

Abstract
Transcription is regulated by acetylation/deacetylation reactions of histone and nonhistone proteins mediated by enzymes called KATs and HDACs, respectively. As a major mechanism of transcriptional regulation, protein acetylation is a key controller of physiological processes such as cell cycle, DNA damage response, metabolism, apoptosis, and autophagy. The deacetylase activity of class III histone deacetylases or sirtuins depends on the presence of NAD+ (nicotinamide adenine dinucleotide), and therefore, their function is closely linked to cellular energy consumption. This activity of sirtuins connects the modulation of chromatin dynamics and transcriptional regulation under oxidative stress to cellular lifespan, glucose homeostasis, inflammation, and multiple aging-related diseases including cancer. Here we provide an overview of the recent developments in relation to the diverse biological activities associated with sirtuin enzymes and stress responsive transcription factors, DNA damage, and oxidative stress and relate the involvement of sirtuins in the regulation of these processes to oncogenesis. Since the majority of the molecular mechanisms implicated in these pathways have been described for Sirt1, this sirtuin family member is more extensively presented in this paper.
1. Introduction
Acetylation is the addition of an acetyl group at the ε-amino group of the lysine residues present within histone and nonhistone proteins and is one of the most extensively studied posttranslational modifications [1]. Acetylation is mediated by enzymes called histone acetyl transferases (HATs), but since a large number of nonhistone proteins are targeted by HATs, these enzymes are also called KATs (K-acetyltransferases) [2]. The removal of the acetyl group is regulated by the activity of histone deacetylases (HDACs). Acetylation of histone tails decreases their net positive charge [3], thereby reducing the chromatin-binding affinity to DNA, which then becomes more accessible to the transcription initiation complexes and the RNA polymerase [4, 5]. The pattern of the N-terminal histone posttranslational modifications mediating transcriptional events is called “histone code” [6].
Transcriptional regulation of gene expression is a complex process involving several posttranslational modifications of histone and nonhistone proteins. The balance between reversible modifications such as acetylation, phosphorylation, methylation, ubiquitination, propionylation, butyrylation, carbonylation, and ADP ribosylation, occurring within specific chromatin domains, controls the expression or silencing of a diverse set of genes [7]. Enzymes regulating the equilibrium of these modifications maintain the chromatin organization and structure, thus fine-tuning the expression of individual genes. Acetylation of the protruding histone tails is generally associated with activation of gene expression whereas deacetylation is linked to inhibition of gene expression [8]. HDACs exert their repressive function on transcription either by condensing the chromatin or as components of large multiprotein complexes, by recruiting inhibitory factors to regulatory DNA elements within gene promoter regions [9]. Transcriptional regulation exerted by HDACs determines vital cellular processes including cell cycle progression, apoptosis, autophagy, response to diverse types of stress, differentiation, and development [10]. Alterations in HDACs-mediated signaling due to overexpression or hyperactivity of these enzymes can lead to disturbed homeostasis and, hence to pathological conditions [11] including systemic autoimmune [12], Huntington's [13], neurodegenerative [14], respiratory [15], and cardiovascular diseases [16], inflammation [17], diabetes [18], cardiac hypertrophy [19, 20], cancer [21], and conditions such as ageing [22, 23].
Eighteen eukaryotic HDACs, bearing a common well-conserved catalytic deacetylase domain, have been identified so far and classified into four classes: I, II, III, and IV [24]. HDAC1, HDAC2, HDAC3, and HDAC8 are members of the class I HDACs similar to Saccharomyces cerevisiae reduced potassium deficiency 3 (Rpd3) deacetylase. They are usually localised in the nucleus and form large multiprotein complexes which confer to these enzymes strict specificity for particular acetylation sites [25]. Class I HDACs can be further divided into HDAC1/HDAC2 and HDAC3 subclasses. Class II members (HDAC 4, 5, 6, 7, 9, and 10) are homologous to the yeast Hda1 deacetylase and can be further subdivided into class IIa (HDAC4, 5, 7 and 9) and IIb (HDAC6 and 10) [26, 27]. Class II HDACs are localized in both the nucleus and the cytoplasm to target histone and nonhistone proteins. HDAC11-related enzymes are considered to form a separate type of HDACs the class IV [28].
The class III HDACs or sirtuins consists of seven members (Sirt1–7) homologous to the yeast HDAC silent information regulator 2 (Sir2) (Figure 1). The common characteristic of this class is that they are nicotinamide adenine dinucleotide (NAD+-) dependent enzymes [29, 30]. The requirement of the NAD+ cofactor and the mitochondrial localisation of some sirtuin family members imply a role of this class of deacetylases in the regulation of the metabolic homeostasis and suggest that histones are not their primary targets. Sirtuins show significant sequence and functional differences from other classes of HDACs in that they carry out deacetylation via a two-step reaction that consumes NAD+ and releases nicotinamide (NAM), O-acetyl-ADP-ribose (AADPR), and the deacetylated substrate [31]. Sirtuins, although relatively similar to each other have divergent biological functions due to distinct cell-type-specific subcellular localisation of each member of the family [31]. In particular, Sirt1 is located in both the nucleus and the cytoplasm, Sirt2 in the cytoplasm, Sirt3, 4, and 5 are mitochondrial, and Sirt6 and 7 are exclusively nuclear [32, 33]. Apart from intracellular localization, Sirt1, 3, and 5 differ from Sirt2, 4, and 6 in the type of reaction they catalyse. Sirt1, 3, and 5 are NAD+-dependent deacetylases catalyzing the deacetylation of histones and nonhistone proteins, whereas Sirt6 is a NAD+-dependent ADP ribosyltransferase (ART) mediating mitochondrial protein ribosylation; Sirt2 and 4 exert both NAD+-dependent HDAC and ART activities [34]. Although the enzymatic activity of Sirt7 as well as its specific substrates have not yet been determined, it has been shown that it resides in the nucleoli and regulates the RNA polymerase I (Pol I) transcriptional machinery [35].
Figure 1.

Schematic representation of human sirtuins family members 1–7, NAD-dependent catalytic domain (gold) (NAD-binding pocket), zinc-binding domain (black), and their intracellular localization.
The uniqueness of sirtuins is that their function as transcriptional regulators is directly linked to intracellular energetics. Accumulating evidence indicates that sirtuins participate in the coordination of several apparently disparate cellular functions such as cell cycle, response to DNA damage, metabolism, apoptosis, and autophagy [29]. These observations suggest that detailed characterization of the function of these enzymes under diverse cellular stress conditions will offer useful information towards designing novel compounds for therapeutic intervention in a wide range of apparently unrelated diseases including diabetes, neurodegenerative disorders, respiratory and cardiovascular diseases, and cancer. This paper will focus on Sirt1 since its physiological role has been more extensively studied, and the evidence regarding the activity of other sirtuins family members is still scarce.
2. Molecular Mechanisms Regulating Sirtuins Activity
Several studies have indicated the ability of the histone deacetylase III family members to deacetylate a wide range of substrates, thus implicating these enzymes in a broad spectrum of biological functions. Hence, considering the molecular circuits regulating sirtuins cellular levels (Figure 2) could provide the basic knowledge towards developing the means to control their accumulation for therapeutic benefit.
Figure 2.
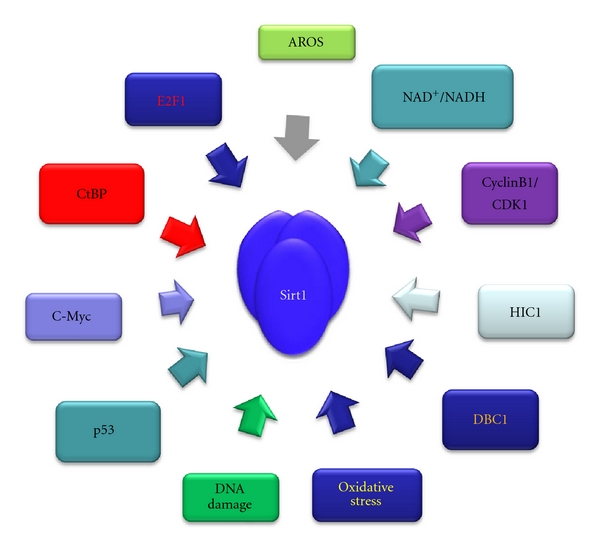
Factors involved in the regulation of Sirt1 gene expression and enzymatic activity.
3. Enzyme Abundance
Sirtuins gene expression has been shown to be under the control of numerous transcription factors involved in the cell cycle regulation and apoptosis. Among them the oxidative stress and DNA damage responsive transcription factor E2F1, which induces cell cycle progression from G1 to S phase, directly binds to the Sirt1 promoter upregulating its gene expression in cells treated with the topoisomerase II inhibitor etoposide [36]. E2F1 phosphorylation by the stress-responsive kinase ataxia telangiectasia mutated (ATM) appears to be a prerequisite for E2F1-mediated regulation of Sirt1 gene expression [36]. In turn, the deacetylation function of Sirt1 inhibits E2F1's transcriptional activity [36].
The tumor suppressor p53, which is one of the most extensively mutated proteins in cancers, is a stress-responsive transcription factor that has also been shown to affect Sirt1 gene expression. Two functional p53-binding sites have been identified in the regulatory region of the Sirt1 promoter, and a complex regulatory network has been described to elucidate the modulation of Sirt1 gene expression in mammalian starved cells [37]. In particular, the activated in nutrient-deprived mammalian cells, forkhead box O transcription factor FOXO3a forms a complex with p53, which is recruited to the two p53-binding sites present within the Sirt1 promoter, thus stimulating Sirt1 gene expression [37]. On the contrary, in normal nutrient conditions, p53 mediates repression of Sirt1 gene expression [37], which is a result of functional cooperation between p53 and the epigenetically regulated repressor hypermethylated in cancer 1 (HIC1) [38]. Thus, transcriptional activity and tumor suppressor functions exerted by p53 are indirectly regulated by HIC1 mediated repression of Sirt1 gene expression [39]. Furthermore, c-Myc upregulates Sirt1 gene expression, and in turn Sirt1-mediated c-Myc deacetylation leads to c-Myc protein degradation [40–42].
