

Abstract
Oncogenic forms of the Abl and Src tyrosine kinases trigger the destruction of the Abi proteins, a family of Abl-interacting proteins that antagonize the oncogenic potential of Abl after overexpression in fibroblasts. The destruction of the Abi proteins requires tyrosine kinase activity and is dependent on the ubiquitin–proteasome pathway. We show that degradation of the Abi proteins occurs through a Ras-independent pathway. Significantly, expression of the Abi proteins is lost in cell lines and bone marrow cells isolated from patients with aggressive Bcr–Abl-positive leukemias. These findings suggest that loss of Abi proteins may be a component in the progression of Bcr–Abl-positive leukemias and identify a novel pathway linking activated nonreceptor protein tyrosine kinases to the destruction of specific target proteins through the ubiquitin–proteasome pathway.
Keywords: Abi, Bcr–Abl, v-Src, ubiquitin-dependent proteolysis, Ph1-positive leukemia
Ubiquitin-dependent proteolysis is a critical component of diverse biological processes including cell cycle progression, the immune response, embryonic development, protein transport, and apoptosis (for review, see Deshaies 1995; Hochstrasser 1995; King et al. 1996; Varshavsky 1997). Here we show that ubiquitin-dependent proteolysis may also play a role in oncogenesis by activated nonreceptor tyrosine kinases.
Oncogenic forms of the Abl tyrosine kinase are linked to the development of human, murine, and feline leukemias (Bergold et al. 1987; Rosenberg and Witte 1988; Laneuville 1995; Gotoh and Broxmeyer 1997). Activation of cellular Abl (cAbl) oncogenic potential may occur as a consequence of chromosomal translocation events that generate chimeric fusion proteins such as Bcr–Abl and Tel–Abl (Gotoh and Broxmeyer 1997). The Bcr–Abl tyrosine kinases are produced by a reciprocal t(9;22)(q34;q11) chromosomal translocation that gives rise to the Philadelphia chromosome (Ph1). The translocation fuses varying amounts of the Bcr gene on chromosome 22 with sequences upstream of the second exon of the c-Abl gene on chromosome 9. Three different Bcr–Abl fusion proteins may be produced. The 210-kD form of Bcr–Abl (p210) is the causative agent of >95% of human chronic myelogenous leukemia (CML) cases (Clarkson et al. 1997; Gotoh and Broxmeyer 1997). The 185-kD Bcr–Abl protein (p185) is associated with a subset of acute lymphocytic leukemia (ALL) (Kurzrock et al. 1987). Recently, a rare 230-kD Bcr–Abl protein (p230) has been detected in patients with chronic neutrophilic leukemia (CNL) (Wada et al. 1995; Melo 1996). The p185 and p210 Bcr–Abl proteins have been shown to elicit ALL- and CML-like syndromes in mice, respectively, and to transform fibroblasts and hematopoietic cells in culture (Daley et al. 1990; Heisterkamp et al. 1990). Several mechanisms have been proposed to explain how Bcr–Abl transforms cells. Among these are increased resistance to apoptosis, enhanced proliferative capacity, defective adhesion, and increased motility of the Bcr–Abl-expressing cells (Clarkson et al. 1997; Cortez et al. 1997; Gotoh and Broxmeyer 1997; Salgia et al. 1997). The biological effects of Bcr–Abl require the constitutive tyrosine kinase activity of the chimeric protein. Multiple proteins have been identified as downstream targets of Bcr–Abl. Among these are proteins involved in the regulation of mitogenic and apoptotic pathways as well as cytoskeletal-associated proteins. Primarily, components of the Ras and phosphatidylinositol 3-kinase (PI3k) pathways are critical for Bcr–Abl-dependent transformation. Dominant interfering mutants of Grb-2, Ras, and c-Jun block Bcr–Abl-mediated transformation (Gishizky et al. 1995; Raitano et al. 1995; Sawyers et al. 1995; Cortez et al. 1996). Grb-2, Ras, and c-Jun are components of the same signaling pathway, thereby emphasizing the essential role of this pathway in the transmission of the Bcr–Abl-transforming signal. Also, inhibition of PI3k and its downstream target, the Akt serine kinase, decreases transformation by Bcr–Abl (Skorski et al. 1995, 1997). In addition, interfering with the function of the transcription factors, c-Myc and NF-κB, abolishes transformation by Bcr–Abl (Sawyers et al. 1992; Reuther et al. 1998).
Less clear is the contribution of other downstream protein targets to Bcr–Abl-dependent transformation. Although the levels of the anti-apoptotic Bcl-2 mRNA and protein are elevated in some Bcr–Abl-expressing cells (Sanchez-Garcia and Grutz 1995), it is unclear whether Bcl-2 is up-regulated in all Bcr–Abl-expressing cells, and whether its up-regulation is necessary and sufficient for the anti-apoptotic activity of Bcr-Abl (Cortez et al. 1996). Likewise, it is not known what role the increased tyrosine phosphorylation of a number of cytoskeletal-associated proteins has on the altered adhesion and the overall transforming properties of Bcr–Abl-positive cells (Salgia et al. 1995).
Although numerous targets for the Abl kinases have been identified, only a few of these have been shown to be important in modulation of the Abl-transforming potential (Gotoh and Broxmeyer 1997). Recently, we and other researchers identified a family of Abl-interactor (Abi) proteins that bind specifically to both the SH3 and carboxy-terminal proline-rich sequences of Abl (Dai and Pendergast 1995; Shi et al. 1995). Two distinct, yet highly related genes, abi-1 and abi-2, were identified and cloned. The corresponding protein products share overall 69% identity with the greatest homology observed in the amino-terminal homeobox-like domain, proline-rich sequences, and the carboxy-terminal SH3 domain. The Abi proteins are substrates of the Abl kinases. Significantly, Abi proteins antagonize the oncogenic activity of Abl in fibroblasts. Overexpression of Abi-1 potently suppresses the transforming activity of viral Abl (v-Abl) in NIH-3T3 fibroblasts (Shi et al. 1995). Furthermore, coexpression of a truncated form of Abi-2 with c-Abl activates the oncogenic potential of c-Abl (Dai and Pendergast 1995). These and other data (Wang et al. 1996; Biesova et al. 1997) suggest that the full-length Abi proteins may function as growth inhibitors in mammalian cells.
Inactivation of molecules that function as growth inhibitors/tumor suppressors is a common event in a large number of cancers (Cordon-Cardo 1995). As illustrated for p53 and pRb, the activity of the tumor suppressors may be abrogated by mutations of the corresponding DNAs or by the sequestration of the tumor suppressor proteins by specific viral or cellular proteins. Increasing evidence is accumulating that implicates selective proteolysis in the functional inactivation of tumor suppressor proteins (Deshaies 1995; Haupt et al. 1997; Kubbutat et al. 1997). In particular, ubiquitin-dependent proteolysis appears to play a role in this process. Protein degradation by the ubiquitin pathway involves the covalent attachment of multiple ubiquitin polypeptides to the substrate protein, followed by the degradation of the polyubiquitinated substrate by the 26S proteasome, a large ATP-dependent multienzyme complex (Varshavsky 1997). Several proteins that function as growth inhibitors/tumor suppressors have been reported to be degraded through ubiquitin-dependent proteolysis. Among these are p53 and the cyclin-dependent kinase inhibitors Sic1p, Far1p, and p27 (Pagano et al. 1995; Feldman et al. 1997; Haupt et al. 1997; Henchoz et al. 1997; Kubbutat et al. 1997; Skowyra et al. 1997; Verma et al. 1997).
