

Abstract
The activity of c-Jun, the major component of the transcription factor AP-1, is potentiated by amino-terminal phosphorylation on serines 63 and 73 (Ser-63/73). This phosphorylation is mediated by the Jun amino-terminal kinase (JNK) and required to recruit the transcriptional coactivator CREB-binding protein (CBP). AP-1 function is antagonized by activated members of the steroid/thyroid hormone receptor superfamily. Recently, a competition for CBP has been proposed as a mechanism for this antagonism. Here we present evidence that hormone-activated nuclear receptors prevent c-Jun phosphorylation on Ser-63/73 and, consequently, AP-1 activation, by blocking the induction of the JNK signaling cascade. Consistently, nuclear receptors also antagonize other JNK-activated transcription factors such as Elk-1 and ATF-2. Interference with the JNK signaling pathway represents a novel mechanism by which nuclear hormone receptors antagonize AP-1. This mechanism is based on the blockade of the AP-1 activation step, which is a requisite to interact with CBP. In addition to acting directly on gene transcription, regulation of the JNK cascade activity constitutes an alternative mode whereby steroids and retinoids may control cell fate and conduct their pharmacological actions as immunosupressive, anti-inflammatory, and antineoplastic agents.
Keywords: AP-1, JNK/SAPK, nuclear hormone receptors, protein phosphorylation, signal transduction
Lipophilic hormones such as steroids, retinoic acid (RA), thyroid hormone (T3), and vitamin D mediate most, if not all, of their actions through specific intracellular receptors that are members of the nuclear receptor superfamily (Mangelsdorf et al. 1995). Nuclear hormone receptors are ligand-regulated sequence-specific transcription factors that may activate or repress gene expression. Ligand-activated gene transcription is generally mediated by binding of nuclear receptors to their cognate DNA elements. Though negative binding elements have been described, repression is mainly conducted by interference with other transcription factors, of which AP-1 is one of the most representative (for review, see Saatcioglu et al. 1994).
AP-1 is a sequence-specific transcription factor composed of either homo- or heterodimers among members within the Jun family (c-Jun, JunB, and JunD) or among proteins of the Jun and Fos (c-Fos, FosB, Fra1, and Fra2) families (for review, see Angel and Karin 1991). Among them, c-Jun is the major component of the AP-1 complex (Bohmann et al. 1987; Angel et al. 1988) and c-Fos is its best known partner. AP-1 is activated by mitogens, oncoproteins, cytokines, and stress agents such as ultraviolet (UV) light. AP-1 activation may be mediated by both transcriptionally independent and -dependent mechanisms, involving post-translational modifications of its components or increases in the expression of their corresponding genes, respectively (Angel and Karin 1991; Karin 1995; Karin et al. 1997).
c-Jun transcriptional activity is enhanced by amino-terminal phosphorylation on Ser-63/73 (Pulverer et al. 1991; Smeal et al. 1991). This inducible phosphorylation is mediated by members of the Jun amino-terminal kinase (JNK, also known as stress-activated protein kinase, SAPK) subfamily (Hibi et al. 1993; Dérijard et al. 1994; Kyriakis et al. 1994; Minden et al. 1994a) and is required to recruit the transcriptional coactivator cAMP response element-binding (CREB) protein (CBP) (Arias et al. 1994; Bannister et al. 1995). Prior to phosphorylation, JNK requires binding to a docking site located in the c-Jun amino-terminal domain (Hibi et al. 1993). This docking site is partially deleted in the v-Jun oncoprotein (deletion of the δ region) and, in consequence, JNK neither binds to nor phosphorylates v-Jun (Hibi et al. 1993). In contrast to c-Jun, however, v-Jun is fully competent to interact with CBP in vivo independently of amino-terminal phosphorylation (Bannister et al. 1995).
c-jun and c-fos are immediate-early genes whose transcription is activated in a protein synthesis-independent manner within a few minutes of cell stimulation. c-jun transcriptional activation is mediated by a TPA response element (TRE) that is bound by the transcriptional activator ATF-2 either as homodimer (van Dam et al. 1995) or as heterodimer with c-Jun (van Dam et al. 1993). In this way, c-Jun may autoregulate expression of its own gene. Similarly, c-fos induction is mediated by the transcriptional activator Elk-1 that binds the serum response element (SRE) in its promoter together with the SRE-binding protein (SRF) (Herrera et al. 1989; Shaw et al. 1989; Hipskind et al. 1991). In addition to c-Jun, both ATF-2 and Elk-1 are phosphorylated and activated by JNK (Cavigelli et al. 1995; Gille et al. 1995; Gupta et al. 1995; Livingstone et al. 1995; van Dam et al. 1995; Whitmarsh et al. 1995).
The JNK subfamily belongs to the mitogen-activated protein kinase (MAPK) group, which in mammals includes several subfamilies such as extracellular signal-regulated kinases (ERKs) and p38/Mpk2/cytokine-supressive anti-inflammatory drug-binding protein (CSBP) among others (for review, see Robinson and Cobb 1997). MAPKs are respectively integrated in different signal transduction pathways and show differences in substrate specificity. However, they are commonly the final step of a protein kinase cascade leading to activation of a MAPK kinase (MAPKK) that activates them by dual phosphorylation on threonine and tyrosine (for review, see Karin and Hunter 1995; Cahill et al. 1996). JNKs, initially identified by their ability to bind and phosphorylate c-Jun amino-terminal domain in response to UV stimulation, are also activated by proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1), and by environmental stress (Hibi et al. 1993; Dérijard et al. 1994; Kyriakis et al. 1994; Minden et al. 1994a).
Though inhibition of AP-1 activity was initially described for the glucocorticoid receptor (GR) (Jonat et al. 1990; Schüle et al. 1990; Yang-Yen et al. 1990), other members of the nuclear receptor superfamily, including RA, T3, vitamin D, and retinoid X receptors, show the same activity (Saatcioglu et al. 1994 and references therein). Recently, it has been reported that transcriptional activation by nuclear receptors requires, similarly to c-Jun, the transcriptional coactivator CBP (Chakravarti et al. 1996; Kamei et al. 1996). Based on these findings, it has been proposed that the mechanism for nuclear receptor–AP-1 antagonism relies on their direct competition for this coactivator (Kamei et al. 1996).
Here we show that hormone-activated nuclear receptors inhibit induction of the JNK signal transduction pathway. As a consequence, nuclear receptors prevent phosphorylation-dependent activation of transcription factors, such as c-Jun, ATF-2, and Elk-1, that are involved in AP-1 induction by either transcriptionally independent or dependent mechanisms. In addition, these findings provide strong evidence for a novel mechanism of AP-1 inhibition by nuclear receptors based on the interference with the c-Jun activation step that is required for CBP interaction.
Results
Dexamethasone blocks AP-1 activation by inhibiting c-Jun amino-terminal phosphorylation
To investigate the mechanism of AP-1 antagonism by nuclear hormone receptor activation we first chose UV light to activate AP-1. Initially, HeLa cells were used because AP-1 activation by UV has been well documented in this cell type and, in addition, HeLa cells harbor endogenous GRs. The initial response to UV irradiation occurs in the absence of protein synthesis and involves post-translational modifications of pre-existing AP-1 complexes such as c-Jun amino-terminal phosphorylation, which leads to AP-1 activation (Stein et al. 1989; Devary et al. 1991, 1992; Radler-Pohl et al. 1993; Sachsenmaier et al. 1994). As shown in Figure 1A, UV irradiation of serum-starved HeLa cells triggers transcription of the AP-1-dependent reporter −73Col–CAT. However, a short pretreatment (45 min) with the synthetic glucocorticoid dexamethasone (Dex) partially inhibits this activation. Moreover, increasing the amount of intracellular GR by cotransfection with pSG5–GR expression vector results in a complete inhibition of UV-induced activity of the −73Col–CAT reporter in response to Dex (Fig. 1A). In contrast, glucocorticoid treatment of uninduced cells did not reduce the basal activity of the AP-1-dependent reporter (Fig. 1A). In agreement with previous data (König et al. 1992), hormone treatment did not decrease the basal AP-1 DNA-binding activity measured by electrophoretic mobility shift assay (EMSA) (Fig. 1B, cf. 0 time points). However, Dex does prevent the increase in AP-1 DNA-binding activity that follows UV stimulation (Fig. 1B). Accordingly, UV induction of c-jun and c-fos gene expression, known to account for this increase in AP-1 DNA-binding activity (Stein et al. 1989; Devary et al. 1991), is severely impaired in Dex-treated cells as deduced from the Western blot analysis shown in Figure 1C.
Figure 1.