Another recently reported pathway regulating Sirt1 gene expression in response to acute metabolic changes involves the fine tuning of the association between the redox sensor carboxy terminal of E1A-binding protein (CtBP) and HIC1 [43]. The transcriptional repression activity of CtBP depends on NADH levels, and in particular high NADH levels promote CtBP dimerization as well as its interaction with other transcriptional repressors such as HIC1 [44–47]. Cellular redox changes sensed by CtBP alter the affinity of the CtBP for HIC1 leading to a reduction of CtBP recruitment to Sirt1 promoter and hence derepression of its gene expression [43].
Sirt1 cellular levels are regulated by both p53 and E2F1 not only at the transcriptional but at the translational level as well. MicroRNA 34a (miR-34a) and miR449a, which are the p53 and E2F1 transcriptional targets, respectively, have been shown to inhibit Sirt1 expression [48, 49] resulting in p53 acetylation and induction of p53-dependent apoptosis. In addition miR199a knockdown during normoxia has been shown to stabilize HIF-1α and Sirt1, whereas miR199a overexpression downregulates prolyl hydroxylase 2 (PHD2) implying that miR199a regulates HIF-1α levels by moderating Sirt1 and hence PHD2 activities [50]. Sirt1 cellular levels are also regulated by the RNA-binding protein HuR, which associates with Sirt1 mRNA leading to increased Sirt1 mRNA stability and, thus elevated Sirt1 protein levels [51].
Regarding other members of the sirtuins family, an estrogen-related receptor (ERRα) responsive element (ERRE) has been mapped within the mouse Sirt3 promoter region, and colocalization of ERRα and peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) has been confirmed in the Sirt3 promoter with chromatin immunoprecipitation assay [52].
4. Catalytic Activity
Sirtuins enzymatic activity is regulated by posttranslational modifications. In vitro evidence indicates that dephosphorylation at specific sites targeted by cyclinB/Cdk1 in a cell-cycle-dependent manner reduces its deacetylase activity [53, 54]. Sirt1 is also phosphorylated by the c-Jun N-terminal kinase 2 (JNK2) [55] and casein kinase 2 (CK2) [56]. JNK2-mediated phosphorylation of Sirt1 is associated with the regulation of its protein stability [55]. Both Sirt1 and CK2 are key regulators of similar biological functions including chromatin remodeling, cell cycle progression, and survival or apoptosis [53]. Multiple conserved phosphorylation sites have been identified within Sirt1 that are potential targets for a variety of kinases such as ATM, casein kinase 1 (CK1), DNA-dependent protein kinase (DNA-PK), extracellular-signal-regulated kinase (ERK1), glycogen synthase kinase 3 (GSK3), IκB kinase (IKK), and mitogen-activated protein kinase (MAPK) [54]. Whether these kinases phosphorylate only Sirt1, other members of the sirtuin class, or Sirt1 phosphorylation by one of these kinases affects other posttranslational modification events, and Sirt1 substrate selectivity is not known. Recently two members of the dual-specificity tyrosine phosphorylation-regulated kinases (DYRK) DYRK1A and DYRK3, which play important role in body growth and brain physiology, have been demonstrated to promote cell survival by phosphorylating Sirt1 and inducing its deacetylase activity [57].
Like Sirt1, Sirt2 is phosphorylated by Cdk1 indicating that this sirtuin family member is also involved in the control of cell cycle. Additional evidence for that is provided by the fact that cyclinE/Cdk2 complex phosphorylates Sirt2 inhibiting its catalytic activity. Since cyclinE/Cdk2 is involved in the regulation of the cell cycle progression from G2 to M phase, inhibition of Sirt2 deacetylase activity mediated by cyclinE/Cdk2 might be a requirement for the cell cycle progression from G2 to M phase [58]. On the other side, overexpression of the CDC14A and CDC14B phosphatases, which are required for efficient DNA repair, inhibits Sirt2 protein degradation and interferes with adhesion and cell migration [59].
The active regulator of Sirt1 (AROS) is a 142 amino acid protein localized in the nucleus that interacts with Sirt1 and activates its deacetylase function [60]. Although the molecular mechanism of AROS-mediated activation of Sirt1 has not been defined, AROS possibly displaces a Sirt1 inhibitor from the deacetylase complex such as the deleted in breast cancer 1 (DBC1) inhibitor, or it recruits another cellular factor which induces a conformational change that activates Sirt1 enzymatic activity [61–63]. It has been shown that the binding affinity between Sirt1 and DBC1 is critical for the determination of cancer cell survival or death [64].
As is the case for other classes of deacetylases, acetylation is another posttranslational modification affecting the activity of various sirtuin members. For example, p300 acetylates Sirt2 and attenuates its deacetylase activity [65]. The molecular mechanism of p300-mediated inactivation of Sirt2 is not clear, but acetylated Sirt2 might acquire different conformation that alters its interaction pattern or affinity with other proteins which facilitate its association with proteins such as the 14-3-3 β/γ. Interaction of Sirt2 with 14-3-3 β/γ might influence its subcellular localization, thereby changing its activity [65].
Sumoylation of Sirt1 has been demonstrated to activate its deacetylase activity and occurs in the absence of DNA damage [66]. Exposure of cells to diverse types of stress conditions such as UV irradiation or hydrogen peroxide results in Sirt1 desumoylation mediated by the desumoylase sentrin-specific protease 1 (SENP1) and inactivation of its deacetylation function. As a consequence the proapoptotic Sirt1 substrates such as p53 are acetylated and hence active and capable to induce cell death [61, 67].
5. Availability of Metabolic Cofactors
The availability of NAD+ in cells is a limiting step in the activation of sirtuins catalytic activity since these enzymes require NAD+ as a cofactor to exert their function [68]. The basal intracellular NAD levels are maintained relatively constant [69] by the NAD biosynthetic and salvage pathways [70]. The precursor of the biosynthetic pathway of NAD synthesis is tryptophan and nicotinic acid (NA) or nicotinamide (NAM) the precursors of the salvage pathway [71, 72]. Human cells produce NAD+ by converting NAM in a two-step reaction catalysed by nicotinamide phosphoribosyltransferase (Nampt) [73]. The first step involves the conversion of NAM to nicotinamide mononucleotide (NMN) by Nampt. NMN is subsequently utilized by nicotinamide/nicotinic acid mononucleotide adenylyltransferase (Nmnat 1, -2, and -3) to regenerate NAD+ [74]. The molecular mechanism of nicotinamide-mediated inhibition of the sirtuins deacetylase activity has been elucidated in recent reports [75, 76]. Deficiency of the NAD+ synthesizing pathways abolishes sirtuins-mediated deacetylation [74] whereas increased NAD+ levels induce their enzymatic function [77]; therefore, by consuming NAD+ in order to exert their effects, sirtuins regulate the fluctuation of the NAD+/NADH ratio, thereby sensing cellular NAD+ concentration and redox status. For more detailed review of the relation between sirtuins NAD+/NADH ratio and oxidative stress see references [77–79].
To summarize, glucose deprivation and metabolic changes associated with calorie restriction alter the NAD+/NADH ratio [80–83]. Since sirtuins associate with chromatin and their function is NAD+-dependent, these enzymes couple changes of the metabolic flux and NAD+ levels with transcription [81].
6. Transcription Factors Associated with Sirtuins
Crucial cellular pathways involved in cell growth, differentiation, stress resistance, migration, and metabolism are modulated by the function of transcription factors whose activity is regulated by sirtuins including p53 [84–86], FOXO proteins [87], peroxisome proliferation-activating receptor-(PPAR-) gamma coactivator-1α (PGC-1α) [88], and nuclear factor-κB (NF-κB) (Figure 3 and Table 1) [34, 89, 90].
Figure 3.
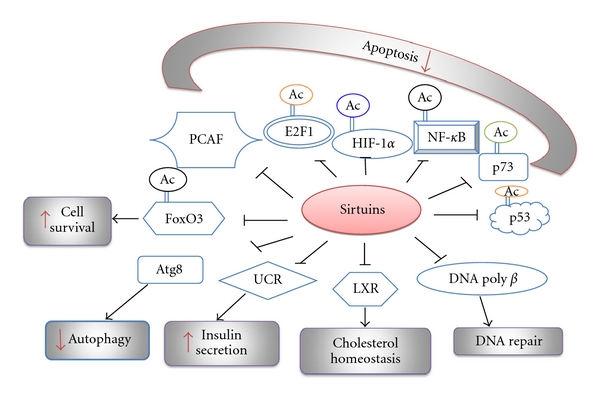
Sirtuins regulate the activity of numerous transcriptional regulators indirectly affecting the outcome of several cellular functions.
Table 1.
Transcription factors associated with sirtuins.
| Sirtuin class | Substrate | Position | Function | Reference |
|---|---|---|---|---|
| K120 | Induction of cell cycle arrest | [100] | ||
| p53 | K372 | Unknown | [86] | |
| K382 | Reduction of apoptosis | [86, 101] | ||
| SIRT1 | HIF-1α | K674 | Negative effect on tumor growth and angiogenesis | [95] |
| FOXO1 | K242, K245 and K262 Not known |
Transcriptional activation Inhibition of FOXO1 activity |
[102] [103] |
|
| FOXO3a | Not known | Induction of cell cycle arrest and resistance to oxidative stress; inhibition of FOXO-mediated induction of apoptosis; inhibition of FOXO transcriptional activity | [104, 105] | |
| E2F1 | Not known | Inhibition of E2F1 transcriptional activity; inhibition of E2F1-mediated apoptosis | [36] | |
| NF-κB | K310 of RelA/p65 subunit | Inhibition of NF-κB transcriptional activity and prevention of the release of proinflammatory mediators | [89] | |
| Sir2α | p53 | Not known | Attenuation of p53-mediated transcriptional activity Inhibition of p53-dependent apoptosis in response to DNA damage | [106] |
| SIRT2 | FOXO3a | Not known | DNA binding and activation of target genes | [43, 107] |
| SIRT3 | AceCS2 | K642 | Activation of the acetyl-CoA synthetase activity of AceCS2 | [108, 109] |
| SIRT5 | PGC-1α | Not known | Unknown | [110] |
| SIRT6 | HIF-1α | Not known | Regulation of glucose homeostasis. Reduction of glycolysis and increase of mitochondrial respiration | [98] |
| SIRT6 | NF-κB | Not known | Reduction of NF-κB-mediated apoptosis and senescence | [99] |
Several lines of evidence converge to the conclusion that sirtuins are integrated in the p53 pathway, and their function depends on the cellular status of p53 [84–86]. Sirtuins and p53 interact at various levels to induce cell cycle progression, senescence, or apoptosis [85] (Figure 3). The interplay between HIC1 and p53 regulates Sirt1 gene expression, and reduction or ablation of HIC1 can lead to tumorigenesis through Sirt1-mediated deacetylation and transcriptional inactivation of p53 [84, 85]. Sirt7 has also been shown to interact with p53 in the mice myocardium [91]. Apart from the acetylation levels, sirtuins regulate the subcellular localization of p53, thus determining the cellular fate under oxidative stress conditions [92].