Here we show that oncogenic forms of the Abl and Src nonreceptor tyrosine kinases elicit the destruction of the Abi proteins by the ubiquitin-dependent proteasome machinery. The elimination of the Abi proteins by the oncogenic tyrosine kinases occurs through a novel Ras-independent pathway that is initiated by the constitutive tyrosine kinase activity of the oncoproteins. Significantly, the expression of Abi proteins is lost in cell lines and bone marrow cells from Ph1-positive leukemia patients. These findings suggest that loss of Abi proteins may play a role in oncogenesis and implicates ubiquitin-dependent proteolysis in tumor progression.
Results
Bcr–Abl down-regulates Abi expression in hematopoietic cells
To understand the role of Abi proteins in Abl-mediated transformation, we investigated the expression of Abi proteins in normal hematopoietic cells as well as in cells transformed by the oncogenic Bcr–Abl fusion protein. The Bcr–Abl gene was introduced into the pro-B BaF3 cell line and the multipotent myeloid progenitor 32D cell line. Both cell types are dependent on interleukin 3 (IL-3) for growth and survival. Expression of Bcr–Abl in BaF3 and 32D cells induces cellular transformation, confers cytokine-independent growth, and blocks apoptosis (Cortez et al. 1995, 1996, 1997). Abi proteins are expressed in BaF3 and 32D cells and migrate as a doublet of 60 and 65 kD (Fig. 1A, lane 1). Because of the strong homology between Abi 1 and Abi 2, the available anti-Abi antibodies cannot distinguish between the two proteins and therefore, we will refer to these protein bands as Abi. Surprisingly, little if any Abi protein could be detected in BaF3 cells transformed with p185 Bcr–Abl (Fig. 1A, lanes 2,3). Similar findings were obtained in 32D cells transformed by Bcr–Abl. These results indicate that the expression of the oncogenic Bcr–Abl tyrosine kinase down-regulates the expression of Abi proteins. Consistent with this notion, the expression of HA-tagged exogenous Abi 2 in 32D cells is also down-regulated by Bcr–Abl in the same manner as that of the endogenous Abi proteins (data not shown). The Bcr–Abl-mediated down-regulation of Abi expression requires Bcr–Abl tyrosine kinase activity, as the expression of the kinase-deficient mutant p185 K671R Bcr–Abl (Cortez et al. 1995) failed to down-regulate Abi expression (Fig. 1A, lane 4). To determine whether the loss of Abi expression in Bcr–Abl-transformed hematopoietic cells is caused by a decrease in the transcription of abi genes, Northern blot analysis and RNase protection assays were performed. BaF3 cells express message for both abi 1 and abi 2 (Fig. 1B, lane 1). No significant change in the abi 1 and abi 2 mRA levels, however, was observed among cells expressing the vector control, wild-type p185 Bcr–Abl, or the p185 K671R Bcr–Abl mutant (Fig. 1B, cf. lanes 1–4). This suggests that Bcr–Abl down-regulates abi expression by a mechanism other than transcriptional regulation.
Figure 1.
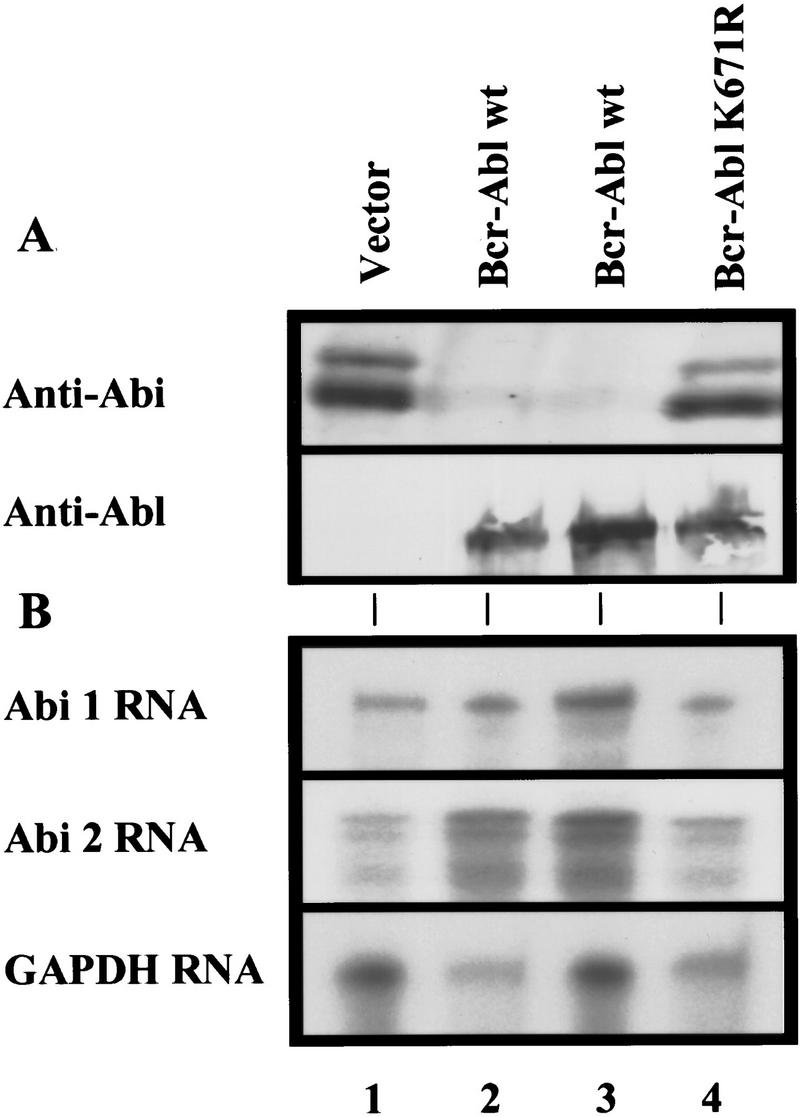
The oncogenic Bcr–Abl tyrosine kinase down-regulates Abi protein expression in BaF3 cells. (A) Loss of Abi protein with p185Bcr–Abl expression. BaF3 cells (2 × 106) were infected (lanes 1,3,4) with the indicated retroviral expression vectors or stably transfected (lane 2) with the indicated plasmid. Abi proteins were detected by immunoblotting with polyclonal 5421 anti-Abi antibodies (top). Bcr–Abl (lanes 2,3) or Bcr–Abl (K671R) (lane 4) protein was detected by immunoblotting with monoclonal anti-Abl antibody (lower). (B) Bcr–Abl expression does not alter abi 1 or abi 2 mRNA levels. abi 1, abi 2, or GAPDH antisense RNA probes were protected against RNase digestion by BaF3 total RNA. 35S-Labeled protected probes were electrophoresed on acrylamide/urea gels and detected by autoradiography.
Bcr–Abl down-regulates Abi expression through the ubiquitin–proteasome pathway
The oncogenic Bcr–Abl proteins down-regulate Abi expression without significantly reducing the level of abi transcripts. This suggests that the down-regulation may occur post-translationally. An increasing number of cellular processes have been shown to be critically dependent on the control of protein abundance catalyzed by the ubiquitin-dependent proteasome pathway (Deshaies 1995). Therefore, we tested whether the down-regulation of Abi expression by Bcr–Abl uses this pathway. First, we examined whether the down-regulation of Abi proteins by Bcr–Abl is caused by increased instability of the proteins. To this end we synthesized 35S-labeled Abi 2 in vitro using a rabbit reticulocyte lysate (RRL) and tested its stability in the presence or absence of in vitro-translated Bcr–Abl. The RRL system has been used commonly as the source of active ubiquitinating enzymes and proteasome complexes (Nielsen et al. 1997; Pagano et al. 1997). In the absence of Bcr–Abl, Abi 2 was relatively stable. In contrast, in the presence of Bcr–Abl Abi 2 was degraded rapidly in the RRL (Fig. 2, cf. lanes 3 and 6). This suggests that Bcr–Abl increases Abi protein instability. Consistent with this, we found that addition of ATPγS, a nonhydrolyzable ATP analog that prevents degradation of ubiquitinated proteins by the proteasome but does not prevent their ubiquitination, blocked Abi 2 degradation in RRL system (Fig. 2, lane 7).
