

Abstract
Receptor tyrosine phosphatases have been implicated in playing important roles in cell signaling events by their ability to regulate the level of protein tyrosine phosphorylation. Although the catalytic activity of their phosphatase domains has been well established, the biological roles of these molecules are, for the most part, not well understood. Here we show that the Caenorhabditis elegans protein CLR-1 (CLeaR) is a receptor tyrosine phosphatase (RTP) with a complex extracellular region and two intracellular phosphatase domains. Mutations in clr-1 result in a dramatic Clr phenotype that we have used to study the physiological requirements for the CLR-1 RTP. We show that the phosphatase activity of the membrane-proximal domain is essential for the in vivo function of CLR-1. By contrast, we present evidence that the membrane-distal domain is not required to prevent the Clr phenotype in vivo. The Clr phenotype of clr-1 mutants is mimicked by activation of the EGL-15 fibroblast growth factor receptor (FGFR) and is suppressed by mutations that reduce or eliminate the activity of egl-15. Our data strongly indicate that CLR-1 attenuates the action of an FGFR-mediated signaling pathway by dephosphorylation.
Keywords: FGF receptor tyrosine kinase, tyrosine phosphatase, C. elegans, CLR-1, EGL-15
Protein tyrosine phosphorylation plays an integral role in cellular signaling events and functions in basic processes such as cell division, differentiation, and cell fate determination. The phosphorylation state of specific tyrosines is regulated by the opposing actions of protein tyrosine kinases and protein tyrosine phosphatases (PTPs; Sun and Tonks 1994; Hunter 1995). PTPs are a diverse family of molecules characterized by the presence of at least one phosphatase domain (Charbonneau and Tonks 1992; Walton and Dixon 1993). This domain spans ∼250 amino acids and contains the core consensus sequence (I/V)HCXAGXXR(S/T)G. Among these conserved residues, the cysteine and arginine have been shown to be essential for phosphatase activity (Streuli et al. 1990; Guan and Dixon 1991; Pot et al. 1991; Cho et al. 1992). The cysteine acts as a nucleophile on the phosphotyrosine-containing substrate and forms a thiophosphate intermediate during the dephosphorylation reaction (Guan and Dixon 1991; Cho et al. 1992; Pot and Dixon 1992). The arginine is also found at the active site and appears to aid in both binding and catalysis via its guanidinium group (for review, see Denu et al. 1996).
PTPs are broadly classified into transmembrane or receptor PTPs (RTPs) and intracellular PTPs (Fischer et al. 1991). The RTPs are further classified on the basis of their extracellular regions (Fischer et al. 1991; Brady-Kalnay and Tonks 1995). Type II isoforms contain amino-terminal immunoglobulin domains followed by fibronectin type III (FN III) repeats. These domains are characteristic of the N-CAM family of cell adhesion molecules, and some type II RTPs have been shown to mediate adhesive interactions (Brady-Kalnay et al. 1993; Gebbink et al. 1993; Sap et al. 1994).
Most RTPs contain two intracellular phosphatase domains. The first domain (D1, membrane-proximal) is catalytically active (Streuli et al. 1989, 1990), whereas the function of the second domain (D2, membrane-distal) is not understood. Some second phosphatase domains are predicted to be catalytically inactive based on the absence of specific conserved residues in their catalytic cores. For example, RPTPβ and HPTPγ are missing the catalytic cysteine known to be essential for phosphatase activity (Krueger et al. 1990; Kaplan et al. 1990). Other second phosphatase domains contain these conserved residues in the catalytic core and can show phosphatase activity (e.g., see Wang and Pallen 1991), but the importance of this domain to the function of the RTP is not clear.
The D1 and D2 domains of RTPs appear to differ structurally as well as functionally. Recent data based on multiple crystal forms of RPTPα identified an amino-terminal segment, termed the amino-terminal wedge, that may be involved in the dimerization of D1 domains (Bilwes et al. 1996). The primary sequence that forms this amino-terminal wedge appears to be found only in D1 domains and not in D2 domains (Bilwes et al. 1996), suggesting that D1 and D2 are inherently different.
To address the physiological functions of RTPs and the roles of their noncatalytic domains, in vivo systems in which RTPs have easily assayable functions are invaluable. The identification of mutations in several Drosophila RTPs has allowed an initial analysis of the roles of RTPs in the nervous system (Desai et al. 1996, 1997; Krueger et al. 1996). Here we identify a receptor tyrosine phosphatase, CLR-1, in the nematode Caenorhabditis elegans. Mutations that compromise the function of CLR-1 confer a distinctive and highly penetrant phenotype. Our analysis of CLR-1 has enabled us to define its role in a fibroblast growth factor receptor (FGFR) signaling pathway in C. elegans and to study the functions of its two intracellular phosphatase domains.
Results
Mutations in clr-1 are isolated as suppressors of egl-15(n1477ts)
egl-15 encodes a C. elegans FGFR that is involved in sex myoblast migration guidance and an essential function (DeVore et al. 1995). Whereas egl-15 mutants homozygous for putative null alleles display a larval arrest phenotype, certain hypomorphic mutations in egl-15 can confer a Scrawny (Scr) body morphology that may be a weaker manifestation of the null phenotype. To identify genes that interact with egl-15, we isolated mutations that could suppress the temperature-sensitive scrawny body morphology of egl-15(n1477ts) animals (Fig. 1B). Eighteen suppressor strains were isolated from animals representing ∼51,000 mutagenized haploid genomes. When observed in a wild-type background, 12 of the 18 suppressor mutations caused various degrees of the distinctive Clr (clear) phenotype originally defined by the temperature-sensitive recessive mutant clr-1(e1745ts) (Fig. 1C; Hedgecock et al. 1990). Consistent with their being clr-1 alleles, all 12 suppressor mutations showed linkage to the dpy-10(e128) marker, which lies 2.2 map units right of clr-1 on linkage group II, and failed to complement clr-1(e1745ts) for the Clr phenotype (data not shown). In addition, we found that clr-1(e1745ts) also suppressed the Scr phenotype of egl-15(n1477ts) animals (Fig. 1D). We conclude that these 12 suppressors are alleles of clr-1. Of the remaining six suppressors, three are likely to be intragenic revertants of egl-15(n1477ts) (see Materials and Methods) and three have not been characterized further.
Figure 1.

Antagonistic roles of egl-15 and clr-1. Bright-field micrographs of adult hermaphrodites. (A) Wild type. (B) egl-15(n1477ts) animal, which displays a temperature-sensitive Scr body morphology. (C) clr-1(e1745ts) animal, which displays a temperature-sensitive Clr phenotype. (D) clr-1(e1745ts); egl-15(n1477ts) double mutant, which displays neither the Clr nor the Scr phenotype, demonstrating a mutual suppression between non-null alleles of clr-1 and egl-15. Because clr-1(e1745ts) is not a potent suppressor of the egg-laying and SM mispositioning defects of egl-15(n1477ts), the animal is beginning to accumulate eggs in her uterus. (E) Clr transgenic animal bearing an extrachromosomal array made with the neu*-activated egl-15 construct. (F) Non-Clr transgenic animal bearing an extrachromosomal array made with a kinase-defective neu*-activated egl-15 construct [egl-15(neu*K672A)]. Animals in A–D were raised at 25°C; transgenic animals shown in E and F were raised at 20°C. Transgenic animals contain the transformation marker plasmid pRF4 that confers a Roller phenotype (Mello et al. 1991). Scale bar (A–F), 300 μm.
From a number of direct visual screens for Clr animals, we collected 15 additional mutations that confer a recessive Clr phenotype in a wild-type background. These mutations all failed to complement clr-1(e1745ts), suppressed the Scr phenotype conferred by egl-15(n1477ts), and were suppressed by egl-15(n1477ts). Because all 15 mutations appeared to be clr-1 alleles, clr-1 is likely to be the only gene that is easily mutable to result in a viable Clr phenotype. These results also provide evidence that the interaction observed between clr-1 and egl-15 does not require a specific type of clr-1 mutation. Although mutations conferring the weakest Clr phenotypes were isolated only as suppressors of egl-15(n1477ts), mutations conferring the stronger Clr phenotypes were obtained in both screens. Certain mutations among those that confer the strongest Clr phenotype appeared to eliminate clr-1 function by a variety of genetic and molecular criteria (see Materials and Methods).
clr-1 appears to play a minor role in guiding the migrations of the hermaphrodite sex myoblasts (SMs). In a wild-type background, mutations in clr-1 do not dramatically affect the ability of the SMs to migrate to their precise final positions (Fig. 2). Furthermore, whereas mutations in clr-1 dramatically suppress the body morphology defects conferred by egl-15(n1477ts) (e.g., see Fig. 1D), they do not rescue the mispositioning of the SMs found in egl-15(n1477ts) mutants (Fig. 2). Nonetheless, clr-1 mutations subtly shift the range of the displaced SMs closer to their wild-type positions and correspondingly decrease the penetrance of the egg-laying defect caused by certain egl-15 mutations, suggesting a minor role in SM migration guidance.
