

Abstract
Malaria is a pathogenic infection caused by protozoa of the genus plasmodium. It is mainly confined to sub-Saharan Africa, Asia and South America. This disease claims the life of over 1.5 to 2.7 million people per year. Owing to such a high incidence of malarial infections, there is an urgent need for the development of suitable vaccines. For the development of ideal vaccines, it is essential to understand the molecular mechanisms of malarial pathogenesis and the factors that lead to malaria infection. Genetic factors have been proposed to play an important role in malarial pathogenesis. Complement receptor 1 (CR1) is an important host red blood cell protein involved in interaction with malarial parasite. Various polymorphic forms of CR1 have been found to be involved in conferring protection or increasing susceptibility to malaria infections. Low-density allele (L) of CR1 gave contradictory results in different set of studies. In addition, Knops polymorphic forms Sl (a+) and McC (a) have been found to contribute more towards the occurrence of cerebral malaria in malaria endemic regions compared to individuals with Sl (a-) / McC (a/b) genotype. This article reviews the research currently going on in this area and throws light on as yet unresolved mysteries of the role of CR1 in malarial pathogenesis
Keywords: Complement receptor 1, Knops polymorphism, Plasmodium falciparum erythrocyte membrane protein 1
Introduction
Malaria caused by protozoa of the genus Plasmodium is the most important parasitic disease in humans. This disease claims the life of over 1.5 to 2.7 million people per year.[1] Owing to such a high incidence of malarial infections, there is an urgent need for the development of suitable vaccines. Development of vaccines is only possible through the identification of major molecular pathways of pathogenesis and immunity for malarial parasite. Parasite virulence phenotype and host genetic factors are the two major foci of research to understand malarial pathogenesis. Plasmodium falciparum virulence phenotype termed as rosetting, causes clumps of erythrocytes and brings about vascular obstruction and impaired tissue perfusion.[2] This property has been associated with severe malaria.[3,4] Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) has been found to be the protein involved in rosetting[5,6] and CR1 is the associated host counterpart.[7]
PfEMP1 is an adhesion protein encoded by the large and diverse var gene family that is involved in clonal antigenic variation and plays a central role in P. falciparum pathogenesis.[8,9] The extra-cellular portion of this protein contains several distinct domains, by virtue of which, it interacts with several types of surface molecules like intercellular adhesion molecule (ICAM-1), type A and Type B blood groups, thrombospondin, E-Selectin, chondroitin sulfate, CR1 and CD36. Previously it has been shown that antibodies to PfEMP1 can confer protection to placental malaria.[10] Bull et al. showed the existence of rare and prevalent isolates that express a subset of variant surface antigens (PfEMP1) and it was suggested that antibodies to the strain specific PfEMP1 can prevent re-infection with previously encountered isolates.[11,12] Based on these observations, it has been suggested that certain regions of PfEMP1 that are involved in binding with CD36/ circumsporozoite antigen (CSA)/ CR1 may be potential vaccine targets.
CR1 has been shown to play a very important role in malaria pathogenesis. Various polymorphic forms of CR1 have been found to have protective or susceptibility effect in malaria. CR1 exhibits three types of polymorphisms: Density polymorphism (determined by alleles H for high and L for low expression on erythrocyte surface),[13,14] Structural polymorphism (differences in molecular weight ranging from 260-250 Kda, determined by alleles A, B, C and D)[13,15,16] and Knops blood group polymorphism.[17]
There have been suggestions that differential expression of this protein on erythrocytes might determine susceptibility of an individual to development of cerebral malaria and severe malaria-associated anemia. Low-density allele of CR1 gave contradictory results in different set of studies.[18,19] Knops polymorphic forms Sl (a-) and McC (b+) have been found to be present at increased frequency in malaria endemic regions. In particular, individuals with Sl (a-) / McC(a/b) genotype were less likely to have cerebral malaria than individuals with Sl (a+)/ McC(a/a).[20] Because PfEMP1 binds to the active site of CR1, it might inhibit CR1 functions. On the other hand modulating the interaction between CR1 and PfEMP1 somehow, can reduce rosetting- associated severity of malaria. It has been shown that monoclonal antibodies against CR1 can block as well as reverse rosette formation.[21] In order to be able to use CR1- PfEMP1 interaction as a therapeutic target, it is essential to understand the role of CR1 and its polymorphic forms in the patho-physiology of malaria. This article reviews the role of various polymorphic forms of CR1 in malarial pathogenesis.
Etiology of malaria
Malaria is an acute and/or chronic infection caused by protozoans of the genus Plasmodium. Four species cause disease in humans namely, Plasmodium falciparum, Plasmodium vivax , Plasmodium ovale and Plasmodium malariae. Out of the four species, majority of the infections are caused by the former two species i.e. P. falciparum and P. vivax.
Characteristics of Plasmodium infection
Plasmodium falciparum causes the most deadly and severe infections.[22,23] It infects all ages of RBCs, leading to a higher parasitemia. Mature trophozoites and schizonts are sequestered in the microvascular system leading to tissue ischemia.[22] A schematic representation of the life cycle of malarial parasite is depicted in Figure 1. The problem of spread of infections becomes all the more acute because of widespread drug resistance in P. falciparum infections.[24,25]
Figure 1.
