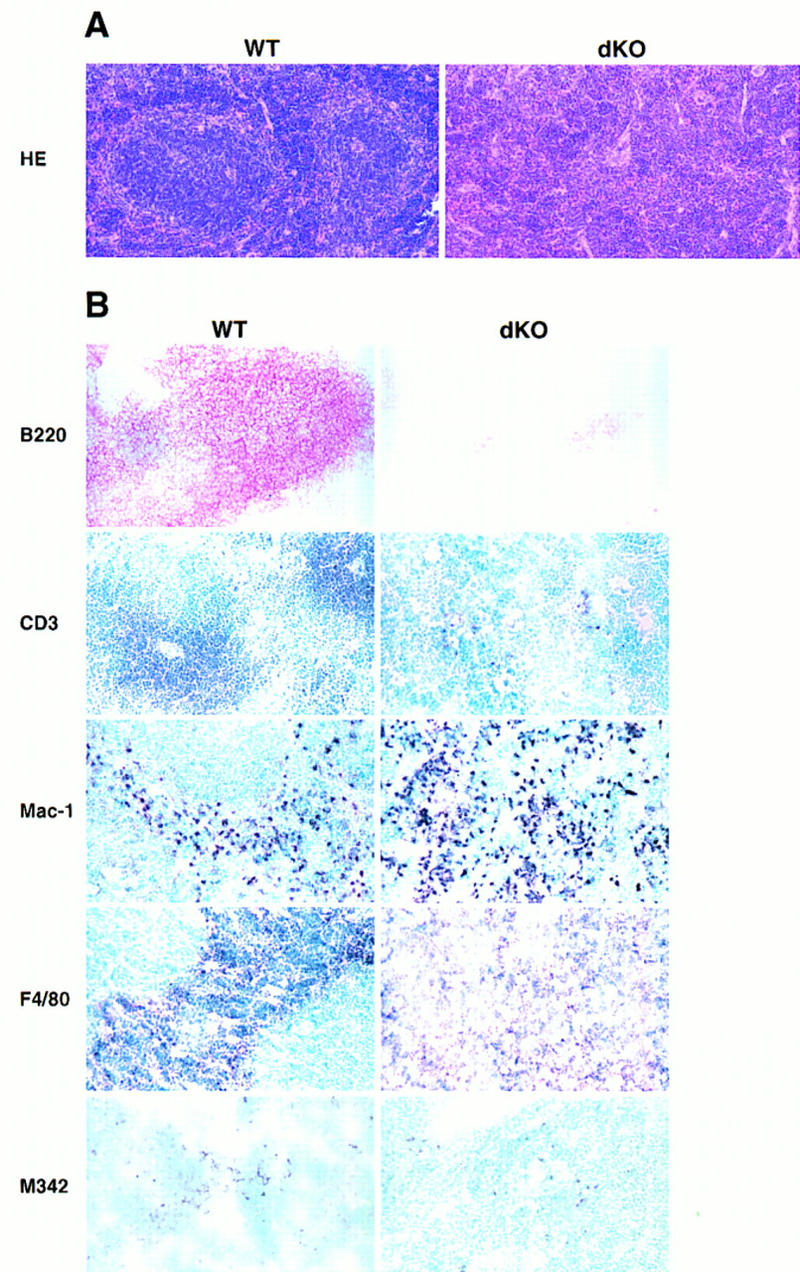
Histological abnormalities in spleens of double knockout mice. (A) Spleens of mutant mice lack white pulp. Bouin’s-fixed paraffin-embedded sections were obtained from spleens of a 10-day-old double knockout mouse (dKO, right panels) and a wild-type littermate (WT, left panels) and stained with HE, as indicated. (B) Absence of B- and T-cell areas in spleens from 2- to 4-week-old double knock-out mice (dKO) and littermate controls (WT) (right and left panels, respectively). Control littermates in A and B were p50(+/+), p52(+/−), except for macrophage marker panels, in which the control was p50(+/−), p52(+/+). Acetone-fixed, frozen sections were processed with anti-B220, anti-CD3, anti-macrophage (Mac-1 and F4/80) or anti-dendritic cell (M342) antibodies, as indicated. HRP-conjugated secondary antibodies were used, except for the B220 staining where the secondary antibody was AP conjugated. Stained cryosections from representative pairs of littermates are shown.
Although macrophage development was not halted at an early stage in double knockout mice, some mature macrophage functions were impaired. Following stimulation with lipopolysaccharide (LPS), thioglycollate-elicited mutant macrophages failed to induce significant levels of granulocyte-macrophage colony-stimulating factor (GM–CSF) and G–CSF mRNA (Fig. 2D). Also, induction of M–CSF mRNA appeared to be defective in mutant macrophages, although the basal level of expression of this mRNA was already increased over that of control macrophages. In addition to these dramatic changes, LPS-mediated induction of mRNAs for several other cytokines appeared to be either partially blocked (IL12/p35, IL10) or partially increased [β-interferon (IFN-β), IFN-γ, and tumor necrosis factor-β (TNF-β)], although more quantitative analyses are required to confirm these less dramatic changes. Together, these data indicate that p50- and p52-containing complexes play important roles in differentiated functions of macrophages, where they may act either directly or indirectly as both transcriptional repressors or activators.
Impaired development of B cells and defects in splenic architecture
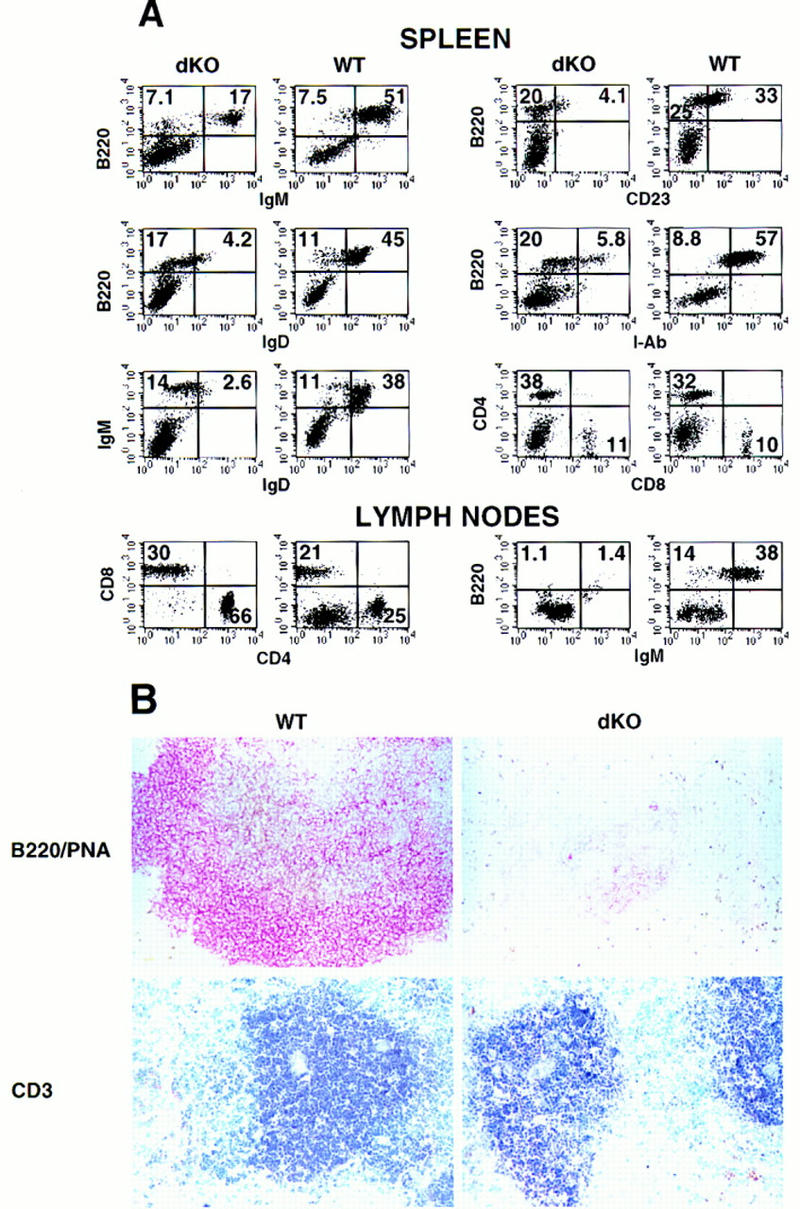
Flow cytometric (FCM) analyses of splenocytes from ∼2-week-old double knockout mice consistently revealed a modest reduction (1.3- to 2.6-fold) in the relative amount of B220+ B cells when compared to wild-type controls (Fig. 3, spleen, left panels). The IgM+ B cells were, however, significantly reduced and IgD+ cells were nearly undetectable (Fig. 3, spleen, left panels), suggesting an absence of mature IgM and IgD double-positive B cells. In support of this notion, splenocytes from double knockout mice exclusively expressed lower levels of B220 and profoundly reduced levels of I-Ab [major histocompatibility complex (MHC) class II] per cell and were nearly negative for CD23 (Fig. 3, spleen, left panels). The data indicate the absence of mature cells and the presence only of pro-B, pre-B, or immature B cells. In contrast, mature B cells were readily observed in littermate controls (B220bright, CD23+, I-Ab bright). We conclude that development of double knockout B cells is halted at an immature stage (CD19+, B220low, CD23−, IgM+, IgD−, and class IIlow expression; for review, see Rajewsky 1996; Duchosal 1997; Melchers 1997).