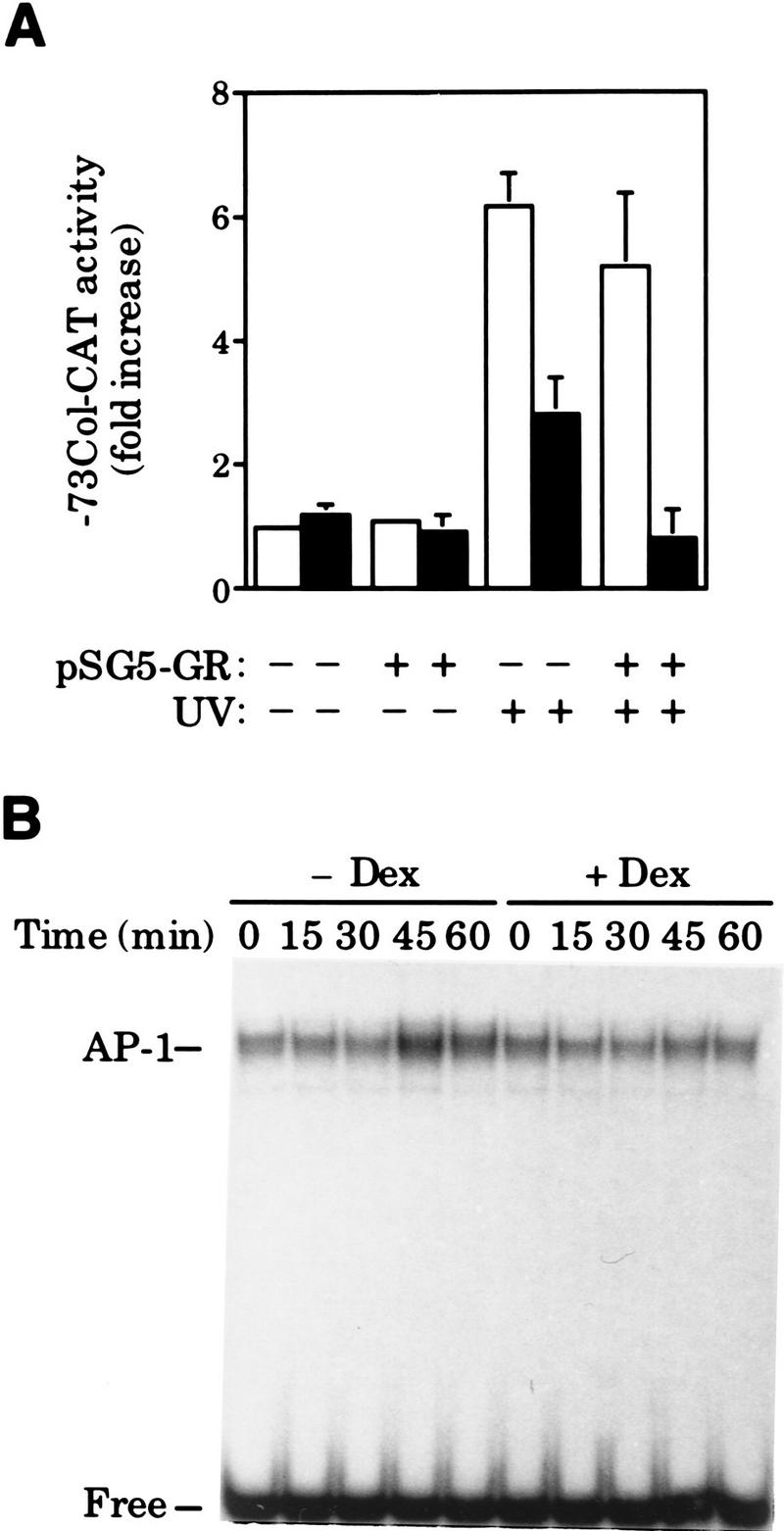
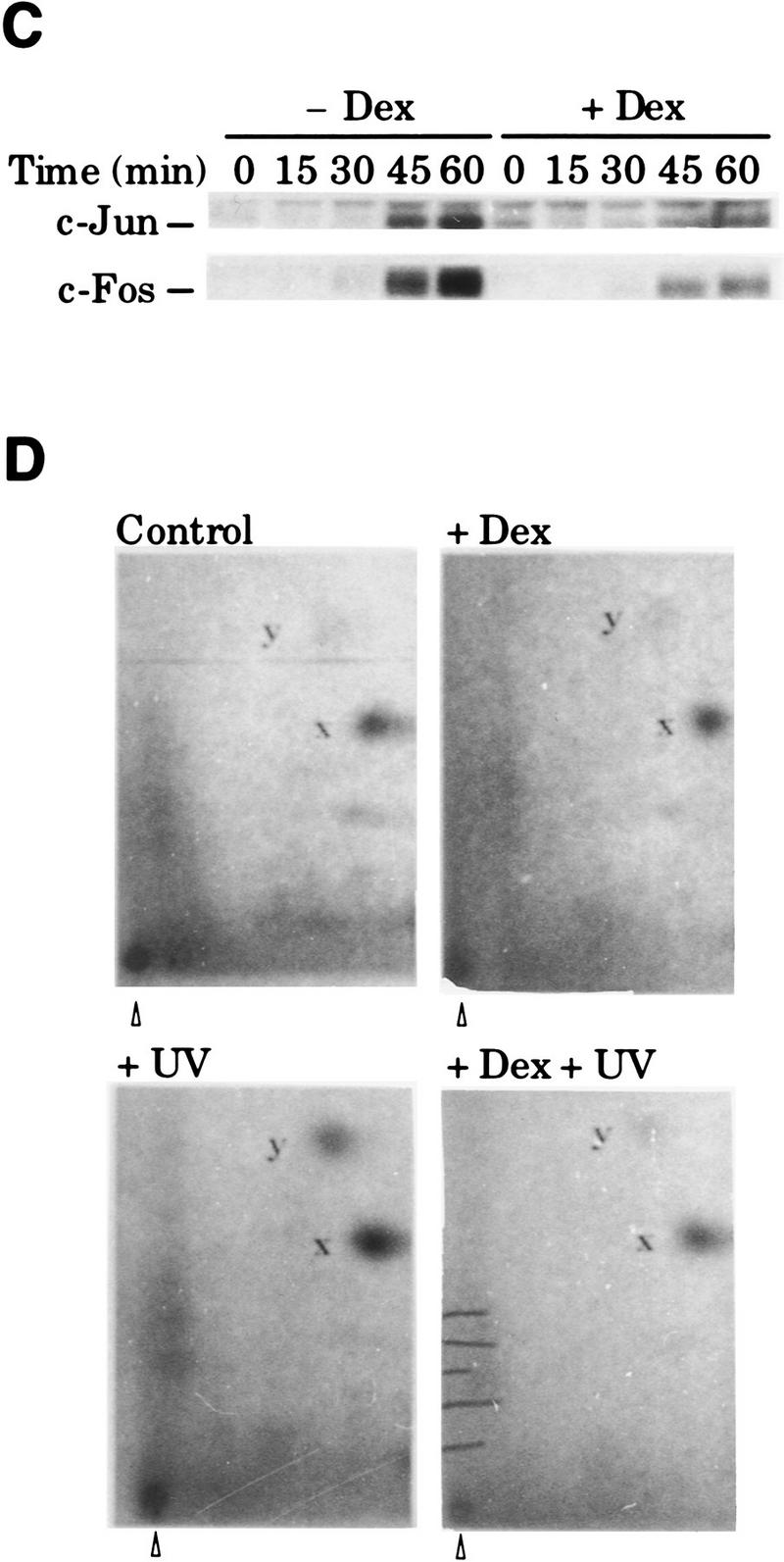
Dex blocks UV-induced AP-1 activation by preventing c-Jun amino-terminal phosphorylation in HeLa cells. (A) Inhibition of UV-induced AP-1 activation by Dex in cells transiently transfected with the −73Col–CAT reporter (3 μg), along with pSG5–GR (0.2 μg), when indicated. (B) Dex inhibits the increase in AP-1 DNA-binding activity induced by UV stimulation. EMSA was performed with nuclear extracts (5 μg/lane) prepared from cells treated with vehicle (open bars) or Dex (solid bars) and harvested at the indicated time points after UV irradiation. Mobility of free and AP-1-complexed probe is indicated. (C) Dex blocks UV induction of c-jun and c-fos gene expression. Same extracts as in B were analyzed by immunoblotting (20 μg of extract/lane) to monitor the accumulation of c-Jun and c-Fos. (D) Dex prevents c-Jun amino-terminal phosphorylation in response to UV irradiation. c-Jun isolated by immunoprecipitation from metabolically-labeled cells (see Materials and Methods) was subjected to tryptic phosphopeptide mapping. y and x spots correspond to phosphorylation of Ser63/73, respectively. The map origin is marked by an arrowhead.
These results suggested that GR interfered with the activation of pre-existing AP-1 complexes in HeLa cells. As mentioned, phosphorylation of c-Jun on Ser-63/73 is one of the earliest UV-induced events that mediates the transcriptionally independent activation of AP-1 (Stein et al. 1989; Devary et al. 1991, 1992; RadlerPohl et al. 1993; Sachsenmaier et al. 1994). Therefore, we investigated the effect of Dex on in vivo UV-induced c-Jun phosphorylation. Quantitation of tryptic phosphopeptide maps of in vivo-labeled c-Jun showed that UV stimulation increased phosphorylation on Ser-63/73 by a factor of 3 and 2, respectively (y and x phosphopeptides in Fig. 1D). This increase was abolished by Dex pretreatment (Fig. 1D). In agreement with transient transfection assays (Fig. 1A), Dex did not significantly affect c-Jun phosphorylation in uninduced conditions (Fig. 1D).
Next, we studied the effect of Dex on the activation of the −73Col–CAT reporter by the cytokine tumor necrosis factor TNF-α, an AP-1 inducer in HeLa cells. Similarly to its action on UV stimulation, Dex also inhibited TNF-α-induced activation of the −73Col–CAT reporter in a receptor-dependent manner (Fig. 2A). The analysis of c-Jun phosphorylation on Ser-63 by immunoblotting using a specific antibody showed that phosphorylation of this serine residue in response to TNF-α was, again, severely impaired by Dex activation of endogenous GR in HeLa cells (Fig. 2B). Because the level of c-Jun increases rapidly in response to this cytokine in a transcription-dependent manner (Brenner et al. 1989), the same experimental procedure was repeated by adding actinomycin D 15 min before TNF-α to keep the amount of c-Jun constant. As shown in Figure 2B, actinomycin D efficiently blocked the transcriptional response of c-jun but did not prevent the inhibition by Dex of c-Jun amino-terminal phosphorylation.
Figure 2.


TNF-α-induced AP-1 activation and c-Jun amino-terminal phosphorylation is inhibited by Dex. (A) Inhibition of TNF-α-induced AP-1 activity by Dex in HeLa cells transiently transfected with the −73Col–CAT reporter (3 μg) along with pSG5–GR (0.2 μg), when indicated. (B) Dex prevents c-Jun amino-terminal phosphorylation in response to TNF-α. Western blot analysis of nuclear extracts (20 μg/lane) prepared from HeLa cells treated with vehicle (open bars) or Dex (solid bars) and collected at the indicated time points after TNF-α stimulation. Additionally, cells were treated with either vehicle (−Act. D) or 1 μg/ml of actinomycin D (+Act. D) 15 min before TNF-α addition. Specific antibodies to detect c-Jun phosphorylated on Ser-63 (P-S63–c-Jun) (top) or total c-Jun (bottom) were subsequently used on the same membrane after stripping.