Acetylation of HIF-1α by arrest-defective protein 1 (ARD1) [93] and p300/CBP-associated factor (PCAF) [94] plays important role in the regulation of the protein stability and transcription target selectivity [94] of this transcription factor. Sirt1 deacetylates and represses HIF-1α transcriptional activity [95] whereas Sirt1-mediated deacetylation of HIF-2α induces its signaling in hypoxic conditions [96] suggesting that sirtuins signaling promotes the distinctive function of HIF-1α and HIF-2α [94, 96, 97]. Given the fact that HIF-1α and HIF-2α play crucial roles in the cellular adaptation to metabolic stress by regulating the expression of several genes involved in glucose metabolism, it is possible that the extent of their acetylation determines the pathway of cellular energy production and redox balance depending on the type of tissue and environmental stress [90, 94–96, 98]. In accord with this perception, Sirt6 is recruited by HIF-1α to histone 3 and deacetylates H3 lysine 9 within the promoter regions of several glycolytic genes repressing their gene expression, thereby regulating glucose homeostasis [98, 99].
The mammalian redox responsive FOXO transcription factors provide another example of the role of sirtuins in the determination of the cellular fate under oxidative stress conditions [104, 111]. Sirtuins target FOXO transcription factors under conditions of oxidative stress and determine their subcellular localisation, protein stability, and transcriptional activity [104, 111]. FOXO are involved in the cell cycle arrest at the G1-S and G2-M checkpoints [112], in scavenging reactive oxygen species (ROS) [113], and in the induction of the expression of genes involved in the DNA damage response, differentiation, glucose metabolism, and apoptosis [112, 114]. Sirt1-mediated deacetylation of FOXO3 and FOXO4 under stress induces cell cycle arrest instead of apoptosis [105]. Sirt2 and Sirt3 have also been shown to associate with FOXO transcription factors modulating their transcriptional activity and subcellular localization [107, 115–119]. Therefore, sirtuins by controlling the function of FOXO transcription factors indirectly exert a pivotal role in the regulation of multiple cellular processes [104] (Figure 3 and Table 1).
The E2F family of transcription factors is involved in the control of cell cycle progression, DNA damage response, and induction of apoptosis [120]. The transition from G1 to S phase checkpoint involves tight regulation of the E2F transcriptional activity by the pocket proteins retinoblastoma (pRb) tumor suppressor, p107 and p130. Acetylation of E2F1 by PCAF facilitates the binding of this transcription factor to its conserved DNA responsive elements and activation of gene expression of its targets, including several proapoptotic factors such as Apaf-1, Bim, caspase 7, and p73 [121]. Sirt1 binds and deacetylates pRb engaging the pRb tumor suppressor pathway in the oxidative stress-response [122–124].
Various subunits of the NF-κB family of transcription factors are acetylated at multiple sites, affecting the DNA-binding and transcriptional activity of these proteins, thus modulating the release of proinflammatory mediators [125]. The p65/RelA subunit physically interacts and is deacetylated by Sirt1 resulting in the inhibition of the NF-κB-mediated transcription [89, 90, 126, 127]. Deacetylation of NF-κB and inhibition of its transcriptional activity by Sirt1 and Sirt6 protects pancreatic β cells from NF-κB inflammatory response and preserves insulin secretion [99, 128].
7. Cellular Response to Stress
Sirtuins substrates are involved in the coordination of cellular responses to diverse stresses including inflammation, hypoxic stress, and heat shock, thereby regulating cell survival or death, differentiation, and endocrine signaling. In particular, sirtuins regulate the transcriptional activity of NF-κB, p53, HIF-1α, HIF-2α, FOXOs, E2F1, and heat shock factor protein1 (HSF1), which are involved in the regulation of aging and aging-related diseases.
8. DNA Damage
In order to protect themselves from the high incidence of damage that could lead to mutations, genomic instability, or cell death and the different types of DNA damage that might occur, eukaryotic cells have developed appropriate mechanisms to detect and repair their damaged DNA. Efficient repair of the damage requires that the DNA repair machinery circumvents the barrier formed by the histone and nonhistone proteins that package DNA into chromatin and accesses the damaged site in a timely manner. In this respect, chromatin-remodeling and histone-modifying enzymes are crucial for the ability of eukaryotic cells to detect and repair DNA breaks. Consistent with this notion, several reports have indicated that acetylases and deacetylases are recruited in the vicinity of DNA breaks [129].
Direct indication for the involvement of sirtuins in DNA damage response has been revealed in Sirt1−/− and Sirt6−/− mice which exhibit increased radiation sensitivity, chromosomal aberrations, and impaired DNA repair [130, 131].The involvement of sirtuins in the DNA damage response was initially suggested by observations demonstrating increased chromatin recruitment of Sirt1 to sites of DNA-DSB in mammalian cells upon diverse types of DNA damage including oxidative stress in a manner involving stress-responsive kinases such as ATM [132–134]. Upon DNA damage, Sirt1 deacetylates the Nijmegen breakage syndrome (NBS1) which is a DNA-DSB sensor and repair protein, thus facilitating the recruitment of other required factors to the sites of damaged DNA and optimal repair through homologous recombination (HR) and nonhomologous end joining (NHEJ) [135]. Sirt1 is also required for the optimal function of the nucleotide excision repair (NER) pathway. In particular, it has been shown that Sirt1 impairs NER by suppressing the xeroderma pigmentosum C (XPC) gene expression which is essential for the recognition of DNA lesions and NER initiation [136]. Overexpression of Sirt1 represses proteins with DNA damage repair functions such as various FOXO family members [105], Ku70 [137], p73 [138], pRb [122], and Werner helicase (WRN) [139]. Further evidence for the involvement of Sirt1 in the DNA damage response is provided by the fact that it is transcriptionally upregulated by breast cancer 1 early onset (BRCA1), which binds to DNA-DSB and plays a significant role in DNA repair and the maintenance of genomic stability [140]. Overall, Sirt1 is involved in DNA damage response by modulating the expression of genes involved in DNA repair and by recruiting to sites of DSBs factors participating in the processing of DNA damage.
Sirt6−/− mice display sensitivities associated with deficiencies in base excision repair (BER) such as genomic instability and enhanced sensitivity to ionizing radiation and DNA damaging agents [131]. The detailed molecular mechanisms by which Sirt6 regulates DNA damage repair has been suggested to involve the function of the DNA-PKcs which is a kinase that takes part in the NHEJ [141]. Sirt6-mediated deacetylation of the H3K9 at sites surrounding DSBs allows DNA-PKcs or other repair factors to access the DNA lesions [131]. Sirt6-dependent deacetylation of the C-terminal-binding protein (CtBP) -interacting protein (CtIP) which promotes DNA end resection and is required for efficient homologous recombination is another proposed mechanism for SIRT6-dependent processing of DNA damage repair [134, 142]. Consistently with the role of other chromatin-modifying enzymes, Sirt6 in response to DNA damage is recruited to DNA breaks either genome-wide or locally contributing directly to DNA damage repair or indirectly by permitting access to the DNA lesions to the DNA damage repair machinery.
Further research is required to characterise the molecular networks linking transcription and chromatin modifications to DNA damage response and repair as well as to elucidate the role of other sirtuin family members in these processes. It will also be interesting to determine whether different sirtuin family members are involved in the same or diverse DNA damage and repair pathways and whether they function in concert or exert antagonistic effects.
9. The NAD+/NADH Ratio
During glycolysis and citric acid cycle, energy from nutrients is transferred to NAD+ which is reduced to NADH. NADH is then oxidized back to NAD+ by transferring its reducing electrons to electron acceptors and ultimately to oxygen, and the energy released during this process is coupled to ATP generation through oxidative phosphorylation (OXPHOS) [77]. Increasing evidence suggests that marked alterations in the NAD+/NADH ratio may have detrimental effects on the cellular fate; therefore, NADH levels are very tightly regulated in cells [143]. High cytosolic NADH levels result in reactive oxygen species (ROS) generation and oxidative damage by several mechanisms including providing substrates for NAD(P)H oxidase and release of iron from ferritin. Furthermore, high levels of cytosolic NADH can either promote OXPHOS by increasing the mitochondrial NADH levels or inhibit OXPHOS by promoting pyruvate to lactate conversion and reducing the permeability of the voltage-dependent anion channel (VDAC) in the mitochondrial outer membrane [143]. Since the effects of altered cytosolic NADH levels on cell injury are complicated, further studies are required to resolve this issue.
Sirtuins consume NAD+ in order to exert their enzymatic functions, and by doing so they alter the NAD+/NADH ratio, thereby modifying the redox status within the cells and hence serving as cellular sensors of reduced glucose and NAD+ availability [23, 67, 74, 75]. Changes in energy or increase of the NAD+/NADH ratio enhances sirtuins activity and protein deacetylation implying that there is a close association between the cellular redox status and the acetylation levels within the cells [68]. Indeed, Sirt1 has been shown to determine survival or apoptosis of renal tubular cells by fine tuning the ROS scavenger catalase gene expression in the presence or absence of intracellular ROS [144]. In particular, in the presence of high levels of intracellular ROS, Sirt1 induces FOXO3a-mediated upregulation of catalase gene expression leading to reduction of oxygen consumption and ROS levels leading to cell survival [144–146]. Reduction of ROS levels by Sirt1 is the mechanism underlying the development of resistance to oxidative stress which is associated with aging and diseases such as type 2 diabetes mellitus [145]. Sirt2-mediated deacetylation of FOXO [107, 116] and Sirt3-dependent decrease of ROS production in brown adipocytes [147] as well as Sirt3 involvement in antioxidants production and NADPH regeneration [148] suggest that several sirtuin family members play important role during oxidative stress. More work is needed to characterise the detailed mechanisms by which sirtuins contribute to the control of cellular redox levels, which potentially will lead to the development of novel therapeutic approaches for the treatment of diseases typified by high-inflammatory states.
