

Abstract
Although the etiology of autism remains largely unknown, cytogenetic and genetic studies have implicated maternal copy number gains of 15q11–q13 in 1–3% of autism cases. In order to understand how maternal 15q duplication leads to dysregulation of gene expression and altered chromatin interactions, we used microcell-mediated chromosome transfer to generate a novel maternal 15q duplication model in a human neuronal cell line. Our 15q duplication neuronal model revealed that by quantitative RT–PCR, transcript levels of NDN, SNRPN, GABRB3 and CHRNA7 were reduced compared with expected levels despite having no detectable alteration in promoter DNA methylation. Since 15q11–q13 alleles have been previously shown to exhibit homologous pairing in mature human neurons, we assessed homologous pairing of 15q11–q13 by fluorescence in situ hybridization. Homologous pairing of 15q11–q13 was significantly disrupted by 15q duplication. To further understand the extent and mechanism of 15q11–q13 homologous pairing, we mapped the minimal region of homologous pairing to a ∼500 kb region at the 3′ end of GABRB3 which contains multiple binding sites for chromatin regulators MeCP2 and CTCF. Both active transcription and the chromatin factors MeCP2 and CTCF are required for the homologous pairing of 15q11–q13 during neuronal maturational differentiation. These data support a model where 15q11–q13 genes are regulated epigenetically at the level of both inter- and intra-chromosomal associations and that chromosome imbalance disrupts the epigenetic regulation of genes in 15q11–q13.
INTRODUCTION
Autism spectrum disorders (ASDs; MIM 209850) include a complex group of neurodevelopmental disorders characterized by impairments in reciprocal social interactions, problems in communication and a restricted range of behaviors and interests. ASD affects individuals of all socio-economic and cultural backgrounds and has a prevalence of 6 per 1000 individuals, with males affected four times more frequently than females (1,2). There is compelling evidence for a genetic etiology of ASD from twin studies that have shown high concordance between monozygotic twins (3–5). To date, cytogenetic studies have identified a number of pathogenic chromosomal anomalies in ASD patients (1). In addition, current microarray-based technologies have enabled the detection of submicroscopic microdeletions and microduplications [copy number variations (CNVs)] and revealed that submicroscopic CNVs can have a pathogenic role in ASD as well (3,4). Human chromosome 15q11–q13 is frequently involved in clinically important genomic rearrangements, including interstitial deletions, duplications and supernumerary marker chromosome, called isodicentric chromosome (idic) (5). After fragile X, maternal 15q11–q13 duplications are the most common cytogenetic cause of autism, occurring in ∼1–3% of individuals with ASD (6–9). A recent analysis of 10K single-nucleotide polymorphism data identified 15q11–q13 CNVs in 17 of 1749 autism patients (10).
Genomic imprinting is an epigenetic mechanism that establishes parent-of-origin-specific gene expression patterns. Deletions of 15q11–q13 on the paternal chromosome 15 cause Prader–Willi syndrome (PWS; MIM 176270), whereas maternal deletions cause Angelman syndrome (AS; MIM 105830) (11). Parental differences in DNA methylation at imprinting control regions have a crucial role in genomic imprinting of 15q11–q13, as well as differences in DNA replication timing, histone modifications, chromosome nuclear organization and mitotic recombination frequencies (12). However, despite progress in molecular characterization of 15q11–q13 rearrangements and imprinting mechanisms, the molecular pathogenesis of the autism phenotype resulting from maternal 15q11–q13 duplications remains largely unknown. The ubiquitin E3 ligase gene UBE3A is the primary candidate gene presumed to be dysregulated in 15q duplication syndrome because of imprinted maternal expression in the brain. In particular, UBE3A has been analyzed due to its role in AS, its pattern of imprinting and its biological function (13–15). However, 15q11–q13 duplications also include the cluster of three gamma-aminobutyric acid A receptor (GABAAR) subunit genes (GABRB3, GABRA5, GABRG3) and paternally expressed genes implicated in splicing (SNRPN), circadian rhythm (MAGEL2), respiration (NDN) and nucleolar function (HBII85, HBII52) (16,17). While increased copy number of 15q11–q13 was predicted to increase expression of the maternally expressed UBE3A and biallelically expressed genes such as GABRB3 and GABRA5, prior investigation of an idic15 brain sample with PWS-like features instead showed reduced levels of GABRB3, GABRA5, GABRG3, SNRPN, HBII85 and HBII52 (18). The striking parent-of-origin pattern of inheritance for 15q11–q13 rearrangements has led to the hypothesis that higher order epigenetic dysregulation of transcription within this region may be involved in ASD.
The higher order, inter-chromosomal association of homologous pairing of maternal and paternal alleles of 15q11–q13 was originally described in lymphocytes and predicted to regulate maintenance of imprinting in the locus (19). Homologous pairing of 15q11–q13 was shown to occur in neurons and be deficient in brains of individuals with Rett syndrome (RTT; MIM 312750), autism and maternal 15q duplication (18,20). Although these observations raise the possibility that homologous pairing of 15q11–q13 is an important spatial organization modulating neuron-specific transcripts within the paired regions, the mechanisms of homologous pairing and its effects on gene expression within the paired regions in neurons has not been previously determined.
Here, we made use of microcell-mediated chromosome transfer (MMCT) technology to generate a 15q11–q13 maternal duplication model in a human neuronal cell line. Using this MMCT method, a maternal copy of human chromosome 15 was successfully transferred into human SH-SY5Y neuronal cells. Our experimental models of 15q duplication syndrome recapitulated the reduced levels of several transcripts resulting from increased maternal 15q11–q13 dosage previously observed in the brain as well as the reduced homologous pairing. We further show that the chromatin regulators MeCP2 and CTCF are required for the homologous pairing of 15q11–q13 during neuronal differentiation. These results provide insight into the inter- and intra-chromosomal spatial interactions influencing 15q11–q13 transcript levels in neurons and provide support for an epigenetic dysregulation pathway by which 15q11–q13 duplications may result in ASD.
RESULTS
Establishment of a maternal 15q duplication model by microcell fusion
We have previously established mouse A9 hybrid cells retaining a single human chromosome of defined parental origin for the investigation of imprinted loci in humans (21). Gene expression and DNA methylation studies demonstrated that the appropriate imprinting status of human loci was maintained in mouse A9 hybrids (22–24). In this study, to generate a novel maternal 15q duplication model in a neuronal cell line, a maternal human chromosome 15 was transferred from mouse A9 hybrids into human SH-SY5Y neuronal cells by MMCT (Fig. 1A). SH-SY5Y cells were selected as a recipient cell line because they can be induced to undergo differentiation within 72 h using PMA, resulting in a morphologic change in the extension of axonal projections and the increased expression of neuron-specific enolase (25). In addition, SH-SY5Y cells are diploid for most chromosomes (26). Our PCR-restriction fragment length polymorphism (RFLP) analysis identified 20 independent SH-SY5Y microcell hybrids that contained an extra maternal copy of human chromosome 15, and therefore named SH(15M) cell lines (Fig. 1B). To further confirm the state of the introduced maternal human chromosome 15 in SH-SY5Y cells, we performed cytogenetic analyses using fluorescence in situ hybridization (FISH) (Fig. 1C). Finally, we confirmed that introduced maternal chromosome 15 was maintained stably in SH(15M) cell lines under appropriate selection conditions. For further analyses, we selected three independent SH(15M) cell lines that morphologically resembled parental SH-SY5Y cells.
Figure 1.