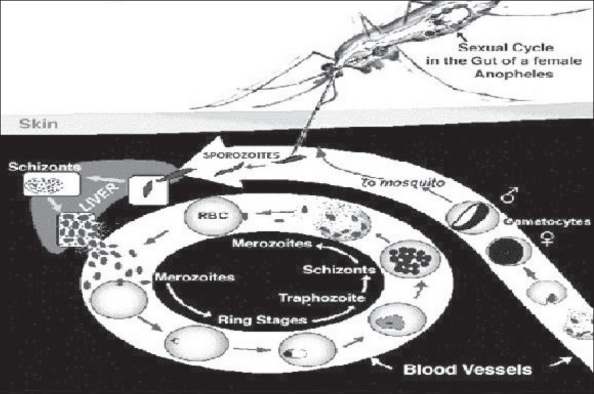
Life cycle of malarial parasite
Plasmodium vivax usually does not cause life-threatening infections. It only infects reticulocytes and produces hypnozoites, which are latent in the liver. Relapses can occur up to 5 years after infection.[26] The parasite uses the Duffy blood receptor to enter RBCs; hence Duffy negative individuals are not infected by this species.[27]
Epidemiology of malaria
Malaria was first documented in 1700 B.C.[28] 40% of the world's population lives in areas where there is a risk of acquiring this disease. Worldwide, an estimated 300-500 million people contract malaria each year, resulting in 1.5-2.7 million deaths annually.[29,30] About 90% of these cases are from tropical Africa; remaining cases are reported from Brazil, India and Sri Lanka. India alone contributes 80% of the victims.[31] 0.1 million of these are children below the age of 5 years.[32] In endemic areas, infected children show symptoms between 4 to 8 months of age. Symptoms include fever, irritability, poor feeding, vomiting, diarrhea and convulsions. In most of the cases the infection is limited to recurring episodes of fever, chills and shakes and can be controlled with proper medication.
However in some of the individuals severe malaria can precipitate due to variety of factors including genetic, environmental, socio-economic status of individuals. It has now been realized that treatment or prevention of malaria is only possible through identification of these factors. In particular, greater emphasis is now being laid on identifying the host genetic factors involved in malaria pathogenesis.
Genetic factors in malaria pathogenesis: Host genes involved in malaria
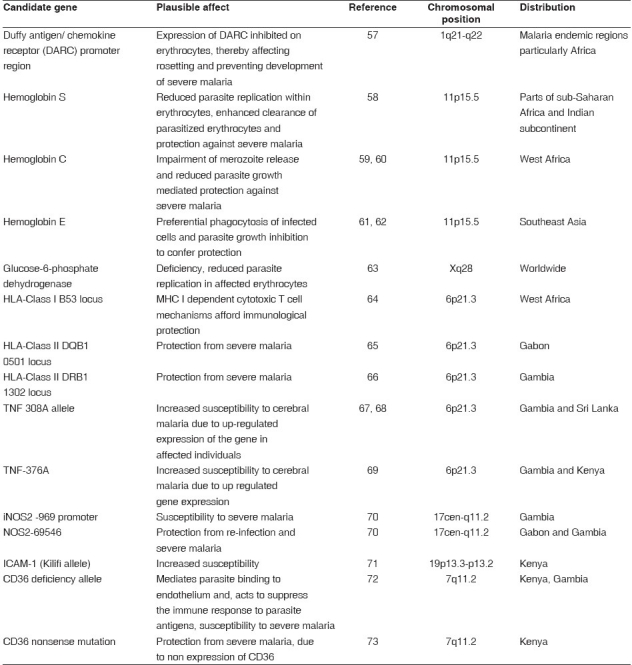
Polymorphic forms of a number of host genes involved in immunity have been associated with protection or susceptibility to malaria. The most plausible candidates include hemoglobin variants, Duffy blood group antigen variants, glucose-6-phosphate dehydrogenase, histocompatibilty genes, tumor necrosis factor-α, ICAM-1, nitric oxide synthase promoter region (NOS 2).[33] Other candidate genes include CR1, which is involved in erythrocyte rosetting and thrombospondin receptor CD36, which mediates parasite binding to endothelium and also possibly to dendritic cells.[33,34] The candidate genes and their plausible role in malaria pathogenesis are listed in Table 1.
Table 1.
The host genes involved in the pathogenesis of malaria
Rosetting of P. falciparum infected erythrocytes
Plasmodium falciparum virulence phenotype is a major area of research these days. This phenotype termed as rosetting[2] causes clumps of cells and brings about vascular obstruction and impaired tissue perfusion. This property has been associated with severe malaria in many studies in Africa.[3,4] Rosetting is mediated by a protein present on the surface of infected RBCs, PfEMP1.[5,6] Plasmodium falciparum secretes protein PfEMP1, which has dual purposes of antigenic variation and cytoadherence.[9] It binds to endothelial receptors (CD36, ICAM-1, CSA on placenta) causing microvascular sequestration, thereby leading to abnormal function of affected tissues [Figure 2].[35,36] Peripheral sequestration also protects Plasmodium falciparum from being sequestered in the spleen.
Figure 2.