Figure 2.

In vitro degradation of Abi 2 is stimulated by p185Bcr–Abl and is ATP dependent. Abi 2 was synthesized in the presence of [35S]methionine using a coupled transcription–translation kit (Promega). The labeled Abi 2 was incubated in a protein-degradation reaction mix with or without unlabeled p185Bcr–Abl, and with or without ATPγS, as indicated. Samples (equal volume) were removed at indicated time points and analyzed by SDS-PAGE and fluorography.
We then tested whether the down-regulation of Abi proteins by Bcr–Abl in hematopoietic cells is dependent on the ubiquitin–proteasome pathway. BaF3 cells transfected with either a control vector or an expression vector for p185 Bcr–Abl were treated with two specific inhibitors of the ubiquitin–proteasome machinery, LLnL (N-acetyl-l-leucinyl-l-leucinyl-l-norleucinal) and lactacystin (Aberle et al. 1997). As shown by Western blot analysis, LLnL and lactacystin inhibited the down-regulation of Abi expression in Bcr–Abl-expressing cells (Fig. 3A, lanes 5,6). Treatment of BaF3 cells expressing p185 Bcr–Abl with LLnL and lactacystin resulted in the accumulation of 60- and 65-kD Abi proteins, as well as immunoreactive Abi proteins with a slower mobility in SDS gels. The slower mobility bands may represent Abi proteins that are modified during the process of ubiquitin-mediated proteolysis. Interestingly, the Abi proteins, in particular the slower mobility forms, also accumulated to higher levels in the control BaF3 cells treated with LLnL and lactacystin (Fig. 3A, lanes 2,3). This suggests that Abi expression in normal cells may also be regulated, at least in part, by ubiquitin-mediated proteolysis. We then examined whether Abi proteins are targets for ubiquitination. A plasmid expressing an HA-tagged Abi 2 was transfected into Bosc 23 cells in the presence of an expression plasmid encoding His6-tagged ubiquitin (Treier et al. 1994). The His6-tagged ubiquitinated proteins were purified by Ni–agarose chromatography (Treier et al. 1994; Aberle et al. 1997) and subjected to Western blot analysis with monoclonal antibody to HA to detect the HA-tagged Abi 2 protein. As shown in Figure 3B, Abi 2 is ubiquitinated.
Figure 3.

Bcr–Abl down-regulates Abi expression through an ubiquitin-dependent proteolysis pathway. (A) Proteasome-specific inhibitors LLnL and lactacystin inhibit Bcr–Abl-induced Abi down-regulation. BaF3 cells transfected by vector alone (lanes 1–3) or expression vector encoding p185 Bcr–Abl (lanes 4–6) were untreated (lanes 1,4) or treated with 50 μm LLnL (lane 2,5) or 10 μm lactacystin (lanes 3,6) for 8 hr. Cells (2 × 106) were lysed in SDS sample buffer and subjected to Western blot analysis with 5421 anti-Abi antibodies. (B) Abi 2 is ubiquitinated in Bosc 23 cells. Bosc 23 cells were cotransfected with pMT107, a plasmid expressing His6-tagged ubiquitin, plus either pCGN control plasmid (lanes 1,3) or pCGN-Abi 2 plasmid encoding HA-tagged human Abi 2 (lanes 2,4). Total cell lysates (lanes 1,2) or ubiquitin substrate conjugates that were affinity precipitated by Ni2+ chelate chromatography (lanes 3,4) were subjected to Western blot analysis with anti-HA monoclonal antibody. The position of ubiquitinated Abi 2 is indicated by an arrowhead.
Oncogenic Src tyrosine kinase down-regulates Abi expression
We then wanted to test whether Abi proteins could also be down-regulated by expression of other oncogenic tyrosine kinases such as v-Src. A BaF3 cell line transfected with a zinc-inducible v-Src expression plasmid (Canman et al. 1995) was used in this experiment. The cells were treated with or without zinc for 8 hr to induce v-Src expression and cell lysates were subjected to Western blot analysis with either Abi (Fig. 4A, top) or v-Src (Fig. 4A, bottom) specific antibodies. Expression of v-Src was increased dramatically with the addition of zinc and the increased expression of v-Src correlated with a dramatic reduction of Abi expression (Fig. 4A, cf. lane 3 to lane 4). Thus, like Bcr–Abl, the oncogenic v-Src tyrosine kinase also down-regulates Abi expression in BaF3 cells. To determine whether the v-Src induced down-regulation of Abi expression is mediated by the ubiquitin–proteasome degradation pathway, we examined the effect of LLnL on Abi expression in v-Src-expressing BaF3 cells. v-Src-transfected cells were incubated with zinc in the presence or absence of LLnL. Zinc induced expression of v-Src (Fig. 4B, lanes 5,6), regardless of the presence or absence of LLnL. Down-regulation of Abi expression in cells treated with LLnL was completely inhibited compared with cells without LLnL treatment (Fig. 4B, cf. lanes 5 and 6). This result demonstrates that v-Src down-regulates Abi expression through ubiquitin-mediated proteolysis.
Figure 4.
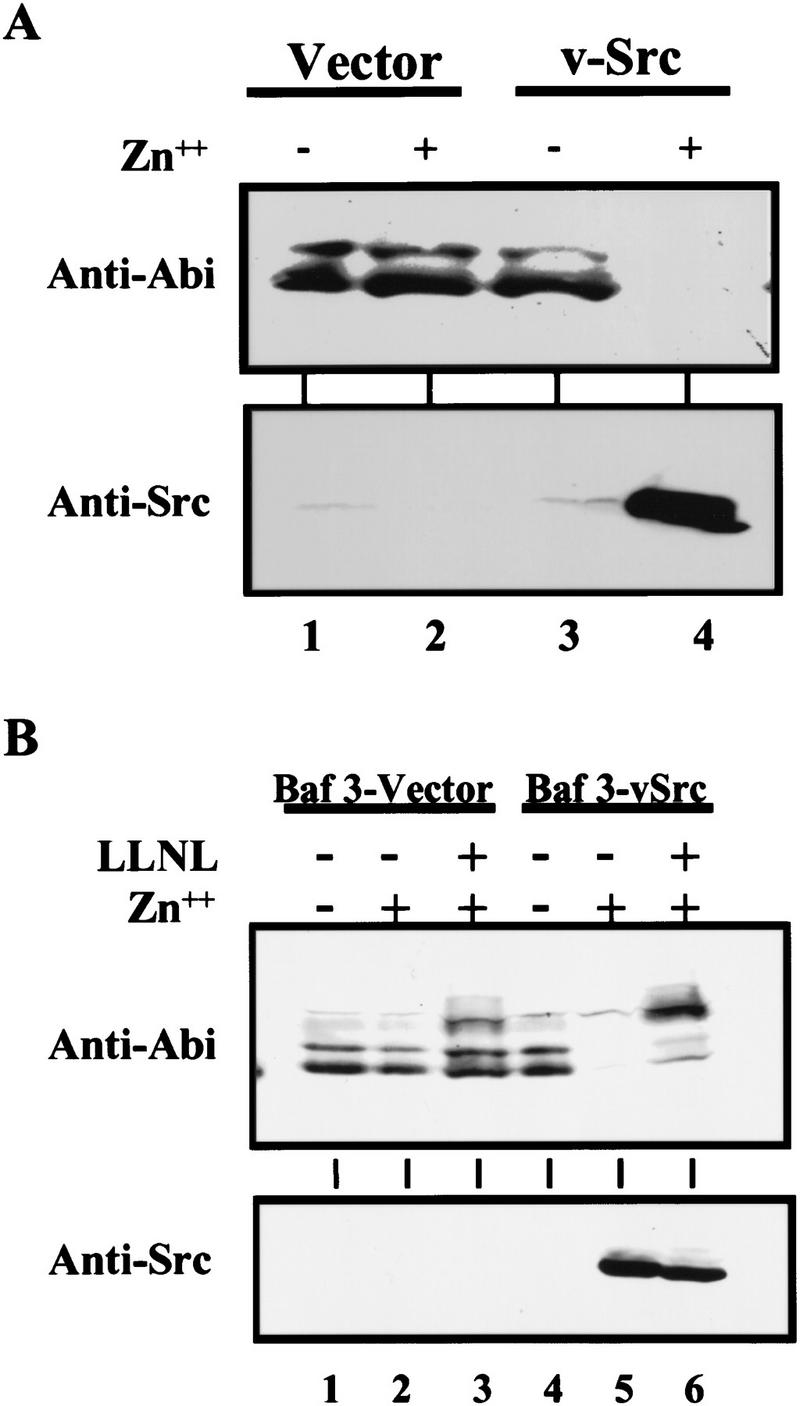
v-Src down-regulates Abi expression through an ubiquitin-dependent proteolysis pathway. (A) Oncogenic v-Src tyrosine kinase down-regulates Abi expression. BaF3 cells transfected by vector alone (lanes 1,2), or zinc-inducible v-Src-expressing vector (lanes 3,4) were treated with or without 75 μm ZnCl2 for 8 hr, as indicated. Total cell lysates (2 × 106) were subjected to Western blot analysis with either 5421 anti-Abi antibodies (top) or anti-v-Src antibodies (bottom). (B) Proteasome inhibitor LLnL inhibits v-Src-induced down-regulation of Abi expression. BaF3 cells transfected by vector alone (lanes 1–3) or zinc-inducible v-Src expression vector (lanes 4–6) were treated with (lanes 3,6) or without (lanes 1,2,4,5) 50 μn LLnL for 1 hr. ZnCl2 (75 μm) was then added as indicated, and cells were incubated for an additional 7 hr. Cells (2 × 106) were lysed in SDS sample buffer and subjected to Western blot analysis with either 5421 anti-Abi (top) or anti-v-Src antibodies (bottom).