Figure 2.

clr-1 mutations have minor effects on SM migration. The positions of individual SMs are indicated by hash marks and are scored relative to the animal diagrammed above, as previously described (Thomas et al. 1990). The ruler indicates positions in units of the distance between Pn.p cells; the arrow marks their normal positions flanking the precise center of the gonad. Many other alleles of clr-1 have similar effects on the SM distribution of egl-15(n1477ts) animals, and clr-1(e1745ts) has similar weak effects on all other egl-15 mutants tested (data not shown). SMs were scored at 20°C for all strains shown.
The Clr phenotype
Animals homozygous for the clr-1(e1745ts) mutation appear completely wild-type at the permissive temperature of 15°C but display a severe Clr phenotype when raised at the nonpermissive temperature of 25°C (Fig. 1C). Clr animals are extremely short, immobile, and infertile, and their pharynx and intestine appear to float within the pseudocoelom. When viewed using Nomarski microscopy, the cells and cell processes in these animals appear to be separated from each other, a phenotype that has been used extensively to visualize neuronal processes and cell boundaries in C. elegans (e.g., see Way and Chalfie 1988; Hedgecock et al. 1990). These abnormalities all appear to result from clear fluid accumulating in the pseudocoelom. Whereas some animals homozygous for the most severe alleles of clr-1 die during larval development with hypodermal ruptures, most remain alive and display an extreme Clr phenotype. The biological basis of the Clr phenotype is not understood, although it mimics the phenotype observed when cells in the C. elegans excretory system (Nelson and Riddle 1984) or the CAN neurons (Forrester and Garriga 1997) are destroyed by laser microsurgery.
Relationship between clr-1 and egl-15
The mutual suppression observed between egl-15(n1477ts) and mutations in clr-1 indicates that these genes act antagonistically. To clarify the nature of the interactions between clr-1 and egl-15, double mutants using putative null alleles of both clr-1 and egl-15 were constructed. Complete loss of egl-15 function results in early larval arrest (DeVore et al. 1995). The clr-1(e2530); egl-15(n1454) double mutant also displays this larval arrest phenotype but does not display the Clr phenotype of similarly staged clr-1(e2530) mutants (Fig. 3). Thus, the Clr phenotype of clr-1 mutants requires egl-15 activity. The finding that both hypomorphic and null alleles of egl-15 can suppress the Clr phenotype conferred by clr-1 null mutations suggests that CLR-1 acts as a negative regulator of EGL-15 function and that the Clr phenotype may be caused by hyperactivity of this FGFR-mediated signaling pathway.
Figure 3.

Interaction between null alleles of egl-15 and clr-1. Nomarski photomicrographs of the pharyngeal and anterior body regions of first larval stage hermaphrodites. (A) Wild type. (B) egl-15(n1454), a null mutant. (C) clr-1(e2530), a null mutant. (D) clr-1(e2530); egl-15(n1454) double mutant. The Clr phenotype is seen only in C, which shows the intestine separating from the body wall. Anterior is left; dorsal is up. The close proximity of the intestine and body wall demonstrates suppression of the Clr phenotype by loss of egl-15. Scale bar (A–D), 30 μm.
To test directly whether EGL-15 hyperactivity can confer a Clr phenotype, we replaced sequences encoding the transmembrane domain of EGL-15 with those encoding the transmembrane domain of the constitutively activated oncogenic form of the neu proto-oncogene (Bargmann et al. 1986). This domain can cause the constitutive dimerization and activation of the Neu receptor tyrosine kinase (Weiner et al. 1989). Introduction of this egl-15(neu*) construct into wild-type animals by germ-line transformation could confer a Clr phenotype similar to that observed in clr-1 mutants (Fig. 1E). By contrast, introduction of a similar construct containing an additional mutation that is predicted to abolish kinase activity had no effect [egl-15(neu*K672A); Fig. 1F]. Thus, we conclude that hyperactivation of the kinase activity of EGL-15 can confer a Clr phenotype.
clr-1 is predicted to encode a receptor tyrosine phosphatase
Cosmids from the region of the C. elegans physical map most likely to contain clr-1 were tested for their abilities to rescue the Clr phenotype of clr-1(e1745ts) animals following germ-line transformation. This assay was used to show that clr-1 activity is contained on cosmid F43G3 and to narrow rescuing activity to a 7.8-kb genomic fragment (data not shown). cDNAs with sequences that correspond to portions of the 7.8-kb rescuing fragment were isolated and analyzed. These cDNAs provide evidence for two alternatively spliced clr-1 transcripts, termed cDNA1 and cDNA2 (see Materials and Methods).
The clr-1 transcript corresponding to cDNA1 contains a single, large ORF that is predicted to encode a protein of 1409 amino acids (Fig. 4A). Within this protein are two stretches of hydrophobic amino acids (residues 1–19 and 719–738) predicted to correspond to a signal sequence and a transmembrane domain. This protein also contains an amino-terminal region of 72 amino acids (residues 64–135) of which 15% are cysteines. Comparative sequence analysis revealed the presence of two phosphatase domains in the presumptive intracellular portion of CLR-1 (Fig. 5), indicating that CLR-1 is a receptor tyrosine phosphatase. In addition, our searches suggested the presence of two immunoglobulin-like domains and one FN III repeat in the presumptive extracellular region of CLR-1 (data not shown). The combination of these domains is characteristic of the type II family of RTPs (Fischer et al. 1991); however, no previously known RTP has the same number and arrangement of domains found in CLR-1. Based on this difference in overall organization and the fact that the CLR-1 sequence has similar levels of identity to the sequences of several RTPs, we conclude that CLR-1 is not the specific homolog of any known RTP.
Figure 4.

Two alternatively spliced clr-1 transcripts. (A) Genomic organization of the 9-kb (StuI–StuI) clr-1 genomic rescuing fragment. (Solid boxes) Coding sequence; (hatched boxes) untranslated regions of cDNA1. The SL1 trans-spliced leader sequence is found at the 5′ end of clr-1 transcripts. Domains identified on the basis of sequence similarity are labeled: (C-rich) Cysteine-rich; (IG) immunoglobulin-like; (FN) fibronectin type III repeat; (TM) transmembrane; (D1,D2) phosphatase domains 1 and 2. The deletion associated with clr-1(e2530) is indicated above the clr-1 genomic structure. (^) Alternate splice-donor sites. (B) The two transcripts corresponding to clr-1 differ in the use of a single splice–donor site. Genomic sequence surrounding the alternative splice is shown on the first line. Exons are boxed; the intron is in parenthesis. (Dashed box) Additional seven nucleotides found in cDNA1, which maintains an ORF that extends beyond the phosphatase domains. The splice that generates cDNA2 creates a UGA stop codon (*) just downstream of the splice–acceptor site. (C) clr-1-rescuing activity of the individual alternatively spliced clr-1 forms. Diagrams of the predicted protein products of the constructs introduced by germ-line transformation are shown. (Horizontal line) Cell membrane; (half circles) immunoglobulin domains; (hatched boxes) FN III repeats; (solid boxes) phosphatase domains. DNA constructs were tested for rescue in both F1 transformants (Rescued F1s) and stable lines (Rescued lines).
Figure 5.

Comparison of the tyrosine phosphatase domains from CLR-1, DPTP (Streuli et al. 1989), HPTPα (Krueger et al. 1990), and HPTPε (Krueger et al. 1990). (Solid boxes) Identities among at least two of the eight domains; (dashes) gaps introduced by the alignment. The membrane-proximal (D1) and membrane-distal (D2) domains are grouped separately. The 11 amino acid core consensus sequence is underlined. Numbers are according to residues in the D1 domain of CLR-1. The amino-terminal wedge of the CLR-1 D1 domain corresponds to amino acids 772–802.
The clr-1 transcript corresponding to cDNA2 is identical to that corresponding to cDNA1 except for seven fewer nucleotides at the 3′ end of exon 13 (Fig. 4B). This difference appears to result from the use of an alternative splice–donor site seven nucleotides upstream of the site used in cDNA1. As a consequence of the use of this alternative splice site, cDNA2 contains a stop codon that truncates its protein product early within the first phosphatase domain. Northern analysis revealed a single band that corresponds to the 4.4-kb transcript size predicted for both cDNAs (data not shown).