Figure 3.
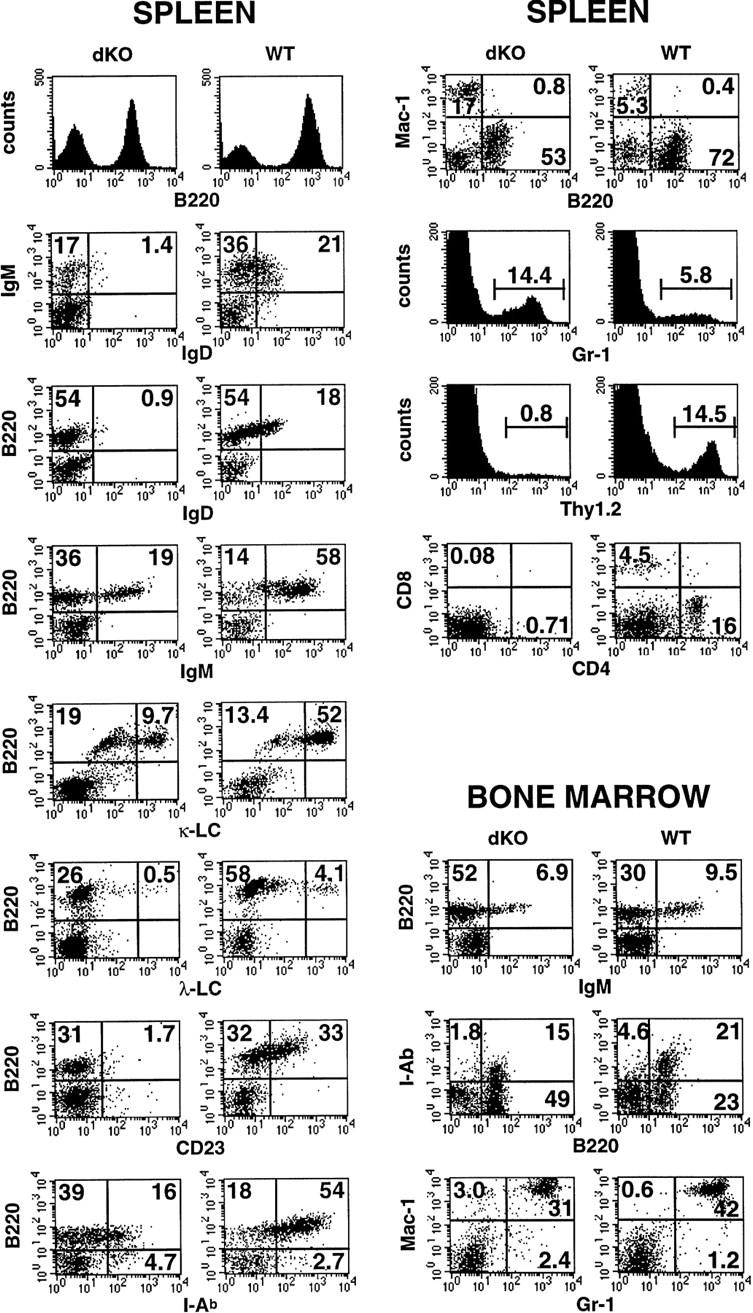
Defects of B and T lymphocytes in double knockout mice. Three-color FCM analysis of spleen and BM cell suspensions from 13- to 15-day-old double knockout mice (dKO, left panels) and p50(+/+), p52(+/−) littermate controls (WT, right panels). Two-color profiles or single-color histograms are displayed. The data are taken from analyses of three representative sets of mice, but the results were confirmed with two additional pairs. The following antibodies (listed top to bottom and left to right) were used. Spleen: B220–biotin, IgD–FITC vs. IgM–biotin; IgD-FITC vs. B220–PE; IgM–biotin vs. B220–PE; κ-light chain (LC)–FITC vs. B220–PE; λ-LC–FITC vs. B220–PE; CD23–FITC vs. B220–biotin; I-Ab-biotin vs. B220–FITC; B220–FITC vs. Mac-1–PE; Gr-1–FITC; Thy1.2–biotin; and CD4–biotin vs. CD8a–PE. Bone marrow: IgM–biotin vs. B220–PE; B220–FITC vs. class II (I-Ab)–biotin; and Gr-1–FITC vs. Mac-1–PE. Numbers reflect the percentage of positively stained cells. The percentage of B220+ BM cells was modestly increased in mutant mice, whereas class II expression was more intense in wild-type BM, even though fully mature cells were nearly absent, as judged by lack of expression of IgD (not shown).
Previous studies with cultured cell lines have suggested that NF-κB is required for expression of the Igκ locus, controlling rearrangement and/or transcription of the locus (Kirillov et al. 1996; Scherer et al. 1996). Nevertheless, κ light chains were expressed on splenocytes of double knockout mice, as determined by FCM analyses, and although the number of positive cells was low, consistent with the reduced number of IgM+ cells, the amount expressed per cell appeared normal (Fig. 3, spleen, left panels). Furthermore, no skewing toward λ light-chain expression was noted. These results were confirmed by immunohistochemical analyses of spleens of double knockout mice (data not shown). Therefore, NF-κB complexes containing p50 or p52 are not essential for κ expression.
FCM analyses of bone marrow (BM) cells revealed no further obvious deficiencies of mutant mice, although considerably fewer cells were recovered from these mice than from littermate controls. As expected, mature B cells were absent in mutant BM, as judged by the markers shown (Fig. 3, bone marrow panels), as well as by lack of expression of IgD (data not shown); however, mature cells were usually present at only low levels in wild-type BM as well, at least in these very young mice (Fig. 3, bone marrow panels). We conclude that absence of p50 and p52 blocks the development of mature B cells at a stage that normally coincides approximately with exit of normal B cells from the BM. B cells appear to progress to an immature stage, as indicated by the presence of IgM+ cells. Progression past the pro-B cell stage in BM was also confirmed by the presence of B220+, CD43− cells, as CD43 is down-regulated as B cells advance past the pro-B-cell stage (data not shown). Although it is possible that progression through early stages of development may have been quantitatively affected in double knockout mice, the data suggest that a definite block occurred only at the immature IgM+ stage of B-cell development.
Spleens of double knockout mice contained a higher proportion of Mac-1+ monocyte/macrophages and GR-1+ granulocytes than those of wild-type littermates, but this was likely a reflection of the relative decrease in other cells: In addition to the noted decrease in B cells, we observed almost no T cells in mutant spleen, a point that will be discussed in more detail below (Fig. 3, spleen, right panels; Thy1.2, CD4, and CD8 markers). No signs of inflammation were noted (data not shown). Unlike spleens, BM of double knockout mice contained near normal proportions of GR-1+ and Mac-1+ cells (Fig. 3, bone marrow panels).
Histologic analysis demonstrated a severely disrupted splenic architecture in double knockout mice, with no white pulp present [Fig. 4A, hematoxylin/eosin (HE) stain]. Furthermore, no lymph node structures could be identified in double knockout mice upon gross anatomic examination. In spleens, weakly staining B220+ B cells were diffusely distributed and only a few rare T cells were noted (Fig. 4B, B220 and CD3 markers; see below). Mac-1+ and F4/80+ macrophages were present throughout the spleen, whereas these cells are normally excluded from the white pulp, as seen in control mice (Fig. 4B). M342+ interdigitating dendritic cells (DCs) were present in mutant spleens, but they were generally fewer in number and were not as organized as those seen in littermate controls (Fig. 4B; Breel et al. 1987; Agger et al. 1992).
Defective maturation of B cells tracks with this cell lineage in adoptive transfers
Mice deficient in RAG-1 (recombination activation gene-1, hereafter referred to as RAG-1 mice) lack both T and B cells (Mombaerts et al. 1992). To test whether the defect in development of B cells is associated with these hematopoietic cells or due to defects in other cells known to interact with B cells, that is, stromal cells, irradiated RAG-1 mice were reconstituted with BM of double knockout mice. FCM analyses of adoptively transferred RAG-1 mice revealed engraftment of hematopoietic cells from double knockout BM. CD3+/T-cell receptor (TCR+) double knockout T cells were abundant in spleen and lymph nodes of chimeric mice (Fig. 5A,B, see below for further discussion). In contrast, B220+ B cells and, in particular, IgM+ B cells were reduced in number in spleens of RAG-1 mice having received mutant BM, as compared to RAG-1 mice having received wild-type BM (Fig. 5A, spleen, left panels). Importantly, the B cells generated from mutant precursors were profoundly lacking in IgD and CD23 and expressed only low levels of B220 and I-Ab (Fig. 5A, spleen, all panels) indicating the absence of mature B cells, whereas B cells of earlier developmental stages were present. These data demonstrate that the developmental arrest of double knockout B cells occurred in the RAG-1 background as well, suggesting that the defect tracks with the B-cell lineage. Development is halted at an immature stage, prior to expression of mature B-cell markers. The immature mutant B cells, though present in spleens of RAG-1-recipient mice, failed to populate lymph nodes, as determined by FCM (Fig. 5A, lymph nodes) and immunohistochemistry (not shown).
Figure 5.