10. Cell Cycle
The involvement of sirtuins in the sensitisation and repair of DNA damage as well as the regulation of the cellular redox state implies that these enzymes are involved in the control of the cell cycle in order to provide the necessary time for the cells to repair their DNA damage under conditions of oxidative stress. Multiple transcription factors playing critical role in the control of cellular proliferation and apoptosis have been identified as sirtuin substrates (Table 1); therefore, changes in sirtuins cellular levels affect the ability of cells to divide. Sirtuins exert both positive and negative effects on cell growth promoting and inhibiting cellular proliferation. Many studies converge to the conclusion that inhibition of sirtuins is beneficial in cancer treatment, which is consistent with the negative effects they mediate mainly on the tumor suppressor p53 preventing senescence and programmed cell death [149]. Furthermore, increased expression of sirtuins in cancer cells coincides with pRb hyperphosphorylation and p16INK4A downregulation [150, 151].
Cell cycle regulation through Sirt1 is carried out by the deacetylation of three members of the FOXO family of transcription factors, namely FOXO1, FOXO3a, and FOXO4 [104, 152]. Deacetylation of FOXO alters their interaction pattern with E2F1 or p53, leading to cell cycle arrest or apoptosis in a manner dependent on environmental conditions, tissue characteristics, and cellular metabolic state [152]. In response to oxidative stress, Sirt1 deacetylates FOXO3a increasing its ability to cause cell cycle arrest and preventing it from inducing apoptosis [104]. Cell cycle arrest is also the result of Sirt1-mediated deacetylation of FOXO1 and FOXO4 which are activated by deacetylation and induce gene expression of the p27Kip1 cyclin/cdk inhibitor. In addition, deacetylation of these transcription factors induces gene expression of the antioxidant manganese superoxide dismutase (MnSOD) and the DNA repair gene growth arrest and DNA damage inducible (GADD45), thereby enhancing the cellular defence to oxidative stress [104, 152]. Taken together, these observations indicate that in response to oxidative stress, Sirt1 selectively targets FOXO transcription factors to their prosurvival subset of transcriptional target genes, thereby inducing cell cycle arrest and resistance to oxidative damage. Apart from FOXO, Sirt1 inhibits cellular proliferation by destabilising c-Myc [40, 153].
The role of Sirt2 in cell cycle regulation has been elucidated in cells overexpressing Sirt2 where it acts as a G2/M checkpoint regulator preventing chromosomal instability [53, 59, 154]. Sirt2 has also been shown to deacetylate tubulin and cause cell cycle arrest prior to entry into mitosis in response to microtubule inhibitors such as nocodazole [155].
Studies in Sirt3 null mouse embryonic fibroblasts have indicated that the main function of this sirtuin family member is the regulation of the mitochondrial ROS levels [156]. Sirt3 is normally localised inside the mitochondria but in response to cellular stress, it can be detected in the nucleus where it relocates in the presence of overexpressed Sirt5 [157]. In addition, Sirt3 and Sirt4 have recently been shown to exert antiapoptotic effects in response to DNA damage when the levels of NAD+ are extremely low [158].
Sirt6 is a key component of the base excision repair, but whether it delays cell cycle progression to allow time for DNA repair to occur, it is not known [131]. Sirt6 has been shown to associate with telomere maintenance and its depletion results in abnormal telomere structures, end-to-end chromosomal fusions, and premature senescence [159].
Sirt7 is localised in the nucleoli and is involved in the regulation of the RNA polymerase I and the transcription of the ribosomal gene (rDNA). During mitosis, Sirt7 is phosphorylated and retains its nucleolar localisation until telophase when it is dephosphorylated and activated to resume rDNA transcription [160].
In summary, the detailed role of all sirtuin family members in the regulation of the cell cycle requires further investigation in order to understand the link between cell cycle control and their ability to regulate transcription and acquire diverse subcellular localisation during different cell cycle phases. Moreover, it is intriguing to investigate whether each member of the sirtuins family exerts its effect on cell cycle individually or in combination with other sirtuin family members or other cell cycle regulators.
11. Apoptosis
Sirt1 interacts, deacetylates, and thereby negatively regulates the transactivation function of various key transcription factors playing central role in the determination of the cellular fate (apoptosis/survival) such as p53 [85], E2F1 [36], members of the FOXO family of transcription factors [111], NF-κB [89], HIF-1α [95] and the DNA-PKcs subunit, and DNA damage end-joining protein Ku70 [161] (Table 1). Sirt1 deacetylates several sites of the tumor suppressor p53 including K320, K372, and K382, which are selectively acetylated by PCAF and p300 under diverse stress conditions, thus antagonising senescence or inducing either cell cycle arrest or apoptosis [94]. Hyperacetylation of p53 enhances its transactivation function consequently leading to the increase of the gene expression of its proapoptotic targets. Sirt1-dependent deacetylation of p53 reduces the ability of p53 to induce gene expression of its proapoptotic targets, thereby suppressing apoptosis in response to oxidative stress and DNA damage ([85] and Figure 4). Besides the control of p53 activity, Sirt1 regulates the proapoptotic function of the proapoptotic p53 target Bax by retaining it in the cytoplasm in complex with the hypoacetylated form of Ku70 and preventing it from translocating to the outer mitochondrial membrane to induce apoptosis [162, 163]. Upon cellular stress conditions such as UV irradiation, Ku70 is acetylated by PCAF and dissociates from Bax which is now able to induce cell death [163].
Figure 4.
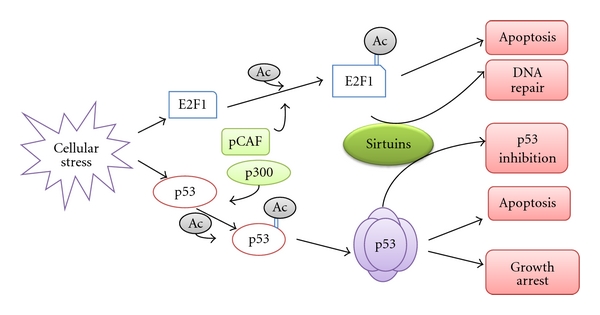
Role of sirtuins in the celluar response to stress.
Similarly to p53, the activity of FOXO transcription factors is regulated by acetylation and deacetylation modifications. Sirt1 inhibits the ability of FOXO3a to promote apoptosis by blocking gene expression of the proapoptotic Bim, Puma, and TRAIL which are FOXO3a transcription targets in a cellular context manner [161, 164]. Furthermore, Sirt1 regulates cellular apoptotic responses through the E2F1/p73 pathway [121], and it is important in controlling cell death in hypoxic and heat shock conditions by increasing the activity of HIF-2α [96] and heat shock factor 1 (HSF1) [165]. Cleavage of Sirt1 by caspase is another possible pathway through which this deacetylase induces apoptosis, but the necessity of this cleavage for the initiation of apoptosis has not been confirmed [166, 167].
It appears that Sirt1 has both negative and positive effects on apoptosis since by deacetylating p53 and FOXO transcription factors, it suppresses apoptosis [168] whereas in response to TNFα signaling by deacetylating RelA/p65, it inhibits the transcriptional activity of the antiapoptotic transcription factor NF-κB [61, 89].
The role of Sirt3 in apoptosis appears to be more complicated since this sirtuin family member has been found to cause cell death in several nonstressed human cancer cell lines in which Bcl-2 had been silenced [169] as well as in myeloid leukemia cell lines treated with the natural flavonoid kaempferol [170], whereas both Sirt3 and Sirt4 have been shown to be required for the maintenance of the mitochondrial NAD+ in genotoxic stress conditions and hence cell survival [109, 158].
It has recently been demonstrated that Sirt5 interacts with cytochrome c in the mitochondrial membrane [148, 171] but the functional consequences of the Sirt5-dependent deacetylation of cytochrome c and in particular whether this modification has any impact on apoptosis have not yet been elucidated.
Sirt7 depletion by RNAi led to inhibition of cell growth and induction of apoptosis in U2OS cells [35] and primary cardiomyocytes [91] postulating that this sirtuin family member is also essential for cell survival [35, 160] although its effects are probably tissue-type specific.
Taken together the aforementioned observations imply that the design of sirtuin inhibitors to promote apoptosis should take into consideration whether the targets deacetylated by sirtuins are deacetylated by members of other HDAC families, that sirtuins might have both positive and negative effects on apoptosis, and that sirtuins do not enhance to the same extent the transcriptional activity of all the genes induced by the same transcription factors, and it might not be sufficient to block their catalytic function to achieve induction of cell death.
12. Autophagy
Autophagy is a cytoprotective process by which eukaryotic cells degrade damaged or dysfunctional organelles and proteins with long half-life. The degradation takes place in the lysosome where the cytoplasmic contents to be degraded are delivered enclosed in double-walled membrane vesicles called autophagosomes which originate from the endoplasmic reticulum [172–174]. The fact that the efficiency of the autophagic degradation declines during aging and the efficient maintenance of autophagy leads to lifespan extension attracted the interest of several research laboratories to investigate the signaling pathways regulating autophagy with the aim to understand the factors involved in the lifetime determination and the causes of age-related degenerative diseases. Recent research efforts have shed light on the biochemical autophagic machinery and the crucial role of several genes called autophagy-related (Atg) genes in the execution of this process [175–177].
Sirtuins are important longevity factors as well; therefore, it was hypothesised that lifespan might be determined by interacting signaling network pathways regulating sirtuins functions and autophagy. Support to this hypothesis was lent by the observation that Sirt1−/− and Atg5−/− deficient mice exhibit partially similar phenotypes as well as that Sirt1−/− mouse embryonic fibroblasts under starved conditions are unable to activate autophagy [178]. In addition, transiently expressed wild type Sirt1 but not its inactive deacetylase mutant could stimulate basal levels of autophagy [178]. The molecular mechanisms entailed in Sirt1-mediated regulation of autophagy have not yet been elucidated, but it has been proposed that Sirt1 induces autophagosome formation by associating and deacetylating the Atg5, Atg7, and Atg8 components of the autophagic machinery in a NAD+-dependent manner, thus facilitating the assembly of Atg complexes [172, 178–183].