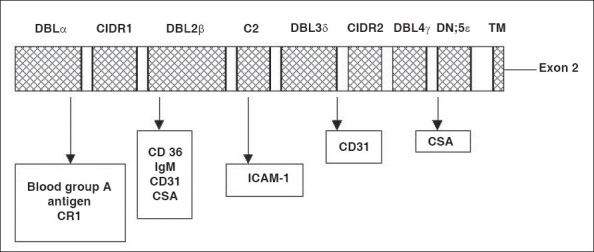
Binding sites for different antigens located on the surface of pFEMP1 DBL - Duffy Binding like, ICAM - Intercellular cell adhesion molecule, CSA - Circumsporozoite antigen, CIDR - Cysteine rich interdomain region, TM - Transmembrane
It has been found that rosetting of erythrocytes is associated with the occurrence of severe malaria, subsequently; a role for PfEMP1 in rosetting was suggested.[5,6] CR1 has been found to be the associated host counterpart for mediating the clumping of uninfected erythrocytes.[7]
Complement receptor 1 and rosetting
CR1 is a 200-kDa single chain membrane bound glycoprotein and a member of the regulators of complement activation (RCA) gene cluster. CR1 possesses complex tri and tetra N-linked oligosaccharides in its mature form and the gene for this protein is located on the q32 arm of chromosome 1.[37,38] It is composed of a number of repeated domains called short consensus repeats (SCRs) each of which is composed of 60 amino acids containing four invariant cysteines.[39] The extra-cellular domain of the CR1 is composed of 30 SCRs, the first 28 of which are arranged in tandem repeats in homologous groups of 7, with each group known as long homologous repeat (LHR).[38] SCRs 8-12 and SCRs 15-18 preferentially bind to C3b and SCRs 1-4 preferentially bind to C4b.[38,39]
The region of CR1 that interacts with infected erythrocytes to form rosettes has been mapped to LHR-B and first three SCRs of LHR-C, SCR 10 and 17 have been particularly found to play an important role in this interaction.[37]
Effect of differential CR1 expression on malarial pathogenesis
Erythrocytes infected with P. falciparum rosette efficiently with uninfected erythrocytes that have normal copy number of CR1, but not with cells in which the number <60.[7] As the number of CR1 molecules on RBCs increases, so does their propensity to form rosettes.[7] Rosette formation is inhibited by sCR1.[7] Differences in the expression of CR1 on erythrocytes might determine susceptibility of an individual towards development of cerebral malaria and severe malaria-associated anemia. In one of the studies it was suggested that young children may be more susceptible to SMA because of their lower levels of RBC complement regulatory proteins, which make them less equipped to handle IC formation and complement activation.[40] Previously same group of researchers had proved that a decline in levels of CR1 and increase in immune complex levels significantly associates with SMA.[41] The mechanism for the loss of CR1 from the surface of erythrocytes is being investigated. A series of experiments indicated that CR1 present in the form of clusters on RBC surface, undergoes unclustering due to the binding of IgM -C3b complexes to glycophorin A.[42] Unclustering might promote rapid loss of CR1 from the surface of erythrocytes infected with the malaria parasite.
CR1 polymorphisms: Are they determinants of malarial susceptibility?
CR1 is a highly polymorphic glycoprotein. Three different polymorphic forms of CR1 have been identified, namely structural (size variation 160-250 kDa), density (high and low expression on RBCs controlled by alleles H and L) and knops blood group (McC (a+)/McC (b+); Sl (a+)/Sl (a-); Kna/ Knb).
a) Structural polymorphism: Four different structural polymorphic forms of CR1 are known, namely A, B, C and D (CR1*1, CR1*2, CR1*3, CR1*4) with respective molecular weights of 190, 220, 160 and 250 kDa (under non-reducing conditions).[15] This polymorphism is regulated by four autosomal co-dominant alleles.[15] A polymorphism in the CR1 transcripts with incremental differences of 1.4 kb in mRNA was present in donors expressing the various polymorphic forms. This difference corresponds to the size of one LHR and 40 kDa difference, seen among allotypic forms of CR1. Therefore on the basis of this observation it was suggested that the insertion or deletion forms the basis of structural polymorphism.[16] Analysis of restriction fragment length polymorphism (RFLP) suggested that intragenic duplication rather than alternate mRNA splicing is responsible for the allotypic differences.[13] Various studies have proved that structural polymorphism of CR1 occurs at different frequencies in distinct populations. Van Dyne et al. had reported the gene frequencies for A and B alleles to be 0.87 and 0.12 for whites, 0.74 and 0.22 for blacks and 0.98 and 0.02 for Orientals.[43] Moulds et al. reported the gene frequencies for A and B alleles to be 0.82 and 0.11 for African-Americans based in Houston and 0.87 and 0.11 for Caucasians based in Houston.[44] Dykman et al . in 1984 had reported the gene frequencies to be 0.83 and 0.16 for A and B alleles respectively.[45] In Indian subjects the percentage distribution of AA, AB and BB structural phenotypes had been previously found to be 94.9%, 5.1% and 0% respectively with relative gene frequencies of 0.975 for A allele and 0.02 for B allele, homozygous BB phenotype had not been identified in this study.[46] In one of our previous studies, we found that out of 117 normal subjects, 82.9% had AA pattern, 16.24% were AB and 0.85% had the BB genotype. The gene frequencies for A and B alleles were found to be 0.911 and 0.088 respectively.[47] Although the gene frequencies for different structural alleles of CR1 differ for distinct populations, but on the whole allele A is the most prevalent in all populations.
b) Density polymorphism: Second type of polymorphism is a Hind III RFLP, which in Caucasians but not in Africans, correlates with CR1 copy number on erythrocytes.[13] Homozygotes for the L (low expression) allele usually express fewer than 200 copies of CR1, homozygotes for the H (high expression) allele express several times this number and heterozygotes are intermediate.[13] This polymorphism arises due to a single base change in the intron of d1d2 segment within the LHR-D (Long homologous repeat) region resulting in the generation of a polymorphic Hind III site within this region.[14] The gene frequencies for H and L alleles of CR1 have been found to be different in different populations. Mitchell et al had shown the gene frequencies to be 0.75 and 0.25 for healthy controls.[48] Wilson et al. found the percentage distribution for HH, HL and LL density polymorphic genotypes to be 65%%, 25% and 10% respectively in case of normal individuals.[49] Moldenhauer et al. had also reported gene frequencies of 0.27 and 0.73 for the L and H allele respectively.[50] Study on the percentage distribution of HH, HL and LL phenotypes in Indian subjects had shown the distribution to be 63%, 29% and 8% respectively. Gene frequencies for H and L alleles in this study were 0.77 and 0.23 respectively. HH had been found to be the predominant form in this study.[51] Genotypic frequencies of HH, HL and LL forms have also been studied in the malaria endemic and non-endemic groups in different populations. In non-endemic Caucasian and Choctaw population groups in USA, the gene frequencies for H and L alleles were found to be 0.82, 0.18 and 0.84, 0.16 respectively. In endemic Black Africans the gene frequencies for H and L alleles were 0.85 and 0.15; in S. Chinese-Taiwanese 0.71 and 0.29; in Pacific Asians 0.42 and 0.58 and in Cambodians 0.53 and 0.47 respectively.[52] Previously we also conducted a study to ascertain the distribution of H and L alleles in 117 normal healthy Indian individuals. In our study, gene frequencies for H and L alleles were 0.53, 0.47 respectively.[47] This indicates that the genotypic frequencies observed for Indian population is similar to that of Pacific Asians and Cambodian population.