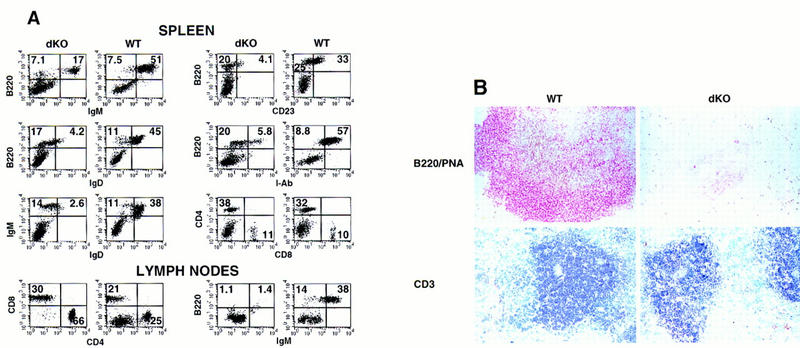
Developmental arrest of B cells is due to defects that track with to the B-cell lineage. Lethally irradiated RAG-1-deficient mice were injected with BM cells isolated from 22-day-old double knockout mice (dKO) or p50(+/+), p52(+/−) or p50(+/−), p52(+/+) (WT; these mice are indistinguishable from wild type) littermate control donor mice, as indicated. Fifteen weeks later mice were challenged i.p. with TNP–KLH (100 μg) adsorbed to alum. Mice were analyzed 9-days after challenge. (A) Three-color FCM analysis of spleen and lymph nodes of adoptively transferred RAG-1-deficient mice. Two-color profiles or single-color histograms are displayed in black and white, and genotypes of donor animals are as indicated. The following antibodies (listed top to bottom and left to right) were used. Spleen: IgM–biotin vs. B220–PE; IgD–FITC vs. B220–PE; IgD–FITC vs. IgM–biotin; CD23–PE vs. B220–biotin; I-Ab–biotin vs. B220–PE, and CD4–biotin vs. CD8a–PE. Lymph nodes: CD4–biotin vs. CD8a–PE and IgM–biotin vs. B220–PE. Numbers in the quadrants reflect the percentage of total spleen and lymph node cells in that quadrant. FCM data are representative of three paired sets of adoptively transferred RAG-1 mice. (B) Stained cryosections from representative double knockout (dKO, right panels) and p50(+/−), p52(+/+) (WT, left panels) control littermate marrow-transferred RAG-1-deficient animals are shown. Splenic cryosections were stained with PNA (brown) and anti-B220 antibodies (red) (top panels) or anti-CD-3 antibodies (blue; bottom panels), as indicated.
Immunohistochemical analyses of spleens of RAG-1 mice adoptively transferred with double knockout BM showed organized T cells (CD3+ T cells; see below) in the periarterial lymphoid sheaths (PALS) but only few, relatively disorganized B220+ B cells (Fig. 5B). In further agreement with the FCM analyses, these B cells did not express IgD+ (not shown); they were also unable to form peanut agglutinin (PNA)+ clusters, a hallmark of germinal centers, following challenge of the chimeric mice with 2, 4, 6-trinitro-phenyl–keyhole limpet hemocyanin (TNP–KLH) (MacLennan 1994; Liu and Banchereau 1996). As expected, PNA+ germinal centers were readily detected in the control RAG-1 mice that had received BM from wild-type mice (Fig. 5B, double staining for B220 and PNA). We conclude that double knockout B cells arrest at an immature stage of development, apparently because of a defect associated with this lineage, and that the arrested cells are not immunocompetent, as they are unable to form germinal centers in response to antigen, a function of mature B cells.
T cells and defective thymic architecture
Thymus glands of double knockout mice yielded between 10- and 30-fold fewer cells than those of their control littermates (Fig. 6A). FCM analyses revealed a relative decrease in CD4+ and CD8+ single-positive T cells in mutant mice, although the extent of this decrease varied somewhat with age and among individual mice (Fig. 6B). We also noted a relative reduction in the population of dull-staining double-positive T cells (CD4dull CD8dull) (Fig. 6B) and less intense staining for TCR, CD5, and CD69 in these thymocytes (data not shown), suggesting that positive selection may be partially impaired (Robey and Fowlkes 1994; Jameson et al. 1995; Anderson et al. 1996). Consistent with this and the exceedingly low absolute numbers of single-positive T cells present in thymus glands, we detected almost no peripheral T cells in double knockout mice, as determined by FCM analysis of spleens or blood (Fig. 3, spleen, right panels; Thy1.2, CD4, and CD8 markers) or by immunohistochemical analysis of spleen (Fig. 4B, CD3 marker).
Figure 6.
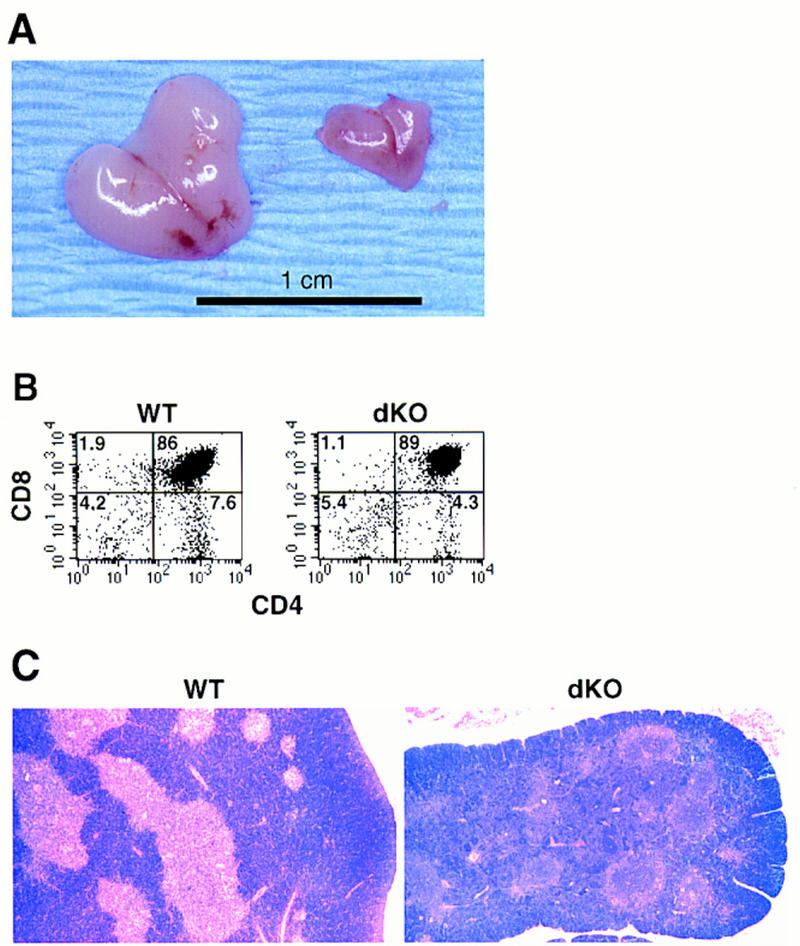
Impaired thymus glands in double knockout mice. (A) Macroscopic appearance of thymic glands obtained from a pair of 17-day-old littermates: (right) double knockout; (left) p50(+/+), p52(+/−). Original magnification was 1.3×. The dKO thymus shown is one of the larger ones seen in mutant mice. (B) FCM analysis of a thymic cell suspension from a 5-day-old double knockout mouse (dKO, right panel) and a littermate control (p50(+/+), p52(+/−); WT, left panel). CD4–biotin vs. CD8a–PE two-color profiles are displayed. Results were confirmed with two additional pairs of mice. Numbers in the quadrants reflect the percentage of total cells in that quadrant. (C) Altered microarchitecture in thymic glands of double knockout mice. Representative, Bouin’s-fixed paraffin-embedded sections were obtained from thymic glands of a 10-day-old double knockout mouse (dKO, right) and a wild-type littermate control (WT, left) and stained with HE, as indicated.
As noted, adoptive transfers of double knockout BM cells failed to generate mature B cells, but they generated abundant amounts of CD4+ and CD8+ T cells in spleen and lymph nodes of RAG-1 mice (Fig. 5A, FCM for CD4 and CD8; Fig. 5B, immunohistochemistry for CD3). This finding argues against the notion that developmental defects intrinsic to T cells might have been responsible for lack of peripheral T cells in double knockout mice. Nevertheless, it remains to be determined whether differentiated functions of double knockout T cells are defective.
Medullary structures of the thymus appeared profoundly defective in double knockout mice (Fig. 6C, HE staining). Although thymic medullary DCs were detected by immunohistochemical analysis [Fig. 7A, M342 marker (Agger et al. 1992)], they were not as clearly organized as those seen in control littermates and appeared to be reduced in numbers. Furthermore, mutant mice completely lacked medullary epithelial cells (MECs), as determined by staining with Ulex europaeus lectin (UEA-1) (Farr and Anderson 1985; Surh et al. 1992) and 4F1 antibodies (Kampinga et al. 1989) (Fig. 7A). Furthermore, the intense medullary staining for I-Ab seen in control littermates was absent in double knockout mice. MHC class II is known to be highly expressed in wild-type MECs and thymic medullary DCs (Burkly et al. 1995; Laufer et al. 1996). This suggests that the thymic DCs of mutant mice express less I-Ab, reflecting perhaps a functional impairment of these cells (see also below). In contrast to MECs, thymic cortical epithelial cells (CECs) were present in double knockout mice (Fig. 7A), although they were more randomly distributed, in line with the loss of the thymic architecture. Thus, p50 and/or p52 are required for normal thymic architecture (in addition to splenic architecture), possibly by contributing to the development, differentiation, and/or localization of MECs and DCs.