Hormone-activated nuclear receptors specifically interfere with transactivation mediated by c-Jun amino-terminal phosphorylation independently of the c-Jun DNA-binding domain
Next, we examined whether the inhibition of c-Jun activation by GR was mediated by the amino-terminal domain of c-Jun. For that purpose, we took advantage of the full UV responsiveness of the c-Jun amino-terminal transactivation domain when linked to unrelated DNA-binding domains such as those from GHF-1 (Smeal et al. 1991; Devary et al. 1992) or Gal4 (Hibi et al. 1993; Radler-Pohl et al. 1993; Black et al. 1994) transcription factors. Cotransfection experiments in F9 cells showed that ligand-activated GR inhibits UV-induced activation of a GHF-1-dependent reporter, 3xGHF-1–Luc (luciferase), by the c-Jun–GHF-1 chimera (Fig. 3A). In agreement with Yang-Yen et al. (1990), basal activity of the c-Jun-GHF-1 chimera is not repressed by glucocorticoids (Fig. 3A). Moreover, another member of the nuclear receptor superfamily, thyroid hormone receptor (TRα-1), also known to antagonize AP-1 (Desbois et al. 1991; Zhang et al. 1991), behaved in a similar fashion (Fig. 3A). Similarly to the c-Jun–GHF-1 activity in uninduced cells, hormone activation of these nuclear receptors also failed to decrease the activity of the UV-unresponsive mutant c-JunA63/73–GHF-1 harboring alanine instead of serine at positions 63 and 73, in any condition (Devary et al. 1992) (Fig. 3A). As with wild-type c-Jun, quantitative analysis of tryptic phosphopeptide maps of in vivo-phosphorylated c-Jun-GHF-1 showed that GR activation caused a 60% reduction in the overall UV-induced phosphorylation of c-Jun amino-terminal domain, including Ser-63/73, as well as threonine-91 and -93 (Thr-91/93), two minor additional UV-induced phosphorylation sites (Dérijard et al. 1994; Papavassiliou et al. 1995) (Fig 3B).
Figure 3.
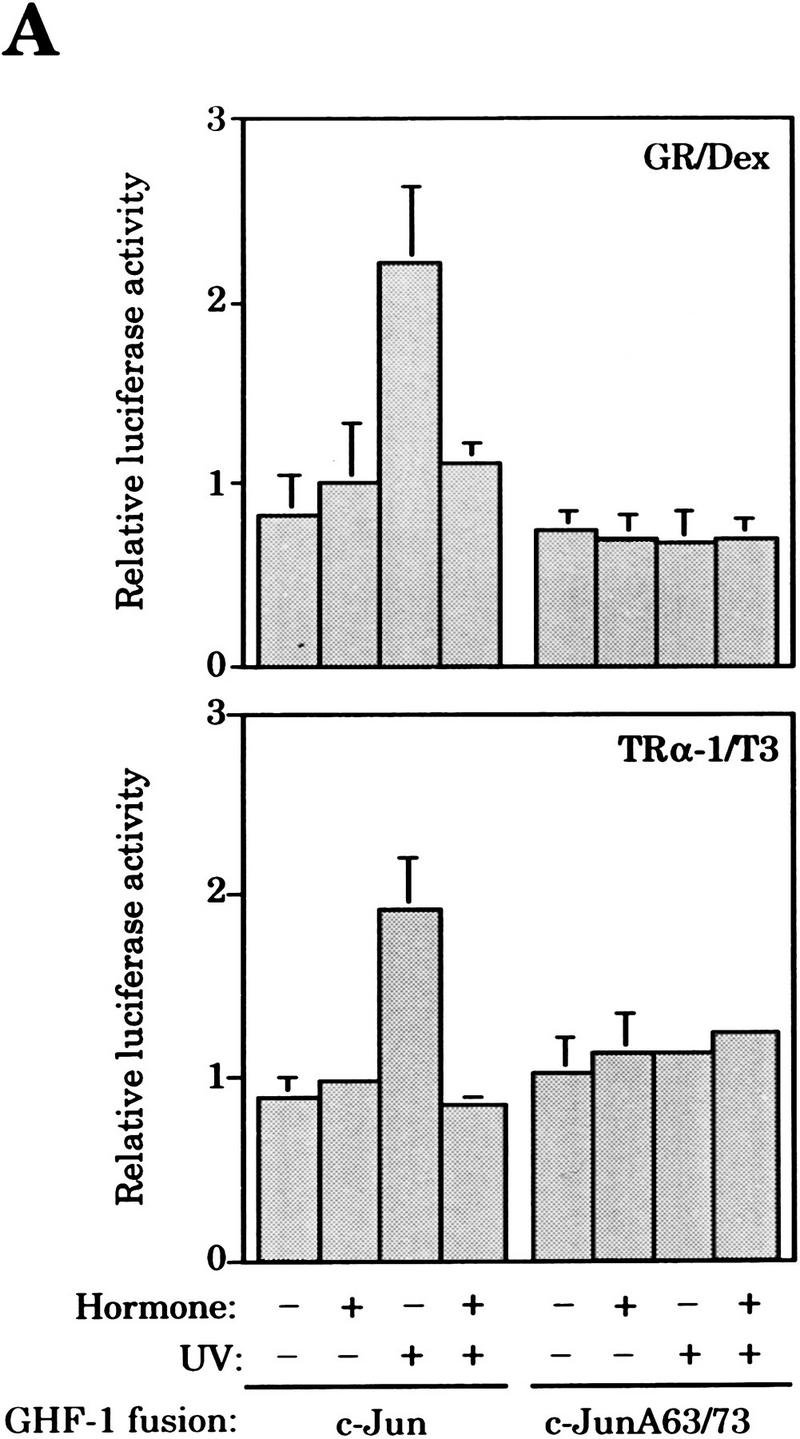
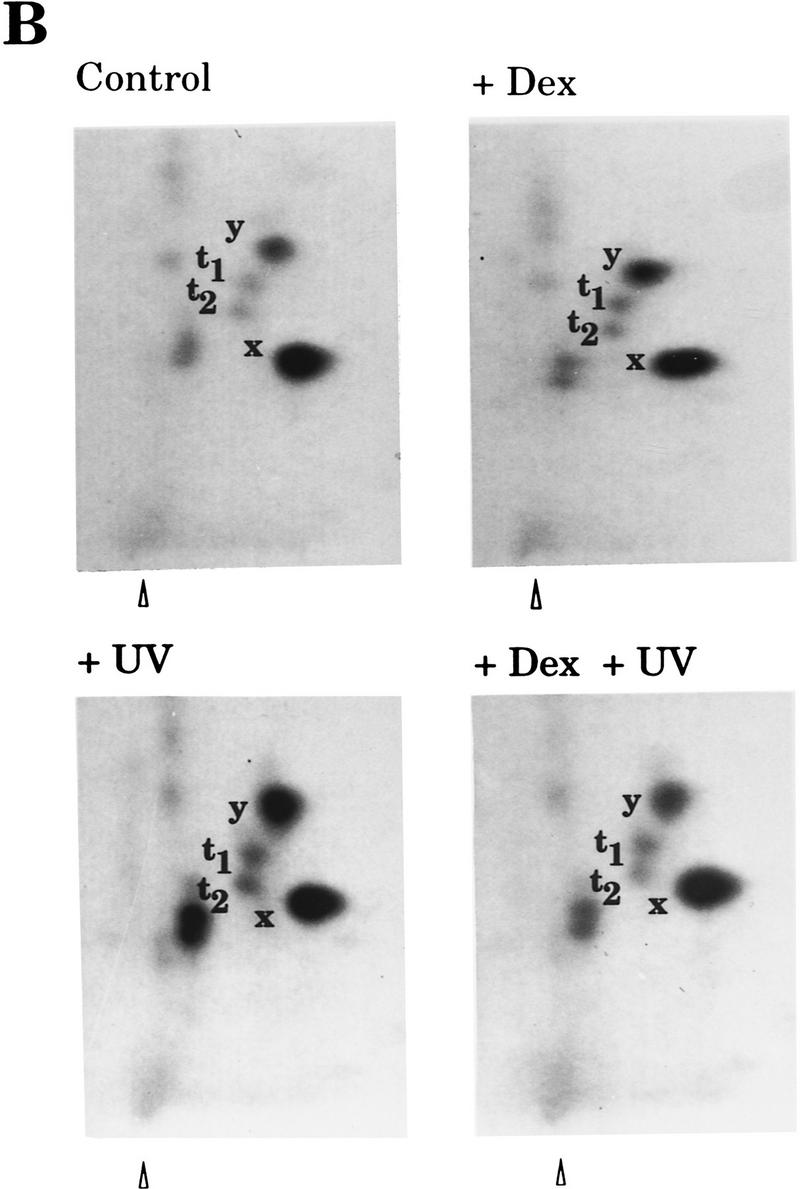
Hormone-activated nuclear receptors block UV-induced c-Jun amino-terminal-mediated transactivation and c-Jun amino-terminal phosphorylation independently of the c-Jun DNA-binding domain. (A) Activity of the 3xGHF-1–Luc reporter (3 μg) transfected in F9 cells along with either pRSV–c-Jun-GHF-1 or pRSV–c-JunA63/73-GHF-1 (0.5 μg) and pSG5–GR or pSG5–TRα-1 (0.2 μg) as indicated. Cells were treated with Dex or T3 and UV irradiated as indicated. (B) Tryptic–phosphopeptide maps of c-Jun–GHF-1 chimeric protein. HeLa cells were cotransfected with pRSV–c-Jun–GHF-1 (10 μg) and pSG5–GR (2 μg). Twelve hours after precipitate removal, cells were split into four plates, serum-starved, and metabolically labeled (see Materials and Methods). UV irradiation was performed 45 min after hormone or vehicle addition, and cells were harvested 20 min later. y, x, t1, and t2 spots corresponding to phosphorylation of Ser-63/73 and Thr-91/-93, respectively, are indicated. The arrowhead marks map origin.
To further confirm these results, expression vectors coding for Gal4–c-Jun and Gal4–c-JunA63/73 chimeras were constructed. Again, both ligand-activated GR and TRα-1 efficiently inhibited UV activation of the Gal4–c-Jun chimera (Fig. 4A, left and right panels, respectively). We also tested the effect of activated RA receptor (RAR). Previous studies have shown that RAR can also repress AP-1 in a ligand-dependent manner (Nicholson et al. 1990; Desbois et al. 1991; Schüle et al. 1991; Yang-Yen et al. 1991). Gal4-derived constructs were used, as interactions between GHF-1 and RAR (Rhodes et al. 1993) make the GHF-1-derived chimeras particulary unsuitable for this study. As shown in Figure 4A (middle panel), liganded RAR also inhibits UV-induced c-Jun amino-terminal-mediated transactivation, behaving analogously to GR and TRα-1.
Figure 4.