Apart from its direct effects on the components of the autophagic apparatus, Sirt1 associates with well-characterized mediators of autophagy and lifespan such as mTOR (target of rapamycin) [184] FOXO transcription factors [111, 183] p53 [185] and E2F1 [186]. The molecular mechanisms by which autophagy is negatively regulated by mTOR are not well established, nevertheless it has been reported that mTOR inhibition or Sirt1 activation prolong, lifespan [173, 187]. Sirt1 and FOXO3 interact in response to oxidative stress, and Sirt1 deacetylates and activates FOXO3 which in turn induces the expression of many autophagy-related genes stimulating autophagy and cellular stress resistance [111]. Several signaling pathways link autophagy and the tumor suppressor p53, which has been shown to exert both negative and positive effects on this process [188, 189]. It appears that only cytoplasmic p53 can induce autophagy in a manner involving the E3 ligase activity of HDM2 [189]. It is not known whether Sirt1 exerts its effects on autophagy through p53 by affecting the tumor suppressor's subcellular localisation or protein stability [190]. E2F1 is a transcriptional regulator of autophagy as it upregulates the expression of several ATGs as well as the damage-regulated autophagy modulator (DRAM) [186]. Since Sirt1 modulates E2F1's transcriptional activity [36, 121], it is possible that Sirt1-determined acetylation status of E2F1 mediates its effects on autophagy.
There are not many reports in the literature that address the role of the other sirtuin family members in autophagy apart from Sirt2 that has been shown to deacetylate FOXO1 and dissociate its complex with Atg7 leading to apoptosis [191]. Other open questions in relation to the regulation of autophagy by sirtuins are whether increasing autophagy by activating sirtuins would have beneficial effects in terms of longevity, or it would lead to side effects by affecting other vital cellular functions in which sirtuins have been implicated.
13. Sirtuins in Pathology
Sirtuins are key components of a broad range of biological processes, which are directly or indirectly linked to aging. Sirtuins can regulate the aging process partly through their ability to connect the nutritional status of the cell to chromatin modifications and regulation of gene expression [192, 193]. Their role as longevity mediators is due to their activity as modulators of several calorie restriction (CR) pathways [192]. CR extends life span by shifting the glucose metabolism toward respiration [68] followed by possible alterations of the NAD+/NADH ratio, modulation of the sirtuins deacetylase or ADP ribosyl-transferase activity [81], and slow rate of ROS generation which correlates with increased longevity [194]. Aging process and several age-related diseases such as diabetes [128, 195], cardiovascular [196], neurodegenerative [197], respiratory [198], and autoimmune diseases [12], and cancer [22, 29, 34, 69, 199] on the other side are accompanied by elevated redox cellular content or low-grade chronic, proinflammatory stress [32]. In view of the fact that sirtuins are involved in the regulation of the aging process and in sensing oxidative stress, it has been suggested that sirtuins could play a vital role in the development of a variety of aging diseases and their function could be targeted for therapeutic benefit in these diseases. Here the role of the most extensively studied member of the sirtuin family, Sirt1, in tumorigenesis and as a target for the development of anticancer therapeutics will be discussed.
14. Sirt1 in Oncogenesis
The role of Sirt1 in tumorigenesis appears to be complex as this protein has been associated with oncogenic and tumor suppressor function, and both elevated and decreased levels have been detected in different types of cancer suggesting that Sirt1 functions in tumorigenesis are tissue-type and context specific [149, 200, 201]. For example, increased Sirt1 levels have been identified in acute myeloid leukaemia (AML) [202] and colon cancer where Sirt1 might promote proliferation and survival [203] or cell growth inhibition [204]. Both increased and reduced levels of Sirt1 have been detected in prostate cancer [130, 205] and decreased levels in ovarian and glioma cancers [37, 130] which is possibly due to defects in the activity of tumor suppressor genes that regulate its gene expression [38].
Sirt1 determines changes in gene transcription and most of its effects in oncogenesis by deacetylating and thus regulating the function of a large number of tumor suppressors or oncogenes (Table 1) such as p53 [86, 106], FOXO transcription factors [104, 105], p73 [206], pRb [122], PML [101], Ku70 [163], hTERT [207] MyoD [208], NF-κB [209], and BCL6 [210].
Sirt1 is a direct effector of p53 transcriptional activity regulating the gene expression of p53 targets involved in cell cycle arrest (CDKN1A encoding p21WAF-1/CIP-1) and apoptosis (Bax) under DNA damage conditions as well as p53 protein stability [84, 94]. In turn Sirt1 gene expression is under the control of the p53-HIC1 loop [85, 211]. The precise role of Sirt1 in tumorigenesis is, therefore, dependent on the presence and activity of p53 [149]. Sirt1-mediated deacetylation of FOXO has been shown to promote ubiquitination and degradation of these transcription factors [212] as well as to induce FOXO-dependent cell cycle arrest and evasion of apoptosis in response to DNA damage [104, 105]. Deacetylation of pRb in a Sirt1-dependent manner has been suggested to reverse cell cycle arrest after DNA damage repair has taken place [122]. Another mechanism by which Sirt1 promotes cell survival is by deacetylating E2F1, thereby repressing its transcriptional activity and preventing the induction of the E2F1/p73 apoptotic pathway in response to DNA damage [121]. One of the major functions of Sirt1 is the regulation of inflammation through deacetylation of the proinflammatory transcription factor NF-κB [209] and consequent modulation of gene expression of various cytokines (TNFα, ICAM-1 (intercellular adhesion molecule-1), IL-6 (interleukin-6), and IL-8) [32, 89, 213]. In addition, tumorigenesis might be affected by Sirt1 through its considerable regulatory effects on metabolic processes and the regulation of oxidative stress [214, 215].
Therefore, specific Sirt1 inhibitors for each type of tissue needs to be considered in order to increase acetylation in the case of inactivation of oncogenes or activation of the expression of proapoptotic genes.
15. Conclusions and Perspectives
Deacetylation reaction catalysed by class III deacetylases requires the consumption of NAD+ and links chromatin epigenetic changes and transcriptional regulation with energy metabolism. Another important difference between sirtuins and the other classes of deacetylases is that apart from deacetylase, some class III family members possess ADP-ribosyltransferase activity. The mitochondrial localisation of these enzymes and their involvement in acetylation/deacetylation processes of mitochondrial proteins is an additional indication of the coupling between metabolic networks and acetylation. These characteristics of the sirtuin family members implicate them in a wide range of diverse cellular processes ranging from glucose homeostasis, to cellular growth, senescence, stress resistance, and metabolism. This in combination with the fact that there are seven members of the sirtuins family raise the theoretical proposal that intervention in each one of the different cellular processes modulated by distinct members of the sirtuins family could increase the specificity of therapeutics by selectively targeting different sirtuins-mediated pathways. Consequently inhibitors specific for each member of the sirtuins family could provide a valuable tool towards understanding the link of individual sirtuin-member-specific biological functions to alternative pathological situations.
Although intensive research in recent years has provided many answers in relation to the role of sirtuins in human physiology, there are still many questions and controversies that require to be addressed. For instance, identification of the factors determining sirtuin members' tissue as well as organelle specific function and whether this is linked to aging or other physiological or pathological conditions could facilitate individualisation of treatment with sirtuins modulators. In addition, it is not known whether specific sirtuin family members can only function within particular pathways, or overlapping synergistic or antagonistic effects between different members of the family could impact on the same pathways. Moreover, in the case of the sirtuin members exerting both deacetylase and ADP ribosyl transferase activity, the conditions determining the one or the other activity and the relative contribution of each one of these activities to the development of disease requires further investigation. Future research will also define the reason that Sirt1 overexpression in some cancer tissues has oncogenic effect and tumor suppressive in others.
Clearly there is a lot of work required to fully understand the complex role of sirtuins in human physiology and to hopefully identify new therapeutic uses of sirtuin activators and inhibitors.