c) Knops polymorphism: The third type of polymorphism represented by Knops blood group system is of particular interest.[53] In this system, Mca and Mcb is one allelic antigen pair and Sla and Vil is another pair. The corresponding phenotypes for the first pair are McC (a+) and McC (b+) and for the second pair are Sl (a+) and Sl (a-).[53] Studies have now established the molecular basis for Knops polymorphism. These antigens have been localized on the LHR-D segment of CR1. Single nucleotide polymorphisms occurring in SCR 25, which lead to amino acid substitutions, result in generation of these polymorphic forms [Figure 3].[17] Population based studies have been carried out to determine the distribution of different types of Knops polymorphic forms in different populations. The gene frequencies for Sl (a+) and Sl (a-) in African American persons are almost equal (0.48 vs. 0.52) wheras Sl (a-) is greatly increased in Africa.[17] On the contrary, Sl (a-) is almost completely absent in Caucasian and Asian persons.[54] Data from India however, is still not available on this aspect of CR1 polymorphism and we are investigating the distribution of Knops polymorphic forms of CR1 in the Indian population.
Figure 3.
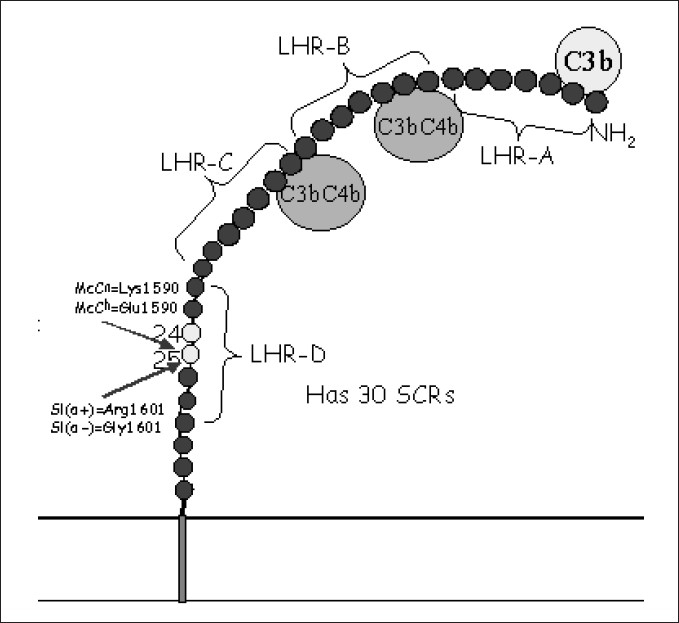
Location of Knops antigen system on complement receptor 1 molecule Sl - Swain Langley blood group, McC - McCoy blood group, LHR - Long homologous repeat, SCR - Short consensus repeat
Out of the three polymorphic forms, size polymorphism has not been found to play a role in determining susceptibility to severe malaria. With regard to density polymorphism, some studies suggest that low-density allele confers protection against malaria[18] whereas another[19] suggested that low- density allele might be a risk factor for severe forms of malaria. Erythrocytes with low CR1 expression (because of the homozygous LL genotype of CR1) have been shown to form reduced number of rosettes with Plasmodium falciparum infected cells.[55] In Africa, rosetting has been shown to correlate with disease severity.[3] Since erythrocytes having low CR1 form fewer rosettes, it has been postulated that low E-CR1 might protect from severe malaria.
Recently, Cockburn et al. suggested that homozygous form of L allele confers protection against severe malaria possibly due to reduced rosetting mediated micro vascular obstruction.[18] On the contrary Nagayasu et al. had observed a significantly higher frequency of the LL genotype in the severe malaria group compared to the uncomplicated malaria group. They reasoned that the presence of circulating immune complexes in the P. falciparum infected individuals contributes to pathogenesis. The authors had suggested that the rate of clearance of immune complexes is thought to be related to the number of CR1 molecules on erythrocytes. Therefore, decreased expression of CR1 (due to L allele) may contribute to the pathogenesis of severe malaria through the deposition of immune complexes and resulting tissue damage.[19] Therefore the role of L allele in the pathogenesis of severe malaria still remains controversial.
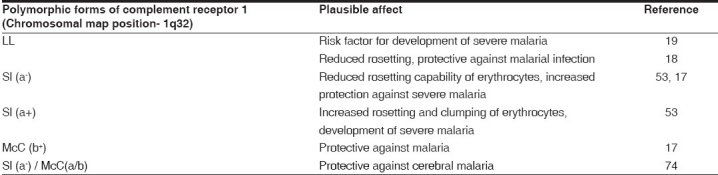
Knops polymorphic forms Sl (a-) and McC (b+) have been found to be present at increased frequency in malaria endemic regions[17] and hence suggested to be protective against rosetting and severe malaria. The role of these polymorphic forms in affecting the rosette formation has also been investigated.[18,53] Knops phenotype Sl (a-) has been found to rosette less efficiently than Sl(a+) and hence suggested to be more protective.[53] Because of less rosetting of uninfected Sl(a-) with infected erythrocytes, there might be less parasite sequestration in the microvasculature in such individuals and consequently protection against obstruction of blood flow. Recently it was found that children with genotype Sl (a-) are less likely to have cerebral malaria, than those children with Sl (a+). In particular, individuals with Sl (a-) / McC(a/b) genotype were less likely to have cerebral malaria than individuals with Sl (a+)/ McC(a/a). This observation led the researchers to hypothesize that the Sl (a-) allele and, possibly, the McCb allele evolved in the context of malaria transmission and that in certain combinations probably confer a survival advantage on these populations[20]Table 2].