Figure 7.
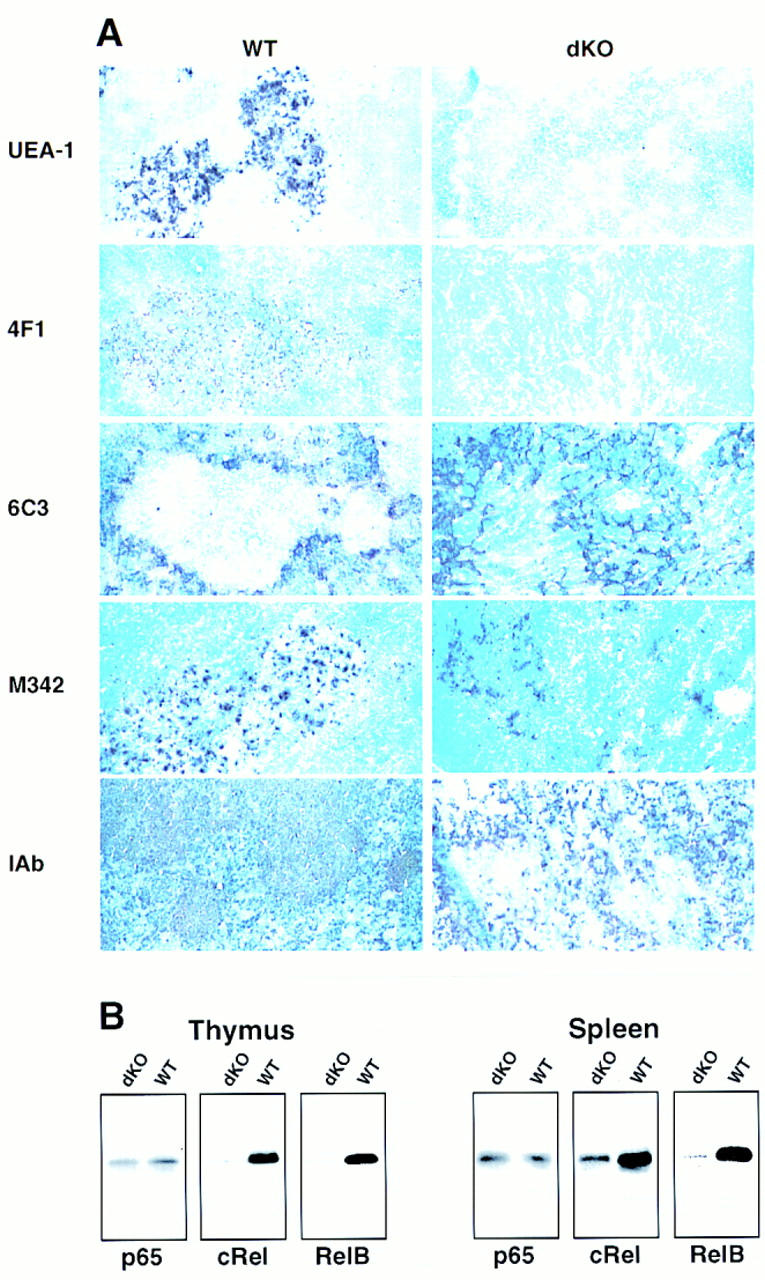
Histologic abnormalities in thymus glands of double knockout mice. (A) Thymic glands were obtained from 15- to 17-day-old double knockout mice (dKO, right) and littermate controls [WT, left; all controls were p50(+/+), p52(+/−), except for the I-Ab staining where the genotype of the control mouse was p50(+/−), p52(+/+)]. Acetone-fixed, frozen sections were processed with UEA-1 (staining MECs) or anti-MEC (4F1), anti-cortical ephitelial cell (6C3), anti-dendritic cell (M342), and anti-class II (I-Ab) antibodies, as indicated. Stained cryosections from representative pairs of littermates are shown. (B) Reduced expression of RelB and c-Rel in spleens and thymus glands of double knockout mice. Western blot analyses of thymus glands (left panels) and spleens (right panels) from 13-day-old double knockout (dKO) and control littermates [WT, p50(+/+), p52(+/−)] mice are shown. Antibodies to various NF-κB family members were used as indicated.
We observed a nearly complete lack of RelB and c-Rel protein in thymus extracts of double knockout mice, whereas expression of p65 was near normal (Fig. 7B, thymus). We also observed a roughly corresponding decrease in mRNA levels as judged by preliminary RT–PCR assays (data not shown). High levels of RelB in wild-type animals have been reported for DCs (Carrasco et al. 1993) and for MECs (Burkly et al. 1995) and high levels of c-Rel have been noted in MECs (Carrasco et al. 1994). The lack of c-Rel is consistent with loss of MECs, whereas lack of RelB may reflect a loss of MECs and a defect in the reduced number of thymic DCs present (see above). Expression of RelB was also nearly undetectable in splenic extracts of double knockout mice and c-Rel was markedly reduced, whereas p65 was near normal (Fig. 7B, spleen). In wild-type spleen, high levels of RelB have been associated primarily with DCs (Carrasco et al. 1993) and folicular dendritic cells (FDCs) (Feuillard et al. 1996), whereas high levels of c-Rel have been associated with FDCs (Feuillard et al. 1996) and mature B cells (Liou et al 1994; Miyamoto et al. 1994). The results with double knockout splenic extracts are therefore consistent with loss of FDCs [previously noted in p52-deficient mice (Franzoso et al. 1998] and lack of mature B cells, and they suggest potential defects in splenic DCs as well, in line with the immunohistochemical analysis.
Discussion
The present study demonstrates that NF-κB transcription factors carry out essential functions in development. Mice lacking both the p50 and p52 subunits of NF-κB have arrested development of osteoclasts and B cells. The defects appear to track with the affected hematopoietic lineages, as demonstrated by adoptive transfer experiments. Mice deficient in either the p50 or p52 subunit did not present with such defects (Sha et al. 1995; Franzoso et al. 1998), implying redundant functions of these two highly homologous subunits in development of cell lineages. The role of NF-κB in osteoclast formation was unanticipated, and a role in B cell maturation had not been shown previously. As a consequence of the osteoclast deficiency, double knockout mice developed osteopetrosis and succumbed within 3–4 weeks of birth.
Loss of both p50 and p52 profoundly reduces the repertoire of NF-κB dimers available in mutant mice. In addition to forming homodimers, which have diverse functions (Franzoso et al. 1992, 1993; Bours et al. 1993; Fujita et al. 1993), these two subunits are usually the primary partners for p65 and c-Rel and likely the exclusive partners for RelB. The only complexes that form in the absence of p50 and p52 are homo- or heterodimers of p65 and c-Rel, which normally represent only a minor proportion of DNA-binding NF-κB complexes (for review, see Baeuerle and Henkel 1994; Siebenlist et al. 1994; Verma et al. 1995; Baldwin 1996) and are present in mutant stimulated splenocytes at levels not dramatically different from those of control cells (save for some reduction of c-Rel complexes because of partial loss of c-Rel protein), at least as judged by electrophoretic mobility shift assays (data not shown). Double knockout mice also lack the precursor forms of the p50 and p52 proteins (p105 and p100, respectively), which contain IκB-like domains and are retained in the cytoplasm until processed (see Baeuerle and Henkel 1994; Siebenlist et al. 1994; Verma et al. 1995; Baldwin 1996).
Impaired osteoclast development
Osteopetrosis, a disease characterized by impaired bone resorption (Popoff and Marks 1995), can result from defects in the bone-forming stromal osteoblasts that are required for osteoclast formation or from defects in the osteoclast lineage itself (Yoshida et al. 1990; Soriano et al. 1991; Takahashi et al. 1991; Wang et al. 1992; Grigoriades et al. 1994; Tondravi et al. 1997). Adoptively transferred hematopoietic precursors from wild-type mice rescued osteopetrosis in double knockout mice, implying that these mice harbored a defect that tracked with the osteoclast lineage rather than with the osteoblast lineage, because the latter cells were not transferred (Lowe et al. 1993). In vitro coculture experiments supported this conclusion: Mature osteoclasts could not be generated from splenic precursors of double knockout mice, even when cocultured with wild-type osteoblasts (see Fig. 2A). Furthermore, histochemical staining for TRAP activity demonstrated that mature multinucleated osteoclasts as well as their immediate mononuclear precursors were absent from bones of double knockout mice. Therefore, the block in osteoclast development occurs prior to the appearance of TRAP-positive mononuclear cells.
Recent studies have shown that NF-κB inhibitors can induce apoptosis of mature osteoclasts and inhibit bone resorption by isolated osteoclasts (Ozaki et al. 1997). However, we found no clear evidence that the failure of osteoclast formation in double knockout mice was due to increased apoptosis of these cells or their precursors. Although it is possible that there was increased apoptosis of TRAP− osteoclast mononuclear precursors, we saw no obvious evidence of this in bone sections of the double knockout mice.