To determine whether either cDNA1 or cDNA2 could provide clr-1 rescuing activity, we subcloned the unique portion of each cDNA into the genomic rescuing fragment and tested the abilities of the resulting constructs to rescue clr-1(e1745ts). As shown in Fig. 4C, the construct encoding only the full-length protein provided clr-1 rescuing activity, whereas the construct encoding only the truncated product did not. Thus, the full-length form of CLR-1 appears to be necessary for clr-1 function in vivo.
The role of the phosphatase domains in CLR-1 function
The core consensus sequence of tyrosine phosphatase domains, (I/V)HCXAGXXR(S/T)G, lies at the active site and contains the cysteine and arginine residues that play essential roles in catalysis (Streuli et al. 1990; Denu et al. 1996). In CLR-1, the membrane-proximal domain (D1) contains this core consensus sequence (Fig. 5). In the distal domain (D2), however, the arginine is replaced with glutamic acid, suggesting that this domain is not catalytically active (Fig. 5). As in other RTPs (Bilwes et al. 1996), only the D1 domain of CLR-1 appears to contain the amino-terminal wedge sequences.
To determine whether CLR-1 has intrinsic tyrosine phosphatase activity, we tested CLR-1 for its ability to release inorganic phosphate from a tyrosine phosphorylated substrate in vitro. This experiment was performed in two different ways. First, full-length CLR-1 was expressed in COS cells, immunoprecipitated using anti-CLR-1 antibodies, and assayed for tyrosine phosphatase activity (Fig. 6A). By this assay, full-length CLR-1 has significant phosphatase activity when compared to the vector control. Because only the first phosphatase domain was predicted to be catalytically active, we similarly tested a CLR-1 construct, CLR-1(C1013S), in which we mutated the active-site cysteine in the first phosphatase domain to serine. The activity of CLR-1(C1013S) was indistinguishable from the vector control, providing strong evidence that CLR-1 itself has phosphatase activity rather than the activity being caused by another molecule that coimmunoprecipitates with CLR-1. Our second assay for CLR-1 phosphatase activity assessed the specific activity of the phosphatase domains of CLR-1 (Fig. 6B). A portion of the intracellular domain of CLR-1 containing both phosphatase domains was expressed as a fusion protein with glutathione S-transferase (GST), purified, and assayed in vitro for phosphatase activity. The phosphatase domains from wild-type CLR-1 showed significant phosphatase activity in this assay, whereas the identical protein containing the C1013S mutation showed no activity. These data support the hypothesis that CLR-1 can function as a tyrosine phosphatase and that only the D1 domain has significant catalytic activity.
Figure 6.
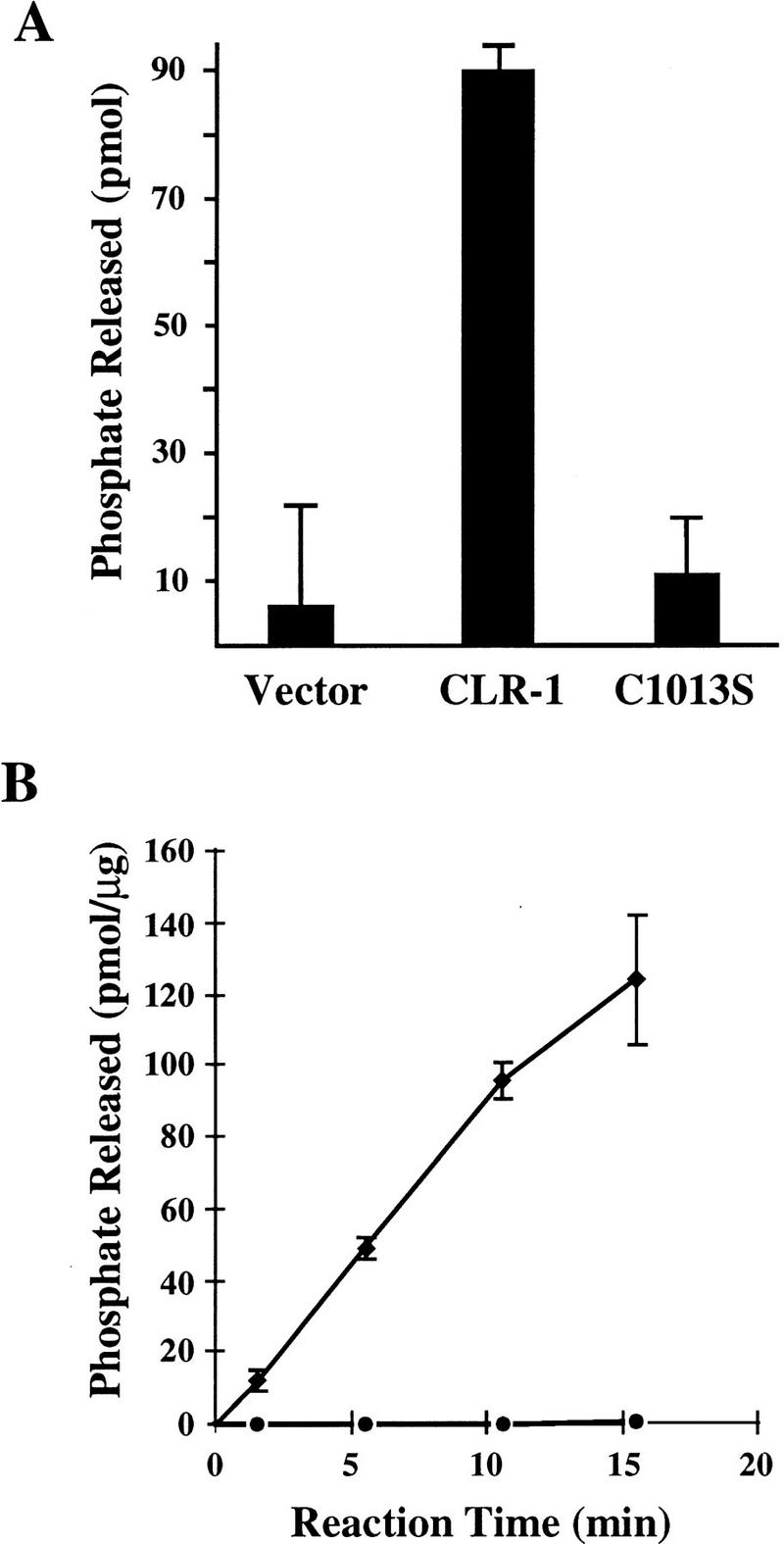
Tyrosine phosphatase activity of CLR-1. (A) Full-length CLR-1 expressed in COS cells. (Solid bars) Amount of phosphate released from a Tyr-32P-labeled RCML substrate by CLR-1 immunoprecipitates from COS cells transiently transfected with the wild-type clr-1 cDNA1 (CLR-1), the clr-1 cDNA1 harboring the C1013S mutation in the membrane-proximal phosphatase domain (C1013S), or the expression vector alone (Vector). (B) The phosphatase domain region of the intracellular domain of CLR-1 expressed in E. coli. Phosphate released was measured for GST fusions to the wild-type phosphatase domains (♦) and the identical protein bearing the C1013S mutation in the D1 domain (•). Error bars represent the standard errors of the mean.
To test whether the observed phosphatase activity of the D1 domain is essential for the physiological function of CLR-1, we introduced the C1013S mutation into the genomic rescuing fragment. This alteration abolished clr-1-rescuing activity (Fig. 7), supporting the hypothesis that CLR-1 functions as a phosphatase in vivo. Further evidence that the catalytic activity of CLR-1 is necessary for function comes from the finding that several endogenous clr-1 mutations affect conserved residues in the D1 domain (Table 1). Three of these mutations affect residues in the catalytic core consensus sequence. These three mutations are among those that confer the strongest Clr phenotypes, consistent with the importance of this region for catalysis.
Figure 7.

Role of CLR-1 phosphatase domains in clr-1-rescuing activity. Injected constructs are diagrammed as in Fig. 4. C1013S has the catalytic cysteine of the membrane-proximal phosphatase domain mutated to serine. Pst FS has a frameshift mutation at a PstI site at the beginning of the region encoding the membrane-distal phosphatase domain. ΔD2 deletes the region between the site of the nonsense mutation in clr-1(e1745ts) and the normal clr-1 stop codon. The rescue results shown are from constructs lacking the GFP insert; similar results were obtained with constructs bearing the clr-1::GFP fusion. Localization and steady-state levels of CLR-1 products from the injected construct were determined by measuring GFP expression in lines obtained from constructs bearing the clr-1::GFP fusion. At least 10 animals from each line were observed for GFP expression. Lines were scored as GFP+ if the intensity and distribution of GFP expression was similar to that observed for transformants bearing the wild-type clr-1::GFP construct. Those lines scored as GFP− had no detectable GFP expression in all animals observed. GFP results are shown for lines obtained in a clr-1(e1745ts) background, except for the ΔD2 construct in which case results were obtained in a wild-type background. Similar GFP expression was observed in both genetic backgrounds wherever tested. GFP expression levels for the ΔD2 construct were slightly greater than for the wild-type clr-1::GFP construct.