Characterization of SH-SY5Y neuronal cells containing an extra maternal human chromosome 15. (A) A schematic diagram showing the construction of SH-SY5Y microcell hybrids [SH(15M) cell] containing an extra maternal copy of human chromosome 15. Transfer of a maternal human chromosome 15 from mouse A9 cells to SH-SY5Y cell was performed by MMCT technology. (B) RFLP analysis to confirm presence of donor chromosome 15 in SH(15M) cells is shown. DNA from introduced maternal human chromosome 15 donor A9 cells (lane 1), SH-SY5Y cells (lane 2), SH(15M)-1, 2, 3 cells (lanes 3–5) and mouse A9 cells (lane 6) was amplified by PCR using primers that span a PvuII polymorphism. Both chromosome 15 copies in the host SH-SY5Y cells were digested by PvuII, but the donor chromosome 15 was undigested. SH(15M) cells contained both digested and undigested fragments. (C) Results from DNA-FISH analysis of SH(15M) cells. Metaphase chromosomes from SH(15M) were hybridized in situ with Vysis LSI GABRB3 (red) and CEP15 (green) probes. The arrowheads indicate three copies of chromosome 15.
Quantitation of 15q11–q13 transcripts in a maternal 15q duplication model
To determine the effect of increased maternal chromosome 15 dosage on transcript levels in SH(15M) cells, quantitative RT–PCR was used to measure the levels of eight transcripts at the 15q11–q13 locus and two non-15q11–q13 housekeeping gene controls, GAPDH and ACTB (Fig. 2A). The non-imprinted genes CYFIP1 and TJP1 revealed the expected 1.5-fold increase in transcript abundance in all three SH(15M) cell lines (Fig. 2B and C). In contrast, expression of non-imprinted GABRB3 and CHRNA7 genes was significantly reduced compared with the parental cell line in all three SH(15M) lines, despite increased 15q11–q13 dosage (Fig. 2D and E). Moreover, paternal expression of NDN and SNRPN transcripts was also significantly reduced despite the increased maternal dosage (Fig. 2F and G). Furthermore, there was no difference between control SH-SY5Y cells and SH(15M) cells in the levels of maternally expressed UBE3A transcript, even though a 2-fold increase was expected (Fig. 2H). In addition, another maternally expressed gene, ATP10A, was also significantly reduced despite the increased maternal dosage (Fig. 2I). While extra copies of genes are expected to result in increased transcript levels, our SH(15M) cell lines recapitulated the direction of transcriptional alterations previously observed in a human brain sample with maternal 15q duplication (18).
Figure 2.

Analysis of 15q11–q13 transcript levels in experimental model of dup15q syndrome. (A) Physical map of the imprinted gene cluster in human chromosome 15q11–q13. PWS-IC is the PWS imprinting center, which is essential for establishment of the paternal epigenetic state of the region. Genes or transcripts (filled boxes) are drawn approximately to scale. Transcriptional direction is indicated by arrowheads and arrows. P, paternally expressed genes; M, maternally expressed genes; B, biallelically expressed genes. (B–I) Summary of quantitative RT–PCR measurements of eight transcripts in 15q11–q13, normalized to the housekeeping genes GAPDH and ACTB. Error bars represent ±SEM. All qRT–PCR analyses were performed on cDNA from PMA-treated differentiated SH-SY5Y cells (WT) and PMA-treated differentiated SH(15M) cells (1–3). (B)–(E) are non-imprinted biallelically expressed genes, (F) and (G) are paternally expressed imprinted genes and (H) and (I) are maternally expressed imprinted genes.
We next assessed the DNA methylation status of NDN, SNRPN, UBE3A, GABRB3 and CHRNA7 promoter CpG islands by bisulfite sequencing to determine whether the DNA methylation status of these promoters was correlated with the observed reductions in transcript levels. GABRB3, CHRNA7 and UBE3A promoters in the SH(15M) cells were almost completely unmethylated and appeared similar to those observed in control SH-SY5Y cells (Supplementary Material, Fig. S1A–C). Similarly, the expected maternal DNA methylation levels were observed for NDN and SNRPN promoter CpG islands in both SH-SY5Y cells and SH(15M) cells (Supplementary Material, Fig. S1D and E). Because these CpG islands are differentially methylated in a parent-of-origin-specific manner, increased DNA methylation levels were expected following chromosome transfer of the methylated maternal allele (27,28). A combined bisulfite restriction analysis (29) showed strong concordance with bisulfite sequencing results (data not shown). These findings indicate that promoter DNA methylation is not correlated with altered transcript levels of NDN, SNRPN, UBE3A, GABRB3 or CHRNA7 in SH(15M) cells.
Disruption of 15q11–q13 homologous pairing in a maternal 15q duplication model
Homologous chromosomal association or pairing of maternal and paternal 15q11–q13 alleles has previously been described in both lymphocytes (19) and neurons (20) and disrupted in the 15q dup brain (18). Therefore, we investigated the impact of an extra maternal human chromosome 15 on normal interactions between maternal and paternal 15q11–q13 alleles. In comparison with SH-SY5Y cells, SH(15M) cells displayed a significant reduction in the percentage of paired alleles for nuclei hybridized with a Vysis LSI GABRB3 probe (Fig. 3A and B). However, there was no difference in the percentage of paired alleles of nuclei hybridized with a control chromosome 15 pericentromeric CEP15 probe. Homologous pairing was not observed in either SH-SY5Y or SH(15M) cells hybridized with a non-15 control, a pericentromeric probe specific for chromosome 11 (CEP11; data not shown). Thus, the extra maternal human chromosome 15 in the SH(15M) cells conferred a specific effect on the nuclear organization of 15q11–q13 and modeled the homologous pairing defect previously observed in the 15q duplication syndrome brain (18). In addition, our observations indicate that homologous pairing of 15q11–q13 could influence gene expression within the paired region.
Figure 3.
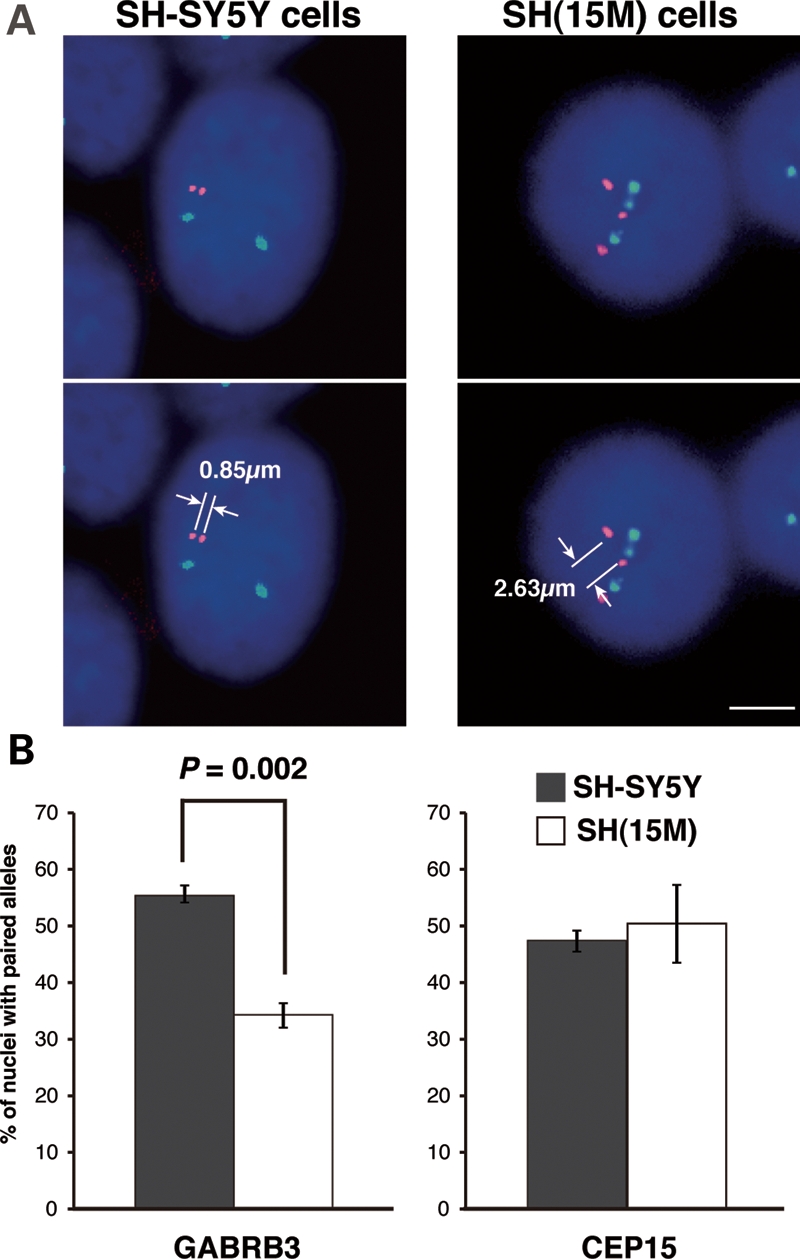
Reduced homologous pairing of GABRB3 alleles as a result of duplicated maternal chromosome 15 in SH-SY5Y cells. (A) Representative image of chr.15–chr.15 localization in SH-SY5Y cells (left panel) and SH(15M) cells (right panel) using Vysis LSI GABRB3 SpectrumOrange/CEP15 SpectrumGreen. The iVision software was used to measure distances between two signals. In case of SH(15M) cells, we measured the shortest inter-chromosomal distances between the three signals. Scale bars: 5 µm. (B) Differences in homologous pairing in SH-SY5Y cells (gray bars) and SH(15M) cells (white bars), as determined by hybridization with Vysis LSI GABRB3 (left panel) and CEP15 (right panel) FISH probes. Pairing was scored as homologous FISH signals with distances ≤2 μm apart. The bars indicate the mean ± SEM of three replicate experiments, in each of which 100–150 nuclei were scored. Significantly fewer SH(15M) cell nuclei showed pairing when the GABRB3 probe was used. In contrast, no significant difference was observed in SH(15M) cells in pairing between alleles detected by the pericentromeric CEP15 probe.