Development along the macrophage/osteoclast lineage requires the activity of transcriptonal factors, PU.1 and c-Fos, as well as production of M-CSF by stromal cells. PU.1 knockout mice have cell-autonomous defects in the myeloid/monocyte lineage. They do not form osteoclasts or macrophages and develop severe osteopetrosis (Tondravi et al. 1997). op/op mice, which lack functional M-CSF (Yoshida et al. 1990; Takahashi et al. 1991), and c-Fos knockout mice also fail to generate osteoclasts and become osteopetrotic, but these animals produce macrophages (Felix et al. 1990; Wang et al. 1992; Grigoriades et al. 1994), suggesting that c-Fos and M-CSF are required for commitment to the osteoclast lineage. Osteopetrosis also occurs in c-Src knockout mice (Soriano et al. 1991), but here osteoclast function, rather than development, is impaired (Boyce et al. 1992). In our mutant mice, Mac-1+ and F4/80+ mature macrophages were detected in both liver and spleen, although differentiated functions of peritoneal macrophages were impaired (see below). While the precise role for NF-κB factors in formation and function of the various tissue macrophages remains to be determined, a PU.1-like defect in an early multipotential precursor is ruled out. NF-κB factors may be required as early as the point of commitment to the osteoclast lineage or as late as the mitotic phase, which occurs just prior to generation of postmitotic TRAP+ mononuclear osteoclasts (Fig. 8).
Figure 8.

Model for osteoclast development indicating stage at which NF-κB appears to be required (see text).
Macrophages of double knockout mice were functionally impaired. Thioglycollate-elicited, mutant macrophages failed to respond normally to LPS. Induction of several cytokine mRNAs appeared to be altered and, in a few cases, dramatically so (decrease in GM–CSF and G–CSF, and increase in IFN-β). Increased IFN-β production has been noted previously in virally challenged p50 knockout mice, possibly because of the release from the transcriptional repression exercised by p50 homodimers on this gene (Sha et al. 1995; Franzoso et al. 1992, 1993). An equally complex, but different, pattern of altered cytokine regulation has been observed previously in c-Rel knockout mice (Kontgen et al. 1995; Gerondakis et al. 1996; Grigoriadis et al. 1996). Together, these and our findings reveal intricate gene-specific uses of NF-κB factors in both positive and negative transcriptional regulation.
Discovery of the role of NF-κB in osteoclast development may open up new avenues for investigation of the pathogenesis and treatment of postmenopausal osteoporosis, the most common bone disease in the developed world (Melton 1995), in which bone is lost due to increased resorbing activities of osteoclasts (Horowitz 1993). Recent studies suggest that the increased activity of osteoclasts is mediated by cytokines interleukin-1 (IL-1), IL-6, TNF, and GM–CSF (Pacifici et al. 1991; Jilka et al. 1992; Kitazawa et al. 1994; Jimi et al. 1995; Kimble et al. 1995). These cytokines are known transcriptional targets of NF-κB in differentiated cells and are induced to high levels during inflammatory reactions; however, with the exception of IL-6, these cytokines are also potent activators of NF-κB (Baeuerle and Henkel 1994; Siebenlist et al. 1994; Verma et al. 1995; Baldwin 1996), and as such they may stimulate osteoclast formation and survival (E. Jimi, I. Nakamura, T. Ikeber, N. Takahashi, and T. Suda, unpubl.). Therefore, these studies, together with our findings, implicate NF-κB and cytokines in both the formation of osteoclasts and in the regulation of their activity, particularly under conditions such as postmenopausal osteoporosis and rheumatoid arthritis, in which increased production of cytokines is associated with bone loss. Consequently, NF-κB may provide a new multifunctional target for therapeutic intervention to reduce abnormally high bone-resorbing activity in human diseases.
Impaired B-cell development
Loss of p50 and p52 also blocks B-cell development, with cells of this lineage progressing only to an immature stage (CD19+, IgM+, B220dull, CD43−, IAb dull, CD23−, IgD−), devoid in expression of any mature B-cell markers (such as CD23, IgD, B220bright, IAb bright; for review, see Rajewsky 1996; Duchosal 1997; Melchers 1997). Whether loss of p50 and p52 affects progression of B cells through earlier stages of development (without blocking it) remains to be explored. Previous studies of mice deficient in various subunits of NF-κB did not uncover an involvement of this transcription factor in maturation of B cells. However, such a role is consistent with the observation that only mature B cells contain constitutive NF-κB activity, whereas primary pre-B/immature B cells do not (Liou et al. 1994; Miyamoto et al. 1994). The constitutive activity consists primarily of c-Rel/p50 complexes, in line with increased levels of expression of c-Rel in these cells. In agreement with the lack of mature B cells in double knockout mice, c-Rel expression in total spleen extracts was markedly reduced. Although little is known about the transition of B cells from an immature to a mature stage, it has been speculated that positive selection occurs, as only few of the numerous, short-lived immature BM B cells are rescued to become longer-lived mature, peripheral B cells (Rajewsky 1996; Duchosal 1997; Melchers 1997). The transition may involve the B-cell receptor-associated Syk kinase (Turner et al. 1995), and maintenance of mature B cells, if not also their generation from immature cells, has been shown to require a functional B-cell receptor (Lam et al. 1997). p50- and p52-containing NF-κB factors could play a direct role in the transition/maintenance and rescue of B cells to become longer-lived cells, consistent with demonstrated anti-apoptotic functions of NF-κB (Beg and Baltimore 1996; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996; Wu et al. 1996). Alternatively, they could play an indirect role, for example, by regulating proper homing of, or signaling within, maturing cells.
In contrast to the critical roles of p50- and p52-containing NF-κB factors in generation of mature B cells, these factors are not absolutely required for expression of κ light chains. A requisite role for NF-κB factors in expression and/or rearrangement of the κ locus has been proposed based on ex vivo studies (Kirillov et al. 1996; Scherer et al. 1996; see also Shaffer et al. 1997). It is possible that p65 and c-Rel complexes that remain present in p50 and p52 double knockout mice are sufficient to execute this function.
The developmental defect of double knockout B cells appears to track with this lineage, as indicated by failure of mutant stem cell precursors to fully mature, even on a RAG-1 mouse background. Recipient RAG-1 mice contained only immature double knockout B cells, which were present in spleen but were unable to populate lymph nodes. In contrast, adoptively transferred RAG-1 mice readily supported the generation of double knockout T cells, as judged by the abundance of single-positive (CD4 or CD8), CD3+ T cells in both spleen and lymph nodes of these mice. It is worth noting that whereas the adoptive transfer experiments clearly suggest defects lying within the osteoclast and B-cell lineages, it remains theoretically possible that other hematopoietic cells are required for development of these lineages and are defective, although no such cells are known to exist. Our experiments furthermore do not address whether the defects are cell autonomous or not, because even if the primary defect lay within the B-cell lineage, for example, it may or may not involve defective production of a B-cell lineage-derived extracellular-acting factor required to promote further B-cell development, acting directly or indirectly.
Impaired thymic and splenic microarchitecture
The splenic white pulp is missing and the thymic medulla is disrupted in double knockout mice. Thymic MECs are absent, and both thymic and splenic DCs appear to be functionally impaired, specific defects that resemble those noted in RelB-deficient mice (Burkly et al. 1995; Weih et al. 1995; DeKoning et al. 1997) but not in p50- or p52-deficient mice. In support of the notion that a defect common to both the double knockout and RelB-deficient mice may be responsible for some shared thymic and splenic abnormalities, RelB function may be completely deficient in double knockout mice, as p50 and p52 are the only known dimeric partners of RelB. Beyond this, expression of RelB was extremely low in extracts of double knockout thymus glands or spleen. Lack of RelB is explained in part by loss of RelB-containing cell types, but in addition, the data imply a defect in the reduced number of double knockout DCs that were present, as such cells normally produce high levels of this protein. Thus, it is possible that expression of RelB in DCs depends on p50- and p52-containing complexes.
Although adoptive transfers of wild-type fetal liver or BM rescued the osteopetrotic phenotype, they did not change the small size of the thymus or the impaired architecture of thymus and spleen. Although the time between adoptive transfer and sacrifice may have been too short to observe rescue, some defects may not be rescued using this approach, because they may lie in nonhematopoietic cells, such as MECs. It is also possible that rescue requires the presence of wild-type cells during an early phase of development, a phase that had been completed prior to transfer. In any case, the thymic impairment is not likely to be due to defects intrinsic to T cells, because adoptive transfers of double knockout BM into RAG-1-deficient mice generated apparently mature T cells from mutant precursors. Consequently, lack of peripheral T cells in double knockout mice may simply reflect the low absolute numbers of single-positive thymic cells and the short time for accumulation prior to sacrifice.
Altogether, the results presented with p50 and p52 double knockout mice reveal an extensive and complex involvement of NF-κB factors in development and function of hematopoietic cells and generation of lymphoid organ architecture. NF-κB factors are thus central to the establishment and responses of the host defense system, perhaps reflecting an intricate, underlying regulatory network. Generation of mice lacking other combinations of NF-κB factors may reveal yet more critical roles of this transcription factor family, especially in development, where utilization of redundant functions of the various factors may be the rule rather than the exception.
Materials and methods
Generation of p52/p100-deficient mice
p52/p100-deficient mice were generated in a manner analogous to that described previously for Bcl-3 knockout mice (Franzoso et al. 1997), as described in detail elsewhere (Franzoso et al. 1998). As a result of the deletion of the exons encoding the p52 dimerization domain and the first seven ankyrin repeats of the p100 precursor, homozygous mice lacked expression of functional protein.
