

Abstract
Different classes of sensory neurons in dorsal root ganglia (DRG) are generated in two waves: large-diameter trkC+ and trkB+ neurons are born first, followed by small-diameter trkA+ neurons. All such neurons require either neurogenin (ngn) 1 or 2, two neuronal determination genes encoding basic helix–loop–helix (bHLH) transcription factors. ngn2 is required primarily if not exclusively for the generation of trkC+ and trkB+ neurons, whereas the generation of most or all trkA+ neurons requires ngn1. Comparison with previous lineage tracing data in the chick suggests that this dichotomy reflects a requirement for the two ngns in distinct sensory precursor populations. The neurogenesis defect in ngn2−/− embryos is transient and later compensated by ngn1-dependent precursors, suggesting that feedback or competitive interactions between these precursors may control the proportion of different neuronal subtypes they normally produce. These data reveal remarkable parallels in the roles of bHLH factors during neurogenesis in the DRG, and myogenesis in the neighboring myotome.
Keywords: Sensory neurons, neurogenesis, neurotrophins, neurogenins, bHLH protein
Transcription factors in the basic helix–loop–helix (bHLH) family play a central and phylogenetically conserved role in the determination of cell type. In vertebrates a family of muscle-specific bHLH factors (called the myogenic regulatory factors, or MRFs) is essential for the determination of myoblast fates (for reviews, see Molkentin and Olson 1996; Yun and Wold 1996). Analogously, in Drosophila a family of bHLH factors, called proneural genes, is essential for the determination of neural fates (for review, see Anderson and Jan 1997). Strikingly, the cell-type specificity and primary sequence of bHLH factors appear coordinately conserved across phylogeny. Thus, mammalian neurogenic bHLH factors are more highly related to Drosophila proneural genes than they are to mammalian myogenic bHLH factors (Johnson et al. 1990), and vice-versa (Michelson et al. 1990).
A puzzling feature of bHLH factors is the apparent multiplication of functionally similar genes expressed within a given tissue. The MRF subfamily, for example, consists of four highly related genes: myoD, myf5, myogenin, and MRF4/herculin (Molkentin and Olson 1996; Yun and Wold 1996). Similarly, the achaete–scute complex of Drosophila contains a tandem array of four highly related proneural genes (Alonso and Cabrera 1988). Some of this multiplication reflects the fact that related bHLH genes act in cascades to control determination and differentiation within both nerve and muscle (Jan and Jan 1993; Weintraub 1993). However, this cannot fully explain the reason for such multiplication, as both loss- and gain-of-function assays have revealed apparent redundancy for genes acting at similar levels in the developmental hierarchy (for reviews, see Weintraub et al. 1991; Campuzano and Modolell 1992).
It has become clear recently that the apparent genetic redundancy of myogenic bHLH factors at the tissue level masks an underlying nonredundant function at the cellular level. The subtle phenotypes of myf5 and myoD single mutants suggested initially that these genes were functionally redundant (Braun et al. 1992; Rudnicki et al. 1992), a conclusion supported by the clear myogenic defects observed in double mutants (Rudnicki et al. 1993). However, more recent analyses have shown that myf5 and myoD are expressed initially by distinct subpopulations of myogenic precursors (Braun and Arnold 1996), each of which may compensate for the loss of the other in single mutants (Patapoutian et al. 1995; Braun and Arnold 1996; for reviews, see Molkentin and Olson 1996; Yun and Wold 1996). These data suggested that, at least in muscle, duplication of highly related bHLH determination genes may reflect their utilization by distinct classes of progenitor cells.