Bcr–Abl-mediated down-regulation of Abi expression is Ras independent
Oncogenic Bcr–Abl proteins elicit cellular transformation through multiple signal transduction pathways (Gotoh and Broxmeyer 1997). Previously, we and other investigators have shown that Ras function is activated in Bcr–Abl-transformed cells and it is a necessary component for Bcr–Abl-mediated transformation (Pendergast et al. 1993; Puil et al. 1994; Cortez et al. 1995, 1996). Studies on photoreceptor cell differentiation of the Drosophila eye have shown that activation of the Ras/Map kinase-signaling cascade results in the ubiquitin-mediated degradation of Tramtrack (TTK) (Li et al. 1997; Tang et al. 1997), a transcriptional repressor of neuronal cell fates, as well as of the transcription factor YAN (Rebay and Rubin 1995), a general inhibitor of differentiation of many cell types in the Drosophila eye. Therefore, we examined whether the Bcr–Abl-mediated down-regulation of Abi expression is Ras dependent. We used p185 and p210 Bcr–Abl-transformed 32D cells that coexpress a dominant-negative form of Ras, Ras Asn 17 (Feig and Cooper 1988), under the control of a glucocorticoid responsive promoter. Previously, we have shown that the inducible expression of dominant- negative Ras Asn 17 blocks Bcr–Abl from activating Ras in these cells (Cortez et al. 1996). Cells were treated with dexamethasone for 24 hr to obtain high-level expression of dominant-negative Ras Asn 17. The expression of Bcr–Abl (data not shown), dominant -negative Ras (Fig. 5A, lanes 1–4), and Abi proteins (Fig. 5A, lanes 5–11) was evaluated by Western blot analysis. Despite overexpression of dominant-negative Ras Asn 17, the expression of Abi proteins in the dexamethasone-treated cells (Fig. 5A, lanes 9,11) is down-regulated to low levels similar to nontreated cells (Fig. 5A, lanes 8,10) or control cells that express Bcr–Abl alone (Fig. 5A, lanes 6,7). Because Raf is an immediate downstream component of Ras in the Ras/Map kinase signaling cascade, we tested whether the enforced expression of an activated Raf protein kinase would elicit the down-regulation of Abi expression in BaF3 cells. A BaF3 cell line that inducibly expresses an activated form of human c-Raf (c-Raf–BXB) (Canman et al. 1995) from a zinc-responsive promoter was grown in the presence or absence of zinc. Zinc induced the expression of c-Raf-BXB (Fig. 5B). Consistent with the findings in Figure 5A, the enforced expression of the activated Raf did not affect the expression of Abi protein (Fig. 5B, cf. lane 3 to lane 4). Taken together, our results demonstrate that down-regulation of Abi expression by oncogenic Bcr–Abl is Ras and Raf independent.
Figure 5.
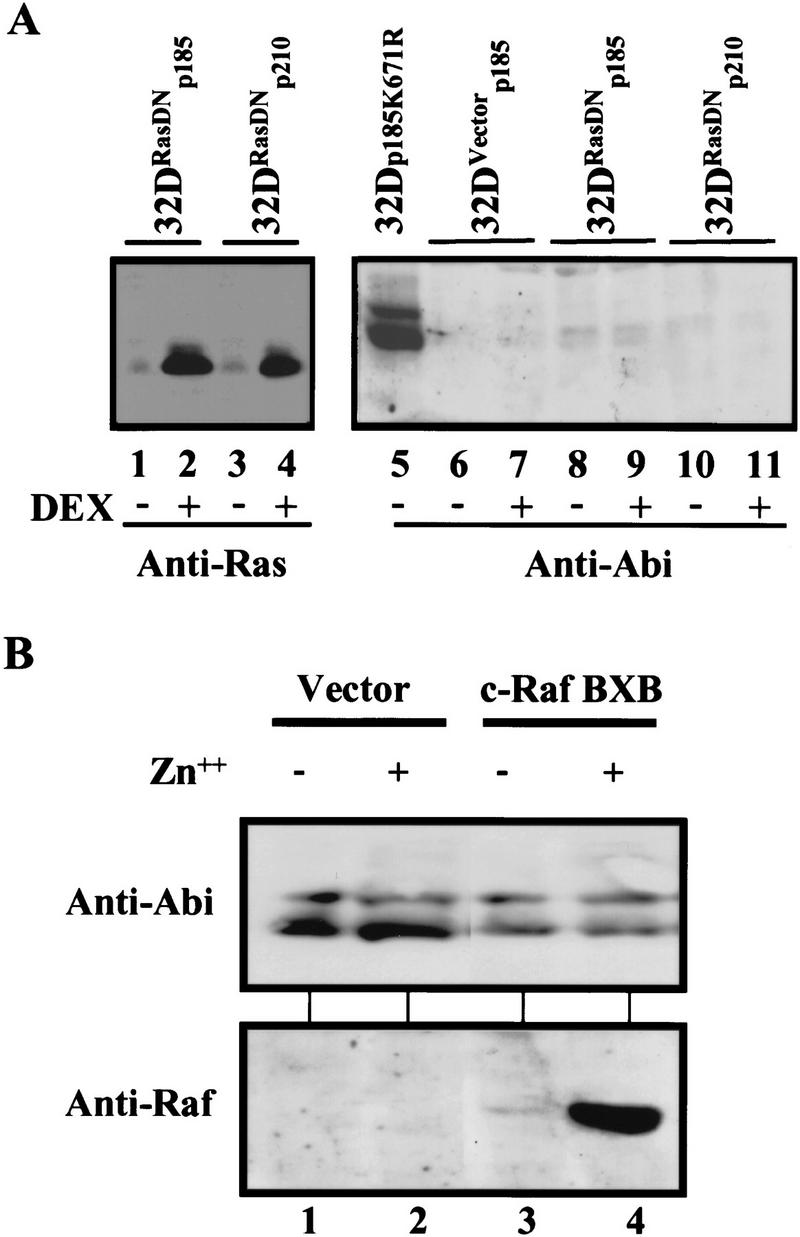
Bcr–Abl-mediated down-regulation of Abi expression is independent of Ras signaling. (A) Expression of a dominant-negative Ras, Ras Asn 17, failed to block Bcr–Abl-mediated down-regulation of Abi expression. 32D cells expressing a control plasmid (32DVector, lanes 6,7) or a plasmid inducibly expressing Asn 17 Ras (32DRasDN, lanes 1–4 and 8–11) were infected with retroviruses carrying either the p185Bcr–Abl (lanes 1,2, and 6–9) or p210Bcr–Abl (lanes 3,4,10,11) oncogenes. The cells were treated with or without 50 nm dexamethasone for 24 hr, as indicated, to induce the expression of Ras Asn 17. A control 32D cell line infected with a retrovirus encoding a kinase-deficient p185Bcr–Abl mutant was also included (lane 5). Cells (2 × 106) were lysed in SDS sample buffer and subjected to Western blot analysis with either anti-Ras antibody (pan Ras, Santa Cruz) (lanes 1–4) or 5421 anti-Abi antibody (lanes 5–11) as indicated. (B) Expression of an activated form of c-Raf does not down-regulate Abi expression. BaF3 cells transfected with either vector alone (lanes 1,2), or an inducible expression vector encoding activated c-Raf, c-Raf–BXB, were treated with or without 75 μm ZnCl2 for 8 hr as indicated. Cells (2 × 106) were lysed in SDS sample buffer and subjected to Western blot analysis using 5421 anti-Abi antibody (top) or anti-Raf antibody (Santa Cruz, bottom).