Table 2.
Complement receptor 1 polymorphisms associated with protection or susceptibility to malaria
DNA sequence comparison of CR1 revealed that single amino acid variations in SCR 25 of LHR D are responsible for different knops phenotypes.[56] It has also been found that it is the SCR 8, 9, 10 and SCR 15, 16 and 17 of LHR B and LHR C, which interact directly with PfEMP1.[2] SCR 25 has not been experimentally been shown to be involved in direct interaction with PfEMP1.
The molecular mechanism for the exact role of Sl(a-) and McCb alleles in conferring protection against malaria has still not been worked out. Various possible explanations have however been given, first being the bringing together of SCR 25 in the vicinity of SCR 8, 9, 10 and/or SCR 15, 16, 17 because of secondary and tertiary folding of CR1 protein. Second explanation that has been given is that changes in SCR 25 might affect binding of C1q or mannan binding lectin to LHR-D and possibly these proteins might be interacting with PfEMP1. Another hypothesis that has been given is the role of SCR 25 in the CR1 clustering which is the process required for clearance of immune complexes by erythrocytes.[53]
Discussion
Many unsolved mysteries still remain. One of these is the potential role of Mc (b+) and Sl (a-) in conferring protection against malaria. PfEMP1 binds to the active site of CR1, it might therefore inhibit CR1 functions i.e. ligand binding, cofactor activity or that of the decay-accelerating activity. This in-turn could lead to excessive complement activation and hence more of inflammation. On the other hand, the CR1-PfEMP1 interaction could be a potential therapeutic target. Development of a vaccine against PfEMP1 may prevent some cases of severe malaria. Monoclonal antibodies can be developed, that can reverse rosette formation in patients of SMA. However, in order to achieve any of these desired targets, it is absolutely essential to get a more thorough understanding of the interaction of CR1 with PfEMP1 and the role of CR1 polymorphisms in malarial pathogenesis.
Footnotes
Source of Support: Department of Science and Technology, Delhi
Conflict of Interest: None declared.
References
- 1.Malaria, 1982-1997. Wkly Epidemiol Rec. 1999;74:265–70. [PubMed] [Google Scholar]
- 2.Kaul DK, Roth EF, Jr, Nagel RL, Howard RJ, Handunnetti SM. Rosetting of Plasmodium falciparum infected red blood cells with uninfected red blood cells enhances microvascular obstruction under flow conditions. Blood. 1991;78:812–9. [PubMed] [Google Scholar]
- 3.Rowe A, Obeiro J, Newbold CI, Marsh K. Plasmodium falciparum rosetting is associated with malaria severity in Kenya. Infect Immun. 1995;63:2323–6. doi: 10.1128/iai.63.6.2323-2326.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heddini A, Pettersson F, Kai O, Shafi J, Obiero J, Chen Q, et al. Fresh isolates from children with severe Plasmodium falciparum malaria bind to multiple receptors. Infect Immun. 2001;69:5849–56. doi: 10.1128/IAI.69.9.5849-5856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez V, Wahlgren M. Rosetting and autoagglutination in Plasmodium falciparum. Chem Immunol. 2002;80:163–87. [PubMed] [Google Scholar]
- 6.Chen Q, Barragan A, Fernandez V, Sundstrom A, Schlichtherle M, Sahlen A, et al. Identification of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) as the rosetting ligand of the malaria parasite P. falciparum. J Exp Med. 1998;187:15–23. doi: 10.1084/jem.187.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowe JA, Moulds JM, Newbold CI, Miller LHP. P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement receptor 1. Nature. 1997;388:292–95. doi: 10.1038/40888. [DOI] [PubMed] [Google Scholar]
- 8.Baruch DI, Pasloske BL, Singh HB, Bi X, Ma XC, Feldman M, et al. Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 1995;82:77–87. doi: 10.1016/0092-8674(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 9.Smith JD, Chitnis CE, Craig AG, Roberts DJ, Hudson Taylor DE, Peterson DS, et al. Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell. 1995;82:101–10. doi: 10.1016/0092-8674(95)90056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Schlichtherle M, Wahlgren M. Molecular aspects of severe malaria. Clin Microbiol Rev. 2000;13:439–50. doi: 10.1128/cmr.13.3.439-450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bull PC, Lowe BS, Kortok M, Marsh K. Antibody recognition of Plasmodium falciparum erythrocyte surface antigens in Kenya: Evidence for rare and prevalent variants. Infect Immun. 1999;67:733–9. doi: 10.1128/iai.67.2.733-739.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bull PC, Kortok M, Kai O, Ndungu F, Ross A, Lowe BS, et al. Plasmodium falciparum-infected erythrocytes: Agglutination by diverse Kenyan plasma is associated with severe disease and young host age. J Infect Dis. 2000;182:252–9. doi: 10.1086/315652. [DOI] [PubMed] [Google Scholar]
