

Abstract
Understanding neural circuits requires methods to record from many neurons simultaneously. For in vitro studies, one currently available technology is planar multielectrode array (MEA) recording. Here we document the use of MEAs to study the mouse vomeronasal organ (VNO), which plays an essential role in the detection of pheromones and social cues via a diverse population of sensory neurons expressing hundreds of types of receptors. Combining MEA recording with a robotic liquid handler to deliver chemical stimuli, the sensory responses of a large and diverse population of neurons can be recorded. The preparation allows us to remove the intact neuroepithelium of the VNO from the mouse and stimulate with a battery of chemicals or potential ligands while monitoring the electrical activity of the neurons for several hours. Therefore, this technique serves as a useful method for assessing ligand activity as well as exploring the properties of receptor neurons. We present the techniques needed to prepare the vomeronasal epithelium, MEA recording, and chemical stimulation.
Protocol
Part 1. Electrical Recording Instrumentation
We use a planar multielectrode array (ALA Scientific Instruments) with 10 μm flat titanium nitride electrodes insulated with silicon nitride. 60 electrodes are arranged in two fields consisting of a 5x6 grid with center-to-center spacing of 30 μm. (Other configurations are also possible; electrodes with a recording surface 10 μm in diameter are better than larger electrodes in terms of isolating the activity of single neurons.)
Electrical signals are amplified (1000x amplification) using a MEA 1060 amplifier (ALA Scientific Instruments). Signals are acquired at 10 kHz using a data acquisition card (National Instruments) and custom data acquisition software (one can also purchase commercial software).
The tissue is held down on the MEA by a nylon mesh (53 μm), which is attached to a custom insert which fits snugly inside the culture tube ring of the MEA.
Part 2. Liquid Delivery Instrumentation
The tissue requires continuous superfusion, which we guarantee by supplying Ringer's solution (115 mM sodium chloride, 5 mM potassium chloride, 2 mM calcium chloride, 2 mM magnesium chloride, 25 mM sodium bicarbonate, 10 mM HEPES, and 10 mM D-(+)-glucose) from an HPLC pump. Stimuli are presented by performing injections into an HPLC valve and switching between flow-through and the loop. Both the HPLC pump and the injection robot's syringe pump are supplied with Ringer's solution.
Ringer's solution is kept in a heated water bath (ISOTEMP 105) at 42° C throughout the experiment and is continuously bubbled with 95% O2 / 5% CO2. A separate flask of Ringer's without glucose is prepared for backing the syringe pump. Ringer's solution is delivered to the tissue using a 307 HPLC pump (Gilson) at a rate of 3 mL/min. In a typical 5-6 hour experiment, 1 L of each Ringer's solution is sufficient.
Throughout the experiment, the tissue is continuously superfused with 37°C Ringer's solution. A heater probe is positioned directly above the tissue.
Stimuli are delivered using the Gilson 215 Liquid Handler robotic arm. Stimuli are stored in rubber-capped glass vials, and under the control of Gilson software, the robotic arm delivers the stimuli to the perfusion line while constant flow rate and pressure are maintained.
The state of the HPLC valve (loop engaged/not engaged) is represented by an electrical voltage, which is recorded by the acquisition computer as an extra channel to ensure synchronization with the neuronal activity.
Part 3. Preparing the MEA and Stimulus Delivery Device
Prior to performing the dissection, fill the MEA well with BSA in dH2O for at least 5 minutes at room temperature.
Rinse the MEA using warm-bubbled Ringer's solution and place the MEA on ice.
Prime the liquid handler and HPLC pump according to manufacturer's instructions.
Part 4. VNO Dissection
Before performing the dissection, fill 2 60x15 mm Petri dishes with bubbled Ringer's solution and place on ice. Also fill 2 Sylgard coated Petri dishes of the same size with Ringer's solution and place on ice. Chill an additional 50mL of warm-bubbled Ringer's for use during the dissection.
The mouse is sacrificed via CO2 asphyxiation followed by cervical dislocation.
Make bilateral incisions from the side of the mouth, running parallel to the jaw line, to the back of the head. Join the 2 cuts along the back of the neck and remove the dorsal part of the head, leaving the lower jaw attached to the mouse.
Using fine scissors, cut between the tissue and either side of the upper jaw bone to separate the tissue from the bone.
Using #3 forceps, peel off the palate tissue to expose the nasal cavity.
Using small scissors or a scalpel, cut between and on either side of the upper 2 front teeth to free the vomeronasal capsule from the bone.
Using #3 forceps, pick up the vomeronasal capsule by the flat bone and place in the Petri dish on ice. The steps up through immersion should be performed in less than 3 minutes.
Place the dish under a stereomicroscope, illuminated with reflected light. Remove the bony capsule from one VNO at a time using #3 forceps. Use one hand to stabilize the bony capsule by gripping from the caudal end and the other hand to peel the bone away from the VNO, taking care not to damage the VNO.