The expression of Abi proteins is lost in cell lines and bone marrow cells from Philadelphia chromosome-positive leukemia patients
The finding that Bcr–Abl down-regulates the expression of Abi proteins in BaF3 cells and 32D cells prompted us to test whether the Abi protein levels are also down-regulated as a consequence of Bcr–Abl expression in primary bone marrow cells, the natural target of the oncogenic Bcr–Abl tyrosine kinase. Mouse bone marrow cells were infected with Bcr–Abl and the expression of Abi proteins was examined by Western blotting (Fig. 6A, bottom). Consistent with the results observed in BaF3 and 32D cells (Fig. 6A, lanes 1,2), infection of bone marrow cells with Bcr–Abl retrovirus results in a loss of Abi expression (Fig. 6A, cf. lanes 3 and 4, bottom) that correlates with the expression of Bcr–Abl protein (Fig. 6A, top). Then we tested whether the expression of Abi proteins is also reduced in bone marrow cells from patients with Ph1-positive human leukemias. The Abi proteins were shown to be expressed in bone marrow cells from either normal human samples or a Ph1-negative leukemia patients (Fig. 6B, lanes 3,4). In contrast, Abi protein expression was lost in bone marrow cells from a Ph1-positive patient with ALL or from a Ph1-positive patient with CML in the blast crisis phase of the disease (Fig. 6B, lanes 1,2). K562 and MEG01 are cell lines derived from patients with CML in the blast crisis phase of the disease. Therefore, we compared these cell lines with a human myeloid cell line (MO7e), a cell line from a Ph1-negative acute myelogenous leukemia patient (KG1), and a cell line from a T-cell leukemia patient (Jurkat), for the expression of Abi proteins. Examination of the mRNAs for abi 1 and abi 2 by reverse-transcriptase PCR revealed that they are present in all of these cell lines (data not shown). As shown in Figure 6C, expression of Abi proteins was undetectable in those cells from the Ph1-positive leukemia patients, whereas Abi proteins are present in the other cell lines (Fig. 6C, lanes 5,6). Consistently, the down-regulation of Abi expresion correlates with the expression of oncogenic Bcr–Abl (Fig. 6C, upper). Taken together, these data suggest that loss of Abi protein expression may be a component in the progression of Bcr–Abl-positive leukemias.
Figure 6.

Expression of Abi proteins is lost in Bcr–Abl-transformed primary bone marrow cells and cells from Ph1-positive leukemia patients. Western blots were performed to compare Abl immunoreactivity (top) with Abi protein expression (bottom) in murine cells, human leukemia samples, and human cell lines. (A) 32D cells were infected with retroviral constructs containing vector alone (32D/v, lane 1) or p210 Bcr–Abl (32D/p210, lane 2). Primary mouse bone marrow (mBM, lane 3) was infected with a retroviral construct containing p185 Bcr–Abl (mBM/p185, lane 4) and analyzed after 18 days of selection with G418. (B) Bone marrow was obtained from patients with Ph1-positive acute lymphoblastic leukemia (ALL, lane 1), or with Ph1-positive chronic myelogenous leukemia in blast crisis (CML/BC, lane 2), and was compared to normal bone marrow (control, lane 3) and bone marrow obtained from a patient with Ph1-negative acute myelogenous leukemia (AML, lane 4). (C) Ph1-negative human leukemia cell lines (Jurkat and KG1, lanes 1,2) and human myeloid cell line MO7 (lane 3) were compared to Ph1-positive cell lines (MEG01 and K562, lanes 5,6) or to a Ph1-negative cell line infected with retrovirus containing p210 Bcr/Abl (MO7/p210, lane 4).
Discussion
We have identified a novel pathway downstream of the oncogenic Abl and Src nonreceptor tyrosine kinases that targets the destruction of the Abl-interacting Abi family of proteins through the ubiquitin–proteasome pathway. The down-regulation of the Abi proteins requires the tyrosine kinase activity of Abl, and it is independent of the Ras–Raf pathway. Significantly, the degradation of the Abi proteins appears to be selective. Other molecules known to be degraded by the ubiquitin-dependent proteolysis pathway in response to extracellular signals or cell cycle progression, such as IκBα and the cyclin-dependent kinase inhibitor p27, are not affected by expression of oncogenic forms of Abl (Reuther et al. 1998; Z. Dai and A.M. Pendergast, unpubl.). It is likely, however, that additional proteins may be targeted for ubiquitin-dependent degradation after expression of the Abl and Src oncogenic tyrosine kinases.