Table 1.
clr-1 mutations in phosphatase domain 1
| Allele
|
Protein alteration
|
Sequence
|
Phenotypic strength
|
|
|---|---|---|---|---|
| wild type
|
mutant
|
|||
| 15°C non-Clr | ||||
| e1745ts | W1072–Opal | TGG | TGA | 20°C intermediate |
| 25°C strong | ||||
| n1635 | P985S | CCG | TCG | intermediate |
| n1641 | A1015V | GCT | GTT | strong |
| n1649 | A862V | GCT | GTT | weak |
| n1660 | G1021R | GGA | AGA | strong |
| gm30 | C1013Y | TGC | TAC | strong |
Sequence alterations found in clr-1 mutants are underlined. Altered amino acids are abbreviated in the single-letter code and are numbered as in Fig. 5. The phenotypic strength of the alleles was judged in homozygous animals according to the following criteria: (non-Clr) Animals are indistinguishable from the wild type; (weak) the only observable defect is a subtle filling of the pseudocoelom; (intermediate) animals display an obvious fluid-filled pseudocoelom but are predominantly non-Dpy and the strain can be propagated; (strong) animals are severely Clr, infertile, Dpy, and immobile.
The failure of the C1013S construct to rescue clr-1(e1745ts) in germ-line transformation rescue experiments could be a consequence of global effects on CLR-1 rather than the intended specific alteration in the function of the D1 phosphatase domain. To monitor the localization and steady-state levels of expression of altered clr-1 constructs introduced by germ-line transformation, we created a construct encoding a protein in which the green fluorescent protein (GFP) is fused to the amino terminus of the predicted mature CLR-1 product. This construct (clr-1::gfp) rescued clr-1(e1745ts), albeit at slightly lower efficiency than the wild-type genomic rescuing fragment (data not shown). In lines transformed with this construct, GFP fluorescence was detected at the periphery of body muscles, consistent with localization of the CLR-1 RTP to the plasma membrane (Fig. 8). Although the C1013S mutation abolished rescuing activity, it did not affect either the localization or expression levels of GFP when introduced into the clr-1::gfp construct (Fig. 7). Thus, the inability of CLR-1(C1013S) to rescue clr-1(e1745ts) is likely caused by the lack of phosphatase activity of the D1 domain.
Figure 8.

GFP expression in animals containing clr-1::GFP transgenes. Wild-type clr-1::GFP at 15°C (A) and 25°C (B); clr-1(e1745ts)::GFP at 15°C (C) and 25°C (D). All lines were generated in a wild-type background using clr-1::GFP constructs injected at 150 ng/μl; additional lines behaved similarly.
The ability to use clr-1::gfp to distinguish specific mutational effects versus more global effects allowed us to perform a structure/function analysis of the D2 phosphatase domain of CLR-1. Several lines of evidence indicate that this phosphatase domain is not essential for clr-1-rescuing activity, although mutational perturbations carboxy-terminal to domain D1 can destabilize the entire protein. First, a frameshift mutation predicted to result in a CLR-1 product truncated at the very beginning of the D2 phosphatase domain (Pst FS) could confer partial rescuing activity (Fig. 7). The reduced rescuing activity of this construct appears to be caused by reduced levels of steady-state product, since no detectable GFP fluorescence was visible in any of the transformed lines (Fig. 7). Second, the temperature-sensitive mutation clr-1(e1745ts) was found to correspond to a nonsense mutation affecting the very carboxyl terminus of the D1 phosphatase domain (W1072–opal; Table 1). We postulated that the same truncated product is encoded at both temperatures. Since clr-1(e1745ts) appears wild-type at the permissive temperature, this product must be functional. However, at the nonpermissive temperature, the premature translational termination might result in destabilization of either the clr-1 message or encoded protein, and steady-state levels of the product could drop below a level critical for normal function. The temperature-sensitivity of steady-state levels of CLR-1(e1745ts) protein was tested by introducing the W1072–opal mutation into the clr-1::gfp construct. Consistent with our model, all lines obtained with this construct displayed a temperature-sensitive GFP fluorescence pattern (Fig. 8).
To test whether a CLR-1 construct completely lacking the D2 phosphatase domain could provide efficient clr-1-rescuing activity, we deleted clr-1 sequences between the site of the e1745ts mutation and the endogenous clr-1 stop codon (ΔD2). This construct was capable of providing clr-1-rescuing activity following germ-line transformation. In addition to rescuing the temperature-sensitive mutant clr-1(e1745ts) (Fig. 7), this construct could also rescue the null mutant clr-1(n1992) (see Materials and Methods), eliminating the possibility of any contribution from the chromosomal copies of clr-1. On the basis of these data, the D2 phosphatase domain of CLR-1 does not appear to be required to prevent the Clr phenotype in vivo. The quantitative decrease in rescuing activity observed for animals transiently transformed with this construct might reflect slightly compromised activity caused by the deletion of the D2 domain or truncation of the final α helix of the D1 domain. It is interesting that the ΔD2 construct apparently produces a temperature-stable product, given that its encoded product is essentially identical to that encoded by clr-1(e1745ts). In fact, levels of GFP fluorescence from the ΔD2 clr-1::gfp construct are more robust than those from the wild-type clr-1::gfp construct (data not shown). It is possible that the additional untranslated portion of clr-1(e1745ts) is responsible for destabilizing the message at the nonpermissive temperature.
Discussion
We have discovered that clr-1 encodes an RTP comprising a large extracellular region containing two immunoglobulin-like domains and a FN III repeat and an intracellular region containing two phosphatase domains. Mutations that compromise or eliminate the function of the CLR-1 protein confer a dramatic translucent or Clr phenotype. The Clr phenotype has provided an assay for CLR-1 function that we have used to assess the roles of its phosphatase domains and to implicate CLR-1 in regulating an FGFR signaling pathway in C. elegans.
The membrane-proximal phosphatase (D1) domain of CLR-1 appears to be catalytically active and essential for its in vivo function. The full-length CLR-1 protein showed in vitro phosphatase activity when expressed in COS cells, as did a bacterially expressed fragment of CLR-1 containing both phosphatase domains. Both activities appeared to be completely dependent upon the catalytic activity of the D1 phosphatase domain, since all phosphatase activity was abolished by mutating the nucleophilic cysteine of this domain to serine. This alteration also destroyed clr-1-rescuing activity without changing the expression or localization of the CLR-1 product. Thus, CLR-1 phosphatase activity appears to be essential for its physiological function. This conclusion is supported by the identification of alterations of key residues of the D1 phosphatase domain in clr-1 mutants.
In contrast, the membrane-distal phosphatase (D2) domain of CLR-1 does not appear to have tyrosine phosphatase catalytic activity and is not essential for clr-1-rescuing activity in vivo. The D2 domain is missing a residue present within the catalytic core of active phosphatase domains and is thought to be essential for activity. Furthermore, the D2 domain did not appear to have any in vitro tyrosine phosphatase activity. The nonessential nature of the D2 phosphatase domain for CLR-1 function is supported by a variety of CLR-1 mutant forms that lack the D2 domain but appear to be functional. These include a temperature-sensitive mutant, clr-1(e1745ts), that is phenotypically wild-type at the permissive temperature. The molecular lesion associated with e1745ts is predicted to truncate CLR-1 prior to the second phosphatase domain at all temperatures, and yet is non-Clr at the permissive temperature. This result is corroborated by the function of a construct that eliminates clr-1 sequences between the position of the e1745ts nonsense mutation and the endogenous clr-1 stop codon. This construct is capable of rescuing even a putative null allele of clr-1. These data demonstrate that CLR-1 can function essentially normally without its D2 phosphatase domain.