When the bone is removed, transfer the VNO to a Sylgard coated Petri dish. Use the remaining dish for the other VNO. Switch the illumination to transmitted light.
Using micro-scissors or #5 forceps, separate the VNO from the blood vessel by cutting along the edge of the VNO. At this point you will have the neural sheet completely separated from the blood vessel.
Place the VNO with the internal (dendritic) side up. Using #3 forceps, anchor the VNO to the Sylgard at a corner of the tissue. Peel the neural epithelium from the basal lamina using a small fragment of a Teflon-coated razor blade held with a clamp. Holding the blade at a shallow angle (< 30° from the dish surface) is best.
Using a glass pipette, transfer the piece of neural epithelium from the dish to the MEA. Position the VNO on top of the electrode field with the dendritic side up. Remove excess Ringer's solution with a pipette. Place the mesh tissue holder into the well of the array to hold the VNO in place, and fill with chilled Ringer's solution.
Part 5. Recording
Place the MEA in a MEA 1060 amplifier and position the heater probe directly above the tissue.
To confirm proper heater probe placement, stimulate with Ringer's solution containing 50 mM potassium (substituted for equimolar sodium) for 2 seconds. Adjust positioning to minimize the latency of the neural response to the potassium solution.
Superfuse the tissue with heated Ringer's solution for 45 minutes to 1 hour prior to recording to allow the tissue to acclimate.
Electrical signals are saved to disk using custom recording software.
Part 6. Data Analysis
Offline data analysis can be done using the raw electrode recordings. For example, we measure the firing rate changes of recorded cells in response to particular ligands. The resulting data sets are large and can be used to address many different questions; familiarity with a programming or analysis language (C, Matlab, R, Python, or similar) is required.
Part 7. Representative Results
This preparation is typically stable enough to record from for 6 hours, yielding a large amount of data (6 hours of recording from 60 electrodes).
 Figure 1. A snapshot of simultaneous activity across one half of the array is shown. The time window spans 400 ms. Please click here to see a larger version of figure 1.
Figure 1. A snapshot of simultaneous activity across one half of the array is shown. The time window spans 400 ms. Please click here to see a larger version of figure 1.
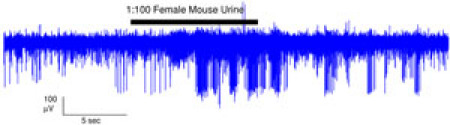 Figure 2. In a typical experiment, we stimulate with particular compounds of interest for 10 seconds. This trace is an example of a response to 100-fold diluted female mouse urine. Please click here to see a larger version of figure 2.
Figure 2. In a typical experiment, we stimulate with particular compounds of interest for 10 seconds. This trace is an example of a response to 100-fold diluted female mouse urine. Please click here to see a larger version of figure 2.
Discussion
Multielectrode array recordings allow us the ability to monitor the simultaneous activity of a large population of neurons. This is a useful tool for probing the properties of a sensory system, specifically the VNO. Unlike the main olfactory system, the VNO detects stimuli in liquid phase. In addition, the VNO is a heterogeneous tissue. There are approximately 300 different types of receptors in the mouse1, presumably with different response profiles. MEA recordings combined with the robotic arm allow us to reliably deliver liquid stimuli to a large and diverse population of neurons and identity a response electrophysiologically. In addition, the stability of the VNO in vitro allows time for multiple repeats of a stimulus.
Previously, this technique has been used to investigate the sensory properties of VSNs in response to mouse urine2 as well as serve as a reliable assay for identifying individual ligands present in urine3. In the future, this technique may be highly useful to further ligand discovery and in characterizing the sensory properties of the VNO.
Acknowledgments
We would like to thank members of the Holy Lab. This work was supported by the National Institute for Deafness and Communication Disorders (R01 DC005964 to T.E.H.) and NSF IGERT 0548890 (H.A.A).
Experiments on animals were performed in accordance with the United States Animal Welfare Acts and National Institutes of Health guidelines and were approved by the Washington University Animal Care and Use Committee.
References
- Dulac C, Torelo AT. Molecular detection of pheromone signals in mammals: from genes to behaviour. Nat Rev Neurosci. 2003;4:551–562. doi: 10.1038/nrn1140. [DOI] [PubMed] [Google Scholar]
- Holy TE. Responses of Vomeronasal neurons to natural stimuli. Science. 2000;289:1569–1572. doi: 10.1126/science.289.5484.1569. [DOI] [PubMed] [Google Scholar]
- Nodari F. Sulfated steroids as natural ligands of mouse pheromone-sensing neurons. J Neurosci. 2008;28:6407–6418. doi: 10.1523/JNEUROSCI.1425-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]