It has become increasingly apparent that ubiquitin-dependent proteolysis of specific proteins is a highly regulated process. Linkage of ubiquitin to proteins that display a distinct degradation signal, results in the destruction of the ubiquitin–protein conjugate by the 26S proteasome (Varshavsky 1997). The conjugation of ubiquitin to the target protein involves a series of steps that begin with the formation of a thioester bond between ubiquitin and the ubiquitin-activating enzyme (E1). Ubiquitin is then transesterified to an ubiquitin-conjugating enzyme (UBC or E2) and subsequently transferred to the target protein, usually with the involvement of an ubiquitin protein ligase (E3). The latter is the component of the ubiquitin conjugation system that is involved in substrate recognition (Varshavsky 1997). Several ubiquitin-dependent degradation signals have been identified to date. Regulated destruction of target proteins is usually dependent on phosphorylation, interaction with specific proteins, or both. Most of the phosphorylation-regulated degradation signals identified to date are mediated by serine/threonine kinases (Rebay and Rubin 1995; Henchoz et al. 1997; Maniatis 1997; Verma et al. 1997) . Although the tyrosine kinase activity of Bcr-Abl is absolutely required for Abi degradation, it is not clear at present whether direct tyrosine phosphorylation of the Abi proteins is critical for their proteolytic degradation. It is possible that the activated tyrosine kinases may induce Abi degradation through the phosphorylation of serine/threonine residues on Abi by protein kinases activated downstream of the oncogenic tyrosine kinases. Alternatively, the activated tyrosine kinases may induce the formation of a complex between Abi proteins and specific cellular proteins that target Abi for degradation. These two types of degradation signals are not mutually exclusive. Multiple serine, threonine, and tyrosine residues are found in the Abi proteins that may be phosphorylated by various protein kinases. Abi protein also contain sequences rich in proline, glutamic acid, serine, and threonine, designated as PEST, which have been found in many proteins that are targeted for ubiquitin-dependent degradation (Deshaies 1995; Rechsteiner and Rogers 1996). However, PEST sequences alone are not sufficient to identify those proteins that are targets of ubiquitin-dependent degradation (Varshavsky 1997). Extensive mutagenesis of the Abi proteins is necessary to identify those residues critical for their ubiquitin-dependent degradation. The availability of an in vitro degradation assay for the Abi proteins (Fig. 2) will facilitate the identification of the residues on Abi important for ubiquitin-dependent degradation and will permit the isolation of the protein recognition complex that targets Abi for degradation by the proteasome.
The finding that Abi protein expression is lost in cells after expression of the transforming Bcr–Abl and v-Src tyrosine kinases, together with the discovery that Abi proteins are absent in cell lines and bone marrow cells isolated from patients with aggressive Bcr–Abl-positive leukemias, suggests that loss of Abi proteins by the ubiquitin–proteasome pathway may be a component in the progression of Bcr–Abl-positive leukemias and possibly other cancers. The irreversible nature of proteolysis makes this process uniquely suited for the elimination of growth inhibitory molecules during tumor progression. Indeed, several tumor-suppressor and growth-inhibitory proteins have been shown to be degraded by ubiquitin-dependent proteolysis. Among these is the p53 tumor-suppressor protein that is targeted for degradation by the human papilloma virus E6 oncoprotein (Scheffner et al. 1990) and the cellular Mdm 2 protein (Haupt et al. 1997; Kubbutat et al. 1997). Similarly, SHP-1, a protein tyrosine phosphatase that is implicated in receptor-mediated inhibitory signals, is targeted for ubiquitin-dependent degradation by an activated form of the Kit receptor tyrosine kinase (Piao et al. 1996). More recently, another link between tumor progression and increased proteasome-dependent degradation was provided by the finding that the cell cycle inhibitor p27 is targeted for ubiquitin-dependent degradation in aggressive colorectal carcinomas (Loda et al. 1997). These examples show that selective degradation of proteins that participate in the control of growth inhibitory pathways represents an alternative mechanism for their inactivation without the involvement of mutations or deletions in the corresponding genes.
Although our findings and those of other published reports are consistent with the hypothesis that Abi proteins function as growth inhibitors/tumor suppressors, an alternative role for these proteins, which cannot be ruled out at the present time, is that Abi proteins are downstream substrates of the oncogenic tyrosine kinases. The phosphorylated Abi proteins may transduce a signal from the oncogenic tyrosine kinases. Abi activation may be coupled to Abi destruction through the ubiquitin–proteasome pathway. The tightly coupled activation and the proteasome-dependent destruction of a protein has been documented for the p58 component of the yeast kinetochore Cbf 3 protein complex (Kaplan et al. 1997). The p58 protein is activated by phosphorylation as assayed by DNA-binding activity and subsequently it is degraded by the proteasome in a ubiquitin-dependent step. The phosphorylation and degradation of p58 are tightly coupled events that require the product of the SKP1 gene p23 Skp1 (Kaplan et al. 1997). It has been proposed that p23 Skp1 functions as an adaptor that recruits a protein kinase by binding to both p58 and the unknown kinase. Also, p23 Skp1 is a component of the E3 ubiquitin ligase complex that targets p58 to the proteasome machinery. In this manner p58 is regulated positively by phosphorylation and regulated negatively by ubiquitin-dependent proteolysis. Linked activation and negative regulation of cellular proteins in response to phosphorylation has also been reported in mammalian cells for the STAT 1 transcription factor (Kim and Maniatis 1996). Activated STAT 1 has been shown to be regulated negatively by the ubiquitin–proteasome pathway. Tyrosine phosphorylation of STAT 1, which is induced by treatment of cells with interferon-γ, is required for its nuclear translocation and activation of transcription and it is also required for STAT 1 ubiquitination and subsequent degradation. A similar role for the phosphorylation of Abi proteins may exist in cells transformed by the activated Abl an Src tyrosine kinases.
The finding that Abi proteins are targeted for degradation by both oncogenic Abl and Src suggests that the activities of these two tyrosine kinases may be linked or alternatively, that they may regulate independently the function of specific common target proteins such as Abi. The data also raise the possibility that Abi proteins may be targeted for degradation in other human cancers where the Src family of nonreceptor tyrosine kinases are activated constitutively. Future work will be geared toward elucidating the cellular components of this novel pathway and the identification of additional targets of ubiquitin-dependent degradation triggered by the activity of oncogenic protein tyrosine kinases.
Materials and methods
Cell culture and retroviral infection
BaF3 cells and 32D cells were grown in RPMI containing 10% fetal calf serum (FCS) and 10% WEHI-conditioned media (WEHI-CM) as a source of IL-3. Stable mass populations of cells expressing bcr–abl transgenes were generated by retroviral infection as previously described (Cortez et al. 1995). BaF3 cell lines expressing v-Src and c-Raf-BXB (Canman et al. 1995) were grown in RPMI containing 10% FCS and 10% WEHI-CM. To induce the expression of v-Src and c-Raf-BXB, cells were treated with 75 μm ZnCl2 for 8 hr. To inhibit ubiquitin-dependent proteolysis, cells were treated with either 10 μm lactacystin or 50 μm LLnL for 8 hr. Cells were washed once with PBS and lysed directly in SDS sample buffer [50 mm Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 5% β-mercaptoethanol] for Western blot analysis.
Anti-Abi antibodies
Rabbit polyclonal antibodies 5421 and 4575 anti-Abi were raised to a recombinant GST–Abi 2Δ1-100 fusion protein and a synthetic Abi 2 peptide (amino acids 318–329), respectively. Antibodies were affinity purified by standard techniques (Harlow and Lane 1988).
RNA analysis
abi 1 and abi 2 mRNA levels were determined by RNase protection assays. Total RNA was isolated from BaF3 cells using TRIzol reagent (GIBCO BRL) or the RNeasy Mini Kit (Qiagen) as directed by each of the manufacturers. The protocols for these two methods were followed as directed. Antisense probes for abi 1 and abi 2 mRNA were generated by in vitro transcription with T3 RNA polymerase (Stratagene). abi 1 probe was transcribed from a linearized template containing the T3 promoter from the pPCR-Script Amp SK(+) plasmid (Stratagene) and a 244-nucleotide fragment of abi 1 cDNA. abi 2 probe was generated from linearized pBlueScript II SK +/− (Stratagene) plasmid containing a 435-bp fragment of abi 2 mouse genomic DNA composed of a 48-bp intronic region and 397 nucleotides from a single exon. The probe for mouse GAPDH RNA was generated from the pTRI–GAPDH mouse linear fragment (Ambion) using T3 RNA polymerase. RNase protection experiments were carried out in accordance with instructions provided by the manufacturer (Ambion). Hybridization of probes to RNA protected the probes from digestion with RNase A plus T1 (Ambion). To detect abi 2 by RNase protection, 30 μg of total RNA was used per lane. Twenty micrograms of RNA was used for abi 1 and 10 μg of RNA was used for GAPDH. After digestion, protected probes for abi 1, abi 2, and GAPDH were analyzed in separate lanes of 6% acrylamide/urea gels. 35S-Labeled protected fragments were detected by autoradiography.