- 13.Wong WW, Cahill M, Rosen MD, Kennedy CA, Bonaccio ET, Morris MJ, et al. Structure of the human CR1 gene.Molecular basis of the structural and quantitative polymorphisms and identification of a new CR1-like allele. J Exp Med. 1989;169:847–63. doi: 10.1084/jem.169.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cornillet P, Philbert F, Kazatchkine MD, Cohen JH. Genomic determination of the CR1 (CD35) density polymorphism on erythrocytes using polymerase chain reaction amplification and HindIII restriction enzyme digestion. J Exp Med. 1991;136:193–7. doi: 10.1016/0022-1759(91)90006-2. [DOI] [PubMed] [Google Scholar]
- 15.Dykman TR, Hatch JA, Aqua MS, Atkinson JP. Polymorphism of the C3b/C4b receptor (CR1): Characterization of a fourth allele. J Immunol. 1985;134:1787–9. [PubMed] [Google Scholar]
- 16.Holers VM, Chaplin DD, Leykam JF. Human complement C3b/C4b receptor (CR1) mRNA polymorphism that correlates with the CR1 allelic molecular weight polymorphism. PNAS. 1987;84:2459–63. doi: 10.1073/pnas.84.8.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moulds JM, Thomas BJ, Doumbo O, Diallo DA, Lyke KE, Plowe CV, et al. Identification of the Kna/ Knb polymorphism and a method for Knops genotyping. Transfusion. 2004;44:164–9. doi: 10.1111/j.1537-2995.2004.00615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cockburn IA, Mackinnon MJ, O’Donnell A, Allen SJ, Moulds JM, Rowe JA, et al. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. PNAS. 2004;101:272–7. doi: 10.1073/pnas.0305306101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagayasu E, Ito M, Akaki M, Nakano Y, Kimura M, Looareesuwan S, et al. CR1 density polymorphism on erythrocytes of falciparum malaria patients in Thailand. Am J Trop Med Hyg. 2001;64:1–5. doi: 10.4269/ajtmh.2001.64.1.11425154. [DOI] [PubMed] [Google Scholar]
- 20.Moulds JM, Kassambara L, Middleton JJ, Baby M, Sagara I, Guindo A, et al. Identification of complement receptor one (CR1) polymorphisms in West Africa. Genes Immun. 2000;1:325–9. doi: 10.1038/sj.gene.6363676. [DOI] [PubMed] [Google Scholar]
- 21.Rowe J, Rogerson SJ, Raza A. Mapping of the region of complement receptor (CR) 1 required for Plasmodium falciparum rosetting and demonstration of the importance of CR1 in rosetting in field isolates. J Immunol. 2000;165:6341–46. doi: 10.4049/jimmunol.165.11.6341. [DOI] [PubMed] [Google Scholar]
- 22.Miller HL, Good MF, Milon G. Malaria pathogenesis. Science. 1994;264:1878–83. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- 23.White NJ, Ho M. The Pathophysiology of malaria. Adv Parasitol. 1992;31:83–173. doi: 10.1016/s0065-308x(08)60021-4. [DOI] [PubMed] [Google Scholar]
- 24.Duraisingh HT, Refour P. Multiple drug resistance genes in malaria-from epistasis to epidemiligy. Mol Microbiol. 2005;57:874–7. doi: 10.1111/j.1365-2958.2005.04748.x. [DOI] [PubMed] [Google Scholar]
- 25.Cowman AF. Functional analysis of drug resistance in Plasmodium falciparum in the post genomic era. Int J Parasitol. 2001;31:871–8. doi: 10.1016/s0020-7519(01)00201-6. [DOI] [PubMed] [Google Scholar]
- 26.Golijan J, Felczak-Korzybska I, Nahorsk WL, Myjak P. Malaria relapse and recrudescence among travelers to the tropics. Int Marit Health. 2003;54:92–100. [PubMed] [Google Scholar]
- 27.Miller H, Mason SJ, Clyde DF, McGinniss MH. The resistance factor to P. vivax in blacks. The Duffy-blood group genotype, FyFy. N Engl J Med. 1976;295:302–4. doi: 10.1056/NEJM197608052950602. [DOI] [PubMed] [Google Scholar]
- 28.Vannozzi F. The triumph of china China. Infez Med. 2000;8:110–11. [PubMed] [Google Scholar]
- 29.Muentener P, Schlagenhauf P, Steffen R. Imported malaria: Trends and perspectives. Bull World Health Organ. 1999;77:560–6. [PMC free article] [PubMed] [Google Scholar]
- 30.Sachs J, Malaney P. The economic and social burden of malaria. Nature. 2002;415:680–5. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 31.Mishra SK, Mohanty S. Clinical presentations of severe and complicated malaria in India. Indian Acad Clin Med. 2001;2:125. [Google Scholar]
- 32.Okelo GB. Cerebral malaria and its management. Afr J Med Pract. 1994;1:11. [PubMed] [Google Scholar]
- 33.Kwiatkowski D. Genetic susceptibility to malaria getting complex. Curr Opin Genet Dev. 2000;10:320–4. doi: 10.1016/s0959-437x(00)00087-3. [DOI] [PubMed] [Google Scholar]
- 34.Weatherall DJ, Miller LH, Baruch DI, Marsh K, Doumbo OK, Casals-Pascual C, et al. Malaria and the red cell. Hematology Am Soc Hematol Educ Program. 2002:35–57. doi: 10.1182/asheducation-2002.1.35. [DOI] [PubMed] [Google Scholar]