The role of the D2 phosphatase domains found in most RTPs has been one of several unresolved issues concerning the functions of RTPs. D2 domains have been found to mediate associations with other molecules (Serra-Pagès et al. 1995; Debant et al. 1996) and to be able to alter the in vitro substrate specificity of the D1 domain (Streuli et al. 1990; Wang and Pallen 1991). Furthermore, domains having striking sequence similarity to tyrosine phosphatase domains, but lacking phosphatase activity, have also been found to have other catalytic activities, suggesting that D2 domains might similarly have other activities (Takagi et al. 1997). Nonetheless, the physiological roles of these domains have been difficult to establish. Using CD45-defective cell lines, the physiological roles of the two intracellular phosphatase domains of the CD45 RTP have been studied. CD45, which is necessary for antigen receptor signal transduction (Trowbridge and Thomas 1994), appears to require the tyrosine phosphatase activity of its D1 domain but not that of its D2 domain (Desai et al. 1994). A single membrane-tethered tyrosine phosphatase domain can even restore some CD45-like activity to CD45-defective cell lines. However, this activity is clearly suboptimal, leaving open the possibility of other important roles for the D2 domain of CD45 (Motto et al. 1994). We have several lines of evidence that suggest that the D2 domain of CLR-1 is not essential for in vivo clr-1-rescuing activity; the absence of this domain appears to confer no obvious phenotypic consequences in otherwise wild-type animals.
CLR-1 affects an FGFR signaling pathway
The isolation of clr-1 alleles as suppressors of hypomorphic mutations affecting the EGL-15 FGFR implicates the CLR-1 RTP in FGF signaling pathways. The mutual suppression observed between clr-1 and egl-15 mutants indicates that the CLR-1 phosphatase and the EGL-15 kinase have opposing actions. Genetic analysis using putative null alleles of egl-15 and clr-1 demonstrated that complete loss of CLR-1 activity produces a phenotype that is dependent upon the functional integrity of the EGL-15 FGFR. In contrast, complete loss of EGL-15 FGFR activity produces a phenotype that is unaffected by CLR-1 RTP function. Based on these observations, we propose that CLR-1 acts as a negative regulator of an EGL-15-mediated signaling pathway. Thus, the Clr phenotype of clr-1 mutants may result from the hyperactivity of this FGFR. Consistent with this hypothesis, an EGL-15 construct bearing a mutation known to constitutively activate a variety of receptor tyrosine kinases could confer a Clr phenotype when introduced into an otherwise wild-type background. Taken together with the requirement for the activity of the D1 phosphatase domain of CLR-1, CLR-1-mediated dephosphorylation seems likely to reduce signaling through the EGL-15 pathway.
Stimulation of FGFRs, like other receptor tyrosine kinases, results in the activation and tyrosine phosphorylation of both the receptor itself and additional proteins thought to act as downstream signaling components (for review, see van der Geer and Hunter 1994). Some of these components include phospholipase C-γ (Burgess et al. 1990), Shc (Spivak-Kroizman et al. 1994; Vainikka et al. 1994; Wang et al. 1994), and the p90 phosphoprotein FRS2 (Coughlin et al. 1988; Kouhara et al. 1997). Although these components have not yet been identified in FGF signaling pathways in C. elegans, the EGL-15 FGFR contains regions that are conserved among many FGFRs and include some of the tyrosine residues known to be phosphorylated in response to FGF in the human type 1 FGFR (Mohammadi et al. 1991, 1996). Although these residues are possible targets of CLR-1 phosphatase activity, our genetic data do not establish that CLR-1 dephosphorylates EGL-15 directly. Rather, CLR-1 could act on any molecule activated by tyrosine phosphorylation in response to the stimulation of EGL-15.
The complete suppressibility of the Clr phenotype by mutations in egl-15 does not necessarily indicate that CLR-1 acts only on a component of the FGFR signaling pathway. It is possible that CLR-1 has roles in other pathways as well, but that its failure to function in these pathways does not result in an obvious phenotypic effect in a wild-type background. In fact, we have observed (Chen et al. 1997; M.J. Stern, unpubl.) that mutations in clr-1 can partially suppress the effect of mutations that compromise the vulval induction pathway mediated by the epidermal growth factor receptor (EGFR) homolog LET-23 (Aroian et al. 1990). Thus, CLR-1 may partially antagonize signaling mediated by both FGFR and EGFR tyrosine kinases. Such other roles of CLR-1 might not be revealed by clr-1 mutations in a wild-type background if the other pathways affected by CLR-1 have different primary negative regulatory elements.
Other genes associated with clr-1
RTPs, including CLR-1, have large regions that are not necessary for tyrosine phosphatase catalytic activity and that might play important regulatory roles by mediating interactions with other components. Such components might be identified by mutations that confer a similar Clr phenotype. We have characterized 15 mutants that were isolated on the basis of their Clr phenotypes, and all have mutations in clr-1. This result indicates that positively acting factors, if they exist, are not easily mutable to give this specific phenotype. Conversely, components that negatively regulate CLR-1 activity might be identified as clr-1 suppressors that restore activity to a partially inactivated clr-1 mutant. We have isolated a large number of clr-1(e1745ts) suppressors that define four genes (DeVore et al. 1995). These genes do not appear to encode components that negatively regulate CLR-1, as representative alleles of each of these four genes suppress the putative null mutant clr-1(e2530) (M. Kokel and M.J. Stern, unpubl.). The behavior of these mutants is more consistent with their requirement for the EGL-15 signal transduction pathway. The identification of sem-5 among these suppressors is consistent with this hypothesis.
The discovery of the large class of protein tyrosine phosphatases led to great excitement in part because it was expected that they would play instrumental roles in the regulation of pathways controlled by tyrosine phosphorylation. Many important developmental processes involve FGF signaling pathways whose activation is mediated by early tyrosine phosphorylation events. Thus, the regulation of these pathways is critical to many aspects of development and might be important in understanding the basis of various human diseases. In fact, a number of human skeletal disorders are associated with the hyperactivation of FGF receptors (De Moerlooze and Dickson 1997). The discovery of the CLR-1 RTP that attenuates the activity of an FGF signaling pathway in C. elegans provides a mechanism that might help control these important pathways as well.
Materials and methods
Genetic manipulations
C. elegans strains were maintained according to Brenner (1974) and grown at 20°C unless otherwise indicated. All work used strains derived from C. elegans var. Bristol, strain N2 (Brenner 1974). Standard genetic manipulations were used for all strain constructions (Herman 1988). clr-1(e2530); egl-15(n1454) double mutants were obtained from clr-1(e2530); egl-15(n1477ts)/egl-15(n1454) animals. Data for the genetic mapping of clr-1 are available from the C. elegans Genetic Stock Center and ACeDB.
The following clr-1 mutations were isolated in a variety of clonal screens on the basis of their abilities to confer a Clr phenotype in live animals: e1745ts, e2530, n1946, n1992, n2151, n2152, n2153, n2158, n2169, ay4, ay67, ay72, q421, gm30, and gm79. In addition to failing to complement clr-1(e1745ts), seven of these mutations (e2530, n1946, n1992, ay72, q421, gm30, and gm79) also showed linkage to markers tightly linked to clr-1.
clr-1(e2530) and clr-1(n1992) belong to a class of clr-1 mutations that confer the strongest Clr phenotype observed and behave as null mutations by a number of genetic criteria. clr-1(n1992) is associated with an amber nonsense mutation affecting Trp6 of CLR-1 and is suppressible by the amber suppressor sup-5(e1464sd) (data not shown). Molecular evidence that clr-1(e2530) is a null allele comes from the identification of a 645-bp deletion predicted to disrupt the D1 phosphatase domain and truncate the intracellular domain encoded by this allele (Fig. 4A).
Isolation and characterization of egl-15(n1477ts) suppressors
We isolated suppressors of the Scr body morphology of egl-15(n1477ts) mutants by mutagenizing egl-15(n1477ts) animals maintained at 20°C with ethyl methanesulfonate (Brenner 1974). L4 hermaphrodites were transferred to separate Petri dishes, three per plate. After the appearance of F2 progeny, the plates were transferred to 25°C and screened by eye for healthy animals after two to three days. True breeding non-Scr animals could be maintained at 25°C. Autosomal suppressors were separated from the X-linked egl-15(n1477ts) mutation by three consecutive backcrosses, and the resulting strains were observed for segregating animals with visible mutant abnormalities. The clr-1 mutations n1632, n1634, n1635, n1640–n1644, n1646, n1647, n1649, and n1650 were isolated in this screen. Three additional clr-1 mutations, n1657, n1660, and n1666, were isolated in a similar screen.
Three suppressors (n1633, n1639, and n1648) appeared to be X-linked during the backcrossing protocol and are candidates for being intragenic pseudorevertants of egl-15(n1477ts) X. First, these mutations are semidominant suppressors of egl-15(n1477ts). Second, n1633, the most strongly semidominant suppressor, was mapped to the region of egl-15 by three-factor mapping: in 58 crossover events in the 12 map unit region from dpy-6 to unc-9 spanning egl-15, n1633 was inseparable from egl-15(n1477ts). Third, a molecular lesion that corresponds to the n1633 mutation has been identified in egl-15 (T692I; D. DeVore and M.J. Stern, unpubl.).