Analysis of Abi expression in bone marrow cells
P185-expressing primary mouse bone marrow cells were generated by retroviral infection as described (Cortez et al. 1995). Mouse bone marrow was obtained by flushing the femurs of 3- to 4-week-old male BALB/c mice (Charles River Labs) with IMDM containing 2% FCS and either was used for retroviral infection followed by G418 selection as described (McLaughlin et al. 1989), or processed through two rounds of ammonium chloride lysis, lysed in 2× SDS sample buffer and used directly for Western blot analysis using antibodies against the Abl kinase domain (8E9, Pharmingen) or Abi-2. Bone marrow samples from leukemia patients were obtained from the SWOG Human Tissue Bank (University of New Mexico) or the Duke Human Tissue Bank (Duke University Medical Center). Normal bone marrow was obtained after informed consent from patients undergoing autologous bone marrow transplantation for nonhematological malignancies. After Ficoll density gradient centrifugation (Sigma), light density human bone marrow cells were resuspended in PBS containing 0.01 mg/ml aprotinin (Boehringer Mannheim), 5 mm benzamidine (Sigma), and 1 mg/ml AEBSF (“Pefabloc SC” Boehringer Mannheim), then lysed with 2× SDS sample buffer for Western blot analysis. Mouse myeloid and mouse and human bone marrow samples were normalized to give roughly equal levels of Abl immunoreactivity.
In vitro degradation assay
35S-Labeled human Abi 2 was produced using RRL and plasmid pT7T3-Abi 2 with a coupled transcription–translation kit (Promega). The transcription–translation reaction was performed at 30°C for 120 min in the presence of [35S]methionine, as indicated by the manufacturer. To produce unlabeled p185Bcr–Abl, the transcription–translation reaction was performed using plasmid pGEMp185Bcr–Abl in the presence of a complete amino acid mix. Protein stability was analyzed by incubating 3 μl of 35S-labeled Abi 2 in 50 μl of degradation mix [33% RRL, 50 mm Tris-HCl (pH 8.0), 5 mm MgCl, 2 mm dithiothreitol, 1 mm ATP, and 2 mm methionine] at 37°C in the presence or absence of 3 μl of p185Bcr–Abl-containing lysate. Where indicated, 1 mm ATP was substituted with 2 mm ATPγS. The reaction was stopped by addition of an equal volume of 2× SDS sample buffer and analyzed by SDS-PAGE.
Affinity precipitation
Purification of His-tagged ubiquitin expressed transiently in Bosc 23 cells was performed following the method of Treier et al. (1994). Briefly, 48 hr after transfection cells were lysed with 1 ml of GTN buffer per 60-mm dish [6 m guanidinium–HCl, 20 mm Tris-HCl (pH 8.0), 200 mm NaCl, 10 mm imidazole, 0.1% TX-100]. The lysate was sonicated with a microtipped sonifier at setting 4 for 20 sec to reduce viscosity. Fifty microliters of Ni2+–NTA–agarose beads (Qiagen) was added and mixed for 4 hr at room temperature. The beads were successively washed with the following solutions (pH 8.0): 1 ml of GTN; 1 ml of 8 m urea, 20 mm Tris-HCl, 200 mm NaCl, 0.1% TX-100; 1 ml of 8 m urea, 20 mm Tris-HCl, 1 m NaCl, 0.1% TX-100; 1 ml of 4 m urea, 20 mm Tris-HCl, 200 mm NaCl, 0.1% TX-100; 1 ml of 1 m urea, 20 mm Tris-HCl, 200 mm NaCl, 0.1% TX-100; and 1 ml of 20 mm Tris-HCl, 200 mm NaCl, 10 mm imidazole, 0.1% TX-100. The bound His6-tagged ubiquitin substrate complexes were analyzed by SDS-PAGE and Western blotting.
Acknowledgments
We thank Dr. T.M. Gilmer for BaF3 cell lines expressing v-Src and c-Raf BXB; Dr. C.L. Willman for the human leukemia samples; Dr. N. Tonks for MO7 and MO7/p210 cell lines; Dr. J.T. Parsons for anti-v-Src antibodies; Dr. D. Bohmann for His-tagged ubiquitin-expressing vector; Dr. H.S. Symonds for advice with the RNase protection assay; and Drs. A.R. Means, X.F. Wang, and J. Nevins for critical review of the manuscript. This work was supported by grants from the National Cancer Institute (CA61033 and CA70940). Z.D. is a Special Fellow of the Leukemia Society of America. R.C.Q. is supported by the Four Schools Physician-Scientist Program, sponsored by the Lucille P. Markey Charitable Trust. K.D.C. is supported by the Medical Scientist Training Program. D.C. was supported by Howard Hughes Medical Institute predoctoral fellowship. G.W.R. was supported by an Environmental Protection Agency Fellowship. A.M.P. is a Whitehead Scholar and a Scholar of the Leukemia Society of America.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL pende014@mc.duke.edu; FAX (919) 681-7148.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. β-Catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergold PJ, Wang JY, Hardy Jr WD, Littau V, Johnson E, Besmer P. Structure and origins of the HZ2-feline sarcoma virus. Virology. 1987;158:320–329. doi: 10.1016/0042-6822(87)90204-2. [DOI] [PubMed] [Google Scholar]
- Biesova Z, Piccoli C, Wong WT. Isolation and characterization of e3B1, an eps8 binding protein that regulates cell growth. Oncogene. 1997;14:233–241. doi: 10.1038/sj.onc.1200822. [DOI] [PubMed] [Google Scholar]
- Canman CE, Gilmer TM, Coutts SB, Kastan MB. Growth factor modulation of p53-mediated growth arrest versus apoptosis. Genes & Dev. 1995;9:600–611. doi: 10.1101/gad.9.5.600. [DOI] [PubMed] [Google Scholar]
- Clarkson BD, Strife A, Wisniewski D, Lambek C, Carpino N. New understanding of the pathogenesis of CML: A prototype of early neoplasia. Leukemia. 1997;11:1404–1428. doi: 10.1038/sj.leu.2400751. [DOI] [PubMed] [Google Scholar]
- Cordon-Cardo C. Mutations of cell cycle regulators. Biological and clinical implications for human neoplasia. Am J Pathol. 1995;147:545–560. [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Kadlec L, Pendergast AM. Structural and signaling requirements for BCR-ABL-mediated transformation and inhibition of apoptosis. Mol Cell Biol. 1995;15:5531–5541. doi: 10.1128/mcb.15.10.5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Stoica G, Pierce JH, Pendergast AM. The BCR-ABL tyrosine kinase inhibits apoptosis by activating a Ras-dependent signaling pathway. Oncogene. 1996;13:2589–2594. [PubMed] [Google Scholar]
- Cortez D, Reuther G, Pendergast AM. The Bcr-Abl tyrosine kinase activates mitogenic signaling pathways and stimulates G1-to-S phase transition in hematopoietic cells. Oncogene. 1997;15:2333–2342. doi: 10.1038/sj.onc.1201400. [DOI] [PubMed] [Google Scholar]
- Dai Z, Pendergast AM. Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl-transforming activity. Genes & Dev. 1995;9:2569–2582. doi: 10.1101/gad.9.21.2569. [DOI] [PubMed] [Google Scholar]
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ. Make it or break it: The role of ubiquitin-dependent proteolysis in cellular regulation. Trends Cell Biol. 1995;5:428–434. doi: 10.1016/s0962-8924(00)89102-3. [DOI] [PubMed] [Google Scholar]
- Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235–3243. doi: 10.1128/mcb.8.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RM, Correll CC, Kaplan KK, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Gishizky ML, Cortez D, Pendergast AM. Mutant forms of growth factor-binding protein-2 reverse BCR-ABL-induced transformation. Proc Natl Acad Sci. 1995;92:10889–10893. doi: 10.1073/pnas.92.24.10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh A, Broxmeyer HE. The function of BCR/ABL and related proto-oncogenes. Curr Opin Hematol. 1997;4:3–11. doi: 10.1097/00062752-199704010-00002. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies, a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oreb M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–298. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukaemia in bcr/abl transgenic mice. Nature. 1990;344:251–253. doi: 10.1038/344251a0. [DOI] [PubMed] [Google Scholar]