(A) Hormone-activated nuclear receptors specifically block UV-induced transcriptional activation mediated by c-Jun amino-terminal phosphorylation. Activity of the 5xGal4–Luc reporter (3 μg) transfected in F9 cells along with the pSG424 derivative construct (2 μg) encoding the Gal4–c-Jun, Gal4–v-Jun, or Gal4–c-JunA63/73 fusion proteins and the expression vector pSG5–GR, pSG5–RARα or pSG5–TRα-1 (0.2 μg), as indicated. Cells were subjected to the indicated treatments. (B) Ligand-activated RAR prevents UV-induced c-Jun amino-terminal phosphorylation. F9 cells were transiently transfected with 20 μg of pRSV–c-Jun and 2 μg of pSG5–RARα. After precipitated removal, cells were split into four plates, serum starved overnight and, when relevant, UV irradiated 45 min after RA or vehicle addition. Nuclear extracts were prepared from cells harvested 25 min after UV stimulation and analyzed by Western blot (80 μg of nuclear extract/lane). Immunodetection of c-Jun phosphorylated on Ser-63 (top) and the total amount of c-Jun (bottom) was subsequently performed on the same filter after stripping. (C) Ligand-activated TRα-1 prevents UV-induced c-Jun amino-terminal phosphorylation. Assays were performed as in B. Cells were transiently transfected with 20 μg of pRSV–c-Jun–GHF-1 and 2 μg of pSG5–TRα-1 and treated when indicated with thyroid hormone (T3). The total amount of c-Jun–GHF-1 was detected using an anti-GHF-1 antibody. Immunodetections of c-Jun–GHF-1 phosphorylated on Ser-63 and the total amount of c-Jun–GHF-1 are showed in the top and bottom panels, respectively.
A role for CBP mediating the antagonistic action in our assays was analyzed by testing the effect of nuclear receptor activation on the activity of the Gal4–v-Jun chimera. It has been shown previously that CBP interacts with c-Jun and v-Jun and stimulates both Gal4–c-Jun and Gal4–v-Jun (Bannister et al. 1995). In contrast to c-Jun, however, v-Jun does not require phosphorylation on Ser-63/73 to interact with CBP in vivo (Bannister et al. 1995). In addition, Gal4–v-Jun is not UV stimulated due to the deletion of the δ region located in the amino-terminal domain of c-Jun (Hibi et al. 1993). As shown in Figure 4A, transcriptional activation mediated by Gal4–v-Jun is not inhibited in any circumstance by hormone addition. This result indicates that under our experimental conditions CBP action is not affected by hormone-activated receptors.
Transfection experiments using either GHF-1- or Gal4-derived chimeras strongly suggested that RA and TRα-1 were likely to exert their inhibitory action on the c-Jun amino-terminal domain by the same mechanism as the GR. To test this, we studied the consequence of ligand activation of these nuclear receptors directly on c-Jun amino-terminal phosphorylation. As shown in Figure 4, B and C, both RA and TRα-1 were able to inhibit UV-induced c-Jun phosphorylation on Ser-63 when overexpressed in F9 cells by transient transfection along with c-Jun or c-Jun–GHF-1, respectively.
Overall, these data demonstrate that at least three nuclear hormone receptors, including those for glucocorticoid, RA, and TR, block c-Jun activation by a mechanism that is both independent of c-Jun DNA-binding domain and relies specifically on the c-Jun amino-terminal phosphorylation step.
Hormone-activated nuclear receptors prevent activation of the JNK signaling pathway
Because UV-activated members of the JNK subgroup of MAPKs are the major mediators of c-Jun amino-terminal phosphorylation (Hibi et al. 1993; Dérijard et al. 1994; Kyriakis et al. 1994; Minden et al. 1994a), we next studied the effect of hormone action on JNK protein and activity levels. Western blot analysis of whole cell extracts showed that Dex treatments as long as 8 hr do not alter the content of JNK protein in HeLa cells (Fig. 5A). In contrast, Dex strongly inhibits the increase in JNK activity that follows UV stimulation in this cell type (Fig. 5B,C). Likewise, hormone-activated endogenous RAR expressed in F9 cells, or exogenous TRα-1 expressed in EpH4 + TRα-1 cells (López-Barahona et al. 1995) also caused a reduction in UV-induced JNK activity (Fig. 5B,C). Hormone treatment affected neither the basal level of JNK activity (compare 0 time points in Fig. 5B) nor the JNK protein content upon UV stimulation in any cell line tested (Fig. 5B). The requirement of hormonal receptors for the observed effects was evident as JNK activity after UV exposure was not significantly affected by TR in parental EpH4 cells, which are essentially devoid of endogenous TRα-1 (López-Barahona et al. 1995) (Fig. 5B,C). In addition, an inhibitory action on the UV-induced JNK activation by 1,25-dihydroxyvitamin D3 was also observed in the GH4 cell line known to harbor endogenous vitamin D receptors (data not shown). In-gel kinase assays gave similar results to solid-phase kinase assays and additionally showed that UV-induced activity of both 46- and 54-kD JNK isoforms (Hibi et al. 1993) was equally affected by hormone treatment (Fig. 5D). Inhibition of the JNK cascade activation is independent of the stimulus used to induce the pathway, as Dex treatment of HeLa cells also inhibited JNK activation by TNF-α (see Fig. 8C, below).
Figure 5.

Hormone-activated nuclear receptors block activation of JNK signaling pathway. (A) Cellular content of JNK is not modified by Dex. JNK Western blot of whole cell extracts (20 μg/lane) from HeLa cells harvested at the indicated time points after Dex addition. (B) Effect of nuclear hormone receptor activation on UV-induced activity of JNK. Serum-starved cells from the cell lines listed aside were pretreated with the indicated hormone (+hormone) or with vehicle (−hormone) and UV stimulated. Extracts were prepared (see Materials and Methods) at the indicated time points after irradiation. For each cell line, the top panel shows JNK activity as measured by solid-phase kinase assay; the bottom panel shows the total JNK amount present in each cell extract (20 μg/lane) as measured by Western blot. (C) Quantitative analysis of the effect of nuclear hormone receptor activation on JNK activity after UV stimulation. Kinetics of JNK activation after UV stimulation in the indicated cell lines pretreated (•) or not (○) with the indicated hormone. JNK activity is represented as fold activation over that present in vehicle-treated cells at the 0 time point. Average results from three independent experiments performed as in B are shown. (D) Effect of Dex on in-gel JNK activity. In-gel kinase assay of cell extracts prepared from HeLa cells pretreated with either vehicle (−Dex) or dexamethasone (+Dex) and harvested at the indicated time points after UV stimulation.
Figure 8.

(A,B) Receptor–dosage dependence of JNK inhibition. COS-7 cells were transiently transfected with a constant amount of pcDNA3–HA–JNK (2 μg) and increasing amounts of pSG5–GR (total DNA brought up to 7 μg with pSG5). Twelve hours after precipitate removal, each plate was split into four, and after allowing attachment, cells were serum starved for 16 hr. In each set, two plates were treated with Dex and the other two with vehicle. After 45 min, cells were UV irradiated or not, as indicated, and harvested 20 min later. HA–JNK activity was determined by immune complex assay using an anti-HA antibody. Autoradiographs are shown in A (top) together with the analysis of HA–JNK protein levels by Western blotting using an anti-HA antibody (bottom). Substrate phosphorylation was quantitated in an Instant Imager and, for each initial pSG5–GR input, the percentage of JNK inhibition by Dex vs. vehicle treatment was determined. Average values of three independent experiments are shown in B. (C) Short Dex pretreatment prevents JNK activation. Solid-phase JNK assay of extracts from HeLa cells stimulated with TNF-α at the indicated time points after Dex addition. Cells were harvested 15 min after cytokine stimulation. (D) Actinomycin D does not block Dex action. Serum-starved HeLa cells were treated with either ethanol (−Act. D) or 1 μg/ml of actinomycin D (+Act. D) 15 min before vehicle (−Dex) or Dex (+Dex) addition and UV stimulated 60 min later. Cells were harvested at the indicated time points after UV irradiation, and JNK activity (top) and protein levels (bottom) were determined by immune complex assay and immunoblotting, respectively, using an anti-JNK antibody. Though similar amounts of cell extracts were analyzed for JNK activity, autoradiographs corresponding to cells untreated with actinomycin D were exposed 24 times longer than those from actinomycin D-treated cells. In contrast, JNK Western blots were equally exposed. (E) GR mutant LS7 is as efficient as the wild-type receptor in JNK signaling pathway interference. COS-7 cells were transiently transfected with pcDNA3–HA–JNK (2 μg) along with pRC/βact–GR–wt or pRC/βact–GR–LS7 (200 ng). After removal of precipitates, cells were split into four plates, serum-starved overnight, and subjected to the indicated treatments. Cells were harvested 25 min after UV stimulation, and HA–JNK activity (top) and protein levels (bottom) were determined by immune complex assay and Western blotting, respectively, using an anti-HA antibody.
Dex-activated GR also prevents JNK-mediated activation of the transcription factors ATF-2 and Elk-1
To extend the validity of our results, we analyzed hormone action on the activation of other JNK substrates, such as ATF-2 (Gupta et al. 1995; Livingstone et al. 1995; van Dam et al. 1995) and Elk-1 (Cavigelli et al. 1995; Gille et al. 1995; Whitmarsh et al. 1995). ATF-2-mediated transactivation was measured using the reporter construct 3xjunTRE–TK–CAT, which contains three copies of the AP-1-binding site present in the c-jun promoter. In F9 cells, UV-induced activation of this reporter is known to be strictly dependent on ATF-2 phosphorylation on threonines 69 and 71 located in the amino-terminal transactivation domain (van Dam et al. 1995). Transfection assays in F9 cells showed that GR inhibits UV-induced activation of the ATF-2-dependent reporter in presence of Dex (Fig. 6A). In addition, activated GR also blocked UV-induced transcription from a 5xGal4–Luc reported by a Gal4-derived chimera containing the amino-terminal domain of ATF-2 (C2, Livingstone et al. 1995), whereas no inhibitory effect was observed when using the equivalent UV-unresponsive Gal4-derived chimera harboring alanine instead of threonine residues at positions 69 and 71 of ATF-2 (C2–T1T2, Livingstone et al. 1995) (Fig. 6B).
Figure 6.

Dex-activated GR inhibits activation of JNK targets other than c-Jun. (A) Dex inhibits UV-induced ATF-2-dependent transactivation in F9 cells. Activity of the 3xjunTRE–TK–CAT reporter transiently cotransfected (3 μg) along with pSG5–GR (0.2 μg). Cells were subjected to the indicated treatments. (B) Dex blocks UV-induced transcriptional activation mediated by the ATF-2 amino-terminal transactivation domain. Activity of the 5xGal4–Luc reporter (3 μg) transfected in F9 cells along with the pSG424-Δ9 derivative construct (0.5 μg) encoding the Gal4–ATF-2(19–96) (C2) or Gal4–ATF-2(19–96)A69/71 (C2–T1T2) fusion proteins and the expression vector pSG5–GR (0.2 μg), as indicated. Cells were subjected to the indicated treatments. (C) Dex inhibits Elk-1mediated transcriptional activation induced by UV irradiation. Activity of the SRE-TK–CAT(Δ4490–183) (3 μg) transiently transfected in HeLa cells subjected to the indicated treatments. (D) UV-induced phosphorylation of Elk-1 is inhibited by Dex. EMSA was performed using nuclear extracts (10 μg/lane) prepared from HeLa cells pretreated with either vehicle (−Dex) or Dex (+Dex), UV irradiated, and harvested at the indicated time points after UV stimulation. Mobility of the uncomplexed (Free) and Elk-1-complexed [(Un.) uninduced; (Ind.) induced] probe is indicated.