The extent to which this mechanism operates more generally is not yet clear. In the vertebrate nervous system, homologs of the Drosophila proneural gene atonal (Jarman et al. 1993) called neurogenins/Math4a-c/NeuroD3 have emerged as determination genes for the neuronal fate analogous to MyoD and myf5 (Gradwohl et al. 1996; Ma et al. 1996, 1998; McCormick et al. 1996; Fode et al. 1998). Like the myogenic determination genes, the neurogenins (ngns) act upstream of a related subfamily of genes exemplified by neuroD (Lee et al. 1995), which appear to act as differentiation factors (for reviews, see Kageyama and Nakanishi 1997; Lee 1997). Initial analysis of ngn1 and ngn2 single mutants has revealed a block at the earliest stages of neurogenesis, in complementary sets of cranial sensory ganglia (Fode et al. 1998; Ma et al. 1998). However in ngn1 mutants there is no obvious phenotype in the CNS (Ma et al. 1998), where ngns are transcribed in highly overlapping patterns (Gradwohl et al. 1996; Sommer et al. 1996; Ma et al. 1997). This suggests that the ngns may act redundantly in some regions of the nervous system.
Such apparent redundancy raises the question of whether the ngns function in the same precursor cells, or rather in distinct precursors that can compensate for one another. Here we have addressed this question by examining the roles of the ngns in the development of trunk dorsal root ganglia (DRG), which contain several different classes of sensory neurons (Snider 1994; Snider and Wright 1996). We find that most or all small-diameter, nociceptive (trkA+) neurons require ngn1, whereas ngn2 is transiently required only for large-diameter trkB+ and trkC+ neurons. The initial requirement for ngn2 is, however, subsequently compensated in an ngn1-dependent manner. Comparison with previous cell lineage studies in chick DRGs (Frank and Sanes 1991) suggests that ngn2 and ngn1 may be required in distinct precursor populations that generate different classes of sensory neurons, analogous to the requirements of myf5 and myoD by distinct subsets of myoblasts. The ability of the ngn1-dependent precursors to compensate for the loss of ngn2-dependent cells further suggests that feedback or competitive interactions between these two populations may control the production of different classes of sensory neurons during normal development.
Results
Sequential expression of ngn2 and ngn1 in the developing DRG
To understand better the sensory neuron phenotypes of ngn1 and ngn2 single mutants, we first re-examined the expression of the ngns during neural crest migration and early dorsal root gangliogenesis in wild-type embryos. The earliest expression of ngn2 was detected in cells at the lateral margins of the neural tube (Fig. 1B,D,F, arrows). Comparison to the expression pattern of Hfh2, a member of the forkhead family of transcription factors identified recently as an early neural crest marker (Labosky and Kaestner 1998), suggested that ngn2 is expressed by neural crest cells very early in their migration (Fig. 1A,C,E, arrows; cf. Fig. 1F, arrowhead). Expression of ngn2 continues into the early stages of DRG condensation, and is then extinguished by ∼E10.5 (Sommer et al. 1996). In contrast, ngn1 expression was not detected in newly emigrating crest cells (Fig. 1, F vs. G and H, arrowheads). Rather, ngn1 expression was detected only after crest cells had migrated to a position between the somite and neural tube and begun their condensation into ganglion primordia (Fig. 1H, arrow).