References
- 1.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363(1-2):15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131(4):633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 3.Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through historic modifications. Cell. 2000;103(2):263–271. doi: 10.1016/s0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 4.Hildmann C, Riester D, Schwienhorst A. Histone deacetylases—an important class of cellular regulators with a variety of functions. Applied Microbiology and Biotechnology. 2007;75(3):487–497. doi: 10.1007/s00253-007-0911-2. [DOI] [PubMed] [Google Scholar]
- 5.Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective. Molecular Cell. 2006;23(3):289–296. doi: 10.1016/j.molcel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 6.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Batta K, Das C, Gadad S, Shandilya J, Kundu TK. Reversible acetylation of non histone proteins: role in cellular function and disease. Sub-Cellular Biochemistry. 2007;41:193–212. [PubMed] [Google Scholar]
- 8.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annual Review of Biochemistry. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 9.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nature Reviews Genetics. 2009;10(3):161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Norris KL, Lee JY, Yao TP. Acetylation goes global: the emergence of acetylation biology. Science Signaling. 2009;2(97):p. pe76. doi: 10.1126/scisignal.297pe76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krämer OH. HDAC2: a critical factor in health and disease. Trends in Pharmacological Sciences. 2009;30(12):647–655. doi: 10.1016/j.tips.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 12.Dieker J, Muller S. Epigenetic histone code and autoimmunity. Clinical Reviews in Allergy and Immunology. 2010;39(1):78–84. doi: 10.1007/s12016-009-8173-7. [DOI] [PubMed] [Google Scholar]
- 13.Gil JM, Rego AC. The R6 lines of transgenic mice: a model for screening new therapies for Huntington’s disease. Brain Research Reviews. 2009;59(2):410–431. doi: 10.1016/j.brainresrev.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends in Neurosciences. 2009;32(11):591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes PJ. Histone deacetylase-2 and airway disease. Therapeutic Advances in Respiratory Disease. 2009;3(5):235–243. doi: 10.1177/1753465809348648. [DOI] [PubMed] [Google Scholar]
- 16.Pons D, de Vries FR, van Den Elsen PJ, Heijmans BT, Quax PHA, Jukema JW. Epigenetic histone acetylation modifiers in vascular remodelling: new targets for therapy in cardiovascular disease. European Heart Journal. 2009;30(3):266–277. doi: 10.1093/eurheartj/ehn603. [DOI] [PubMed] [Google Scholar]
- 17.Blanchard F, Chipoy C. Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases? Drug Discovery Today. 2005;10(3):197–204. doi: 10.1016/S1359-6446(04)03309-4. [DOI] [PubMed] [Google Scholar]
- 18.Villeneuve LM, Natarajan R. The role of epigenetics in the pathology of diabetic complications. American Journal of Physiology. 2010;299(1):F14–F25. doi: 10.1152/ajprenal.00200.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kook H, Lepore JJ, Gitler AD, et al. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. Journal of Clinical Investigation. 2003;112(6):863–871. doi: 10.1172/JCI19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature Reviews Genetics. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villagra A, Sotomayor EM, Seto E. Histone deacetylases and the immunological network: implications in cancer and inflammation. Oncogene. 2010;29(2):157–173. doi: 10.1038/onc.2009.334. [DOI] [PubMed] [Google Scholar]
- 22.Anastasiou D, Krek W. SIRT1: linking adaptive cellular responses to aging-associated changes in organismal physiology. Physiology. 2006;21(6):404–410. doi: 10.1152/physiol.00031.2006. [DOI] [PubMed] [Google Scholar]
- 23.Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26(37):5489–5504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- 24.Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chemistry & Biology. 2002;9(1):3–16. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- 25.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. Journal of Cellular Biochemistry. 2005;96(2):293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 26.Fischle W, Kiermer V, Dequiedt F, Verdin E. The emerging role of class II histone deacetylases. Biochemistry and Cell Biology. 2001;79(3):337–348. [PubMed] [Google Scholar]
- 27.Verdin E, Dequiedt F, Kasler HG. Class II histone deacetylases: versatile regulators. Trends in Genetics. 2003;19(5):286–293. doi: 10.1016/S0168-9525(03)00073-8. [DOI] [PubMed] [Google Scholar]
- 28.Voelter-Mahlknecht S, Ho AD, Mahlknecht U. Chromosomal organization and localization of the novel class IV human histone deacetylase 11 gene. International Journal of Molecular Medicine. 2005;16(4):589–598. [PubMed] [Google Scholar]
- 29.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460(7255):587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochemical and Biophysical Research Communications. 2000;273(2):793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 31.Haigis MC, Guarente LP. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes and Development. 2006;20(21):2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 32.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annual Review of Pathology: Mechanisms of Disease. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochemical Journal. 2007;404(1):1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westphal CH, Dipp MA, Guarente L. A therapeutic role for sirtuins in diseases of aging? Trends in Biochemical Sciences. 2007;32(12):555–560. doi: 10.1016/j.tibs.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes and Development. 2006;20(9):1075–1080. doi: 10.1101/gad.1399706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang C, Chen L, Hou X, et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nature Cell Biology. 2006;8(9):1025–1031. doi: 10.1038/ncb1468. [DOI] [PubMed] [Google Scholar]
- 37.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306(5704):2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 38.Wen YC, Wang DH, RayWhay CY, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123(3):437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 39.Chen W, Cooper TK, Zahnow CA, et al. Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell. 2004;6(4):387–398. doi: 10.1016/j.ccr.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 40.Yuan J, Minter-Dykhouse K, Lou Z. A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. The Journal of Cell Biology. 2009;185(2):203–211. doi: 10.1083/jcb.200809167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zschoernig B, Mahlknecht U. SIRTUIN 1: regulating the regulator. Biochemical and Biophysical Research Communications. 2008;376(2):251–255. doi: 10.1016/j.bbrc.2008.08.137. [DOI] [PubMed] [Google Scholar]
- 42.Kwon HS, Ott M. The ups and downs of SIRT1. Trends in Biochemical Sciences. 2008;33(11):517–525. doi: 10.1016/j.tibs.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Q, Wang SY, Fleuriel C, et al. Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(3):829–833. doi: 10.1073/pnas.0610590104. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Kumar V, Carlson JE, Ohgi KA, et al. Transcription corepressor CtBP is an NAD+-regulated dehydrogenase. Molecular Cell. 2002;10(4):857–869. doi: 10.1016/s1097-2765(02)00650-0. [DOI] [PubMed] [Google Scholar]
- 45.Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9202–9207. doi: 10.1073/pnas.1633591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mani-Telang P, Sutrias-Grau M, Williams G, Arnosti DN. Role of NAD binding and catalytic residues in the C-terminal binding protein corepressor. FEBS Letters. 2007;581(27):5241–5246. doi: 10.1016/j.febslet.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 47.Nardini M, Spanò S, Cericola C, et al. CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. The EMBO Journal. 2003;22(12):3122–3130. doi: 10.1093/emboj/cdg283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lizé M, Pilarski S, Dobbelstein M. E2F1-inducible microRNA 449a/b suppresses cell proliferation and promotes apoptosis. Cell Death and Differentiation. 2010;17(3):452–458. doi: 10.1038/cdd.2009.188. [DOI] [PubMed] [Google Scholar]
- 50.Rane S, He M, Sayed D, et al. Downregulation of MiR-199a derepresses hypoxia-inducible factor-1α and sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circulation Research. 2009;104(7):879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abdelmohsen K, Pullmann R, Lal A, et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Molecular Cell. 2007;25(4):543–557. doi: 10.1016/j.molcel.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kong X, Wang R, Xue Y, et al. Sirtuin 3, a new target of PGC-1α, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One. 2010;5(7) doi: 10.1371/journal.pone.0011707. Article ID e11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.North BJ, Verdin E. Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. The Journal of Biological Chemistry. 2007;282(27):19546–19555. doi: 10.1074/jbc.M702990200. [DOI] [PubMed] [Google Scholar]
- 54.Sasaki T, Maier B, Koclega KD, et al. Phosphorylation regulates SIRT1 function. PLoS One. 2008;3(12) doi: 10.1371/journal.pone.0004020. Article ID e4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ford J, Ahmed S, Allison S, Jiang M, Milner J. JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle. 2008;7(19):3091–3097. doi: 10.4161/cc.7.19.6799. [DOI] [PubMed] [Google Scholar]
- 56.Zschoernig B, Mahlknecht U. Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2. Biochemical and Biophysical Research Communications. 2009;381(3):372–377. doi: 10.1016/j.bbrc.2009.02.085. [DOI] [PubMed] [Google Scholar]
- 57.Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. The Journal of Biological Chemistry. 2010;285(17):13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pandithage R, Lilischkis R, Harting K, et al. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. The Journal of Cell Biology. 2008;180(5):915–929. doi: 10.1083/jcb.200707126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dryden SC, Nahhas FA, Nowak JE, Goustin AS, Tainsky MA. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Molecular and Cellular Biology. 2003;23(9):3173–3185. doi: 10.1128/MCB.23.9.3173-3185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Molecular Cell. 2007;28(2):277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 61.Kong XX, Wang R, Liu XJ, et al. Function of SIRT1 in physiology. Biochemistry. 2009;74(7):703–708. doi: 10.1134/s0006297909070013. [DOI] [PubMed] [Google Scholar]
- 62.Di Marcotullio L, Canettieri G, Infante P, Greco A, Gulino A. Protected from the inside: endogenous histone deacetylase inhibitors and the road to cancer. Biochimica et Biophysica Acta. 2011;1815(2):241–252. doi: 10.1016/j.bbcan.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 63.Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451(7178):587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim JE, Lou Z, Chen J. Interactions between DBC1 and SIRT1 are deregulated in breast cancer cells. Cell Cycle. 2009;8(22):3784–3785. doi: 10.4161/cc.8.22.10055. [DOI] [PubMed] [Google Scholar]
- 65.Han Y, Jin YH, Kim YJ, et al. Acetylation of Sirt2 by p300 attenuates its deacetylase activity. Biochemical and Biophysical Research Communications. 2008;375(4):576–580. doi: 10.1016/j.bbrc.2008.08.042. [DOI] [PubMed] [Google Scholar]
- 66.Yang Y, Fu W, Chen J, et al. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nature Cell Biology. 2007;9(11):1253–1262. doi: 10.1038/ncb1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mellert HS, McMahon SB. Biochemical pathways that regulate acetyltransferase and deacetylase activity in mammalian cells. Trends in Biochemical Sciences. 2009;34(11):571–578. doi: 10.1016/j.tibs.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annual Review of Biochemistry. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 69.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends in Biochemical Sciences. 2007;32(1):12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 70.Ghosh S, George S, Roy U, Ramachandran D, Kolthur-Seetharam U. NAD: a master regulator of transcription. Biochimica et Biophysica Acta. 2010;1799(10-12):681–693. doi: 10.1016/j.bbagrm.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 71.Magni G, Orsomando G, Raffelli N, Ruggieri S. Enzymology of mammalian NAD metabolism in health and disease. Frontiers in Bioscience. 2008;13:6135–6154. doi: 10.2741/3143. [DOI] [PubMed] [Google Scholar]
- 72.Rongvaux A, Andris F, van Gool F, Leo O. Reconstructing eukaryotic NAD metabolism. BioEssays. 2003;25(7):683–690. doi: 10.1002/bies.10297. [DOI] [PubMed] [Google Scholar]