Preparation and staining of bone sections
Bones (calvariae, fore- and hindlimbs, and lumbar vertebrae) were fixed in 10% buffered formalin, decalcified in 10% EDTA, and embedded in paraffin. Sections (4 μm thick) were then stained either with HE or for TRAP activity, as described previously (Hughes et al. 1996).
Generation of osteoclasts in coculture experiments
Primary osteoblasts were isolated from calvariae of 3- to 5-day-old mice using a sequential collagenase/protease digestion (Lowe et al. 1993). Primary osteoblasts (104 cells/well) and spleen cells (5 × 105 cells/well) from either wild-type or p50(−/−) p52(−/−) mutant mice were cocultured on dentine slides in 48-well plates for 20 days in α-minimum essential medium containing 10% fetal calf serum (FCS) and 10−8 m 1,25 dihydroxyvitamin D3. Cells on dentine slides were then fixed in culture wells with 3% paraformaldehyde and 2% sucrose and stained for TRAP activity (Lowe et al. 1993). TRAP+ cells containing three or more nuclei were counted as osteoclasts. After counting, cells were removed and the number of resoption pits formed quantified as described previously (Lowe et al. 1993).
Fetal liver adoptive transfer
Liver cell suspensions were prepared from 14-day wild-type embryos. Cells were then injected intraperitoneally (i.p.) into 4-day-old double knockout and wild-type control animals that had been lethally irradiated (800 rads) 24 hr earlier (see Soriano et al. 1991). Bones were collected and analyzed 3–4 weeks after the transfer. As discussed in the text, recipient double knockout mice were rescued from osteopetrosis, and recipient controls showed no obvious deterioration as a result of the procedure.
Bone marrow adoptive transfer
Bone marrow cells (107), isolated from femora of 22-day-old p50(−/−) p52(−/−) double knockout or p50(+/+) p52(+/−) (indistinguishable from wild type) donor mice were injected intravenously into RAG-1 mice (Jackson Laboratory) that had been lethally irradiated at 900 rads 24 hr earlier. Fifteen weeks later, adoptively transferred RAG-1 mice were injected i.p. with TNP–KLH (100 μg) adsorbed to alum. For cryosections, spleens and lymph nodes were collected 9 days after injection.
Preparation of TNP conjugates
TNP–KLH was prepared as described previously (Franzoso et al. 1998). Briefly, 20 mg of lyophilized KLH (Pierce Chemical Co.) was dissolved in 4 ml of potassium borate buffer (0.25 m at pH 9.2), and 3 mg of TNBS (2,4,6-trinitro-benzyl sulfonic acid, Sigma Chemical Co.) was added along with 16 μl of sodium carbonate (1 m). The reaction was allowed to take place overnight, after which the protein derivatives were dialyzed against PBS (pH 7.4). TNP–KLH conjugates were frozen at −20°C until use.
Preparation and culture of thioglycollate-elicited peritoneal macrophages
Mice were sacrificed 5 days after i.p. injection of 1 ml of a 2.95% thioglycollate solution in water. Peritoneal exudate cells were harvested by washing the peritoneal cavity with 2 ml of cold RPMI medium supplemented with 2% FCS (Hyclone), β-mercaptoethanol (50 mm), penicillin G (100 U/ml), and streptomycin (100 μg/ml). Cells were then incubated in 24-well plates (106/well) at 37°C for 2 hr. Nonadherent cells were removed by washing with complete RPMI medium, and the remaining cells treated with 50 μg/ml of LPS (Escherichia coli serotype 0111:B4, Sigma Chemical Co.) or left untreated for an additional 2 hr before harvesting.
RT–PCR analysis of gene expression
RNA was isolated from adherent thioglycollate-elicited macrophages obtained as described above. The peritoneal exudates were analyzed by FCM analysis and found to be similar for wild-type and double knockout mice (not shown). RNA was isolated from the adherent monolayer of thioglycollate-elicited peritoneal macrophages with or without stimulation of LPS by using Trizol reagent (GIBCO BRL), as instructed by the manufacturer. cDNA synthesis was performed in 25 μl from 1 μg of total RNA using the Superscript II Moloney murine leukemia virus reverse transcriptase (GIBCO BRL) and random primers (Promega). The amount of 0.1–2 μl of the cDNA product was amplified with the Amplitaq DNA polymerase (Perkin Elmer) in 50 μl, and conditions were experimentally assessed for each pair of primers to allow for an analysis in the exponential range. The annealing temperature was 62°C, except for G–CSF (56°C) and IFN-γ (58°C); and the MgCl2 concentration was 1.5 mm, except for G–CSF and GM–CSF (2 mm), and IL-1β (2.8 mm). The number of cycles varied between 30 and 35 (21 cycles for GAPDH). The following primers were used: G–CSF (5′-CCAACTTTGCCACCACCATCTG-3′ and 5′-GGAGCAGCAGCAGGAATCAATA-3′); GM–CSF (5′-TGTGGTCTACAGCCTCTCAGCAC-3′ and 5′-CAAAGGGGATATCAGTCAGAAAGGT-3′); M–CSF (5′-CAGATCAAGGAAGACAACCG-3′ and 5′-ATGGTACATCCACGCTGCGT-3′); TNF-α (5′-ATGAGCACAGAAAGCATGATCCGC-3′ and 5′-CCAAAGTAGACCTGCCCGGACTC-3′); IL-6 (5′-ATGAAGTTCCTCTCTGCAAGAGACT-3′ and 5′-CACTAGGTTTGCCGAGTAGATCTC-3′); IFN-β (5′-CTCCAGCTCCAAGAAAGGACG-3′ and 5′-GAAGTTTCTGGTAAGTCTTCG-3′); IFN-γ (5′-GACTTCAAAGAGTCTGAGG-3′ and 5′-AACGCTACACACTGCATCTTGG-3′); TNF-β (5′-TGACACTGCTCGGCCGTCTCCA-3′ and 5′-GTTGCTCAAAGAGAAGCCATGTCG-3′); IL-12/p40 (5′-CGTGCTCATGGCTGGTGCAAAG-3′ and 5′-GAACACATGCCCACTTGCTG-3′); IL-12/p35 (5′-GGCTACTAGAGAGACTTCTTCC-3′ and 5′-GTGAAGCAGGATGCAGAGCTTC-3′); IL-10 (5′-CACTACCAAAGCCACAAAGC-3′ and 5′-CATGGCCTTGTAGACACCTT-3′); IL-1α (5′-ATGGCCAAAGTTCCTGACTTGTTT-3′ and 5′-CCTTCAGCAACACGGGCTGGTC-3′); IL-1β (5′-ATGGCAACTGTTCCTGAACTCAACT-3′ and 5′-CAGGACAGGTATAGATTCTTTCCTTT-3′); i-NOS (5′-CAAAGTCAAATCCTACCAAAGTGACCTG-3′ and 5′-TGCTACAGTTCCGAGCGTCAAAGACCTG-3′); GAPDH(5′-GGTGAAGGTCGGTGTGAACGGA-3′ and 5′-TGTTAGTGGGGTCTCGCTCCTG-3′).
FCM analysis
Three-color FCM analysis of single cell suspensions of spleen, lymph nodes, and BM was performed with the following antibodies: anti-B220–phycoerythrin (PE), anti-Mac-1–PE, anti-CD8a–PE, anti-CD23–PE, anti-B220–fluorescein isothiocyanate (FITC), anti-IgD–FITC, anti-CD23–FITC, Gr-1–FITC, anti-κ-light chain (LC)–FITC, anti-λ-LC–FITC, anti-B220–biotin, anti-IgM–biotin, anti-class II (I-Ab)–biotin, anti-Thy1.2–biotin, and anti-CD4–biotin; the biotin-conjugated antibodies were followed by addition of streptavidin–Red 670 (GIBCO BRL), as described previously (Shores et al. 1994). All antibodies were purchased from Pharmingen, except for the anti-κ–LC and anti-λ–LC antibodies, which were obtained from Southern Biotechnology.
HE staining of paraffin-embedded tissues
Spleens and thymic glands were fixed in Bouin’s fixative for 24 hr, rinsed, and transferred into 70% ethanol. Tissues were then processed through alcohol and xylene, embedded in paraffin, sectioned at 5 μm, and stained with HE.
Immunoperoxidase staining of frozen sections
Spleens and thymic glands were extracted, placed in OCT freezing medium (Miles Laboratories Inc.), and flash frozen. Ten micrometers of acetone-fixed sections were stained as described previously (Franzoso et al. 1997). Briefly, tissue sections were rehydrated in PBS containing 0.1% BSA (fraction V, PBS/BSA; Sigma Chemical Co.) and blocked for 30 min with 10 μg/ml of 2.4G2 antibody (anti-Fc receptor, Pharmingen) diluted in Dako antibody diluent (Dako Corporation) for staining of spleen sections or with Dako protein block for staining of thymus sections. After blocking, sections were incubated for 60 min with the primary antibody prepared in Dako antibody diluent, washed in PBS/BSA, and incubated for additional 30 min with the biotinylated secondary antibody in Dako antibody diluent (except for those stained with biotin- and HRP-conjugated antibodies and with HRP-conjugated UEA-1). After quenching endogenous peroxidase activity, tissue sections were incubated for 30 min with streptavidin-conjugated HRP (Vector Laboratories), except for the CD45RB/B220 staining where the secondary antibody was alkaline phosphatase (AP)-conjugated (see below). Slides were then washed in PBS/BSA, and the avidin/biotin complexes revealed with the DAB tetrahydrochloride chromogen (Vector Laboratories), according to the manufacturer’s instructions. Finally, slides were rinsed, counterstained with methyl green (Vector Laboratories), and mounted with Permount (Fisher Scientific) (Franzoso et al. 1997). HRP-conjugated UEA-1 (Sigma Chemical Co) and the following primary antibodies were used: Mac-1 (anti-macrophages, BoehringerMannheim), M342 (anti-dendritic cells, a kind gift of Dr. B. Kelsall, National Institutes of Health, Bethesda, MD), 4F1 (anti-thymic MECs, Biosource International), 6C3 (anti-thymic cortical epithelial cells, Pharmingen), anti-I-Ab (Pharmingen), biotinylated anti-CD45RB/B220 (Pharmingen), biotinylated anti-CD3 (Pharmingen), and biotinylated F4/80 (anti-macrophages, Caltag).