- 35.Smith JD, Craig AG, Kriek N, Hudson Taylor D, Kyes S, Fagan T, et al. Identifi cation of a Plasmodium falciparum intercellular adhesion molecule 1 binding domain: A parasite adhesion trait implicated in cerebral malaria. PNAS. 2000;97:1766–71. doi: 10.1073/pnas.040545897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buffet PA, Gamain B, Scheidig C, Baruch D, Smith JD, Hernandez-Rivas R, et al. Plasmodium falciparum domain mediating adhesion to chondroitin sulfate A: A receptor for human placental infection. PNAS. 1999;96:12743–8. doi: 10.1073/pnas.96.22.12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lublin DM, Krsek-Staples J, Pangburn MK, Atkinson JP. Biosynthesis and glycosylation of the human complement regulatory protein decay-accelerating factor. J Immunol. 1986;137:1629–35. [PubMed] [Google Scholar]
- 38.Klickstein LB, Bartow TJ, Miletic V, Rabson LD, Smith JA, Fearon DT. Identification of distinct C3b and C4b recognition sites in the human C3b/C4b receptor (CR1, CD35) by deletion mutagenesis. J Exp Med. 1988;168:1699–717. doi: 10.1084/jem.168.5.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krych M, Hourcade D, Atkinson JP. Sites within the complement C3b/C4b receptor important for the specificity of ligand binding. PNAS. 1991;88:4353–7. doi: 10.1073/pnas.88.10.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.John NW, Béatrice D, Aymric K, Cohen JH, Stoute JA. Age-Related Changes in Red Blood Cell Complement Regulatory Proteins and Susceptibility to Severe Malaria. J Infect Dis. 2004;190:1183–91. doi: 10.1086/423140. [DOI] [PubMed] [Google Scholar]
- 41.Stoute JA, Odindo AO, Owuor BO, Mibei EK, Opollo MO, Waitumbi JN. Loss of red blood cell-complement regulatory proteins and increased levels of circulating immune complexes are associated with severe malarial anemia. J Infect Dis. 2003;187:522–5. doi: 10.1086/367712. [DOI] [PubMed] [Google Scholar]
- 42.Craig ML, Waitumbi JN, Taylor RP. Processing of C3b-opsonized immune complexes bound to non-complement receptor 1 (CR1) sites on red cells: Phagocytosis, transfer and associations with CR1. J Immunol. 2005;174:3059–66. doi: 10.4049/jimmunol.174.5.3059. [DOI] [PubMed] [Google Scholar]
- 43.Dyne SV, Holers VM, Lublin DM, Atkinson JP. The polymorphism of the C3b/C4b receptor in the normal population and in patients with systemic lupus erythematosus. Clin Exp Immunol. 1987;68:570–9. [PMC free article] [PubMed] [Google Scholar]
- 44.Moulds JM, Revielle JD, Arnett FC. Structural polymorphisms of complement receptor 1 (CR1) in systemic lupus erythematosus (SLE) patients and normal controls of three ethnic groups. Clin Exp Immunol. 1996;105:302–05. doi: 10.1046/j.1365-2249.1996.d01-748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dykman TR, Hatch A, Atkinson JP. Polymorphism of the human C3b/C4b receptor.Identification of a third allele and analysis of receptor phenotypes in families and patients with systemic lupus erythematosus. J Exp Med. 1984;159:691–703. doi: 10.1084/jem.159.3.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Panchamoorthy G G, Tiwari SC, Srivastava LM. Inherited structural and quantitative polymorphisms of C3b receptor (CR1) in normal and patients with glomerular diseases. Asian Pac J Allergy Immunol. 1993;11:123–9. [PubMed] [Google Scholar]
- 47.Katyal M, Sivasankar B, Ayub S, Das N. Genetic and structural polymorphism of complement receptor 1 in normal Indian subjects. Immunol Lett. 2003;89:93–8. doi: 10.1016/s0165-2478(03)00155-x. [DOI] [PubMed] [Google Scholar]
- 48.Mitchell J JA, Sim RB, Sim E. CR1 polymorphism in hydralazine-induced systemic lupus erythematosus: DNA restriction fragment length polymorphism. Clin Exp Immunol. 1989;78:354–8. [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson J JG, Wong WW, Murphy EE, Schur PH, Fearon DT. Deficiency of the C3b/C4b receptor (CR1) of erythrocytes in SLE: Analysis of the stability of the defect and of a restriction fragment length polymorphism of the CR1 gene. J Immunol. 1987;138:2708–10. [PubMed] [Google Scholar]
- 50.Moldenhauer F, David J, Fielder AH, Lachmann PJ, Walport MJ. Inherited deficiency of erythrocyte complement receptor type 1 does not cause susceptibility to systemic lupus erythematosus. Arth Rheum. 1987;30:961–6. doi: 10.1002/art.1780300901. [DOI] [PubMed] [Google Scholar]
- 51.Kumar A, Malaviya AN, Sinha S, Khandekar PS, Banerjee K, Srivastava LM. C3b receptor (CR1) genomic polymorphism in rheumatoid arthritis: Low receptor levels on erythrocytes are an acquired phenomenon. Immunol Res. 1994;13:61–71. doi: 10.1007/BF02918226. [DOI] [PubMed] [Google Scholar]
- 52.Thomas BN, Donvito B, Cockburn I, Fandeur T, Rowe JA, Cohen JH, et al. A complement receptor 1 polymorphism with high frequency in malaria endemic regions of Asia but not Africa. Genes Immun. 2005;6:31–6. doi: 10.1038/sj.gene.6364150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krych-Goldberg M, Moulds JM, Atkinson JP. Human complement receptor type 1 (CR1) binds to a major malarial adhesin. Trends Mol Med. 2002;8:531–7. doi: 10.1016/s1471-4914(02)02419-x. [DOI] [PubMed] [Google Scholar]
