

Abstract
Mowat-Wilson syndrome is a mental retardation-multiple congenital anomaly syndrome characterized by a typical facies, developmental delay, epilepsy, and variable congenital malformations, including Hirschsprung disease, urogenital anomalies, congenital heart disease, and agenesis of the corpus callosum. This disorder is sporadic and is caused by heterozygous mutations or deletions of the ZFHX1B gene located in the 2q22 region. We report here the first Moroccan patient, born to consanguineous parents, with Mowat-Wilson syndrome, due to a de novo, unreported mutation of the ZFHX1B gene.
Keywords: Dysmorphia, Mowat-Wilson syndrome, severe mental retardation, ZFHX1B gene
Introduction
Mowat-Wilson syndrome (MWS) (MIM#235730) is characterized by a typical facies, severe mental retardation, epilepsy, and variable congenital malformations, including Hirschsprung's disease (HSCR), genitourinary abnormalities, congenital heart disease, and agenesis of the corpus callosum.[1] The majority of the reports in the literature originate from Northern Europe and Australia.[2] MWS is caused by de novo heterozygous mutations or deletions in the zinc finger homeobox 1B gene (ZFHX1B) located in the 2q22 region. ZFHX1B encodes the Smad-interacting protein-1 (SMADIP1 or SIP1), a transcriptional corepressor involved in the transforming growth factor-beta signaling pathway.[1] Over 100 mutations have been described in patients with typical features of MWS; these have essentially been truncating mutations (nonsense or frameshift) or large deletions of the ZFHX1B gene, suggesting a haploinsufficiency mechanism.[1]
We present the clinical data and the molecular analysis of a 2½-year-old Moroccan boy with Mowat-Wilson syndrome who was born to consanguineous parents.
Case Report
The patient was the first boy born to healthy consanguineous parents, both 29 years old, with no relevant familial history. The pregnancy and delivery were normal and the child was born at term, with normal weight, length, and head circumference. He had normal passing of meconium in the first 24 h of life, and no history of chronic constipation was reported. In the neonatal period, he developed seizures. Hypotonia and psychomotor and developmental delay were observed. He could hold his head up at 10 months, sit by himself at 24 months, and was not yet walking.
At 2½ years of age he presented with striking dysmorphic features: square-shaped face with a prominent but narrow triangular chin, plagiocephaly, thick eyebrows, sunken eyes, hypertelorism, broad nasal bridge, saddle nose, prominent columella, open mouth, and large uplifted ear lobes [Figure 1]. He had a smiling face, with a happy behavioural phenotype. His head circumference was normal and no genital malformations were observed.
Figure 1.
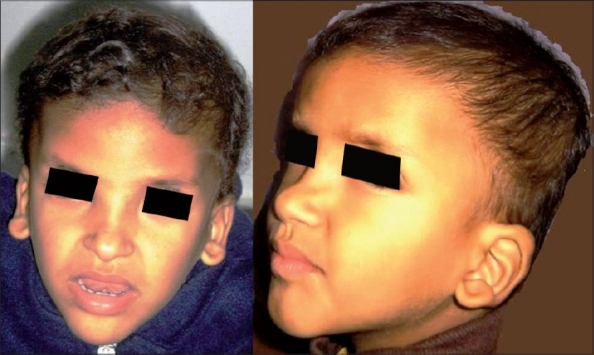
Facial phenotype of the patient
MRI scan of the brain revealed agenesis of the corpus callosum and cerebral hypotrophy. Electroencephalogram (EEG), echocardiography, and genitourinary ultrasound examinations were normal.
Since the patient presented with typical dysmorphic features, severe developmental delay, epilepsy, and agenesis of the corpus callosum we suspected a MWS, and molecular analysis of the ZFHX1B gene was performed. This analysis led to the identification of a novel heterozygous nonsense mutation, c.1165A>T (p.Lys389X) in exon 8 of the ZFHX1B gene.
At the time of diagnosis, the mother of the patient was 8 months pregnant.
Discussion
Mowat-Wilson syndrome, first clinically delineated by Mowat et al. in 1998,[3] is a rare mental retardation-multiple congenital anomalies syndrome associated with typical facial dysmorphism, including hypertelorism, medially flared and broad eyebrows, enophthalmia, prominent columella, pointed chin, and uplifted earlobes, which typically prompts the clinician to consider the diagnosis.[2] Patients can present a variety of other anomalies, such as short stature (50%); microcephaly (84%); HSCR (50%); chronic constipation (25%); malformations of the brain, particularly agenesis of the corpus callosum (60%); seizures (75%), with no predilection for any particular seizure type; congenital heart defects (75%); and urogenital anomalies,[2,4,5] particularly hypospadias (55%).[2,6] In our patient, suspicion of MWS was based essentially on the dysmorphic features associated with severe mental retardation and seizures. Absence of microcephaly, HSCR, congenital heart defect, and hypospadias did not preclude the diagnosis.