- Henchoz S, Chi Y, Catarin B, Herskowitz I, Deshaies RJ, Peter M. Phosphorylation- and ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor Far1p in budding yeast. Genes & Dev. 1997;11:3046–3060. doi: 10.1101/gad.11.22.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr Opin Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- Kaplan KB, Hyman AA, Sorger PK. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell. 1997;91:491–500. doi: 10.1016/s0092-8674(00)80435-3. [DOI] [PubMed] [Google Scholar]
- Kim TK, Maniatis T. Regulation of interferon-γ-activated STAT1 by the ubiquitin-proteasome pathway. Science. 1996;273:1717–1719. doi: 10.1126/science.273.5282.1717. [DOI] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- Kubbutat MHG, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Kurzrock R, Shtalrid M, Romero P, Kloetzer WS, Talpas M, Trujillo JM, Blick M, Beran M, Gutterman JU. A novel c-abl protein product in Philadelphia-positive acute lymphoblastic leukaemia. Nature. 1987;325:631–635. doi: 10.1038/325631a0. [DOI] [PubMed] [Google Scholar]
- Laneuville P. Abl tyrosine protein kinase. Semin Immunol. 1995;7:255–266. doi: 10.1006/smim.1995.0030. [DOI] [PubMed] [Google Scholar]
- Li S, Li Y, Carthew RW, Lai ZC. Photoreceptor cell differentiation requires regulated proteolysis of the transcriptional repressor Tramtrack. Cell. 1997;90:469–478. doi: 10.1016/s0092-8674(00)80507-3. [DOI] [PubMed] [Google Scholar]
- Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nature Med. 1997;3:231–234. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- Maniatis T. Catalysis by a multiprotein IκB kinase complex. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- McLaughlin J, Chianese E, Witte ON. Alternative forms of the BCR-ABL oncogene have quantitatively different potencies for stimulation of immature lymphoid cells. Mol Cell Biol. 1989;9:1866–1874. doi: 10.1128/mcb.9.5.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JV. The diversity of Bcr–Abl fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375–2384. [PubMed] [Google Scholar]
- Nielsen KH, Papageorge AG, Vass WC, Willumsen BM, Lowy DR. The Ras-specific exchange factors mouse Sos (mSos1) and mSos2 are regulated differently: mSos2 contains ubiquitination signals absent in mSos1. Mol Cell Biol. 1997;17:7132–7138. doi: 10.1128/mcb.17.12.7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Sal GD, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulation abundance of the cyclin-dependent kinase inhibitor p27. Science. 1997;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, Batzer A, Rabun KM, Der CJ, Schlessinger J, Gishizky ML. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–185. [PubMed] [Google Scholar]
- Piao X, Paulson R, van der Geer P, Pawson T, Bernstein A. Oncogenic mutation in the Kit receptor tyrosine kinase alters substrate specificity and induces degradation of the protein tyrosine phosphatase SHP-1. Proc Natl Acad Sci. 1996;93:14665–14669. doi: 10.1073/pnas.93.25.14665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puil L, Liu J, Gish G, Mbamalu G, Bowtell D, Pelicci PG, Arlinghaus R, Pawson T. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. EMBO J. 1994;13:764–773. doi: 10.1002/j.1460-2075.1994.tb06319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitano AB, Halpern JR, Hambuch TM, Sawyers CL. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci. 1995;92:11746–11750. doi: 10.1073/pnas.92.25.11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebay I, Rubin GM. Yan functions as a general inhibitor of differentiation and is negatively regulated by activation of the Ras1/MAPK pathway. Cell. 1995;81:857–866. doi: 10.1016/0092-8674(95)90006-3. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- Reuther JY, Reuther GW, Cortez D, Pendergast AM, Baldwin Jr AS. A requirement for NF-κB activation in Bcr-Abl-mediated transformation. Genes & Dev. 1998;12:968–981. doi: 10.1101/gad.12.7.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N, Witte ON. The viral and cellular forms of the Abelson (abl) oncogene. Adv Virus Res. 1988;35:39–81. doi: 10.1016/s0065-3527(08)60708-3. [DOI] [PubMed] [Google Scholar]
- Salgia R, Brunkhorst B, Pisick E, Li JL, Lo SH, Chen LB, Griffin JD. Increased tyrosine phosphorylation of focal adhesion proteins in myeloid cell lines expressing p210BCR/ABL. Oncogene. 1995;11:1149–1155. [PubMed] [Google Scholar]
- Salgia R, Li JL, Ewaniuk DS, Pear W, Pisick E, Burky SA, Ernst T, Sattler M, Chen LB, Griffin JD. BCR/ABL induces multiple abnormalities of cytoskeletal function. J Clin Invest. 1997;100:46–57. doi: 10.1172/JCI119520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Garcia I, Grutz G. Tumorigenic activity of the BCR-ABL oncogenes is mediated by BCL2. Proc Natl Acad Sci. 1995;92:5287–5291. doi: 10.1073/pnas.92.12.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell. 1992;70:901–910. doi: 10.1016/0092-8674(92)90241-4. [DOI] [PubMed] [Google Scholar]
- ————— Genetic requirement for Ras in the transformation of fibroblasts and hematopoietic cells by the Bcr-Abl oncogene. J Exp Med. 1995;181:307–313. doi: 10.1084/jem.181.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Shi Y, Alin K, Goff SP. Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity. Genes & Dev. 1995;9:2583–2597. doi: 10.1101/gad.9.21.2583. [DOI] [PubMed] [Google Scholar]
- Skorski T, Kanakaraj P, Nieborowska-Skorska M, Ratajczak MZ, Wen SC, Zon G, Gewirtz AM, Perussia B, Calabretta B. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood. 1995;86:726–736. [PubMed] [Google Scholar]
- Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, Trotta R, Wlodarski P, Perrotti D, Chan TO, Wasik MA, Tsichlis PN, Calabretta B. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Tang AH, Neufeld TP, Kwan E, Rubin GM. PHYL acts to down-regulate TTK88, a transcriptional repressor of neuronal cell fates, by a SINA-dependent mechanism. Cell. 1997;90:459–467. doi: 10.1016/s0092-8674(00)80506-1. [DOI] [PubMed] [Google Scholar]
- Treier M, Staszewski LM, Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. The ubiquitin system. Trends Biochem Sci. 1997;22:383–387. doi: 10.1016/s0968-0004(97)01122-5. [DOI] [PubMed] [Google Scholar]
- Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- Wada H, Mizutani S, Nishimura J, Usuki Y, Kohsaki M, Komai M, Kaneko H, Sakamoto S, Delia D, Kanamaru A, Kakishita E. Establishment and molecular characterization of a novel leukemic cell line with Philadelphia chromosome expressing p230 BCR/ABL fusion protein. Cancer Res. 1995;55:3192–3196. [PubMed] [Google Scholar]
- Wang B, Mysliwiec T, Krainc D, Jensen RA, Sonoda G, Testa JR, Golemis EA, Kruh GD. Identification of ArgBP1, an Arg protein tyrosine kinase binding protein that is the human homologue of a CNS-specific Xenopus gene. Oncogene. 1996;12:1921–1929. [PubMed] [Google Scholar]