Physical mapping and germ-line transformation
A contig consisting of an overlapping series of yeast artificial chromosomes (YACs) and cosmids assembled by the C. elegans genome project consortium was used to clone clr-1 (Coulson et al. 1988). The position of clr-1 was estimated based on genetic mapping data and the physical positions of lin-31, hlh-1, lin-4, and dpy-10, and localized by germ-line transformation assays (Mello et al. 1991) that relied on the ability of clr-1-containing DNA fragments to rescue the Clr phenotype of clr-1(e1745ts). Strains bearing extrachromosomal arrays formed from cosmids from this region and the cotransformation marker plasmid pRF4 were obtained from R. Predy and J. McGhee. pRF4 confers a Roller (Rol) phenotype to animals bearing this plasmid in extrachromosomal arrays (Mello et al. 1991). An array carrying the cosmid F43G3 was shown to confer a non-Clr phenotype in clr-1(e1745ts) homozygotes raised at 25°C. To narrow the region containing clr-1, transformation mixes containing DNA fragments from cosmid F43G3 (8 ng/μl) and the cotransformation plasmid pRF4 (100 ng/μl) were injected into clr-1(e1745ts) hermaphrodites grown at 15°C. Transformed (Rol) progeny were either shifted to 25°C and scored for rescue of the Clr phenotype (F1 rescuing activity) or maintained at 15°C for a second generation to establish transformed lines. Rescued animals no longer displayed the Clr phenotype when viewed with a dissecting microscope after growth at 25°C. Rescued lines were sufficiently non-Clr to propagate at 25°C. A 7.8-kb StuI–SpeI subclone of F43G3 was the smallest fragment that showed efficient rescuing activity in both F1 transformants and stable lines. This fragment was subsequently found to lack the 3′ terminus of the 3′ untranslated region of clr-1. For all subsequent analyses, we used derivatives of a 9-kb StuI–StuI genomic fragment containing the entire clr-1 gene (shown in Fig. 4A) injected at 30 ng/μl unless otherwise noted.
Germ-line transformation rescue of the clr-1 null mutant clr-1(n1992) was performed by injecting clr-1 constructs to be tested along with pRF4 into clr-1(n1992)+/+bli-2(e768) balanced heterozygotes. Rol lines were obtained and checked to see whether Rol clr-1(n1992) homozygotes could be established. Using this protocol, for one line of three that contained the clr-1(n1992) mutation we could establish a strain that was homozygous for clr-1(n1992) and contained the ΔD2 construct. Wild-type animals from the other two lines could also be found that segregated only Clr animals, suggesting that they, too, were homozygous for clr-1(n1992) and rescued by the ΔD2 construct.
Constructs of activated egl-15 [egl-15(neu*)] and the kinase-defective derivative [egl-15(neu*K672A)] were introduced into wild-type hermaphrodites at 20 ng/μl along with pRF4 as the cotransformation marker (100 ng/μl). F1 Rol transformants were scored for displaying the Clr and other phenotypes.
Isolation of cDNA clones
Six cDNA clones corresponding to different portions of the 7.8-kb rescuing fragment were isolated from a mixed-stage cDNA library (Barstead and Waterston 1989). Two of these cDNAs contain the SL1 splice-leader sequence found at the 5′ end of many C. elegans mRNAs (Krause and Hirsh 1987), suggesting that these cDNAs correspond to the 5′ end of the clr-1 message. One cDNA that does not overlap with the other five contains a poly-A tail and corresponds to the opposite end of the clr-1-rescuing fragment. Four pieces of evidence indicate that the two types of cDNAs represent the two ends of a single clr-1 message: (1) A PCR product bridging these two classes of cDNAs and representing a spliced message can be amplified from a second cDNA library; (2) a single mRNA of the predicted size can be detected on a northern blot; (3) the protein sequence predicted by joining these cDNAs as described below encodes a full-length RTP; and (4) a cDNA/genomic chimera containing a fragment of the full-length cDNA at the joint area could rescue clr-1(e1745ts), whereas one truncated by alternative splicing downstream of this joint failed to rescue.
A full-length clr-1 cDNA2 was assembled from these cDNAs and a genomic NdeI–BsrFI exon fragment that bridged these cDNAs. Two types of PCR products, corresponding to the alternatively spliced portions of cDNA1 and cDNA2, were obtained from each of two independent cDNA libraries (obtained from R. Barstead and L. Miller) using the primers 5′-CTGCCGTTCCTTCTGTTC-3′ and 5′-GGAAGTTGTCATTGATTCTC-3′. We assembled cDNA1 by replacing a BsrFI–EcoRI fragment in cDNA2 with the analagous fragment from a PCR product corresponding to cDNA1. The predicted amino acid sequence of cDNA1 was compared with published sequences using the program BLASTp (Altschul and Lipman 1990). The GenBank accession numbers for cDNA1 and cDNA2 are AF047880 and AF047881, respectively. The complete genomic sequence of clr-1 was obtained from the cosmid F56D1 (accession no. U39997) by the C. elegans Genome Sequencing Consortium.
Constructs
DNA manipulations were performed using standard procedures (Sambrook et al. 1989). Sequences of regions subjected to mutagenesis, PCR, or extension/deletion of restriction enzyme-generated ends were confirmed by sequence determination. Mutations were identified by sequencing exons encoding the very amino-terminal portion of CLR-1 and the D1 phosphatase domain. Genomic DNA from each clr-1; egl-15(n1477ts) strain was used to amplify the clr-1 coding region by PCR. Sequence was determined from PCR pools and confirmed from independently derived pools.
Genomic-cDNA chimeras (constructs NH#197 and NH#198) were constructed by inserting a 2.3-kb BamHI–PstI cDNA fragment spanning exons 9–16 into the corresponding sites in the 9-kb genomic rescuing fragment.
The C1013S mutation was introduced into a fragment of the full-length clr-1 cDNA1 using the Clonetech Transformer site-directed mutagenesis kit with the mutagenic primer 5′-CTCCAGCACTGGAATGTATCAC-3′. The BspEI–PstI fragment of this portion of the cDNA was introduced into the corresponding sites of the genomic rescuing fragment. The wild-type BspEI–PstI cDNA fragment did not affect clr-1-rescuing activity in this context (data not shown). The e1745ts mutation was introduced into the genomic rescuing fragment in a similar manner using the mutagenic primer 5′-CACCATACATGTGTCATTCCGCAAGTGC-3′.
The Pst FS construct (NH#228) was made by deleting 4 bp at the PstI site of the 9-kb genomic rescuing fragment by first cutting with PstI and then digesting the ends with T4 DNA polymerase.
The clr-1::GFP construct was made by adding EagI sites to both ends of GFP(S65C) from pPD95.02 (kindly provided by A. Fire, J. Ahnn, G. Seydoux, and S. Xu) using PCR and subcloning into the EagI site of the genomic rescuing fragment.
The ΔD2 construct was made by first introducing an XbaI site in the genomic rescuing fragment to replace the codon affected by the e1745ts mutation using the Clonetech Transformer site-directed mutagenesis kit with the mutagenic primer 5′-CACCATACATTCTAGATTCCGCAAGTGC-3′. An XbaI site was also added just 5′ to the endogenous stop codon by PCR using the primer 5′-CCTCTAGATAGCTGTTGTTATATTTTTCC-3′. Ligation of the XbaI ends of these fragments results in a construct encoding the putative e1745ts truncated product followed by two amino acids encoded by the inserted XbaI site, the endogenous TAG stop codon, and the 3′ UTR of clr-1.
The egl-15(neu*) construct was made by replacing the transmembrane domain of EGL-15 with that of the oncogenic form of NEU. Unique MluI and KpnI sites were added upstream and downstream, respectively, of sequences encoding the EGL-15 transmembrane domain using the mutagenic primers 5′-GCCATACATTTGCTCACGCGTGACGAACCG-3′ and 5′-GTTTATGGTGTGCAAAGGTACCCTACACAAAAAAGGATTC-3′. MluI and KpnI sites were added to flank sequences encoding the activated NEU transmembrane domain by using the primers 5′-GATACGCGTGGCTGCCCAGCAGAGCAGAG-3′ and 5′-GGGGTACCTCTCCTTCGTTTGATTAGGATTCCAACGACC-3′ to amplify this region from plasmid SRαneu* (kindly provided by C. Burke and D. Stern). The kinase-defective derivative egl-15(neu*K672A) was constructed using the mutagenic primer 5′-CATTTTCAATTTTGCGACAGCAACTGCG-3′.