- 73.Revollo JR, Grimm AA, Imai SI. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Current Opinion in Gastroenterology. 2007;23(2):164–170. doi: 10.1097/MOG.0b013e32801b3c8f. [DOI] [PubMed] [Google Scholar]
- 74.Sauve AA. Sirtuin chemical mechanisms. Biochimica et Biophysica Acta. 2010;1804(8):1591–1603. doi: 10.1016/j.bbapap.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cen Y. Sirtuins inhibitors: the approach to affinity and selectivity. Biochimica et Biophysica Acta. 2010;1804(8):1635–1644. doi: 10.1016/j.bbapap.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 76.Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin nhibition by nicotinamide: altering the NAD+ cosubstrate specificity of a Sir2 enzyme. Molecular Cell. 2005;17(6):855–868. doi: 10.1016/j.molcel.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 77.Houtkooper RH, Cantó C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocrine Reviews. 2010;31(2):194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Naïmi M, Arous C, van Obberghen E. Energetic cell sensors: a key to metabolic homeostasis. Trends in Endocrinology and Metabolism. 2010;21(2):75–82. doi: 10.1016/j.tem.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 79.Yu J, Auwerx J. Protein deacetylation by SIRT1: an emerging key post-translational modification in metabolic regulation. Pharmacological Research. 2010;62(1):35–41. doi: 10.1016/j.phrs.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes and Development. 2004;18(1):12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen D, Bruno J, Easlon E, et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes and Development. 2008;22(13):1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fulco M, Cen Y, Zhao P, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Developmental Cell. 2008;14(5):661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137(3):560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nature Reviews Cancer. 2009;9(2):123–128. doi: 10.1038/nrc2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yi J, Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochimica et Biophysica Acta. 2010;1804(8):1684–1689. doi: 10.1016/j.bbapap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vaziri H, Dessain SK, Eaton EN, et al. hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell. 2001;107(2):149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 87.Shiota M, Yokomizo A, Kashiwagi E, et al. Foxo3a expression and acetylation regulate cancer cell growth and sensitivity to cisplatin. Cancer Science. 2010;101(5):1177–1185. doi: 10.1111/j.1349-7006.2010.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434(7029):113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 89.Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. The EMBO Journal. 2004;23(12):2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bendinelli P, Matteucci E, Maroni P, Desiderio MA. NF-κB activation, dependent on acetylation/deacetylation, contributes to HIF-1 activity and migration of bone metastatic breast carcinoma cells. Molecular Cancer Research. 2009;7(8):1328–1341. doi: 10.1158/1541-7786.MCR-08-0548. [DOI] [PubMed] [Google Scholar]
- 91.Vakhrusheva O, Smolka C, Gajawada P, et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circulation Research. 2008;102(6):703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- 92.Han MK, Song EK, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008;2(3):241–251. doi: 10.1016/j.stem.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bilton R, Trottier E, Pouysségur J, Brahimi-Horn MC. ARDent about acetylation and deacetylation in hypoxia signalling. Trends in Cell Biology. 2006;16(12):616–621. doi: 10.1016/j.tcb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 94.Xenaki G, Ontikatze T, Rajendran R, et al. PCAF is an HIF-1α cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene. 2008;27(44):5785–5796. doi: 10.1038/onc.2008.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Molecular Cell. 2010;38(6):864–878. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 96.Dioum EM, Chen R, Alexander MS, et al. Regulation of hypoxia-inducible factor 2α signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009;324(5932):1289–1293. doi: 10.1126/science.1169956. [DOI] [PubMed] [Google Scholar]
- 97.Kim SH, Jeong JW, Park JA, et al. Regulation of the HIF-1alpha stability by histone deacetylases. Oncology Reports. 2007;17(3):647–651. [PubMed] [Google Scholar]
- 98.Zhong L, D’Urso A, Toiber D, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1α. Cell. 2010;140(2):280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kawahara TLA, Michishita E, Adler AS, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell. 2009;136(1):62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang J, Chen J. SIRT1 regulates autoacetylation and histone acetyltransferase activity of TIP60. The Journal of Biological Chemistry. 2010;285(15):11458–11464. doi: 10.1074/jbc.M109.087585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Langley E, Pearson M, Faretta M, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. The EMBO Journal. 2002;21(10):2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Daitoku H, Hatta M, Matsuzaki H, et al. Silent information regulator 2 potentiates Foxo 1-mediated transcription through its deacetylase activity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(27):10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. The EMBO Journal. 2005;24(5):1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 105.Motta MC, Divecha N, Lemieux M, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116(4):551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 106.Luo J, Nikolaev AY, Imai SI, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107(2):137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 107.Wang F, Nguyen M, Qin FXF, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6(4):505–514. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 108.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(27):10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ahuja N, Schwer B, Carobbio S, et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. The Journal of Biological Chemistry. 2007;282(46):33583–33592. doi: 10.1074/jbc.M705488200. [DOI] [PubMed] [Google Scholar]
- 110.Patel JH, Du Y, Ard PG, et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Molecular and Cellular Biology. 2004;24(24):10826–10834. doi: 10.1128/MCB.24.24.10826-10834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends in Cell Biology. 2004;14(8):408–412. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 112.Huang H, Tindall DJ. Dynamic FoxO transcription factors. Journal of Cell Science. 2007;120(15):2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 113.Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metabolism. 2007;6(2):105–114. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vogt PK, Jiang H, Aoki M. Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle. 2005;4(7):908–913. doi: 10.4161/cc.4.7.1796. [DOI] [PubMed] [Google Scholar]
- 115.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459(7243):113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang F, Tong Q. SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1’s repressive interaction with PPARγ. Molecular Biology of the Cell. 2009;20(3):801–808. doi: 10.1091/mbc.E08-06-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Black JC, Mosley A, Kitada T, Washburn M, Carey M. The SIRT2 deacetylase regulates autoacetylation of p300. Molecular Cell. 2008;32(3):449–455. doi: 10.1016/j.molcel.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jacobs KM, Pennington JD, Bisht KS, et al. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. International Journal of Biological Sciences. 2008;4(5):291–299. doi: 10.7150/ijbs.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. Journal of Clinical Investigation. 2009;119(9):2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.DeGregori J, Johnson DG. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Current Molecular Medicine. 2006;6(7):739–748. doi: 10.2174/1566524010606070739. [DOI] [PubMed] [Google Scholar]
- 121.Pediconi N, Guerrieri F, Vossio S, et al. hSirT1-dependent regulation of the PCAF-E2F1-p73 apoptotic pathway in response to DNA damage. Molecular and Cellular Biology. 2009;29(8):1989–1998. doi: 10.1128/MCB.00552-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wong S, Weber JD. Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochemical Journal. 2007;407(3):451–460. doi: 10.1042/BJ20070151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nature Cell Biology. 2001;3(7):667–674. doi: 10.1038/35083062. [DOI] [PubMed] [Google Scholar]
- 124.Macleod KF. The role of the RB tumour suppressor pathway in oxidative stress responses in the haematopoietic system. Nature Reviews Cancer. 2008;8(10):769–781. doi: 10.1038/nrc2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang R, Chen HZ, Liu JJ, et al. SIRT1 suppresses activator protein-1 transcriptional activity and cyclooxygenase-2 expression in macrophages. The Journal of Biological Chemistry. 2010;285(10):7097–7110. doi: 10.1074/jbc.M109.038604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Salminen A, Kauppinen A, Suuronen T, Kaarniranta K. SIRT1 longevity factor suppresses NF-κB-driven immune responses: regulation of aging via NF-κB acetylation? BioEssays. 2008;30(10):939–942. doi: 10.1002/bies.20799. [DOI] [PubMed] [Google Scholar]
- 127.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor Perspectives in Biology. 2009;1(4) doi: 10.1101/cshperspect.a000034. Article ID a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee JH, Song MY, Song EK, et al. Overexpression of SIRT1 protects pancreatic β-cells against cytokine toxicity by suppressing the nuclear factor-κB signaling pathway. Diabetes. 2009;58(2):344–351. doi: 10.2337/db07-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes and Development. 2011;25(5):409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang RH, Sengupta K, Li C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14(4):312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mostoslavsky R, Chua KF, Lombard DB, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124(2):315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 132.Oberdoerffer P, Michan S, McVay M, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135(5):907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genetics. 2008;4(8) doi: 10.1371/journal.pgen.1000155. Article ID e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010;329(5997):1348–1353. doi: 10.1126/science.1192049. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 135.Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Molecular Cell. 2007;27(1):149–162. doi: 10.1016/j.molcel.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ming M, Shea CR, Guo X, et al. Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(52):22623–22628. doi: 10.1073/pnas.1010377108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Uhl M, Csernok A, Aydin S, Kreienberg R, Wiesmüller L, Gatz SA. Role of SIRT1 in homologous recombination. DNA Repair. 2010;9(4):383–393. doi: 10.1016/j.dnarep.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 138.Dai Y, Faller DV. Transcription regulation by class III histone deacetylases (HDACs)—sirtuins. Translational Oncogenomics. 2008;2008(3):53–65. doi: 10.4137/tog.s483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Li K, Casta A, Wang R, et al. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. The Journal of Biological Chemistry. 2008;283(12):7590–7598. doi: 10.1074/jbc.M709707200. [DOI] [PubMed] [Google Scholar]
- 140.Wang RH, Zheng Y, Kim HS, et al. Interplay among BRCA1, SIRT1, and survivin during BRCA1-associated tumorigenesis. Molecular Cell. 2008;32(1):11–20. doi: 10.1016/j.molcel.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Weterings E, Chen DJ. DNA-dependent protein kinase in nonhomologous end joining: a lock with multiple keys? The Journal of Cell Biology. 2007;179(2):183–186. doi: 10.1083/jcb.200705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sartori AA, Lukas C, Coates J, et al. Human CtIP promotes DNA end resection. Nature. 2007;450(7169):509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ying W. NAD+ and NADH in cellular functions and cell death. Frontiers in Bioscience. 2006;11(supplement 3):3129–3148. doi: 10.2741/2038. [DOI] [PubMed] [Google Scholar]
- 144.Hasegawa K, Wakino S, Yoshioka K, et al. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochemical and Biophysical Research Communications. 2008;372(1):51–56. doi: 10.1016/j.bbrc.2008.04.176. [DOI] [PubMed] [Google Scholar]
- 145.Liang F, Kume S, Koya D. SIRT1 and insulin resistance. Nature Reviews Endocrinology. 2009;5(7):367–373. doi: 10.1038/nrendo.2009.101. [DOI] [PubMed] [Google Scholar]