Likewise, UV-induced activation of an Elk-1-dependent reporter containing the SRE of the c-fos gene, SRE–TK–CAT(Δ4490–183), is inhibited by Dex in HeLa cells (Fig. 6C). In addition, Elk-1-phosphorylation was analyzed by its ability to bind the Drosophila E74 Ets-binding site independently of accessory proteins and to show a reduction on the Elk-1–E74 complex mobility when phosphorylated by JNK (Zinck et al. 1993, 1995; Sachsenmaier et al. 1994). As shown in Figure 6D, Dex prevents the formation of slow-mobility Elk-1–E74 complexes (Ind. vs. Un.) that follow UV stimulation. ATF-2 and Elk-1 are involved in the UV induction of c-jun (van Dam et al. 1995) and c-fos (Cavigelli et al. 1995; Gille et al. 1995; Whitmarsh et al. 1995) gene expression, respectively. Together, these results are in agreement with the repression of UV-induced c-jun and c-fos gene expression by Dex in HeLa cells shown in Figure 1C.
Dex interferes with the JNK signal transduction pathway acting downstream of the MEKK activation step through a receptor–dosage-dependent mechanism
The next set of experiments were aimed at gaining insight into the mechanism of nuclear receptor interference with the JNK signaling cascade. Because the steroid hormone estradiol has been found to activate the ERK signal transduction pathway (Migliaccio et al. 1996), we explored the possibility that the JNK signaling cascade was activated upon hormone treatment. This situation might render a down-regulated pathway unable to respond to the second stimuli, UV or TNF-α, similarly to the described UV-response inhibition by growth factor prestimulation (Sachsenmaier et al. 1994). Nevertheless, this possibility was discarded, as Dex treatment of HeLa cells does not lead to JNK activation at any incubation time point (Fig. 7A).
Figure 7.

(A) Pretreatment with Dex does not activate JNK. In-gel kinase assay of whole cell extracts from HeLa cells were incubated with Dex at the indicated time points. Control extracts from HeLa cells pretreated or not with Dex for 45 min and UV irradiated were included. (B) Dex inhibits JNK activation by interfering with a step downstream of MEKK activation. HeLa cells were transiently cotransfected with pcDNA3–HA–JNK (2 μg) and pSG5–GR (2 μg). Increasing amounts of pCEV29–ΔMEKK and pEBG–SEK-1 (1 μg) were also included as indicated. Twelve hours after precipitate removal, each plate was split into two. Cells were serum-starved for 16 hr, treated with Dex (or vehicle) for 45 min, and harvested. Activity of HA–JNK was determined by immune complex kinase assay using an anti-HA antibody (top). Levels of HA–JNK in these cell extracts were analyzed by Western blot using an anti-HA antibody (bottom). (C) MKP-1 levels are not increased by glucocorticoids. Immunodetection of MKP-1 in whole cell extracts (50 μg/lane) from HeLa cells treated with Dex for the indicated period of time.
To locate the step along the JNK pathway that is the target of hormone action, we reconstituted the final part of the cascade taking advantage of the ability of the constitutively active amino-terminal truncated version of MEKK (ΔMEKK) to phosphorylate SEK-1 and subsequently activate JNK when overexpressed (Minden et al. 1994b; Yan et al. 1994). As expected, JNK activity increased proportionally to ΔMEKK overexpression. Within the dose range of ΔMEKK used (50–200 ng of expression vector), Dex inhibited the final JNK activity output to a similar 50% extent (Fig. 7B; data not shown), which strongly suggests that the step inhibited by hormone is not directly MEKK activity but another step downstream in the pathway. Studies activating the pathway downstream of MEKK were not conclusive because of the low level of JNK activation achieved by transfecting an SEK-1 expression vector (Fig. 7B) and the unavailability of a constitutively active mutant of SEK-1. In addition, this experiment also shows that Dex is able to inhibit an already activated JNK pathway. Up-regulation of negative regulators of the stress signaling cascade, such as the dual protein phosphatase MKP-1 (Liu et al. 1995; Chu et al. 1996), was also investigated. However, Western blot analysis of cell extracts prepared from HeLa cells exposed to Dex showed that the level of MKP-1 remained essentially constant within the incubation time points used in our experiments (Fig. 7C).
We initiated experiments to determine the functional requirements of the nuclear receptors to inhibit the JNK pathway. Similarly to EpH4 cells (Fig. 5B), Cos-7 cells that lack significant levels of endogenous GR only showed inhibition of UV-induced JNK activity in response to Dex when transiently transfected with a GR expression vector. As shown in Figure 8, A and B, increasing amounts of GR gradually augmented the inhibition of JNK activation. Moreover, at high receptor dosage, Dex efficiently inhibits UV-induced JNK activity to basal level (Fig. 8A,B). Therefore, hormone effect on JNK signaling pathway greatly depends on receptor dosage.
We also analyzed the kinetics whereby the inhibitory capacity induced by Dex is achieved. As shown in Figure 8C, a Dex pretreatment for 10 min is sufficient to inhibit the TNF-α activation of JNK activity by 50%, and this inhibition is already maximum 20 min after hormone addition. This result prompted us to analyze the transcription requirement for the interference mechanism. As shown in Figure 8D, actimonycin D does not prevent the inhibitory action of Dex on UV-induced JNK activity in HeLa cells at doses sufficient to block transcriptional activation (Fig. 2B), which caused a superinduction of the pathway in response to UV irradiation (see legend to Fig. 8D). In agreement with these results indicating a transcriptional-independent mechanism, the GR mutant LS7 is as competent as the wild-type receptor in interfering with the induction of the JNK pathway (Fig. 8E). This mutant receptor was reported previously to be severely defective in transcriptional activation while efficient as the wild-type receptor in AP-1 transrepression (Schena et al. 1989; Helmberg et al. 1995).
Discussion
Cells integrate a variety of signals triggered by multiple extracellular stimuli that modulate their physiology. Cross talk between different signaling pathways that regulate gene expression programs is crucial to control cellular homeostasis and cell fate. Frequently, proliferative signals activate the transcription factor AP-1 whereas, in contrast, glucocorticoids and other lipophilic hormones, such as RA, T3, and vitamin D are able to down-modulate this activation in a receptor-dependent manner (for review, see Saatcioglu et al. 1994). Therefore, AP-1–nuclear hormone receptor antagonism provides a model system to study cross-talk mechanisms between two major signal transduction pathways. In addition, for glucocorticoids and retinoids this antagonistic property appears to be responsible for their pharmacological actions as anti-inflammatory, immunosupressive, and antineoplastic agents. A major drawback associated with glucocorticoid and retinoid therapy is, however, the undesirable side effects related to ligand-activated gene transcription. Because both functions, AP-1 antagonism and gene activation, are mechanistically distinguishable, substantial efforts have been focused on the isolation of new ligands able to activate each one selectively (Fanjul et al. 1994; Chen et al. 1995), as well as on the elucidation of the molecular mechanism(s) underlying the antagonistic action (Jonat et al. 1990; Nicholson et al. 1990; Schüle et al. 1990, 1991; Yang-Yen et al. 1990, 1991; Desbois et al. 1991; Zhang et al. 1991; Kamei et al. 1996). Obviously, this will contribute to finding new targets for drug design and pharmacological strategies.
In this paper we propose a novel mechanism whereby nuclear hormone receptors antagonize AP-1 activity. Despite their differences, both nuclear receptors and AP-1 require interaction with the transcriptional coactivator CBP to induce gene expression (Arias et al. 1994; Bannister et al. 1995; Chakravarti et al. 1996; Kamei et al. 1996). Furthermore, in both cases, CBP interaction relies on a critical and previous activation step: hormone binding for nuclear receptors, and c-Jun amino-terminal phosphorylation for AP-1. We have shown that at least three different nuclear hormone receptors, including those for glucocorticoids, RA, and T3, inhibit induction of c-Jun amino-terminal-mediated transactivation by preventing phosphorylation of c-Jun on Ser-63/73. Moreover, this property correlates with their ability to negatively interfere with the final output of the JNK signaling pathway activated by extracellular stimuli. Therefore, we propose that nuclear receptors block AP-1 activation by interfering with induction of JNK, the major mediator of c-Jun activation step (see Fig. 9 for a scheme). Our findings add a novel mechanism by which hormone-activated receptors may block AP-1 activity and, in consequence, expression of AP-1-target genes. This mechanism does not exclude the previously proposed competition for CBP (Kamei et al. 1996). Rather, it may constitute an additional mode to blunt AP-1 action reducing the amount of AP-1 complexes suitable to compete for and interact with CBP. The requirement for CBP to inhibit activation of the JNK signaling pathway by nuclear receptors remains to be elucidated. Nuclear receptors also use this same mechanism to inhibit the activation of other transcription factors targets of JNK, such as ATF-2 and Elk-1, which mediate induction of c-jun and c-fos gene expression, respectively. Thus, using a single strategy nuclear receptors may block both transcription-independent and -dependent activation of AP-1. Additionally, the anti-inflammatory and immunosupressive actions of glucocorticoids have also been related to transrepression of the transcription factor NF-κB (Auphan et al. 1995; Scheinman et al. 1995a,b). Because a link between JNK activation by MEKK and NF-κB induction have been reported (Hirano et al. 1996; Meyer et al. 1996), the inhibition of the JNK pathway activation by glucocorticoids may presumably affect NF-κB activity. According to our findings, the JNK signaling pathway may have to be considered as a potential target for some of the pharmacological actions of glucocorticoids and retinoids.
Figure 9.

Model for the antagonistic action of hormone-activated nuclear receptors on AP-1 activity through the inhibition of the JNK signal transduction pathway. Activation of gene transcription by either nuclear hormone receptors or AP-1 relays on an activation step: hormone (▴) binding for nuclear receptors and c-Jun phosphorylation on Ser-63/73 (▪) for AP-1. According to our data, upon hormone activation nuclear receptors block the induction of the JNK signaling pathway and, in consequence, c-Jun amino-terminal phosphorylation. By this mechanism, steroid/thyroid hormones and RARs may antagonize AP-1 activity and inhibit expression of AP-1 target genes.