Antibodies, COS cell expression, and phosphatase assay
A fragment encoding the amino-terminal portion of the extracellular domain of CLR-1 (the EagI–EcoRI fragment of cDNA1, corresponding to amino acids 23–154) was subcloned into pGEX4T-1 using a short BamHI–EagI linker. The fusion protein was induced with 1 mm IPTG for 4 hr at 30°C, purified by binding to glutathione–agarose beads (Sigma), and used to generate polyclonal anti-CLR-1 antibodies in rabbits (anti-258).
CLR-1 and CLR-1(C1013S) were expressed in COS cells using cDNA1 derivatives in the mammalian expression vector pFLAG-CMV3 (Kodak). NH#318 [CLR-1] and NH#319 [CLR-1(C1013S)] were transfected into COS cells in 10-cm dishes using lipofectamine (GIBCO BRL). Forty-eight hours after transfection, cells were washed once in PBS and lysed for 20 min at 4°C in 1 ml of ice-cold phosphatase lysis buffer (50 mm HEPES at pH 7.4, 150 mm NaCl, 1% Triton X-100 with 5 mm DTT, 1 μg/ml aprotinin, 100 μg/ml PMSF, and 1 μg/ml leupeptin). Protein A-purified αCLR-1 antibodies (anti-258) were used to precipitate CLR-1 from transiently transfected COS cell lysates at 4°C. Immunoprecipitates were washed four times in ice cold phosphatase lysis buffer with 5 mm DTT and two additional times in ice cold phosphatase assay buffer (50 mm HEPES at pH 7.4, 0.1 mg/ml BSA, 2 mm DTT).
Phosphatase assay buffer (120 μl) containing 5 μm Tyr-32P-labeled RCM-lysozyme (Charles et al. 1993) was added to each immunoprecipitate, incubated at 30°C for 20 min, and stopped by addition of 480 μl stop solution (10% suspension of Norit-A charcoal in 0.9 m HCl, 90 mm Na4P2O7, 2 mm Na2HPO4). Reactions were vortexed briefly, incubated at room temperature for 5 min, and centrifuged for 5 min. Phosphate release was determined by counting 500 μl of supernatant in 2 ml of scintillation fluid. Total possible counts were determined by counting substrate added directly to the scintillation fluid; the negative control reaction was performed in the absence of immunoprecipitated material.
Phosphatase activity of domains expressed in Escherichia coli
The region of CLR-1 containing the phosphatase domains was amplified by PCR using the primers 5′-CGCCCGGGTGCAATTCCAGTTGATGATCCTCC-3′ and 5′-GGCGGCCGTATATGTCTTAGAGATATAGTCAGCTAGGG-3′ and cDNA1 or cDNA1(C1013S) as template. These fragments were subcloned between the SmaI and NotI sites of pGEX4T-1 using the flanking SmaI and EagI sites in the primers. Phosphatase domain fusion proteins were induced with 20 μm IPTG for 2 hr at 30°C. Fusion proteins were purified by binding to glutathione agarose and eluting with 50 mm glutathione in 25 mm Tris at pH 7.4, 150 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, 5 mm DTT. Phosphatase assays were performed as described above in a 60 μl volume of assay buffer containing 2 μm Tyr-32P-labeled RCM– lysozyme for the reaction times indicated. The amount of fusion protein assayed was assessed by comparison to a BSA standard.
Acknowledgments
We thank E. Hedgecock for providing clr-1(e1745ts) and the following people for providing additional Clr mutants obtained from their genetic screens: E. Lambie, J. Kaplan, P. Kuwabara, J. Hodgkin, J. Plenefisch, P. Larsen, W. Forrester, G. Garriga, and L. Selfors. We thank R. Barstead and L. Miller for cDNA library aliquots, R. Feinbaum for providing maDf4 prior to publication, B. Williams for some PCR primers and protocols, N. Tsung for preliminary RFLP mapping, J. Schlessinger for mammalian expression vectors and advice, Andy Fire and colleagues for making available unpublished expression vectors, and A. Hudson for making the egl-15(K672A) mutation. Special thanks to R. Predy and J. McGhee for providing strains bearing extrachromosomal arrays with cosmids in the region of clr-1, H. Sun for preparation of Tyr[32P]-RCM-lysozyme and assistance with the phosphatase assay, and T. Chu for many helpful discussions. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources (NCRR). This work was supported by grants from The American Cancer Society (PF-2800), the Lucille P. Markey Charitable Trust, and The Patrick and Catherine Weldon Donaghue Medical Research Foundation. L.D. was supported by a postdoctoral fellowship from the Muscular Dystrophy Association. M.J.S. was a Research Associate and H.R.H. is an Investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL michael.stern@yale.edu; FAX (203) 737-2286.
References
- Altschul SF, Lipman DJ. Protein database searches for multiple alignments. Proc Natl Acad Sci. 1990;87:5509–5513. doi: 10.1073/pnas.87.14.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroian RV, Koga M, Mendel JE, Ohshima Y, Sternberg PW. The let-23 gene necessary for Caenorhabditis elegans vulval induction encodes a tyrosine kinase of the EGF receptor subfamily. Nature. 1990;348:693–699. doi: 10.1038/348693a0. [DOI] [PubMed] [Google Scholar]
- Bargmann CI, Hung MC, Weinberg RA. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986;45:649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- Barstead RJ, Waterston RH. The basal component of the nematode dense-body is vinculin. J Biol Chem. 1989;264:10177–10185. [PubMed] [Google Scholar]
- Bilwes AM, der Hertog J, Hunter T, Noel JP. Structural basis for inhibition of receptor protein-tyrosine phosphatase-α by dimerization. Nature. 1996;382:555–559. doi: 10.1038/382555a0. [DOI] [PubMed] [Google Scholar]
- Brady-Kalnay SM, Tonks NK. Protein tyrosine phosphatases as adhesion receptors. Curr Opin Cell Biol. 1995;7:650–657. doi: 10.1016/0955-0674(95)80106-5. [DOI] [PubMed] [Google Scholar]
- Brady-Kalnay SM, Flint AJ, Tonks NK. Homophilic binding of PTPμ, a receptor-type protein tyrosine phosphatase, can mediate cell-cell aggregation. J Cell Biol. 1993;122:961–972. doi: 10.1083/jcb.122.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess WH, Dionne CA, Kaplow J, Mudd R, Friesel R, Zilberstein A, Schlessinger J, Jaye M. Characterization and cDNA cloning of phospholipase c-γ, a major substrate for heparin-binding growth factor 1 (acidic fibroblast growth factor)-activated tyrosine kinase. Mol Cell Biol. 1990;10:4770–4777. doi: 10.1128/mcb.10.9.4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonneau H, Tonks NK. 1002 Protein phosphatases? Annu Rev Cell Biol. 1992;8:463–493. doi: 10.1146/annurev.cb.08.110192.002335. [DOI] [PubMed] [Google Scholar]
- Charles CH, Sun H, Lau LF, Tonks NK. The growth factor-inducible immediate-early gene 3CH134 encodes a protein-tyrosine-phosphatase. Proc Natl Acad Sci. 1993;90:5292–5296. doi: 10.1073/pnas.90.11.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EB, Branda CS, Stern MJ. Genetic enhancers of sem-5 define components of the gonad-independent guidance mechanism controlling sex myoblast migration in C. elegans hermaphrodites. Dev Biol. 1997;182:88–100. doi: 10.1006/dbio.1996.8473. [DOI] [PubMed] [Google Scholar]
- Cho H, Krishnaraj R, Kitas E, Bannwarth W, Walsh CT, Anderson KS. Isolation and structural elucidation of a novel phosphocysteine intermediate in the LAR protein tyrosine phosphatase enzymatic pathway. J Biol Chem. 1992;114:7296–7298. [Google Scholar]
- Coughlin SR, Barr PJ, Cousens LS, Fretto LJ, Williams LT. Acidic and basic fibroblast growth factors stimulate tyrosine kinase activity in vivo. J Biol Chem. 1988;263:988–993. [PubMed] [Google Scholar]
- Coulson A, Waterson R, Kiff J, Sulston J, Kohara Y. Genome linking with yeast artificial chromosomes. Nature. 1988;335:184–186. doi: 10.1038/335184a0. [DOI] [PubMed] [Google Scholar]
- De Moerlooze L, Dickson C. Skeletal disorders associated with fibroblast growth factor receptor mutations. Curr Opin Genet Dev. 1997;7:378–385. doi: 10.1016/s0959-437x(97)80152-9. [DOI] [PubMed] [Google Scholar]
- Debant A, Serra-Pagès C, Seipel K, O’Brien S, Tang M, Park S-H, Streuli M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci. 1996;93:5466–5471. doi: 10.1073/pnas.93.11.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denu JM, Stuckey JA, Saper MA, Dixon JE. Form and function in protein dephosphorylation. Cell. 1996;87:361–364. doi: 10.1016/s0092-8674(00)81356-2. [DOI] [PubMed] [Google Scholar]