- 146.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. The Journal of Biological Chemistry. 2005;280(16):16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 147.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. The Journal of Biological Chemistry. 2005;280(14):13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 148.Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CFW, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. Journal of Molecular Biology. 2008;382(3):790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 149.Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Research. 2005;65(22):10457–10463. doi: 10.1158/0008-5472.CAN-05-1923. [DOI] [PubMed] [Google Scholar]
- 150.Huang J, Gan Q, Han L, et al. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS One. 2008;3(3) doi: 10.1371/journal.pone.0001710. Article ID e1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vijg J, Maslov AY, Suh Y. Aging: a sirtuin shake-up? Cell. 2008;135(5):797–798. doi: 10.1016/j.cell.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 152.van der Horst A, Burgering BMT. Stressing the role of FoxO proteins in lifespan and disease. Nature Reviews Molecular Cell Biology. 2007;8(6):440–450. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 153.Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? International Journal of Biological Sciences. 2009;5(2):147–152. doi: 10.7150/ijbs.5.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.North BJ, Verdin E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS One. 2007;2(8, article e784) doi: 10.1371/journal.pone.0000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Inoue T, Hiratsuka M, Osaki M, et al. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene. 2007;26(7):945–957. doi: 10.1038/sj.onc.1209857. [DOI] [PubMed] [Google Scholar]
- 156.Huang J-Y, Hirschey MD, Shimazu T, Ho L, Verdin E. Mitochondrial sirtuins. Biochimica et Biophysica Acta. 2010;1804(8):1645–1651. doi: 10.1016/j.bbapap.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 157.Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes and Development. 2007;21(8):920–928. doi: 10.1101/gad.1527307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang H, Yang T, Baur JA, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130(6):1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Michishita E, McCord RA, Berber E, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452(7186):492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Grob A, Roussel P, Wright JE, McStay B, Hernandez-Verdun D, Sirri V. Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. Journal of Cell Science. 2009;122(4):489–498. doi: 10.1242/jcs.042382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gorospe M, de Cabo R. AsSIRTing the DNA damage response. Trends in Cell Biology. 2008;18(2):77–83. doi: 10.1016/j.tcb.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jeong J, Juhn K, Lee H, et al. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Experimental and Molecular Medicine. 2007;39(1):8–13. doi: 10.1038/emm.2007.2. [DOI] [PubMed] [Google Scholar]
- 163.Cohen HY, Lavu S, Bitterman KJ, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Molecular Cell. 2004;13(5):627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 164.You H, Pellegrini M, Tsuchihara K, et al. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. Journal of Experimental Medicine. 2006;203(7):1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Westerheide SD, Anckar J, Stevens SM, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT. Science. 2009;323(5917):1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ohsawa S, Miura M. Caspase-mediated changes in Sir2α during apoptosis. FEBS Letters. 2006;580(25):5875–5879. doi: 10.1016/j.febslet.2006.09.051. [DOI] [PubMed] [Google Scholar]
- 167.Jin Q, Yan T, Ge X, Sun C, Shi X, Zhai Q. Cytoplasm-localized SIRT1 enhances apoptosis. Journal of Cellular Physiology. 2007;213(1):89–97. doi: 10.1002/jcp.21091. [DOI] [PubMed] [Google Scholar]
- 168.Peck B, Chen CY, Ho KK, et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Molecular Cancer Therapeutics. 2010;9(4):844–855. doi: 10.1158/1535-7163.MCT-09-0971. [DOI] [PubMed] [Google Scholar]
- 169.Allison SJ, Milner J. SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle. 2007;6(21):2669–2677. doi: 10.4161/cc.6.21.4866. [DOI] [PubMed] [Google Scholar]
- 170.Marfe G, Tafani M, Indelicato M, et al. Kaempferol induces apoptosis in two different cell lines via Akt inactivation, bax and SIRT3 activation, and mitochondrial dysfunction. Journal of Cellular Biochemistry. 2009;106(4):643–650. doi: 10.1002/jcb.22044. [DOI] [PubMed] [Google Scholar]
- 171.Nakamura Y, Ogura M, Tanaka D, Inagaki N. Localization of mouse mitochondrial SIRT proteins: shift of SIRT3 to nucleus by co-expression with SIRT5. Biochemical and Biophysical Research Communications. 2008;366(1):174–179. doi: 10.1016/j.bbrc.2007.11.122. [DOI] [PubMed] [Google Scholar]
- 172.Salminen A, Kaarniranta K. SIRT1: regulation of longevity via autophagy. Cellular Signalling. 2009;21(9):1356–1360. doi: 10.1016/j.cellsig.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 173.Morselli E, Mariño G, Bennetzen MV, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. The Journal of Cell Biology. 2011;192(4):615–629. doi: 10.1083/jcb.201008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mehrpour M, Esclatine A, Beau I, Codogno P. Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. American Journal of Physiology. 2010;298(4):C776–C785. doi: 10.1152/ajpcell.00507.2009. [DOI] [PubMed] [Google Scholar]
- 175.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual Review of Genetics. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Klionsky DJ, Cregg JM, Dunn WA, et al. A unified nomenclature for yeast autophagy-related genes. Developmental Cell. 2003;5(4):539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 177.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature Reviews Molecular Cell Biology. 2009;10(7):458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- 178.Lee IH, Cao L, Mostoslavsky R, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(9):3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nature Reviews Molecular Cell Biology. 2001;2(3):211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 180.Close P, Creppe C, Gillard M, et al. The emerging role of lysine acetylation of non-nuclear proteins. Cellular and Molecular Life Sciences. 2010;67(8):1255–1264. doi: 10.1007/s00018-009-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology. 2008;23(5):248–262. doi: 10.1152/physiol.00013.2008. [DOI] [PubMed] [Google Scholar]
- 182.Goligorsky MS. SIRTing out the link between autophagy and ageing. Nephrology Dialysis Transplantation. 2010;25(8):2434–2436. doi: 10.1093/ndt/gfq348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circulation Research. 2010;107(12):1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hands SL, Proud CG, Wyttenbach A. mTOR’s role in ageing: protein synthesis or autophagy? Aging. 2009;1(7):586–597. doi: 10.18632/aging.100070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Current Opinion in Cell Biology. 2010;22(2):181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 186.Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 2008;27(35):4860–4864. doi: 10.1038/onc.2008.117. [DOI] [PubMed] [Google Scholar]
- 187.Blagosklonny MV. Linking calorie restriction to longevity through sirtuins and autophagy: any role for TOR. Cell Death and Disease. 2010;1(1, article e12 ) doi: 10.1038/cddis.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tasdemir E, Maiuri MC, Morselli E, et al. A dual role of p53 in the control of autophagy. Autophagy. 2008;4(6):810–814. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- 189.Tavernarakis N, Pasparaki A, Tasdemir E, Maiuri MC, Kroemer G. The effects of p53 on whole organism longevity are mediated by autophagy. Autophagy. 2008;4(7):870–873. doi: 10.4161/auto.6730. [DOI] [PubMed] [Google Scholar]
- 190.van der Veer E, Ho C, O’Neil C, et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. The Journal of Biological Chemistry. 2007;282(15):10841–10845. doi: 10.1074/jbc.C700018200. [DOI] [PubMed] [Google Scholar]
- 191.Zhao Y, Yang J, Liao W, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nature Cell Biology. 2010;12(7):665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 192.Wenzel U. Nutrition, sirtuins and aging. Genes & Nutrition. 2006;1:85–93. doi: 10.1007/BF02829950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wolf NS, Smith DL, Jr., Smith JS. The Comparative Biology of Aging. Dordrecht, The Netherlands: Springer; 2010. Sirtuin function in longevity; pp. 123–146. [Google Scholar]
- 194.Merry BJ. Oxidative stress and mitochondrial function with aging—the effects of calorie restriction. Aging Cell. 2004;3(1):7–12. doi: 10.1046/j.1474-9728.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- 195.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes. 2009;58(12):2718–2725. doi: 10.2337/db09-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Borradaile NM, Pickering G. NAD+, sirtuins, and cardiovascular disease. Current Pharmaceutical Design. 2009;15(1):110–117. doi: 10.2174/138161209787185742. [DOI] [PubMed] [Google Scholar]
- 197.Gan L, Mucke L. Paths of convergence: sirtuins in aging and neurodegeneration. Neuron. 2008;58(1):10–14. doi: 10.1016/j.neuron.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2008;177(8):861–870. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 200.Kojima K, Ohhashi R, Fujita Y, et al. A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochemical and Biophysical Research Communications. 2008;373(3):423–428. doi: 10.1016/j.bbrc.2008.06.045. [DOI] [PubMed] [Google Scholar]
- 201.Lawson M, Uciechowska U, Schemies J, Rumpf T, Jung M, Sippl W. Inhibitors to understand molecular mechanisms of NAD+-dependent deacetylases (sirtuins) Biochimica et Biophysica Acta. 2010;1799(10-12):726–739. doi: 10.1016/j.bbagrm.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 202.Bradbury CA, Khanim FL, Hayden R, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19(10):1751–1759. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 203.Stünkel W, Peh BK, Tan YC, et al. Function of the SIRTI protein deacetylase in cancer. Biotechnology Journal. 2007;2(11):1360–1368. doi: 10.1002/biot.200700087. [DOI] [PubMed] [Google Scholar]
- 204.Firestein R, Blander G, Michan S, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS One. 2008;3(4) doi: 10.1371/journal.pone.0002020. Article ID e2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Huffman DM, Grizzle WE, Bamman MM, et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Research. 2007;67(14):6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 206.Jin MD, Zhi YW, Dao CS, Ru XL, Sheng QW. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. Journal of Cellular Physiology. 2007;210(1):161–166. doi: 10.1002/jcp.20831. [DOI] [PubMed] [Google Scholar]
- 207.Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113(7):881–889. doi: 10.1016/s0092-8674(03)00430-6. [DOI] [PubMed] [Google Scholar]
- 208.Fulco M, Schiltz RL, Iezzi S, et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Molecular Cell. 2003;12(1):51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- 209.Qiu X, Brown KV, Moran Y, Chen D. Sirtuin regulation in calorie restriction. Biochimica et Biophysica Acta. 2010;1804(8):1576–1583. doi: 10.1016/j.bbapap.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 210.Heltweg B, Gatbonton T, Schuler AD, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Research. 2006;66(8):4368–4377. doi: 10.1158/0008-5472.CAN-05-3617. [DOI] [PubMed] [Google Scholar]
- 211.Tseng RC, Lee CC, Hsu HS, Tzao C, Wang YC. Distinct HIC1-SIRT1-p53 loop deregulation in lung squamous carcinoma and adenocarcinoma patients. Neoplasia. 2009;11(8):763–770. doi: 10.1593/neo.09470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27(16):2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 213.Sequeira J, Boily G, Bazinet S, et al. sirt1-null mice develop an autoimmune-like condition. Experimental Cell Research. 2008;314(16):3069–3074. doi: 10.1016/j.yexcr.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 214.Braidy N, Guillemin G, Grant R. Promotion of cellular NAD+ anabolism: therapeutic potential for oxidative stress in ageing and alzheimer’s disease. Neurotoxicity Research. 2008;13(3-4):173–184. doi: 10.1007/BF03033501. [DOI] [PubMed] [Google Scholar]
- 215.Chung S, Yao H, Caito S, Hwang J-W, Arunachalam G, Rahman I. Regulation of SIRT1 in cellular functions: role of polyphenols. Archives of Biochemistry and Biophysics. 2010;501(1):79–90. doi: 10.1016/j.abb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]