We have shown that Dex is able to inhibit the kinase cascade activated by MEKK overexpression. Thus, nuclear receptor interference would rely on the inhibition of MEKK activity or a downstream step in the pathway. However, direct inhibition of MEKK seems unlikely, as a similar degree of inhibition is found when overexpressing increasing amounts of MEKK. Also, we show that Dex not only interferes with the subsequent activation of the JNK pathway but can also inhibit a constitutively activated pathway. This is important, as oncogenes such as Ha-ras, v-src, ost, dbl, or vav are potential upstream activators of the JNK pathway (Cosso et al. 1995; Minden et al. 1995; Crespo et al. 1997).
Interference with the JNK signal transduction cascade occurs in a receptor–dosage-dependent manner. Despite the fact that the inhibitory status is acquired shortly after Dex addition, this result does not rule out the involvement of transcriptional mechanisms in mediating this interference, as during this short period the GR has already accumulated inside the nuclei (Czar et al. 1995; Htun et al. 1996), allowing ligand-activated transcription to start (Ucker and Yamamoto 1984). Nevertheless, the fast kinetics whereby the inhibitory status is achieved, together with the fact that similar inhibitory action on the JNK signaling cascade is accomplished by different hormones such as Dex, RA, and T3 is compatible with a transcriptional-independent mechanism. Moreover, the fact that Dex is still able to inhibit induction of the JNK pathway even when actinomycin D is added 15 min before the hormone, and also that the transcriptional activation-defective GR mutant LS7 is fully efficient interfering with the JNK cascade (Schena et al. 1989; Helmberg et al. 1995) further support the involvement of a transcription-independent mechanism. However, we do not exclude alternative modes of action involving both transcription-independent and -dependent mechanisms, as it has already been reported for glucocorticoids in the transrepression of the NF-κB transcription factor (Auphan et al. 1995; Scheinman et al. 1995a,b). In this context, compelling evidence shows that some lipophilic hormones including glucocorticoids and vitamin D induce the expression of the p21Waf-1 gene in several cell types (Jiang et al. 1994; Wang et al. 1996; Ramalingam et al. 1997; Rogatsky et al. 1997). Actually, we have observed an increase in p21Waf-1 mRNA and protein in HeLa cells after 90 min of dexamethasone treatment (C. Caelles and A. Muñoz, unpubl.). Since p21Waf-1 has been reported to be able to inhibit JNK (Shim et al. 1996), the possibility arose that p21Waf-1 might mediate the inhibitory action of Dex in these cells. However, although the involvement of p21Waf-1 has not been excluded, a mechanism other than transcriptional activation of p21Waf-1 is probably primary conducting the interference, as the inhibitory status induced by Dex is actinomycin D-resistant and takes place earlier than the induction of p21Waf-1 (10–20 min vs. 90 min, respectively).
Different studies have shown that differences in the magnitude and duration of ERK signaling pathway activation lead to distinct cell fate, proliferation, or differentiation (Qiu and Green 1992; Dikic et al. 1994; Traverse et al. 1994). Our data show that endogenous nuclear receptors such as GR in HeLa cells or RAR in F9 cells modulate the magnitude of JNK induction. Therefore, glucocorticoids, RA, and T3 may exert their homeostatic action regulating cellular responses to extracellular stimuli by controlling the extent and/or duration of JNK signaling pathway activation.
Materials and methods
Plasmids, cell culture, treatments, and transfections
The pSG424 Gal4–c-Jun, Gal4–c-JunA63/73, and Gal4–v-Jun constructs initially encoding amino acids 1–257 of chicken c-Jun and 1–230 of v-Jun (Black et al. 1994), respectively, were modified by cloning the corresponding HindIII–Eco47III fragments between the HindIII and SmaI sites of pSG424. The AP-1 binding site present in the pBLCAT2 backbone (Yang-Yen et al. 1990) was eliminated from the SRE–TK–CAT reporter by deleting the DraII–NdeI fragment yielding the SRE–TK–CAT(Δ4490–183) reporter construct. Other plasmids have already been described elsewhere (Angel et al. 1987; Zenlet et al. 1989; Smeal et al. 1991; Saatcioglu et al. 1993; Sánchez et al. 1994; Cosso et al. 1995; Helmberg et al. 1995; Livingstone et al. 1995).
HeLa, Cos-7, EpH4, and EpH4 + TRα-1 cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS). F9 cells were grown in Ham’s F12–DMEM (1:1) supplemented with 10% FCS. Cells were serum starved by changing the culture medium to DMEM plus 0.5% FCS 16 hr before treatment. Thereafter, vehicle (ethanol) or hormone (10−6 m Dex, 10−6 m RA, and 10−7 m T3) was added 45 min before UV irradiation (40 J/m2, UV-C) or TNF-α addition (10 ng/ml). Cells were transiently transfected by standard calcium phosphate precipitation method (Ausubel et al. 1994), and CAT or luciferase activity determined 6 hr after UV stimulation. Rous sarcoma virus (RSV)–β-galactosidase and β-actin–Luc constructs (0.5 μg) were used as internal controls in transfection assays performed in F9 and HeLa cells, respectively. Average results from three independent experiments run in duplicates are shown.
Nuclear extracts and EMSAs
Nuclear extracts from HeLa cells were prepared by suspending PBS-washed cell pellets in NPBT [10 mm Tris-HCl at pH 7.4, 140 mm NaCl, 2 mm MgCl2, 0.1% Triton X-100, 1 mm dithiothreitol (DTT), 20 mm β-glycerophosphate, 100 μm sodium orthovanadate, 0.5 mm PMSF, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin). After 10 min on ice, nuclei were recovered by 10 min of centrifugation through a 50% sucrose cushion in NPB (10 mm Tris-HCl at pH 7.4, 140 mm NaCl, 2 mm MgCl2, 1 mm DTT, 20 mm β-glycerophosphate, 100 μm sodium orthovanadate, 0.5 mm PMSF, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin) at 12,000 rpm and 4°C. Nuclear pellets were suspended in DC (20 mm HEPES–NaOH at pH 7.9, 25% glycerol, 420 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 1 mm DTT, 20 mm β-glycerophosphate, 100 mm sodium orthovanadate, 0.5 mm PMSF, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin) and rotated at 4°C for 30 min. Nuclear extracts were cleared by 10 min of centrifugation at 12,000 rpm and 4°C.
EMSAs were performed on ice in 20-μl reactions containing the appropriated amount of nuclear extract in 10 mm HEPES–KOH at pH 7.9, 10% glycerol, 50 mm KCl, 5 mm MgCl2, 0.1 mm EDTA, 1 mm DTT, 2.5 μg of poly[d(I–C)] and 0.1 ng of 32P-labeled oligonucleotide. DNA–protein complexes and free probe were resolved at room temperature on a 6% nondenaturating polyacrylamide gel with 0.25× TBE. Oligonucleotides corresponding to the collagenase TRE (5′-TCGACGGTATCGATAAGCTATGACTCATTCCGGGGGATC-3′) and the E74 oligonucleotide (Zinck et al. 1993) were used for AP-1 and Elk-1 EMSAs, respectively.
Western blotting
c-Jun, c-Jun phosphorylated on Ser-63, c-Fos, JNK, MKP-1, and HA-tagged proteins were detected in cell extracts by immunoblotting using the specific antibodies sc-045, sc-822, sc-052, sc-474, and sc-1199 from Santa Cruz and 12CA5 from BabCO, respectively. Immunoblots were performed and developed using the ECL detection system (Amersham).
Metabolic labeling and tryptic phosphopeptide mapping
HeLa cells were incubated with [32P]-orthophosphate (1 mCi/ml) in phosphate-free DMEM for 2 hr before Dex (or vehicle) addition and harvested in RIPA buffer 20 min after UV stimulation. Immunoprecipitations were performed using specific antibodies against c-Jun and c-Jun–GHF-1 sc-045 from Santa Cruz, and an anti-GHF-1 described previously (Caelles et al. 1995), respectively. Immunoprecipitates were separated by SDS–12%PAGE, electroblotted onto a nitrocellulose membrane and autoradiographed. Specific bands were excised and subjected to tryptic phosphopeptide mapping as described (Caelles et al. 1995).
Protein kinase assays
Solid-phase kinase and in-gel kinase assays were performed as described previoulsly using glutathione S-transferase (GST)–c-Jun1–223 (GST–c-Jun) as susbtrate (Hibi et al. 1993). Briefly, 5 × 106 cells were lysed in 200 μl of WCE300 (25 mm HEPES–NaOH at pH 7.7, 300 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.1% Triton X-100, 1 mm DTT, 20 mm β-glycerophosphate, 100 μm sodium orthovanadate, 0.5 mm PMSF, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin) and rotated at 4°C for 30 min. Extracts were cleared by 10 min of centrifugation at 12,000 rpm and 4°C. In solid-phase kinase assays, 3 vol of DB (20 mm HEPES–NaOH at pH 7.7, 2.5 mm MgCl2, 0.1 mm EDTA, 0.05% Triton X-100, 1 mm DTT, 20 mm β-glycerophosphate, 100 μm sodium orthovanadate, 0.5 mm PMSF, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin) were added to the extracts. Afterward, extracts were incubated on ice for 10 min, cleared by 10 min of centrifugation at 12,000 rpm and 4°C, and mixed with 10 μg of GST–c-Jun bound to glutathione–agarose beads. Beads were recovered by centrifugation and washed five times with HBIB (20 mm HEPES–NaOH at pH 7.7, 50 mm NaCl, 2.5 mm MgCl2, 0.1 mm EDTA, 0.05% Triton X-100, 1 mm DTT, 20 mm β-glycerophosphate, 100 μm sodium orthovanadate, 0.5 mm PMSF, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin). Phosphorylation was performed in 30-μl reactions containing KB (20 mm HEPES–NaOH at pH 7.6, 2.5 mm MgCl2, 2 mm DTT, 20 mm β-glycerophosphate, 100 μm sodium orthovanadate), 20 μm ATP, and 0.5 μCi [γ-32P]ATP at 30°C for 20 min, stopped by the addition of 4× Laemmli sample buffer, and resolved on a 10% SDS–polyacrylamide gel.