Homologous pairing of 15q11–q13 in the 3′ region of the GABRB3 gene
The mechanism of homologous pairing of 15q11–q13 in neurons is poorly understood, but critical for understanding the changes in transcription that accompany the loss of homologous pairing in 15q duplication syndrome. To map the precise location and extent of 15q11–q13 homologous pairing upon neuronal differentiation, we performed additional FISH analyses using nine different fluorescently labeled bacterial artificial chromosome (BAC) probes distributed over 9Mb of 15q11–q13 (Fig. 4A). We measured the inter-chromosomal distances between FISH signals from each BAC probe both before (untreated) and 72 h after differentiation with PMA (PMA treated) (20). In comparison with undifferentiated SH-SY5Y cells, a high percentage of nuclei displayed close inter-chromosomal distances (2 µm or less) at the GABRB3 locus following neuronal differentiation, as shown by a significant leftward shift in the distribution (Fig. 4B–E). In contrast to the significant decrease observed in inter-chromosome distances at GABRB3, the CYFIP locus showed the opposite pattern, with a significant increase in inter-chromosome distance following differentiation (Fig. 4B–D).
Figure 4.

Increased homologous pairing of GABRB3 alleles during neuronal differentiation is associated with active transcription. (A) Physical map of the imprinted gene cluster in human chromosome 15q11–q13. PWS-IC is the PWS imprinting center, which is essential for establishment of the paternal epigenetic state of the region. Genes or transcripts (filled boxes) are drawn approximately to scale. Transcriptional direction is indicated by arrowheads and arrows. The horizontal bars indicate BAC probes used in the pairing analyses. GABRB3(1) probe indicates the Vysis LSI GABRB3 Spectrum Orange probe. (B) Frequency distribution of distances between homologous alleles at multiple sites along 15q11–q13 for undifferentiated SH-SY5Y cell nuclei, as measured on each BAC probe. Mean distance, open triangle. (C) Frequency distribution of distances between homologous alleles at multiple sites along 15q11–q13 for PMA-treated differentiated SH-SY5Y cell nuclei. Mean distance, open triangle. A high proportion of PMA-treated differentiated SH-SY5Y cells displayed close homologous distances (≤2 µm) at GABRB3 upon neuronal differentiation, as shown by a leftward shift in the distribution. (D) Cumulative distribution of distances between homologous alleles at 0–2 µm. The solid line indicates the undifferentiated SH-SY5Y cells. The dotted line indicates the PMA-treated differentiated SH-SY5Y cells. The closed and open triangles indicate cumulative frequency at 2 µm, respectively. The statistical relevance was assessed by a comparison of the entire histogram of measurement distributions from (B) and (C) using two non-parametric tests, namely Mann–Whitney's U-test and Kolmogorov–Smirnov test. P-values from Mann–Whitney's U-test are as indicated. Sample sizes for each experiment ranged from 100 to 210. (E) Differences in homologous pairing in the undifferentiated SH-SY5Y cells (gray bars) and the PMA-treated differentiated SH-SY5Y cells (white bars), as determined by hybridization with Vysis LSI GABRB3 FISH probes. Pairing was scored as homologous FISH signals with distances ≤2 μm apart. The bars indicate the mean ± SEM of three replicate experiments, in each of which 100–150 nuclei were scored. (F) The PMA-treated differentiated SH-SY5Y cells treated for 4 h with the transcriptional inhibitor, α-amanitin, were hybridized with Vysis LSI GABRB3 (left panel) and CEP15 (right panel) FISH probes and signals measured. Transcriptional inhibition resulted in a significant reduction in the homologous pairing at GABRB3, but not at CEP15. Student's t-test.
To narrow down the genomic region in which the homologous inter-chromosomal association was observed, we performed detailed frequency analyses of homologous allele distances across the GABAAR subunit gene cluster (Fig. 5A). Homologous pairing of 15q11–q13 was observed in the 3′ region of the GABRB3 gene, but not in GABRA5 and GABRG3 (Fig. 5B–D). This in-depth analysis of multiple FISH probes therefore narrowed the locus of homologous interaction to a ∼500 kb region at the 3′ end of GABRB3, with significant changes in homologous pairing during SH-SY5Y differentiation limited to the region detected by a single BAC probe, GABRB3(2).
Figure 5.

Homologous pairing of 15q11–q13 maps to the 3′ region of GABRB3. (A) Physical map of the cluster of GABAAR subunit genes in 15q11–q13. The genes are drawn approximately to scale. The direction of transcription is indicated by arrows. The BAC probes used in the pairing analyses are shown by horizontal bars. (B) Frequency distributions of distances between homologous alleles at multiple sites along 15q11–q13 for undifferentiated SH-SY5Y cell nuclei, as measured on each BAC probe. Mean distance, open triangle. (C) Frequency distributions of distances between homologous alleles at multiple sites along 15q11–q13 for PMA-treated differentiated SH-SY5Y cell nuclei. A high proportion of PMA-treated differentiated SH-SY5Y cells displayed close homologous distances (≤2 μm) at GABRB3 upon neuronal differentiation, as shown by a leftward shift in the distribution. Mean distance, open triangle. (D) Cumulative distance distribution for homologous alleles at 0–2 µm. The solid line indicates the undifferentiated SH-SY5Y cells. The dotted line indicates the PMA-treated differentiated SH-SY5Y cells. The closed and open triangles indicate cumulative frequency at 2 µm, respectively. The statistical relevance was assessed by a comparison of the entire histogram of measurement distributions from (B) and (C) using two non-parametric tests, namely Mann–Whitney's U-test and Kolmogorov–Smirnov test. P-values from Mann–Whitney's U-test are as indicated. Sample sizes for each experiment ranged from 100 to 210. One probe from the 3′ region of GABRB3 showed significant differences in homologous intrachromosomal distance frequencies between undifferentiated SH-SY5Y cells (solid line) and the PMA-treated differentiated SH-SY5Y cells (dotted line). In contrast, one probe from the GABRB3 gene body and two probes overlapping GABRA5 and GABRG3 did not show significant homologous pairing. The more centromeric GABRB3(1) probe showed a trend towards homologous pairing that was not significant.