- Desai DM, Sap J, Silvennoinen O, Schlessinger J, Weiss A. The catalytic activity of the CD45 membrane-proximal phosphatase domain is required for TCR signaling and regulation. EMBO J. 1994;13:4002–4010. doi: 10.1002/j.1460-2075.1994.tb06716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai CJ, Gindhart Jr JG, Goldstein LSB, Zinn K. Receptor tyrosine phosphatases are required for motor axon guidance in the Drosophila embryo. Cell. 1996;84:599–609. doi: 10.1016/s0092-8674(00)81035-1. [DOI] [PubMed] [Google Scholar]
- Desai CJ, Krueger NX, Saito H, Zinn K. Competition and cooperation among receptor tyrosine phosphatases control motoneuron growth cone guidance in Drosophila. Development. 1997;124:1941–1952. doi: 10.1242/dev.124.10.1941. [DOI] [PubMed] [Google Scholar]
- DeVore DL, Horvitz HR, Stern MJ. An FGF receptor signaling pathway is required for the normal cell migrations of the sex myoblasts in C. elegans hermaphrodites. Cell. 1995;83:611–620. doi: 10.1016/0092-8674(95)90101-9. [DOI] [PubMed] [Google Scholar]
- Fischer EH, Charbonneau H, Tonks NK. Protein tyrosine phosphatases: A diverse family of intracellular and transmembrane enzymes. Science. 1991;253:401–406. doi: 10.1126/science.1650499. [DOI] [PubMed] [Google Scholar]
- Forrester WC, Garriga G. Genes necessary for C. elegans cell and growth cone migrations. Development. 1997;124:1831–1843. doi: 10.1242/dev.124.9.1831. [DOI] [PubMed] [Google Scholar]
- Gebbink M, Zondag G, Wubbolts R, Beijersbergen R, Van Etten I, Moolenaar WH. Cell-cell adhesion mediated by a receptor-like protein tyrosine phosphatase. J Biol Chem. 1993;268:16101–16104. [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J Biol Chem. 1991;266:17026–17030. [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Hall DH. The unc-5, unc-6 and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron. 1990;2:61–85. doi: 10.1016/0896-6273(90)90444-k. [DOI] [PubMed] [Google Scholar]
- Herman RK. In: The nematode Caenorhabditis elegans. Wood WB, editor. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1998. pp. 17–45. [Google Scholar]
- Hunter T. Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- Kaplan R, Morse B, Huebner K, Croce C, Howk R, Ravera M, Ricca G, Jaye M, Schlessinger J. Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proc Natl Acad Sci. 1990;87:7000–7004. doi: 10.1073/pnas.87.18.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- Krause M, Hirsh D. A trans-spliced leader sequence on actin mRNA in C. elegans. Cell. 1987;49:753–761. doi: 10.1016/0092-8674(87)90613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger NX, Streuli M, Saito H. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J. 1990;9:3241–3252. doi: 10.1002/j.1460-2075.1990.tb07523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger NX, Van Vactor D, Wan HI, Gelbart WM, Goodman CS, Saito H. The transmembrane tyrosine phosphatase DLAR controls motor axon guidance in Drosophila. Cell. 1996;84:611–622. doi: 10.1016/s0092-8674(00)81036-3. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, Honegger AM, Rotin D, Fischer R, Bellot F, Li W, Dionne CA, Jaye M, Rubinstein M, Schlessinger J. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-γ1. Mol Cell Biol. 1991;11:5068–5078. doi: 10.1128/mcb.11.10.5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, Dikic I, Sorokin A, Burgess WH, Jaye M, Schlessinger J. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol Cell Biol. 1996;16:977–989. doi: 10.1128/mcb.16.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motto DG, Musci MA, Koretzky GA. Surface expression of a heterologous phosphatase complements CD45 deficiency in a T cell clone. J Exp Med. 1994;180:1359–1366. doi: 10.1084/jem.180.4.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson FK, Riddle DL. Functional study of the Caenorhabditis elegans secretory-excretory system using laser microsurgery. J Exp Zool. 1984;231:45–56. doi: 10.1002/jez.1402310107. [DOI] [PubMed] [Google Scholar]
- Pot DA, Dixon JE. Active site labeling of a receptor-like protein tyrosine phosphatase. J Biol Chem. 1992;267:140–143. [PubMed] [Google Scholar]
- Pot DA, Woodford TA, Remboutsika E, Haun RS, Dixon JE. Cloning, bacterial expression, purification, and characterization of the cytoplasmic domain of rat LAR, a receptor-like protein tyrosine phosphatase. J Biol Chem. 1991;226:19688–19696. [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sap J, Jiang YP, Friedlander D, Grumet M, Schlessinger J. Receptor tyrosine phosphatase R-PTP-kappa mediates homophilic binding. Mol Cell Biol. 1994;14:1–9. doi: 10.1128/mcb.14.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Pagès C, Kedersha NL, Fazikas L, Medley Q, Debant A, Streuli M. The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR-interacting protein co-localize at focal adhesions. EMBO J. 1995;14:2827–2838. doi: 10.1002/j.1460-2075.1995.tb07282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak-Kroizman T, Mohammadi M, Hu P, Jaye M, Schlessinger J, Lax I. Point mutation in the fibroblast growth factor receptor eliminates phosphatidylinositol hydrolysis without affecting neuronal differentiation of PC12 cells. J Biol Chem. 1994;269:14419–14423. [PubMed] [Google Scholar]
- Streuli M, Krueger NX, Tsai AYM, Saito H. A family of receptor-linked tyrosine phosphatases in humans and Drosophila. Proc Natl Acad Sci. 1989;86:8698–8702. doi: 10.1073/pnas.86.22.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli M, Krueger NX, Thai T, Tang M, Saito H. Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. EMBO J. 1990;9:2399–2407. doi: 10.1002/j.1460-2075.1990.tb07415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Tonks NK. The coordinated action of protein tyrosine phosphatases and kinases in cell signaling. Trends Biochem Sci. 1994;19:480–485. doi: 10.1016/0968-0004(94)90134-1. [DOI] [PubMed] [Google Scholar]
- Takagi T, Moore CR, Deihn F, Buratowski S. An RNA 5′-triphosphatase related to the protein tyrosine phosphatases. Cell. 1997;89:867–873. doi: 10.1016/s0092-8674(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Thomas JH, Stern MJ, Horvitz HR. Cell interactions coordinate the development of the C. elegans egg-laying system. Cell. 1990;62:1041–1052. doi: 10.1016/0092-8674(90)90382-o. [DOI] [PubMed] [Google Scholar]
- Trowbridge IS, Thomas ML. CD45: An emerging role as a protein tyrosine phosphatase required for lymphocyte activation and development. Annu Rev Immunol. 1994;12:85–116. doi: 10.1146/annurev.iy.12.040194.000505. [DOI] [PubMed] [Google Scholar]
- Vainikka S, Joukov V, Wennström S, Bergman M, Pelicci PG, Alitalo K. Signal transduction by fibroblast growth factor receptor-4 (FGFR-4) J Biol Chem. 1994;269:18320–18326. [PubMed] [Google Scholar]
- van der Geer P, Hunter T. Receptor tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol. 1994;10:251–337. doi: 10.1146/annurev.cb.10.110194.001343. [DOI] [PubMed] [Google Scholar]
- Walton KM, Dixon JE. Protein tyrosine phosphatases. Annu Rev Biochem. 1993;62:101–120. doi: 10.1146/annurev.bi.62.070193.000533. [DOI] [PubMed] [Google Scholar]
- Wang J-K, Gao G, Goldfarb M. Fibroblast growth factor receptors have different signaling and mitogenic potentials. Mol Cell Biol. 1994;14:181–188. doi: 10.1128/mcb.14.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Pallen CJ. The receptor-like protein tyrosine phosphatase HPTPα has two active catalytic domains with distinct substrate specifications. EMBO J. 1991;10:3231–3237. doi: 10.1002/j.1460-2075.1991.tb04886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way JC, Chalfie M. mec-3, a homeobox-containing gene that specifies differentiation of the touch receptor neurons in C. elegans. Cell. 1988;54:5–16. doi: 10.1016/0092-8674(88)90174-2. [DOI] [PubMed] [Google Scholar]
- Weiner DB, Liu J, Cohen JA, Williams WV. A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature. 1989;339:230–231. doi: 10.1038/339230a0. [DOI] [PubMed] [Google Scholar]