For in-gel kinase assays, 20–50 μg of whole cell extracts obtained as described above were applied onto a 10% SDS–polyacrylamide gel containing 100 μg/ml of GST–c-Jun. After eletrophoresis, gels were sequentially subjected to washes of 30 min at room temperature in the following solutions: 50 mm HEPES–NaOH at pH 7.6, 20% 2-propanol; 50 mm HEPES–NaOH at pH 7.6, 5 mm β-mercaptoethanol (β-ME); 50 mm HEPES–NaOH at pH 7.6, 5 mm β-ME, 6 m urea. Renaturation was performed at 4°C by serial washes of 15 min in 50 mm HEPES–NaOH at pH 7.6, 5 mm β-ME, 0.05% Tween, and 3, 1.5, 0.75, or 0.37 m urea, respectively followed by several washes of 30 min in 50 mm HEPES–NaOH at pH 7.6, 5 mm β-ME, 0.05% Tween. Phosphorylation reaction was performed in KB plus 20 μm ATP and 100 μCi [γ-32P]ATP at 30°C for 2 hr. Reaction was stopped and the excess of radioactivity removed by extensive washes in 5% TCA and 1% sodium pyrophosphate at room temperature. In figures 5D and 7A, bands corresponding to substrate phosphorylated by 46- and 54-kD JNK isoforms are indicated.
For immune complex kinase assays, cells were lysed in 20 mm HEPES–NaOH at pH 7.6, 10 mm EGTA, 2.5 mm MgCl2, 1% NP-40, 1 mm DTT, 40 mm β-glycerophosphate, 100 μm sodium orthovanadate, 0.5 mm PMSF, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin, and lysates were cleared by 10-min centrifugation at 12,000 rpm at 4°C. Extracts were immunoprecipitated with an anti-HA antibody (BabCO) or with an anti-JNK1 antibody from Santa Cruz (sc-474), and immune complexes were recovered with protein A–Sepharose. Beads were sequentially washed three times with 1% NP-40 and 2 mm sodium orthovanadate in PBS, once with 100 mm Tris-HCl at pH 7.5 and 500 mm LiCl, and once with KB. Phosphorylation reactions were performed in 30-μl volume containing KB, 20 μm ATP, 0.5 μCi [γ-32P]ATP, and 1 μg of GST–c-Jun at 30°C for 20 min, stopped by the addition of 4× Laemmli sample buffer and resolved on a 10% SDS–polyacrylamide gel.
Acknowledgments
We thank J. Bernal and H. Beug for critically reading the manuscript; M. Karin, H.G. Stunnenberg, D. Barettino, D.A.F. Gillespie, M. Zenke, A. Alonso, A. Helmberg, L.I. Zon, P. Crespo, N. Jones, R. Perona, and J.R. Naranjo for plasmids and reagents; J. Martin and P. Santisteban for allowing us to use their phosphopeptide mapping equipment; M. González-Monge for excellent technical assistance, and L.F. García-Fernández for help with preparation of figures. C.C. and J.M.G.-S. were supported by a postdoctoral contract from Ministerio de Educación y Ciencia of Spain and a predoctoral fellowship from the Comunidad Autónoma de Madrid, Spain. This work was supported by a grant (SAF95-0738) from Comisión Interministerial de Ciencia y Tecnología, Plan Nacional de Investigación y Desarrollo of Spain.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL amunoz@Biomed.iib.uam.es; FAX 34 1 585 45 87.
References
- Angel P, Karin M. The role of Jun, Fos, and the AP-1 complex in cell proliferation and transformation. Biochem Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, Rahmsdor HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis element recognised by a TPA-modulated trans-acting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- Angel P, Allegretto EA, Okino ST, Hattori K, Boyle WJ, Hunter T, Karin M. Oncogene jun encodes a sequence-specific trans-activator similar to AP-1. Nature. 1988;332:166–171. doi: 10.1038/332166a0. [DOI] [PubMed] [Google Scholar]
- Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M, Feramisco J, Montminy M. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature. 1994;370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosupression by glucocorticoids: Inhibition of NK-κB activity through induction of IκB synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Strul K. Current protocols in molecular biology. New York, NY: Greene Publishing Associates/Wiley-Interscience; 1994. [Google Scholar]
- Bannister AJ, Oehler T, Wilhelm D, Angel P, Kouzarides T. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene. 1995;11:2509–2514. [PubMed] [Google Scholar]
- Black EJ, Catling AD, Woodgett JR, Kilbey A, Gillespie DAF. Transcriptional activation by the v-Jun oncoprotein is independent of positive regulatory phosphorylation. Oncogene. 1994;9:2363–2368. [PubMed] [Google Scholar]
- Bohmann D, Bos TJ, Admon A, Nishimura T, Vogt PK, Tjian R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science. 1987;238:1386–1392. doi: 10.1126/science.2825349. [DOI] [PubMed] [Google Scholar]
- Brenner DA, O’Hara M, Angel P, Chojkier M, Karin M. Prolonged activation of jun and collagenase genes by tumor necrosis factor α. Nature. 1989;337:661–663. doi: 10.1038/337661a0. [DOI] [PubMed] [Google Scholar]
- Caelles C, Hennemann H, Karin M. M-phase-specific phosphorylation of the POU transcription factor GHF-1 by a cell cycle-regulated protein kinase inhibits DNA binding. Mol Cell Biol. 1995;15:6694–6701. doi: 10.1128/mcb.15.12.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill MA, Janknecht R, Nordheim A. Signaling pathways: Jack of all cascades. Curr Biol. 1996;6:16–19. doi: 10.1016/s0960-9822(02)00410-4. [DOI] [PubMed] [Google Scholar]
- Cavigelli M, Dolfi F, Claret FX, Karin M. Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J. 1995;14:5957–5964. doi: 10.1002/j.1460-2075.1995.tb00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti D, LaMorte VJ, Nelson MC, Nakajima T, Schulman IG, Juguilon H, Montminy M, Evans RM. Role of CBP/p300 in nuclear receptor signaling. Nature. 1996;383:99–103. doi: 10.1038/383099a0. [DOI] [PubMed] [Google Scholar]
- Chen JY, Penco S, Ostrowski J, Balaguer P, Pons M, Starrett JE, Reczek P, Chambon P, Gronemeyer H. RAR-specific agonist/antagonists which dissociate transactivation and AP1 transrepression inhibit anchorage-independent cell proliferation. EMBO J. 1995;14:1187–1197. doi: 10.1002/j.1460-2075.1995.tb07102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Solski PA, Khosravi-Far R, Der CJ, Kelly K. The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J Biol Chem. 1996;271:6497–6591. doi: 10.1074/jbc.271.11.6497. [DOI] [PubMed] [Google Scholar]
- Cosso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Crespo P, Schuebel KE, Ostrom AA, Gutkind JS, Bustelo XR. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature. 1997;385:169–172. doi: 10.1038/385169a0. [DOI] [PubMed] [Google Scholar]
- Czar MJ, Lyons RH, Welsh MJ, Renoir JM, Pratt WB. Evidence that the FK506-binding immunophilin heat shock protein 56 is required for trafficking of the glucocorticoid receptor from the cytoplasm to the nucleus. Mol Endocrinol. 1995;9:1549–1560. doi: 10.1210/mend.9.11.8584032. [DOI] [PubMed] [Google Scholar]
- Dérijard B, Hibi M, Wu IH, Barret T, Su B, Deng T, Karin M, Davis RJ. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Desbois C, Aubert D, Legrand C, Pain B, Samarut J. A novel mechanism of action for v-ErbA: Abrogation of the inactivation of transcription factor AP-1 by retinoic acid and thyroid hormone receptors. Cell. 1991;67:731–740. doi: 10.1016/0092-8674(91)90068-a. [DOI] [PubMed] [Google Scholar]
- Devary Y, Gottlieb RA, Lau LF, Karin M. Rapid and preferential activation of the c-jun gene during the mammalian UV response. Mol Cell Biol. 1991;11:1804–2811. doi: 10.1128/mcb.11.5.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devary Y, Gottlieb RA, Smeal T, Karin M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell. 1992;71:1081–1091. doi: 10.1016/s0092-8674(05)80058-3. [DOI] [PubMed] [Google Scholar]
- Dikic I, Schlessinger J, Lax I. PC12 cell overexpressing the insulin receptor undergo insulin-dependent neuronal differentiation. Curr Biol. 1994;4:702–708. doi: 10.1016/s0960-9822(00)00155-x. [DOI] [PubMed] [Google Scholar]
- Fanjul A, Dawson MI, Hobbs PD, Jong L, Cameron JF, Harlev E, Graupner G, Lu XP, Pfahl M. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature. 1994;372:107–110. doi: 10.1038/372107a0. [DOI] [PubMed] [Google Scholar]
- Gille H, Strahl T, Shaw PE. Activation of ternary complex factor Elk-1 by stress-activated protein kinases. Curr Biol. 1995;5:1191–1200. doi: 10.1016/s0960-9822(95)00235-1. [DOI] [PubMed] [Google Scholar]
- Gupta S, Campbell D, Dérijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- Helmberg A, Auphan N, Caelles C, Karin M. Glucocorticoid-induced apoptosis of human leukemic cells is caused by the repressive function of the glucocorticoid receptor. EMBO J. 1995;14:452–460. doi: 10.1002/j.1460-2075.1995.tb07021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera RE, Shaw PE, Nordheim A. Occupation of the c-fos serum response element in vivo by a multi-protein complex is unaltered by growth factor induction. Nature. 1989;340:68–70. doi: 10.1038/340068a0. [DOI] [PubMed] [Google Scholar]
- Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes & Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Hipskind RA, Rao VN, Mueller CG, Reddy ESP, Nordheim A. Ets-related protein Elk-1 is homologous to the c-fos regulator factor p62TCF. Nature. 1991;354:531–534. doi: 10.1038/354531a0. [DOI] [PubMed] [Google Scholar]
- Hirano M, Osada S, Aoki T, Hirai S, Hosaka M, Inoue J, Ohno S. MEK kinase is involved in tumor necrosis factor α-induced NF-κB activation and degradation of IκB-α. J Biol Chem. 1996;271:13234–13238. doi: 10.1074/jbc.271.22.13234. [DOI] [PubMed] [Google Scholar]
- Htun H, Barsony J, Renyi I, Gould DL, Hager GL. Visualization of glucocorticoid receptor translocation and intranuclear organization in living cells with a green fluorescent protein chimera. Proc Natl Acad Sci. 1996;93:4845–4850. doi: 10.1073/pnas.93.10.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Lin J, Zao-zohong S, Collart FR, Huberman E, Fisher PB. Induction of differentiation in human promyelocytic HL-60 leukemia cells activates p21, WAF1/CIP1, expression in the absence of p53. Oncogene. 1994;9:3397–3346. [PubMed] [Google Scholar]