Because clustering of GABAAR subunit genes is well conserved on human chromosomes 4, 5, 15 and X, it has previously been hypothesized that the physical proximity of GABAAR subunit genes facilitates temporally and spatially coordinated expression (30). Therefore, we wished to test whether the homologous chromosomal association is also conserved in other clusters of GABAAR subunit genes. To this end, the homologous pairing analyses were extended to GABAAR subunit genes on 4p12 and 5q34 (Supplementary Material, Fig. S2). Unlike 15q11–q13, homologous pairings of 4p12 and 5q34 were not observed during neuronal differentiation. However, transcript levels for GABRG1 and GABRA4 on 4p12, and GABRG2 and GABRA6 on 5q34 were barely detectable in differentiated SH-SY5Y cells (data not shown). Cumulatively, our findings suggest that homologous chromosomal pairing in 15q11–q13 coincides with increased expression of GABAAR subunit genes specifically in this locus during neuronal differentiation, as GABRB3 is highly expressed in differentiated SH-SY5Y cells (data not shown). Therefore, in order to investigate whether active transcription is necessary for homologous chromosomal pairing in 15q11–q13, differentiated SH-SY5Y cells were treated with the transcriptional inhibitor α-amanitin. The observation that α-amanitin caused significant decreases in the percentage of paired alleles for nuclei hybridized with GABRB3 probe suggests that active transcription was required for homologous chromosomal pairing in 15q11–q13 (Fig. 4F).
Involvement of chromatin proteins MeCP2 and CTCF in regulation of 15q11–q13 homologous pairing
Our observations raise the question as to which factors control the homologous pairing of 15q11–q13. Notably, we observed multiple binding sites for MeCP2 and CTCF in a ∼500 kb region between at the 3′ end of GABRB3. We used a short-interfering RNA (siRNA)-mediated knockdown approach to directly test the role of MeCP2 in the homologous pairing of 15q11–q13 by transiently depleting MeCP2 expression. MeCP2 protein depletion in differentiated SH-SY5Y cells was confirmed by western blot analysis and immunostaining (Fig. 6A–C) and chr.15–chr.15 interchromosomal distances were measured between FISH signals of GABRB3 and CEP15 probes (Fig. 6D). Consistent with previous findings, homologous pairing at GABRB3 was significantly reduced in MeCP2 knockdown cells compared with non-targeting siRNA-treated cells (20). Specifically, the two GABRB3 FISH signals were ≤2 µm apart in only 18.5% of MeCP2-deficient nuclei compared with 48.9% in control cells.
Figure 6.

Disruption of GABRB3 homologous pairing via MeCP2 and CTCF knockdown. (A and E) Results from western blot analysis confirming knockdown of MeCP2 (A) and CTCF (E) proteins in SH-SY5Y cells. (B and F) Results from western blot analysis of cell lysates after siRNA-mediated gene silencing. Both MeCP2 (B) and CTCF (F) proteins experienced ∼80% knockdown. (C and G) Expression of MeCP2 (C) and CTCF (G) proteins in siRNA-treated differentiated SH-SY5Y cells. Scale bars: 10 µm. (D and H) Chr.15–Chr.15 distribution profiles and cumulative frequency curves of MeCP2 (D) and CTCF (H) in siRNA-treated differentiated SH-SY5Y cell nuclei. Mann–Whitney's U-test and Kolmogorov–Smirnov tests were used to determine whether the differences between the curves of the siRNA-treated cells (dotted lines) and the non-targeting siRNA-treated SH-SY5Y cells (solid lines) were significant. The statistical relevance was assessed by comparing whole histograms. P-values from Mann–Whitney's U-test are as indicated. Sample sizes for each experiment ranged from 103 to 105.
CTCF is also required for inter- and intra-chromosomal associations during genomic imprinting and X inactivation (31–34). A chromatin immunoprecipitation assay using a CTCF-specific antibody demonstrated that there are three CTCF-binding sites at the 3′ region of the GABRB3 gene (Supplementary Material, Fig. S3). Therefore, we examined whether CTCF deficiency affected homologous pairing at the GABRB3 locus in differentiated SH-SY5Y cells. Western blot and immunostaining analyses showed that CTCF was predominantly depleted at the protein level (Fig. 6E–G). Interestingly, depletion of CTCF resulted in a significant reduction in the homologous pairing at GABRB3, and at CEP15 (Fig. 6H). In contrast, knocking down RAD21 had no obvious effect on the homologous pairing of 15q11–q13 (Supplementary Material, Fig. S4), although this protein has been reported to frequently accumulate at CTCF-binding sites (35). These results suggest that MeCP2 and CTCF are essential mediators for the homologous pairing of 15q11–q13.
DISCUSSION
Autism has been linked to the region on chromosome 15q11–q13 that is responsible for the imprinting disorders PWS and AS (1,3,5). Maternal 15q11–q13 duplications are the most frequent cytogenetically detectable mutation associated with ASD (6–9), but the molecular impact of 15q duplication in neurons has been difficult to assess because of the lack of an appropriate model system. While overexpression of maternally expressed UBE3A was predicted to be the major cause of the ASD phenotype in 15q duplication syndrome (36), Hogart and LaSalle (18) revealed that 15q11–q13 transcripts in maternal 15q11–q13 duplication brain samples were altered in a direction not predicted from maternal copy number gains. This led to the suggestion that an imbalance of 15q11–q13 dosage can disrupt normal epigenetic pathways such as parental homologous pairing and DNA methylation status, changing gene expression patterns within 15q11–q13. Indeed, our prior study showed that various autosomal imbalances commonly affect neuronal differentiation through the dysregulation of gene expression (37).
In this study, we made use of MMCT to generate a 15q11–q13 maternal duplication model in a neuronal cell line to explore the molecular basis of epigenetic dysregulation in 15q dup syndrome. Using this MMCT method, a maternal copy of human chromosome 15 was successfully transferred into human SH-SY5Y neuronal cells. Currently, CNVs, including the 15q duplication, are beginning to provide some insights into the underlying genetic causes of neurodevelopmental disorders, in particular schizophrenia and ASD. However, the impact of CNVs on phenotypic expression remains largely unknown (38). Therefore, this study takes an important step forward by using MMCT as an important tool to define the molecular effects of CNVs in a human neuronal cell line and demonstrating that a chromosome imbalance disrupts normal homologous pairing and alters gene expression patterns.
Quantitative RT–PCR analyses revealed significant alterations in gene expression of NDN, SNRPN, UBE3A, ATP10A, GABRB3 and CHRNA7 in SH(15M) cells, as has previously been observed in the brains of autistic patients (18). We did not find evidence for aberrant promoter DNA methylation of these genes to explain reduced transcription. Instead, we hypothesize that higher order epigenetic alterations at the level of inter- or intra-chromosomal associations lead to transcriptional down-regulation of individual genes in the 15q11–q13 region in 15q duplication syndrome.
15q11–q13 homologous pairing appears to be a recently evolved higher order epigenetic regulatory mechanism, as it does not occur in the syntenic region in the mouse brain (20) or in lymphocytes of gorilla (39). In this study, we also found no obvious evidence for homologous pairing at the Gabrb3 locus in mouse neurons (Supplementary Material, Fig. S5). As previously suggested elsewhere, the most likely explanation for this discrepancy is that mouse 7qC (which is homologous to the 15q11–q13 region) is not adjacent to ribosomal DNA (rDNA) genes as it is for the acrocentric human chromosome 15 (20). In addition, the GABRB3 minimal pairing region defined in this study is highly conserved in chimp but not in mouse. These mouse–human discrepancies could begin to explain why the mouse model of 15q duplication syndrome unexpectedly showed an autism-like phenotype upon paternal transmission in opposition to what is observed in human 15q duplication patients (40). Cumulatively, these findings suggest that there has been some divergence in the genetic and/or epigenetic mechanisms relevant to homologous pairing in the human 15q11–q13 and mouse 7qC regions with potential relevance to autism.