Double staining of frozen sections
Double immunohistochemical staining was performed on acetone-fixed cryosections as described previously (Franzoso et al. 1998). Briefly, tissue sections were rehydrated in PBS/BSA, blocked for 20 min with Dako protein block, and subsequently incubated for 1 hr with biotinylated anti-CD45R/B220 antibodies (Pharmingen) followed by a 30-min incubation with AP-conjugated streptavidin (Vector Laboratories). Sections were then incubated for 1 hr with HRP-conjugated PNA (Dako Corporation), and endogenous peroxidase activity quenched with 1% H2O2 in PBS for 15 min. Washings were as described above. AP and HRP enzymatic activities were finally revealed with the Fast Red (Dako Corporation) and DAB (Vector Laboratories) chromogens, respectively, and specimens mounted in aqueous mounting medium (Dako Corporation).
Western blot and antibodies
Whole-cell extracts were prepared as described previously (Dignam et al. 1983) and proteins separated by SDS-PAGE. Proteins were then blotted onto Immobilon-P membranes (Millipore) and analyzed by the ECL Western blotting detection system (Amersham), as instructed by the manufacturer. The antibodies used were a rabbit anti-peptide antibody directed to the amino terminus of human p65 (Franzoso et al. 1992), a commercial rabbit antibody recognizing amino acids 152–176 of murine c-Rel (Santa Cruz) and a commercial rabbit anti-murine Rel-B carboxy-terminal peptide antibody (Santa Cruz).
Acknowledgments
We thank K. Kelly for review of the manuscript and A.S. Fauci for continued support. We are grateful to W. Sha and D. Baltimore for the p50(−/−) mice, B. Kelsall for the M342 antibody, B. Story and A. Farias for technical assistance with bone histology, R. Germain for discussion, P. Schwartzberg for helpful advice, and M. Rust for help with the preparation of this manuscript. This project was partly funded by a grant from the National Institutes of Health (AR-43510 to B.F.B.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL US3N@NIH.GOV; FAX (301) 402-0070.
References
- Agger R, Witmer-Pack M, Romani N, Stossel H, Swiggard WJ, Metlay JP, Storozynsky E, Freimuth P, Steinman RM. Two populations of splenic dendritic cells detected with M342, a new monoclonal antibody to an intracellular antigen of interdigitating dendritic cells and some B lymphocytes. J Leukocyte Biol. 1992;52:34–42. doi: 10.1002/jlb.52.1.34. [DOI] [PubMed] [Google Scholar]
- Anderson G, Moore NC, Owen JJ, Jenkinson EJ. Cellular interactions in thymocyte development. Annu Rev Immunol. 1996;14:73–99. doi: 10.1146/annurev.immunol.14.1.73. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Baldwin AS. The NF-κB and IκB proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An esssential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes & Dev. 1995a;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995b;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Belvin MP, Anderson KV. A conserved signaling pathway: The Drosophila toll-dorsal pathway. Annu Rev Cell Dev Biol. 1996;12:393–416. doi: 10.1146/annurev.cellbio.12.1.393. [DOI] [PubMed] [Google Scholar]
- Boothby MR, Mora AL, Scherer DC, Brockman JA, Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive respressor of nuclear factor (NF)-κB. J Exp Med. 1997;185:1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 directly transactivates through IκB motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR. Requirement of pp60c-scr expression for osteoclasts to form ruffled borders and resorb bone in mice. J Clin Invest. 1992;90:1622–1627. doi: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breel M, Mebius RE, Kraal G. Dendritic cells of the mouse recognized by two monoclonal antibodies. Eur J Immunol. 1987;17:1555–1559. doi: 10.1002/eji.1830171105. [DOI] [PubMed] [Google Scholar]
- Burkly L, Hession C, Ogata L, Relly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of RelB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- Carrasco D, Ryseck RP, Bravo R. Expression of relB transcripts during lymphoid organ development: Specific expression in dendritic antigen-presenting cells. Development. 1993;118:1221–1231. doi: 10.1242/dev.118.4.1221. [DOI] [PubMed] [Google Scholar]
- Carrasco D, Weih F, Bravo R. Developmental expression of the mouse c-rel proto-oncogene in hematopoietic organs. Development. 1994;120:2991–3004. doi: 10.1242/dev.120.10.2991. [DOI] [PubMed] [Google Scholar]
- DeKoning J, DiMolfetto L, Reilly C, Wei Q, Havran WL, Lo D. Thymic cortical epithelium is sufficient for the development of mature T cells in relB-deficient mice. J Immunol. 1997;158:2558–2566. [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RC. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y. NF-κB RelA-deficient lymphocytes: Normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med. 1997;185:953–961. doi: 10.1084/jem.185.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchosal MA. B-cell development and differentiation. Semin Hematol. 1997;34:2–12. [PubMed] [Google Scholar]
- Farr AG, Anderson SK. Epithelial heterogeneity in the murine thymus: Fucose-specific lectins bind medullary epithelial cells. J Immunol. 1985;134:2971–2977. [PubMed] [Google Scholar]
- Felix R, Cecchini MG, Hofstetter W, Elford PR, Stutzer A, Fleisch H. Impairment of macrophage colony-stimulating factor production and lack of resident bone marrow macrophages in the osteopetrotic op/op mouse. J Bone Miner Res. 1990;5:781–789. doi: 10.1002/jbmr.5650050716. [DOI] [PubMed] [Google Scholar]
- Feuillard J, Koerner M, Israel A, Vassy J, Raphael M. Differential nuclear localization of p50, p52 and RelB proteins in human accessory cells of the immune response in situ. Eur J Immunol. 1996;26:2547–2551. doi: 10.1002/eji.1830261102. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-κB-mediated inhibition. Nature. 1992;359:339–342. doi: 10.1038/359339a0. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Bours V, Azarenko V, Park S, Tomita-Yamaguchi M, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 can facilitate NF-κB-mediated transactivation by removing inhibiting p50 homodimers from select κB sites. EMBO J. 1993;12:3893–3901. doi: 10.1002/j.1460-2075.1993.tb06067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran T, Schacter E, Leonardi A, Anver M, Love P, Sher A, Siebenlist U. Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity. 1997;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- Franzoso, G., L. Carlson, L. Poljak, E.W. Shores, S. Epstein, A. Leonardi, A. Grinberg, T. Tran, T. Scharton-Kersten, M. Anver, P. Love, K. Brown, and U. Siebenlist. 1998. Mice deficient in NF-κB/p52 present with deficits in humoral responses, germinal center reactions and splenic microarchitecture. J. Exp. Med. (in press). [DOI] [PMC free article] [PubMed]
- Fujita T, Nolan GP, Liou H-C, Scott ML, Baltimore D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator through NF-κB p50 homodimers. Genes & Dev. 1993;7:1354–1363. doi: 10.1101/gad.7.7b.1354. [DOI] [PubMed] [Google Scholar]
- Gerondakis S, Strasser A, Metcalf D, Grigoriadis G, Scheerlinck J-P, Grumont RJ. Rel-deficient T cells exhibit defects in production of interleukin 3 and granulocyte-macrophage colony-stimulating factor. Proc Natl Acad Sci. 1996;93:3405–3409. doi: 10.1073/pnas.93.8.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriades AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, Wagner EF. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–448. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- Grigoriadis G, Zhan Y, Grumont RJ, Metcalf D, Handman E, Cheers C, Gerondakis S. The Rel subunit of NF-κB-like transcription factors is a positive and negative regulator of macrophage gene expression: Distinct roles for Rel in different macrophage populations. EMBO J. 1996;15:7099–7107. [PMC free article] [PubMed] [Google Scholar]
- Horowitz MC. Cytokines and estrogen in bone: Anti-osteoporotic effects. Science. 1993;260:626–627. doi: 10.1126/science.8480174. [DOI] [PubMed] [Google Scholar]
- Horwitz BH, Scott ML, Cherry SR, Bronson RT, Baltimore D. Failure of lymphopoiesis after adoptive transfer of NF-κB-deficient fetal liver cells. Immunity. 1997;6:765–772. doi: 10.1016/s1074-7613(00)80451-3. [DOI] [PubMed] [Google Scholar]
- Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGFβ. Nature Med. 1996;2:1132–1136. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- Jameson SC, Hogquist KA, Bevan MJ. Positive selection of thymocytes. Annu Rev Immunol. 1995;13:93–126. doi: 10.1146/annurev.iy.13.040195.000521. [DOI] [PubMed] [Google Scholar]
- Jilka R, Hangoc G, Girasole G, Passeri G, Williams DC, Abrams JS, Boyce B, Boxmeyer H, Manolagas SC. Increased osteoclast development after estrogen loss: Mediation by interleukin-6. Science. 1992;257:88–91. doi: 10.1126/science.1621100. [DOI] [PubMed] [Google Scholar]