- Jonat C, Rahmsdorf HJ, Park KK, Cato ACB, Gebel S, Ponta H, Herrlich P. Antitumor promotion and anti-inflammation: Down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. 1990;62:1189–1204. doi: 10.1016/0092-8674(90)90395-u. [DOI] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose D, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Karin M, Hunter T. Transcriptional control by protein phosphorylation: Signal transmission from the cell surface to the nucleus. Curr Biol. 1995;5:747–757. doi: 10.1016/s0960-9822(95)00151-5. [DOI] [PubMed] [Google Scholar]
- Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- König H, Ponta H, Rahmsdorf HJ, Herrlich P. Interference between pathway-specific transcription factors: glucocorticoids antagonize phorbol ester-induced AP-1 activity without altering AP-1 site occupation in vivo. EMBO J. 1992;11:2241–2246. doi: 10.1002/j.1460-2075.1992.tb05283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gorospe M, Yang C, Holbrook NJ. Role of mitogen-activated protein kinase phosphatase during the cellular response to genotoxic stress. Inhibition of c-Jun amino-terminal kinase activity and AP-1-dependent gene activation. J Biol Chem. 1995;270:8377–8380. doi: 10.1074/jbc.270.15.8377. [DOI] [PubMed] [Google Scholar]
- Livingstone C, Patel G, Jones N. ATF-2 contains a phosphorylation-dependent transcriptional activation domain. EMBO J. 1995;14:1785–1797. doi: 10.1002/j.1460-2075.1995.tb07167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barahona M, Fialka I, González-Sancho JM, Asunción M, González M, Iglesias T, Bernal J, Beug H, Muñoz A. Thyroid hormone regulates stromelysin expression, protease secretion and the morphogenetic potential of normal polarized mammary epithelial cells. EMBO J. 1995;14:1145–1155. doi: 10.1002/j.1460-2075.1995.tb07098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: The second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer CF, Wang X, Chang C, Templeton D, Tan TH. Interaction between c-Rel and the mitogen-activated protein kinase kinase kinase 1 signaling cascade in mediating κB enhancer activation. J Biol Chem. 1996;271:8971–8976. doi: 10.1074/jbc.271.15.8971. [DOI] [PubMed] [Google Scholar]
- Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, Auricchio F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- Minden A, Lin A, Smeal T, Dérijard B, Cobb M, Davis R, Karin M. c-Jun amino-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol Cell Biol. 1994a;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minden A, Lin A, McMahon M, Lange-Carter GL, Dérijard B, Davis RJ, Johnson GL, Karin M. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994b;266:1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPase Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Nicholson RC, Mader S, Nagpal S, Leid M, Rochette-Egly C, Chambon P. Negative regulation of the rat stromelysin gene promoter by retinoic acid is mediated by an AP-1 binding site. EMBO J. 1990;9:4443–4454. doi: 10.1002/j.1460-2075.1990.tb07895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papavassiliou AG, Treier M, Bohmann D. Intramolecular signal transduction in c-Jun. EMBO J. 1995;14:2014–2019. doi: 10.1002/j.1460-2075.1995.tb07193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulverer BJ, Kyriakis JM, Avurch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-Jun mediated by MAP kinases. Nature. 1991;353:670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- Qiu MS, Grenn SH. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron. 1992;9:705–717. doi: 10.1016/0896-6273(92)90033-a. [DOI] [PubMed] [Google Scholar]
- Radler-Pohl A, Sachsenmaier C, Gebel S, Auer HP, Bruder JT, Rapp U, Angel P, Rahmsdorf HJ, Herrlich P. UV-induced activation of AP-1 involves obligatory extranuclear steps including Raf-1 kinase. EMBO J. 1993;12:1005–1012. doi: 10.1002/j.1460-2075.1993.tb05741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam A, Hirai A, Thompson A. Glucocorticoid inhibition of fibroblasts proliferation and regulation of the cyclin kinase inhibitor p21Cip1. Mol Endocrinol. 1997;11:577–586. doi: 10.1210/mend.11.5.9923. [DOI] [PubMed] [Google Scholar]
- Rhodes SJ, Chen R, DiMattia GE, Scully KM, Kalla KA, Lin SC, Yu VC, Rosenfeld MG. A tissue-specific enhancer confers Pit-1-dependent morphogen inducibility and autoregulation of the pit-1 gene. Genes & Dev. 1993;7:913–932. doi: 10.1101/gad.7.6.913. [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathway. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Trowbridge JM, Garabedian MJ. Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol Cell Biol. 1997;17:3181–3193. doi: 10.1128/mcb.17.6.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatcioglu F, Bartunek P, Deng T, Zenke M, Karin M. A conserved C-terminal sequence that is deleted in v-ErbA is essential for the biological activities of c-ErbA (the thyroid hormone receptor) Mol Cell Biol. 1993;13:3675–3685. doi: 10.1128/mcb.13.6.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatcioglu F, Claret FX, Karin M. Negative transcriptional regulation by nuclear receptors. Semin Cancer Biol. 1994;5:347–359. [PubMed] [Google Scholar]
- Sachsenmaier C, Radler-Pohl A, Zinck R, Nordheim A, Herrlich P, Rahmsdorf HJ. Involvement of growth factor receptors in the mammalian UVC response. Cell. 1994;78:963–972. doi: 10.1016/0092-8674(94)90272-0. [DOI] [PubMed] [Google Scholar]
- Sánchez I, Hughes RJ, Mayer BJ, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Gualberto A, Jewel CM, Cidlowski JA, Baldwin AS., Jr Characterization of mechanisms involved in transrepression of NF-κB by activated glucocorticoid receptors. Mol Cell Biol. 1995a;15:943–953. doi: 10.1128/mcb.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS., Jr Role of transcriptional activation of IκBα in mediation of immunosupression by glucocorticoids. Science. 1995b;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- Schena M, Freedman LP, Yamamoto KR. Mutations in the glucocorticoid receptor zinc finger region that distinguish interdigitated DNA binding and transcriptional enhancement activities. Genes & Dev. 1989;3:1590–1601. doi: 10.1101/gad.3.10.1590. [DOI] [PubMed] [Google Scholar]
- Schüle R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, Yang N, Verma IM, Evans RM. Funtional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell. 1990;62:1217–1226. doi: 10.1016/0092-8674(90)90397-w. [DOI] [PubMed] [Google Scholar]
- Schüle R, Rangarnjan P, Yang N, Kliewer S, Ransone LJ, Bolado J, Verma IM, Evans RM. Retinoic acid is a negative regulator of AP-1-responsive genes. Proc Natl Acad Sci. 1991;88:6092–6096. doi: 10.1073/pnas.88.14.6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PE, Schröter H, Nordheim A. The ability of a ternary complex to form over the serum response element correlates with serum inducibility of the human c-fos promoter. Cell. 1989;56:563–572. doi: 10.1016/0092-8674(89)90579-5. [DOI] [PubMed] [Google Scholar]
- Shim J, Lee H, Park J, Kim H, Choi E-J. A non-enzymatic p21 protein inhibitor of stress-activated protein kinases. Nature. 1996;381:804–807. doi: 10.1038/381804a0. [DOI] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- Stein B, Rahmsdorf HJ, Steffetn A, Liftin M, Herrlich P. UV-induced DNA damage is an intermediate step in UV-induced expression of human immunodeficiency virus type 1, collagenase, c-fos, and metallothionein. Mol Cell Biol. 1989;9:5169–5181. doi: 10.1128/mcb.9.11.5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse S, Seedorf K, Paterson H, Marshall CJ, Cohen P, Ullrich A. EGF triggers neuronal differentiation of PC12 cells that overexpress the EGF receptor. Curr Biol. 1994;4:694–701. doi: 10.1016/s0960-9822(00)00154-8. [DOI] [PubMed] [Google Scholar]
- Ucker DS, Yamamoto KR. Early events in the stimulation of mammary tumor virus RNA synthesis by glucocorticoids. J Biol Chem. 1984;259:7416–7420. [PubMed] [Google Scholar]
- van Dam H, Duyndam M, Rottier R, Bosch A, de Vries-Smits L, Herrlich P, Zantema A, Angel P, van der Eb AJ. Heterodimer formation of c-Jun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J. 1993;12:479–487. doi: 10.1002/j.1460-2075.1993.tb05680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995;14:1798–1811. doi: 10.1002/j.1460-2075.1995.tb07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QM, Jones JB, Studzinski GP. Cyclin-dependent kinase inhibitor p27 as a mediator of the G1-S phase block induced by 1,25-dihydroxyvitamin D3 in HL60 cells. Cancer Res. 1996;56:264–267. [PubMed] [Google Scholar]
- Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- Yan M, Dai T, Deak JC, Kyriakis JM, Zon LI, Woodgett JR, Templeton DJ. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature. 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]
- Yang-Yen HF, Chambard JC, Sun YL, Smeal T, Schmidt TJ, Drouin J, Karin M. Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. 1990;62:1205–1215. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- Yang-Yen HF, Zhang X, Graupner G, Tzukerman M, Sakamoto B, Karin M, Pfahl M. Antagonism between retinoic acid receptors and AP-1: Implications for tumor promotion and inflammation. New Biol. 1991;3:1206–1219. [PubMed] [Google Scholar]
- Zelent A, Krust A, Petkovich M, Kastner P, Chambon P. Cloning of murine α and β retinoic acid receptors and a novel receptor γ predominantly expressed in skin. Nature. 1989;339:714–717. doi: 10.1038/339714a0. [DOI] [PubMed] [Google Scholar]
- Zhang XK, Wills KN, Husmann M, Hermann T, Pfahl M. Novel pathway for thyroid hormone receptor action through interaction with jun and fos oncogene activities. Mol Cell Biol. 1991;11:6016–6025. doi: 10.1128/mcb.11.12.6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinck R, Hipskind RA, Pingoud V, Nordheim A. c-fos transcriptional activation and repression correlate temporally with the phosphorylation status of TCF. EMBO J. 1993;12:2377–2387. doi: 10.1002/j.1460-2075.1993.tb05892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinck R, Cahill MA, Kracht M, Sachsenmaier C, Hipskind RA, Nordheim A. Protein synthesis inhibitors reveal differential regulation of mitogen-activated protein kinase and stress-activated protein kinase pathways that converge on Elk-1. Mol Cell Biol. 1995;15:4930–4938. doi: 10.1128/mcb.15.9.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]