While the proximity of GABRB3 to the rDNA repeats on chromosome 15 may have been important in the recent evolution of homologous pairing of this locus, our study has shown that GABRB3 pairing cannot simply be explained as a byproduct of the perinucleolar organization of acrocentric chromosomes in humans, as has been previously suggested (39). Interestingly, while both CEP15 and GABRB3 regions showed pairing, the effect of additional copy number of chromosome 15 on disrupting pairing was specific to GABRB3, not the rDNA adjacent CEP15, suggesting specificity to the GABRB3 pairing beyond the acrocentric effect. Furthermore, the single gene locus most proximal to the rDNA repeats, CYFIP, actually exhibited increased interchromosomal distances with SH-SY5Y differentiation, suggesting that neuronal differentiation induced dynamic locus-dependent chromatin changes that were not simply rDNA proximity effects. Lastly, while transcriptional inhibition reduced GABRB3 pairing, it had no significant effect on pairing at CEP15, demonstrating that GABRB3 pairing was an active occurrence independent from rDNA organization on human 15q11–q13.
The loss of homologous pairing in the SH(15M) neuronal model led us to investigate the mechanism of homologous pairing in the 15q11–q13 region and its relation to autism candidate genes. We narrowed down the region of homologous pairing of 15q11–q13 to the 3′ region of the GABRB3 gene. The GABAAR subunit genes, which encode for subunits of the receptor for the neurotransmitter GABA, are important autism candidate genes (30). Interestingly, while GABAAR subunit genes are biallelically expressed in normal brain samples, at least one of these transcripts showed a gain of monoallelic or biased expression in half (4/8) of brain samples from autistic individuals, suggesting underlying epigenetic dysregulation (41). The pairing region likely contains some key regulatory elements for homologous pairing of 15q11–q13 which epigenetically control gene expression in the region. Our analyses revealed discrete MeCP2 and CTCF neuronal-binding sites within the GABRB3 pairing region were observed in this minimal homologous pairing region required for optimal 15q11–q13 pairing. Differentiated SH-SY5Y cells showed dynamic changes in chromatin structure compared with undifferentiated cells, consistent with a model of active pairing and increased transcription of GABRB3 with differentiation. Interestingly, previous linkage analyses have shown a significant association between GABRB3 polymorphisms and autism (42,43). One such GABRB3 microsatellite has been localized to ∼60 kb beyond the 3′ end of the GABRB3 gene (44) that lies within the GABRB3 minimal pairing region and interacts with the maternal PWS-IC (Supplementary Material, Fig. S6). Interestingly, maternal transmission of a rare GABRB3 signal peptide variant has been recently observed in autism (45).
Recent findings showed that CTCF and OCT4 together can mediate X-chromosome pairing, and the results of our homologous pairing analyses also suggest that CTCF can mediate specific inter-chromosomal associations in the 3′ region of the GABRB3 gene (34). Indeed, we identified three potential CTCF-binding sites bordering the homologous pairing region. On the basis of our RNAi-mediated knockdown studies, we conclude that CTCF controls not only X chromosome pairing but also autosomal 15q11–q13 pairing. Although our study highlights CTCF as a central molecule in inter-chromosomal association, its exact role in the etiology of neurodevelopmental disorders remains uncertain. One recent study revealed that CTCF and cohesin complexes are necessary for establishing the chromatin structure required for brain-derived neurotrophic factor transcription (46). Thus, there is already some evidence indicating how epigenetic factors such as CTCF and MeCP2 may play a role in complex psychiatric and neurodevelopmental disorders (16).
It is plausible that MeCP2, such as CTCF, also controls the homologous pairing of 15q11–q13. Mutations in MECP2 cause RTT, a neurodevelopmental disorder characterized by the loss of speech and acquired motor skills, stereotypical hand movements, and seizures during early childhood (47). In addition, MECP2 mutations have been found in a few patients diagnosed with AS and autism, suggesting overlap in the pathogenesis of these distinct genetic syndromes (48,49). Consistent with phenotypic and genetic overlap among RTT, AS and autism patients, one previous study demonstrated significant defects in the expression of UBE3A and GABRB3 in brain samples from RTT, AS and autism patients (50). Consistent with results of a previous study utilizing an oligonucleotide decoy approach, we demonstrated that homologous pairing at the GABRB3 locus was significantly impacted by siRNA-mediated MeCP2 knockdown (20). This finding suggests that inter-chromosomal associations such as homologous pairing are essential for precise gene expression of GABRB3 during neuronal differentiation. Further experiments are needed to determine how homologous pairing controls the expression of GABRB3 transcripts. In a simple model, homologous 15q11–q13 pairing might provide a transcriptionally positive environment by recruiting homologous alleles at the same transcription factory to increase neuronal gene expression by recycling positive factors at both alleles. Indeed, the transcriptional inhibitor α-amanitin caused a significant decrease in the homologous 15q11–q13 pairing, further demonstrating the requirement of active transcription for homologous 15q11–q13 pairing.
Recently, development of new techniques such as the chromosomal conformation capture (3C) assay has enabled description of long-range intra-chromosomal associations, such as those at the H19-Igf2 and Dlx5/Dlx6 loci (51,52). Similar intra-chromosomal associations have been proposed in the regulation of 15q11–q13 genes because PWS-IC acts in cis to regulate paternal expression of MKRN3, MAGEL2, NDN and SNRPN genes within 15q11–q13 (53). In support of this hypothesis, our DNA-RNA FISH results showed that the PWS-IC makes an allele-specific association with the homologous pairing region of the GABRB3 locus in differentiated SH-SY5Y cells (Supplementary Material, Fig. S6). These results may indicate that the intra-chromosomal association between the PWS-IC and GABRB3 occurs on the maternal 15q11–q13 allele. On the other hand, disassociation of the PWS-IC and GABRB3 is consistent with paternal allele-specific decondensation of chromatin at this locus during neuronal maturation (54). We speculate that maternal 15q duplication might alter the intrachromosomal association between the PWS-IC and GABRB3, thus disrupting the coordinated gene regulation of 15q11–q13.
In conclusion, 15q11–q13 appears to be a useful model region for studying epigenetic pathways and mechanisms such as long-range chromatin organization and homologous pairing. Our observations imply that CNVs such as 15q duplication alter such epigenetic mechanisms, thus disrupting regulation of individual genes at the level of inter- and intra-chromosomal associations. In order to fully understand how CNVs affect the nucleus on a molecular level, it will be important to define chromosomal regulatory elements using diverse genetic approaches, such as MMCT. Furthermore, the characterization of MeCP2 as well as CTCF can reveal additional partners involved in regulation of inter- or intra-chromosomal associations. The investigation of such interplaying partners will help in designing potential drug targets, enabling the development of a therapeutic approach for the treatment of dup15q syndrome.
MATERIALS AND METHODS
Cell lines
A9 hybrids containing a maternal human chromosome 15 tagged with pSTneo were constructed as previously described (55) and were cultured in Dulbecco's modified Eagle medium (DMEM) (WAKO, Tokyo, Japan) supplemented with 10% calf serum (Hyclone, Thermo Scientific, Waltham, MA, USA) and 800 µg/ml G418 (Nakarai, Kyoto, Japan). SH-SY5Y neuroblastoma cells (ATCC, USA) were cultured in the DMEM/F12 medium (WAKO) supplemented with 15% fetal bovine serum (Hyclone, Thermo Scientific). For FISH analysis, cells were seeded onto Lab-Tek™ II-CC2™ Chamber Slides (Nalge Nunc, Penfield, NY, USA) and grown until they reached 30–50% confluency. Cells were fixed either before (untreated) or 72 h after the addition of 16 nm PMA (PMA treated) for 15 min in Histochoice (Ameresco, Solon, OH, USA) and then washed in 1× PBS/0.5% Tween 20 for 5 min and stored in 70% ethanol at −20°C. For α-amanitin experiments, PMA-treated SH-SY5Y cells were treated with 20 or 30 µg/ml α-amanitin for 4 h.