Several differential diagnoses of MWS can be evoked. In patients with mental retardation (MR), microcephaly, and HSCR, born to consanguineous parents, the Goldberg-Shprintzen syndrome (GOSHS), a rare autosomal recessive disorder, can be suspected if associated with specific dysmorphia.[4] Severe MR, seizures, ataxia, microcephaly, a prominent jaw, and a happy behaviour phenotype are also features of Angelman syndrome.[4] Finally, Pitt-Hopkins syndrome can be considered in patients with MR, characteristic facial gestalt, and episodes of hyperventilation.[7,8]
In MWS, the ZFHX1B gene mutations are most often truncating (nonsense, frameshift, or deletions) and no obvious genotype-phenotype correlation has been identified so far. In a few cases, atypical phenotypes have been described with missense or splice mutations of the ZFHX1B gene.[9] Few recurrent mutations (6/100) have been identified.[1] As this disorder is sporadic, with the mutations occurring de novo, the risk of recurrence of MWS is low. However, a case of germline mosaicism has been reported.[10] As the genetic counseling of MWS is reassuring, it is important to evoke this diagnosis, particularly in consanguineous families. In our case, the mother gave birth to a healthy girl.
Acknowledgments
We thank Dr. Imane Jaouad Cherkaoui for her contribution. Nathalie Collot is thanked for valuable technical assistance. Dr. Wahlen Sandra is acknowledged for re-reading the manuscript.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared.
References
- 1.Dastot-Le Moal F, Wilson M, Mowat D, Collot N, Niel F, Goossens M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum Mutat. 2007;28:313–21. doi: 10.1002/humu.20452. [DOI] [PubMed] [Google Scholar]
- 2.Adam MP, Schelley S, Gallagher R, Brady AN, Barr K, Blumberg B, et al. Clinical features and management issues in Mowat-Wilson syndrome. Am J Med Genet A. 2006;140:2730–41. doi: 10.1002/ajmg.a.31530. [DOI] [PubMed] [Google Scholar]
- 3.Mowat DR, Croaker GD, Cass DT, Kerr BA, Chaitow J, Ades LC, et al. Hirschsprung disease, microcephaly, mental retardation and characteristic facial features: Delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet. 1998;35:617–23. doi: 10.1136/jmg.35.8.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mowat DR, Wilson MJ, Goossens M. Mowat-Wilson syndrome. J Med Genet. 2003;40:305–10. doi: 10.1136/jmg.40.5.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zweier C, Thiel CT, Dufke A, Crow YJ, Meinecke P, Suri M, et al. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur J Med Genet. 2005;48:97–111. doi: 10.1016/j.ejmg.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Garavelli L, Cerruti-Mainardi P, Virdis R, Pedori S, Pastore G, Godi M, et al. Genitourinary anomalies in Mowat-Wilson syndrome with deletion/mutation in the zinc finger homeo box 1B gene (ZFHX1B): Report of three Italian cases with hypospadias and review. Horm Res. 2005;63:187–92. doi: 10.1159/000085894. [DOI] [PubMed] [Google Scholar]
- 7.Amiel J, Rio M, de Portugal L, Redon R, Malan V, Boddaert N, et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80:988–93. doi: 10.1086/515582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zweier C, Peippo MM, Hoyer J, Sousa S, Bottani A, Clayton-Smith J, et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome) Am J Hum Genet. 2007;80:994–1001. doi: 10.1086/515583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinritz W, Zweier C, Froster UG, Strenge S, Kujat A, Syrbe S, et al. A missense mutation in the ZFHX1B gene associated with an atypical Mowat-Wilson syndrome phenotype. Am J Med Genet A. 2006;140:1223–7. doi: 10.1002/ajmg.a.31267. [DOI] [PubMed] [Google Scholar]
- 10.McGaughran J, Sinnott S, Dastot-Le Moal F, Wilson M, Mowat D, Sutton B, et al. Recurrence of Mowat-Wilson syndrome in siblings with the same proven mutation. Am J Med Genet A. 2005;137:302–4. doi: 10.1002/ajmg.a.30896. [DOI] [PubMed] [Google Scholar]