- 54.Zimmerman PA, Fitness J, Moulds JM, NcMara DT, Kasehagen LJ, Rowe JA, et al. CR1 Knops blood group alleles are not associated with severe malaria in the Gambia. Genes Immun. 2003;4:368–73. doi: 10.1038/sj.gene.6363980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rowe JA, Rogerson SJ, Raza A, Moulds JM, Kazatchkine MD, Marsh K, et al. Mapping of the region of complement receptor 1 (CR1) required for Plasmodium falciparum rosetting and demonstration of the importance of CR1 in rosetting in field isolates. J Immunol. 2000;165:6341–46. doi: 10.4049/jimmunol.165.11.6341. [DOI] [PubMed] [Google Scholar]
- 56.Moulds JM, Zimmerman PA, Doumbo OK, Kassambara L, Sagara I, Diallo DA, et al. Molecular identification of Knops blood group polymorphisms found in long homologous region D of complement receptor 1. Blood. 2001;97:2879–85. doi: 10.1182/blood.v97.9.2879. [DOI] [PubMed] [Google Scholar]
- 57.Tournamille C, Le Van Kim C, Gane P, Cartron JP, Colin Y. Molecular basis and PCR-DNA typing of the Fya/fyb blood group polymorphism. Hum Genet. 1995;95:407–10. doi: 10.1007/BF00208965. [DOI] [PubMed] [Google Scholar]
- 58.Pasvol G, Weatherall DJ, Wilson RJ. Cellular mechanism for the protective effect of haemoglobin S against P. falciparum malaria. Nature. 1978;274:701–03. doi: 10.1038/274701a0. [DOI] [PubMed] [Google Scholar]
- 59.Friedman MJ. Erythrocytic mechanism of sickle cell resistance to malaria. Proc Natl Acad Sci USA. 1978;75:1994–7. doi: 10.1073/pnas.75.4.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Modiano D, Luoni G, Sirima BS, Simpore J, Verra F, Konate A, et al. Hemoglobin C protects against clinical Plasmodium falciparum malaria. Nature. 2001;414:305–08. doi: 10.1038/35104556. [DOI] [PubMed] [Google Scholar]
- 61.Bunyaratvej A, Butthep P, Yuthavong Y, Fucharoen S, Khusmith S, Yoksan S, et al. Increased phagocytosis of Plasmodium falciparum infected erythrocytes with hemoglobin E by peripheral blood monocytes. Acta Haematol. 1986;76:155–8. doi: 10.1159/000206041. [DOI] [PubMed] [Google Scholar]
- 62.Hutagalung R, Wilairatana P, Looareesuwan S, Brittenham GM, Aikawa M, Gordeuk VR. Influence of hemoglobin E trait on the severity of Falciparum malaria. J Infect Dis. 1999;179:283–6. doi: 10.1086/314561. [DOI] [PubMed] [Google Scholar]
- 63.Usanga EA, Luzzatto L. Adaptation of Plasmodium falciparum to glucose 6-phosphate dehydrogenase-deficient host red cells by production of parasite-encoded enzyme. Nature. 1985;313:793–5. doi: 10.1038/313793a0. [DOI] [PubMed] [Google Scholar]
- 64.Hill AV, Elvin J, Willis AC, Aidoo M. Molecular analysis of the association of HLA-B53 and resistance to severe malaria. Nature. 1992;360:434–9. doi: 10.1038/360434a0. [DOI] [PubMed] [Google Scholar]
- 65.May J, Lell B, Luty AJ, Meyer CG, Kremsner PG. HLA-DQB1*0501-restricted Th1 type immune responses to Plasmodium falciparum liver stage antigen 1 protect against malaria anemia and reinfections. J Infect Dis. 2001;183:168–72. doi: 10.1086/317642. [DOI] [PubMed] [Google Scholar]
- 66.Hill AV, Allsopp CE, Kwiatkowski D. Common west African HLA antigens are associated with protection from severe malaria. Nature. 1991;352:595–600. doi: 10.1038/352595a0. [DOI] [PubMed] [Google Scholar]
- 67.McGuire W, Hill AV, Allsopp CE. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature. 1994;371:508–10. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 68.Knight JC, Udalova I, Hill V. A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nature Genet. 1999;22:145–50. doi: 10.1038/9649. [DOI] [PubMed] [Google Scholar]
- 69.Hill AV. Genetic susceptibility to malaria and other infectious diseases: From the MHC to the whole genome. Parasitology. 1996;112:S75–84. doi: 10.1017/s003118200007668x. [DOI] [PubMed] [Google Scholar]
- 70.Burgner D, Xu W, Rockett K, Gravenor M, Charles IG, Hill AV, et al. Inducible nitric oxide synthase polymorphism and fatal cerebral malaria. Lancet. 1999;352:1193–4. doi: 10.1016/S0140-6736(05)60531-4. [DOI] [PubMed] [Google Scholar]
- 71.Reyes DF, Craig AG, Kyes SA, Peshu N, Snow RW, Berendt AR, et al. A high frequency African coding polymorphism in the N-terminal domain of ICAM-1 predisposing to cerebral malaria in Kenya. Hum Mol Genet. 1997;6:1357–60. doi: 10.1093/hmg/6.8.1357. [DOI] [PubMed] [Google Scholar]
- 72.Aitman TJ, Cooper LD, Norsworthy PJ, Wahid FN, Gray JK, Curtis BR, et al. Malaria susceptibility and CD36 mutation. Nature. 2000;405:1015–6. doi: 10.1038/35016636. [DOI] [PubMed] [Google Scholar]
- 73.Pain A, Urban BC, Kai O, Casals-Pascual C, Shafi J, Marsh K, et al. A non-sense mutation in cd36 gene is associated with protection from severe malaria. Lancet. 2001;357:1502–03. doi: 10.1016/S0140-6736(00)04662-6. [DOI] [PubMed] [Google Scholar]
- 74.Thathy V, Moulds JM, Guyah B, Otieno W, Stoute JA. Complement receptor 1 polymorphisms associated with resistance to severe malaria in Kenya. Malar J. 2005;4:54. doi: 10.1186/1475-2875-4-54. [DOI] [PMC free article] [PubMed] [Google Scholar]