Microcell-mediated chromosome transfer
Introduction of an extra maternal human chromosome 15 into human SH-SY5Y neuronal cells was performed via MMCT, as previously described (56). Briefly, microcells purified from 1 × 108 donor A9 cells containing a maternal human chromosome 15 were fused with SH-SY5Y cells using 47% polyethylene glycol 1000 (PEG1000: Mr 1000; Nakarai). SH-SY5Y microcell hybrid clones were selected individually in the DMEM/F12 medium supplemented with 15% FBS and 600 µg/ml of G418 for 2–3 weeks. These clones were usually maintained stably in culture under appropriate selective conditions. The introduction of maternal human chromosome 15 in SH-SY5Y cells was confirmed by PCR-RFLP analysis and cytogenetic analysis. The primers for RFLP analysis were 5′-AGTGGGCTTCCCTCACTTCT-3′ (forward) and 5′-CAGACAGGCTCCACTTACCC-3′ (reverse). Three independent SH-SY5Y cells with an extra maternal human chromosome 15 obtained from each chromosome transfer were selected for further expression and pairing analyses.
Cytogenetic analysis
To confirm the presence of the transferred human chromosome 15 in SH-SY5Y cells, FISH analysis was performed on fixed metaphase spreads of each SH-SY5Y clone using the Vysis LSI Prader-Willi/Angelman Region Probe (GABRB3) (Abbott, North Chicago, IL, USA), as described previously (21). Chromosomes were counterstained with DAPI (Sigma, St Louis, MO, USA).
FISH and pairing assays
DNA FISH analysis was performed as described previously (54). Briefly, FISH was conducted in SH-SY5Y neuroblasts and SH-SY5Y neurons differentiated by 72 h treatment with 16 nm PMA (20). FISH probes for GABRB3 and CEP15 were obtained commercially from Vysis, but to determine precise locations of pairing, BAC probes were obtained (CHORI, Oakland, CA, USA). BAC DNA was labeled with Green-dUTP or Orange-dUTP (Abbott). BAC clones for FISH are as follow; RP11-69H14 (CYFIP1), RP11-373J1 (NDN), RP11-125E1 (SNRPN), RP11-171C8 (HBII-85), RP11-1081A4 (UBE3A), RP11-339C21 (ATP10A), RP11-92F7 (TJP1), RP11-714E8 (CHRNA7), RP11-638J6 (GABRB3(1)), RP11-345N11 (GABRB3(2)), RP11-974L14 (GABRB3(3)), RP11-243J20 (GABRA5), RP11-89E18 (GABRG3), RP11-79M7 (GABRG1), RP11-905L4 (GABRA2), RP11-620L1 (GABRA4), RP11-1059P8 (GABRB1), RP11-315P17 (GABRB2(1)), RP11-833C4 (GABRB2(2)), RP11-348M17 (GABRA1), RP11-204E3 (GABRG2), RP23-24D4 (Gabrb3, I), RP23-143O21 (Gabrb3, II) and RP23-459J11 (Gabrb3, III). A nick translation kit (Roche, Penzberg, Germany) was used to create probes, which were then hybridized to cells fixed in Histochoice (Amresco), then dehydrated in 70, 90 and 100% ethanol (10 min each) and dried at 50°C. FISH probes were denatured with the fixed cells at 80°C for 2 min and then hybridized overnight at 37°C on cover-slipped slides. Cells were washed three times in 50% formamide/50% 2× SSC for 5 min, 2× SSC for 5 min and 2× SSC/0.1% IGEPAL for 5 min at 46°C and then mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA) containing 5 µg/µl DAPI. To analyze pairing, we took digital images with a BX51 fluorescence microscope (Olympus, Tokyo, Japan) equipped with a RETIGA EXi CCD camera (Qimaging, Surrey, Canada); these were processed in iVision 4.0 software (BioVision Technologies, Exton, PA, USA). The iVision software was also used to measure distances between two signals. All FISH distances are measured in 2D in this study because 3D is less of a concern for neurons, which are relatively flat compared with round nuclei seen in lymphocytes. Although 2D may change some measurements, signals that are significantly closer than expected of random were likely to be close in 3D space. To simplify the analysis of homologous association or ‘pairing’ as has been described previously (19,20), nuclei with FISH signals ≤2 µm apart were scored as ‘paired’, while nuclei with two FISH signals >2 µm apart were scored as ‘unpaired’. In case of SH(15M) cells, we measured the shortest inter-chromosomal distances between the three signals. Measurement and scoring of FISH signals were performed manually and results are averages of at least three independent scores per sample.
Gene expression analysis
Total RNA was extracted using RNeasy columns, according to the manufacturer's protocols (Qiagen, Hilden, Germany) and then treated with RNase-free DNase I (Takara, Kyoto, Japan). First-strand cDNA synthesis was carried out with random primers and SuperScript® III reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Quantitative RT–PCR was performed with GoTaq® qPCR Master Mix (Promega, Fitchburg, WI, USA) on a CFX384 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Three replicate reactions were conducted for each gene analyzed. Melting curve analysis was performed to ensure that a single product was amplified with each primer set. Relative expression levels of 15q11–q13 transcripts were normalized by using the comparative Ct (ΔΔCt) method and the geometric average of a set of two housekeeping genes (GAPDH and ACTB) by the Bio-Rad CFX manager software (version 1.5). Primer sequences are available on request.
RNAi knockdown
We carried out siRNA-mediated knockdown of MeCP2, CTCF and RAD21 proteins using Accell SMART pool siRNAs, according to the manufacturer's protocols (Dharmacon, Lafayette, CO, USA). Accell non-targeting siRNA was used in control knockdowns. Briefly, we plated SH-SY5Y cells onto Lab-Tek™ II-CC2™ Chamber Slides (Nalge Nunc) and then incubated them for 72 h in serum-free Accell SMART pool transfection medium containing 1 µm of MeCP2, CTCF or RAD21 siRNAs. After 72 h of siRNA treatment, we replaced the medium with 1% serum-supplemented Accell medium containing 16 nm PMA and a pool of each siRNA, as appropriate. For pairing analysis and knockdown assessment, cells were fixed in Histochoice (Amresco) or harvested after an additional 72 h of incubation. MeCP2, CTCF and RAD21 knockdowns were confirmed by western blot analysis using rabbit anti-MeCP2 (Diagenode, Sparta, NJ, USA), rabbit anti-CTCF (CST, Danvers, MA, USA), rabbit anti-RAD21 (CST) and rabbit anti-GAPDH (CST), with the appropriate rabbit secondary antibodies conjugated to horseradish peroxidase for detection (Bio-Rad). Immunostaining was conducted with anti-MeCP2, anti-CTCF and anti-RAD21, and detection was achieved with secondary Alexa Fluor 555-goat anti-rabbit or Alexa Fluor 488-goat anti-rabbit (Invitrogen).
DNA methylation analysis
We performed DNA methylation analysis using EpiTect® Bisulfite Kits (Qiagen), according to the manufacturer's protocols. Specific primers for the amplification of bisulfite-treated DNA were designed using MethPrimer (http://www.urogene.org/methprimer). PCR was performed using GoTaq® Master Mix (Promega). PCR conditions were as follows: 2 min hot start at 95°C, followed by 32 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 30 s and extension at 72°C for 30 s. The PCR products were directly cloned into the pGEM®-T Easy vector (Promega). Twenty individual clones were sequenced from both ends using a 310 Genomic Analyzer (Applied Biosystems Inc., Foster City, CA, USA) according to the standard protocols. Primer sequences are available on request.