- Jimi E, Shuto T, Koga T. Macrophage colony-stimulating factor and interleukin 1a maintain the survival of osteoclast-like cells. Endocrinology. 1995;136:808–811. doi: 10.1210/endo.136.2.7835314. [DOI] [PubMed] [Google Scholar]
- Kampinga J, Berges S, Boyd RL, Brekelmans P, Colic M, vanEwijk W, Kendall MD, Ladyman H, Nieuwenhusi P, Ritter MA. Thymic epithelial antibodies: Immunohistochemical analysis and introduction of nomenclature. Thymus. 1989;13:165–173. [PubMed] [Google Scholar]
- Kimble RB, Matayoshi AB, Vannice JL, Williams C, Pacfici R. Simultaneous block of interleukin-1 and tumor necrosis factor is required to completely prevent bone loss in the early postovariectomy period. Endocrinology. 1995;136:3054–3061. doi: 10.1210/endo.136.7.7789332. [DOI] [PubMed] [Google Scholar]
- Kirillov A, Kistler B, Mostoslavsky R, Cedar H, Wirth T, Bergman Y. A role for nuclear NF-κB in B-cell specific demethylation of the Igκ locus. Nature Genet. 1996;13:435–441. doi: 10.1038/ng0895-435. [DOI] [PubMed] [Google Scholar]
- Kitazawa R, Kimble RB, Vannice JL, Kung VT, Pacifici R. Interleukin-1 receptor antagonist and tumor necrosis factor binding protein decrease osteoclast formation and bone resorption in ovariectomized mice. J Clin Invest. 1994;94:2397–2406. doi: 10.1172/JCI117606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement JF, Rice NR, Car BD, Abbondanzo SJ, Powers GD, Bhatt PH, Chen CH, Rosen CA, Stewart CL. IκBα deficiency results in sustained NF-κB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes & Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- Lam K-P, Kühn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- Laufer TM, DeKoning J, Markowitz JS, Lo D, Glimcher LH. Unopposed positive selection and autoreactivity in mice expressing class II MHC only on thymic cortex. Nature. 1996;383:81–85. doi: 10.1038/383081a0. [DOI] [PubMed] [Google Scholar]
- Leenen PJ, DeBruijn MF, Voerman JS, Campbell PA, vanEwijk W. Markers of mouse macrophage development detected by monoclonal antibodies. J Immunol Methods. 1994;174:5–19. doi: 10.1016/0022-1759(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Liou HC, Sha WC, Scott ML, Baltimore D. Sequential induction of NF-κB /Rel family proteins during B-cell terminal differentiation. Mol Cell Biol. 1994;14:5349–5359. doi: 10.1128/mcb.14.8.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-J, Banchereau J. Mutant mice without B lymphocyte follicles. J Exp Med. 1996;184:1207–1211. doi: 10.1084/jem.184.4.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Lowe C, Yoneda T, Boyce BF, Chen H, Mundy GR, Soriano P. Osteopetrosis in src deficient mice is due to an autonomous defect of osteoclasts. Proc Natl Acad Sci. 1993;90:4485–4489. doi: 10.1073/pnas.90.10.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan ICM. Germinal centers. Annu Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- Melchers F. The development of lymphocytes. Int Arch Allergy Immunol. 1997;113:11–13. doi: 10.1159/000237496. [DOI] [PubMed] [Google Scholar]
- Melton LJ. How many women have osteoporosis now? J Bone Miner Res. 1995;10:175–177. doi: 10.1002/jbmr.5650100202. [DOI] [PubMed] [Google Scholar]
- Michaelson JS, Singh M, Snapper CM, Sha WC, Baltimore D, Birshtein BK. Regulation of 3′IgH enhancers by a common set of factors, including κB-binding proteins. J Immunol. 1996;156:2828–2839. [PubMed] [Google Scholar]
- Minkin C. Bone acid phosphatase: Tartrate-resistant acid phosphatse as a marker of osteoclast function. Calcif Tissue Int. 1982;34:285–290. doi: 10.1007/BF02411252. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Schmitt MJ, Verma IM. Qualitative changes inthe subunit composition of κB-binding complexes during murine B-cell differentiation. Proc Natl Acad Sci. 1994;91:5056–5060. doi: 10.1073/pnas.91.11.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaionannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Ozaki K, Takeda H, Iwahashi H, Kitano A, Hanazawa S. NF-κB inhibitors stimulate apoptosis of rabbit mature osteoclasts and inhibit bone resorption by these cells. FEBS Lett. 1997;410:297–300. doi: 10.1016/s0014-5793(97)00653-4. [DOI] [PubMed] [Google Scholar]
- Pacifici R, Brown C, Puschck E, Friederick E, McCracken R, Maggio D, Slatopolsky E, Avioli LV. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci. 1991;88:5134–5138. doi: 10.1073/pnas.88.12.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoff SN, Marks SC., Jr The heterogeneity of the osteopetroses reflects the diversity of cellular influences during skeletal development. Bone. 1995;17:437–445. doi: 10.1016/8756-3282(95)00347-4. [DOI] [PubMed] [Google Scholar]
- Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- Robey E, Fowlkes BJ. Selective events in T cell development. Annu Rev Immunol. 1994;12:675–705. doi: 10.1146/annurev.iy.12.040194.003331. [DOI] [PubMed] [Google Scholar]
- Scherer DC, Brockman JA, Bendall HH, Zhang GM, Ballard DW, Oltz EM. Corepression of RelA and c-Rel inhibits immunoglobulin kappa gene transcription and rearrangement in precursor B lymphocytes. Immunity. 1996;6:563–574. doi: 10.1016/s1074-7613(00)80271-x. [DOI] [PubMed] [Google Scholar]
- Schwarz EM, Krimpenfort P, Berns A, Verma IM. Immunological defects in mice with a argeted disruption in Bcl-3. Genes & Dev. 1997;11:187–197. doi: 10.1101/gad.11.2.187. [DOI] [PubMed] [Google Scholar]
- Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disurption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Peng A, Schlissel MS. In vivo ococupancy of the κ light chain enhancers in primary pro- and pre-B cells: A model for κ locus activation. Immunity. 1997;6:131–143. doi: 10.1016/s1074-7613(00)80420-3. [DOI] [PubMed] [Google Scholar]
- Shores EW, Huang K, Tran T, Lee E, Grinberg A, Love PE. Role of TCR chain in T cell development and selection. Science. 1994;266:1047–1050. doi: 10.1126/science.7526464. [DOI] [PubMed] [Google Scholar]
- Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- Snapper CM, Rosas FR, Zelazowski R, Moorman MA, Kehry MR, Bravo R, Weih F. B cells lacking RelB are defective in proliferative responses, but undergo normal B cell maturation to Ig secretion and Ig class switching. J Exp Med. 1996a;184:1537–1541. doi: 10.1084/jem.184.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper CM, Zelazzowski P, Rosas FR, Kehry MR, Tian M, Baltimore D, Sha WC. B cells from p50/NF-κB knock-out mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol. 1996b;156:183–191. [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Surh CD, Gao EK, Kosaka H, Lo D, Ahn C, Murphy DB, Karlsson L, Peterson P, Sprent J. Two subsets of epithelial cells in the thymic medulla. J Exp Med. 1992;176:495–505. doi: 10.1084/jem.176.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Udagawa N, Akatsu T, Tanaka H, Isogai Y, Suda T. Deficiency of osteoclasts in osteopetrotic mice is due to a defect in the local microenviornment provided by osteoblastic cells. Endocrinology. 1991;128:1792–1796. doi: 10.1210/endo-128-4-1792. [DOI] [PubMed] [Google Scholar]
- Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, Teitelbaum SL. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997;386:81–84. doi: 10.1038/386081a0. [DOI] [PubMed] [Google Scholar]
- Turner M, Mee PJ, Costello PS, Williams O, Price AA, Duddy LP, Furlong MT, Geahlen RL, Tybulewicz VL. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature. 1995;378:298–302. doi: 10.1038/378298a0. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family intimate tales of association and dissociation. Genes & Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MY, Baldwin AS. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wang Z-Q, OVitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF. Bone and haematopoietic defects in mice lakcing c-Fos. Nature. 1992;360:741–745. doi: 10.1038/360741a0. [DOI] [PubMed] [Google Scholar]
- Weih F, Carrasco D, Durham SK, Barton DS, Risszo CA, Ryseck R-P, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- Weih F, Durham SK, Barton DS, Sha WC, Baltimore D, Bravo R. Both multiorgan inflammation and myeloid hyperplasia in RelB-deficient mice are T cell dependent. J Immunol. 1996;157:3974–3979. [PubMed] [Google Scholar]
- ————— p50-NF-κB complexes partially compensate for the absence of RelB: Severely increased pathology in p50(−/−)relB(−/−) double-knockout mice. J Exp Med. 1997;185:1359–1370. doi: 10.1084/jem.185.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz K, FitzGerald MJ, Rothstein TL, Sherr DH, Sonenshein GE. Inhibition of NF-κB/Rel induces apoptosis of murine B cells. EMBO J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Schultz LD, Nihikawa S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]