Chromatin immunoprecipitation
Chromatin was prepared from SH-SY5Y cells and purified by urea gradient centrifugation, as previously described (52). Immunoprecipitation, reverse crosslinking and PCR amplification were also performed as previously described (52), with some modifications. Briefly, a panel of restriction enzymes (NEB, Ipswich, MA, USA) was used to digest chromatin into 100–700 bp DNA fragments. Digested chromatin was pre-cleared with Protein G Dynabeads (Invitrogen) alone, then with normal rabbit IgG (CST) and then finally with Protein G Dynabeads. Pre-cleared chromatin was incubated overnight with rabbit anti-CTCF or with equivalent amounts of normal rabbit IgG as a control for non-specific binding. PCR amplification was performed for 35 cycles with one-twentieth of the IP products. Primer sequences are available on request.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported in part by Grants-in-Aid for JSPS Fellows and a Short-term Fellowship from the International Human Frontier Science Program (M.M.-H.), the Program for Improvement of Research Environment for Young Researchers from the Special Coordination Funds for Promoting Science and Technology (S.H.), Grant-in-Aid for Scientific Research on Priority Areas (S.H.), Grants-in-Aid for Scientific Research on Innovative Areas (S.H.), Grants-in-Aid for Young Scientists (B) (S.H.), The Cell Science Research Foundation, Daiichi-Sankyo Foundation of Life Science, Mitsubishi Pharma Research Foundation and Life Science Foundation of Japan (S.H.), the Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (H.I., S.H.) and the National Institutes of Health (J.M.L., R01HD048799 and R01HD41462).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Karen N. Leung for her advice on FISH protocols and Masako Tada, Hiroyuki Kugoh for critical reading of the manuscript. We would also like to thank Sachiyo Akagi and Miwa Miyano for their help with the MMCT and PCR analysis.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Folstein S.E., Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat. Rev. Genet. 2001;2:943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- 2.Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. J. Autism Dev. Disord. 2003;33:365–382. doi: 10.1023/a:1025054610557. [DOI] [PubMed] [Google Scholar]
- 3.Cook E.H., Jr., Scherer S.W. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 4.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S., et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang N.J., Liu D., Parokonny A.S., Schanen N.C. High-resolution molecular characterization of 15q11-q13 rearrangements by array comparative genomic hybridization (array CGH) with detection of gene dosage. Am. J. Hum. Genet. 2004;75:267–281. doi: 10.1086/422854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cook E.H., Jr, Lindgren V., Leventhal B.L., Courchesne R., Lincoln A., Shulman C., Lord C., Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 1997;60:928–934. [PMC free article] [PubMed] [Google Scholar]
- 7.Schroer R.J., Phelan M.C., Michaelis R.C., Crawford E.C., Skinner S.A., Cuccaro M., Simensen R.J., Bishop J., Skinner C., Fender D., et al. Autism and maternally derived aberrations of chromosome 15q. Am. J. Med. Genet. 1998;76:327–336. doi: 10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 8.Lauritsen M., Mors O., Mortensen P.B., Ewald H. Infantile autism and associated autosomal chromosome abnormalities: a register-based study and a literature survey. J. Child Psychol. Psychiatry. 1999;40:335–345. [PubMed] [Google Scholar]
- 9.Veenstra-Vanderweele J., Christian S.L., Cook E.H., Jr. Autism as a paradigmatic complex genetic disorder. Annu. Rev. Genomics Hum. Genet. 2004;5:379–405. doi: 10.1146/annurev.genom.5.061903.180050. [DOI] [PubMed] [Google Scholar]
- 10.Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J., Liu X.Q., Vincent J.B., Skaug J.L., Thompson A.P., Senman L., et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knoll J.H., Nicholls R.D., Magenis R.E., Graham J.M., Jr, Lalande M., Latt S.A. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am. J. Med. Genet. 1989;32:285–290. doi: 10.1002/ajmg.1320320235. [DOI] [PubMed] [Google Scholar]
- 12.Reik W., Walter J. Genomic imprinting: parental influence on the genome. Nat. Rev. Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 13.Kishino T., Lalande M., Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 14.Matsuura T., Sutcliffe J.S., Fang P., Galjaard R.J., Jiang Y.H., Benton C.S., Rommens J.M., Beaudet A.L. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 15.Jiang Y.H., Sahoo T., Michaelis R.C., Bercovich D., Bressler J., Kashork C.D., Liu Q., Shaffer L.G., Schroer R.J., Stockton D.W., et al. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am. J. Med. Genet. A. 2004;131:1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- 16.Schanen N.C. Epigenetics of autism spectrum disorders. Hum. Mol. Genet. 2006;15:R138–R150. doi: 10.1093/hmg/ddl213. [DOI] [PubMed] [Google Scholar]
- 17.Grafodatskaya D., Chung B., Szatmari P., Weksberg R. Autism spectrum disorders and epigenetics. J. Am. Acad. Child Adolesc. Psychiatry. 2010;49:794–809. doi: 10.1016/j.jaac.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 18.Hogart A., LaSalle J.M. Chromosome 15q11–13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J. Med. Genet. 2009;46:86–93. doi: 10.1136/jmg.2008.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LaSalle J.M., Lalande M. Homologous association of oppositely imprinted chromosomal domains. Science. 1996;272:725–728. doi: 10.1126/science.272.5262.725. [DOI] [PubMed] [Google Scholar]
- 20.Thatcher K.N., Peddada S., Yasui D.H., Lasalle J.M. Homologous pairing of 15q11–13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum. Mol. Genet. 2005;14:785–797. doi: 10.1093/hmg/ddi073. [DOI] [PubMed] [Google Scholar]
- 21.Kugoh H., Mitsuya K., Meguro M., Shigenami K., Schulz T.C., Oshimura M. Mouse A9 cells containing single human chromosomes for analysis of genomic imprinting. DNA Res. 1999;6:165–172. doi: 10.1093/dnares/6.3.165. [DOI] [PubMed] [Google Scholar]
- 22.Mitsuya K., Meguro M., Lee M.P., Katoh M., Schulz T.C., Kugoh H., Yoshida M.A., Niikawa N., Feinberg A.P., Oshimura M. LIT1, an imprinted antisense RNA in the human KvLQT1 locus identified by screening for differentially expressed transcripts using monochromosomal hybrids. Hum. Mol. Genet. 1999;8:1209–1217. doi: 10.1093/hmg/8.7.1209. [DOI] [PubMed] [Google Scholar]
- 23.Meguro M., Mitsuya K., Nomura N., Kohda M., Kashiwagi A., Nishigaki R., Yoshioka H., Nakao M., Oishi M., Oshimura M. Large-scale evaluation of imprinting status in the Prader-Willi syndrome region: an imprinted direct repeat cluster resembling small nucleolar RNA genes. Hum. Mol. Genet. 2001;10:383–394. doi: 10.1093/hmg/10.4.383. [DOI] [PubMed] [Google Scholar]
- 24.Nakabayashi K., Bentley L., Hitchins M.P., Mitsuya K., Meguro M., Minagawa S., Bamforth J.S., Stanier P., Preece M., Weksberg R., et al. Identification and characterization of an imprinted antisense RNA (MESTIT1) in the human MEST locus on chromosome 7q32. Hum. Mol. Genet. 2002;11:1743–1756. doi: 10.1093/hmg/11.15.1743. [DOI] [PubMed] [Google Scholar]
- 25.Pahlman S., Hoehner J.C., Nanberg E., Hedborg F., Fagerstrom S., Gestblom C., Johansson I., Larsson U., Lavenius E., Ortoft E., et al. Differentiation and survival influences of growth factors in human neuroblastoma. Eur. J. Cancer. 1995;31A:453–458. doi: 10.1016/0959-8049(95)00033-f. [DOI] [PubMed] [Google Scholar]
- 26.Spengler B.A., Biedler J.L., Ross R.A. A corrected karyotype for the SH-SY5Y human neuroblastoma cell line. Cancer Genet. Cytogenet. 2002;138:177–178. doi: 10.1016/s0165-4608(02)00523-x. [DOI] [PubMed] [Google Scholar]
- 27.El-Maarri O., Buiting K., Peery E.G., Kroisel P.M., Balaban B., Wagner K., Urman B., Heyd J., Lich C., Brannan C.I., et al. Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat. Genet. 2001;27:341–344. doi: 10.1038/85927. [DOI] [PubMed] [Google Scholar]
- 28.Jay P., Rougeulle C., Massacrier A., Moncla A., Mattei M.G., Malzac P., Roëckel N., Taviaux S., Lefranc J.L., Cau P., et al. The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat. Genet. 1997;17:357–361. doi: 10.1038/ng1197-357. [DOI] [PubMed] [Google Scholar]
- 29.Xiong Z., Laird P.W. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hogart A., LaSalle J.M. The Neurochemical Basis of Autism. Springer; 2010. Epigenetic Dysregulation of 15q11–13 GABAA Receptor Genes in Autism; pp. 113–127. [Google Scholar]
- 31.Kurukuti S., Tiwari V.K., Tavoosidana G., Pugacheva E., Murrell A., Zhao Z., Lobanenkov V., Reik W., Ohlsson R. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc. Natl Acad. Sci. USA. 2006;103:10684–10689. doi: 10.1073/pnas.0600326103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohlsson R., Bartkuhn M., Renkawitz R. CTCF shapes chromatin by multiple mechanisms: the impact of 20 years of CTCF research on understanding the workings of chromatin. Chromosoma. 2010;119:351–360. doi: 10.1007/s00412-010-0262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams A., Flavell R.A. The role of CTCF in regulating nuclear organization. J. Exp. Med. 2008;205:747–750. doi: 10.1084/jem.20080066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donohoe M.E., Silva S.S., Pinter S.F., Xu N., Lee J.T. The pluripotency factor Oct4 interacts with Ctcf and also controls X-chromosome pairing and counting. Nature. 2009;460:128–132. doi: 10.1038/nature08098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wendt K.S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T., et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 36.Herzing L.B., Cook E.H., Jr., Ledbetter D.H. Allele-specific expression analysis by RNA-FISH demonstrates preferential maternal expression of UBE3A and imprint maintenance within 15q11-q13 duplications. Hum. Mol. Genet. 2002;11:1707–1718. doi: 10.1093/hmg/11.15.1707. [DOI] [PubMed] [Google Scholar]
- 37.Kai Y., Wang C.C., Kishigami S., Kazuki Y., Abe S., Takiguchi M., Shirayoshi Y., Inoue T., Ito H., Wakayama T., et al. Enhanced apoptosis during early neuronal differentiation in mouse ES cells with autosomal imbalance. Cell Res. 2009;19:247–258. doi: 10.1038/cr.2008.305. [DOI] [PubMed] [Google Scholar]
- 38.Lee C., Scherer S.W. The clinical context of copy number variation in the human genome. Expert Rev. Mol. Med. 2010;12:e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- 39.Teller K., Solovei I., Buiting K., Horsthemke B., Cremer T. Maintenance of imprinting and nuclear architecture in cycling cells. Proc. Natl Acad. Sci. USA. 2007;104:14970–14975. doi: 10.1073/pnas.0704285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakatani J., Tamada K., Hatanaka F., Ise S., Ohta H., Inoue K., Tomonaga S., Watanabe Y., Chung Y.J., Banerjee R., et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11–13 duplication seen in autism. Cell. 2009;137:1235–1246. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hogart A., Nagarajan R.P., Patzel K.A., Yasui D.H., Lasalle J.M. 15q11–13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum. Mol. Genet. 2007;16:691–703. doi: 10.1093/hmg/ddm014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cook E.H., Jr, Courchesne R.Y., Cox N.J., Lord C., Gonen D., Guter S.J., Lincoln A., Nix K., Haas R., Leventhal B.L., et al. Linkage-disequilibrium mapping of autistic disorder, with 15q11–13 markers. Am. J. Hum. Genet. 1998;62:1077–1083. doi: 10.1086/301832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buxbaum J.D., Silverman J.M., Smith C.J., Greenberg D.A., Kilifarski M., Reichert J., Cook E.H., Jr, Fang Y., Song C.Y., Vitale R. Association between a GABRB3 polymorphism and autism. Mol. Psychiatry. 2002;7:311–316. doi: 10.1038/sj.mp.4001011. [DOI] [PubMed] [Google Scholar]
- 44.Martin E.R., Menold M.M., Wolpert C.M., Bass M.P., Donnelly S.L., Ravan S.A., Zimmerman A., Gilbert J.R., Vance J.M., Maddox L.O., et al. Analysis of linkage disequilibrium in gamma-aminobutyric acid receptor subunit genes in autistic disorder. Am. J. Med. Genet. 2000;96:43–48. doi: 10.1002/(sici)1096-8628(20000207)96:1<43::aid-ajmg9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 45.Delahanty R.J., Kang J.Q., Brune C.W., Kistner E.O., Courchesne E., Cox N.J., Cook E.H., Jr., Macdonald R.L., Sutcliffe J.S. Maternal transmission of a rare GABRB3 signal peptide variant is associated with autism. Mol. Psychiatry. 2011;16:86–96. doi: 10.1038/mp.2009.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang J., Zhang B., Heath H., Galjart N., Wang X., Milbrandt J. Nicotinamide adenine dinucleotide (NAD)-regulated DNA methylation alters CCCTC-binding factor (CTCF)/cohesin binding and transcription at the BDNF locus. Proc. Natl Acad. Sci. USA. 2010;107:21836–21841. doi: 10.1073/pnas.1002130107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 48.Watson P., Black G., Ramsden S., Barrow M., Super M., Kerr B., Clayton-Smith J. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J. Med. Genet. 2001;38:224–228. doi: 10.1136/jmg.38.4.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lam C., Yeung W., Ko C., Poon P., Tong S., Chan K., Lo I., Chan L., Hui J., Wong V., et al. Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J. Med. Genet. 2000;37:E41. doi: 10.1136/jmg.37.12.e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samaco R.C., Hogart A., LaSalle J.M. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murrell A., Heeson S., Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 2004;36:889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 52.Horike S., Cai S., Miyano M., Cheng J.F., Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 53.Yang T., Adamson T.E., Resnick J.L., Leff S., Wevrick R., Francke U., Jenkins N.A., Copeland N.G., Brannan C.I. A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat. Genet. 1998;19:25–31. doi: 10.1038/ng0598-25. [DOI] [PubMed] [Google Scholar]
- 54.Leung K.N., Vallero R.O., DuBose A.J., Resnick J.L., LaSalle J.M. Imprinting regulates mammalian snoRNA-encoding chromatin decondensation and neuronal nucleolar size. Hum. Mol. Genet. 2009;18:4227–4238. doi: 10.1093/hmg/ddp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inoue J., Mitsuya K., Maegawa S., Kugoh H., Kadota M., Okamura D., Shinohara T., Nishihara S., Takehara S., Yamauchi K., et al. Construction of 700 human/mouse A9 monochromosomal hybrids and analysis of imprinted genes on human chromosome 6. J. Hum. Genet. 2001;46:137–145. doi: 10.1007/s100380170101. [DOI] [PubMed] [Google Scholar]
- 56.Koi M., Shimizu M., Morita H., Yamada H., Oshimura M. Construction of mouse A9 clones containing a single human chromosome tagged with neomycin-resistance gene via microcell fusion. Jpn. J. Cancer Res. 1989;80:413–418. doi: 10.1111/j.1349-7006.1989.tb02329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.