Figure 1.
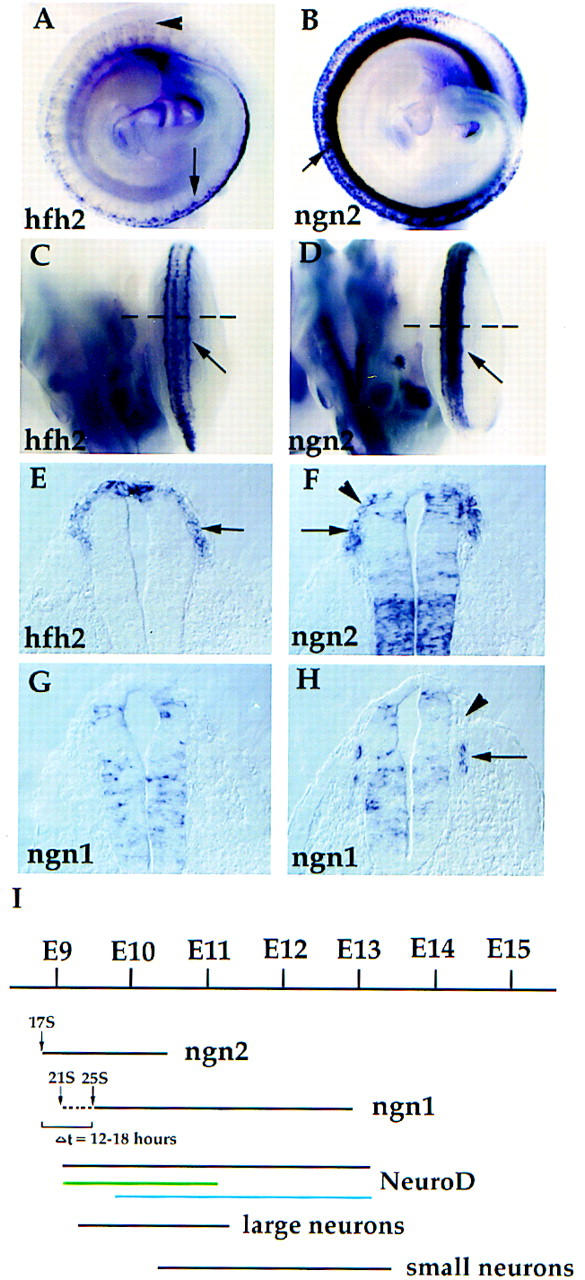
Sequential expression of ngn1 and ngn2 in the developing DRG. Whole-mount in situ hybridizations were performed with Hfh2, ngn2, and ngn1 as probes. (A,B) Lateral view of E9.75 (27 somites) embryo. (Arrowhead and arrow in A) Rostral and caudal parts of the embryo, respectively. Heads of the embryos are not shown (downward direction). (C,D) Dorsal view of caudal halves of the same embryos. Expression of Hfh2 and ngn2 was detected in emigrating crest cells (A–D, arrows), which is clearly seen in transverse sections (E,F, arrows). Positions of the sections in E and F are indicated by the broken lines in C and D. ngn1 expression was not detected in a section (G) at an axial level equivalent to E and F but was detected in sections through a more rostral part of the embryo (H, arrow) (DRG development shows a rostral to caudal gradient). Unlike ngn2, ngn1 was not expressed in newly emigrating crest cells (cf. H and F, arrowheads). Expression of ngn2 in cervical DRG was first detected at E8.75–E9 (17 somites) and disappeared at E10.5; expression of ngn1 started at E9–E9.25 (20–21 somites) and lasted all the way to E13 (data not shown) (summarized in I). There is a short period of ngn2-dependent ngn1 expression (I, broken line, Δt; see Fig. 6A). Although separate waves of ngn2-dependent (green line) and ngn1-dependent (blue line) neuroD expression can be distinguished clearly in mutants (see Fig. 2B), the end and start points of these two respective waves are approximate. The time line of large and small neuron generation in cervical DRG is adapted from Lawson and Biscoe (1979).
Additional examination extended the previous observation that expression of ngn2 and ngn1 spans two distinct periods in the developing DRG (Sommer et al. 1996) (summarized in Fig. 1I). For example in cervical DRGs, expression of ngn2 starts at E8.75–E9 (17 somites) and ends at E10.5, whereas expression of ngn1 starts at E9–E9.25 (20–21 somites) and extends until E13 (data not shown). The onset of ngn2 expression precedes that of ngn1 by about four somites of development. However, analysis of ngn2 mutants (see below) indicates that the earliest expression of ngn1 likely reflects cross-regulation by NGN2. Therefore the actual onset of NGN2-independent ngn1 expression follows that of ngn2 by ∼12–18 hr (Fig. 1I, dashed line). From E10.5 to E13, only ngn1 is expressed in cervical DRG (Fig. 1I).
Two transient but overlapping phases of neuroD expression revealed by analysis of ngn1 and ngn2 mutant embryos
Throughout the rostrocaudal axis, expression of neuroD follows that of ngn2 (Sommer et al. 1996) and spans the period during which ngn1 expression is initiated and extinguished (Fig. 1I, NeuroD). Analysis of ngn1 and ngn2 single mutants revealed, however, that the apparently continuous expression of neuroD in developing wild-type DRG (Fig. 2A,C,E,G, +/+) is apparently a composite of two distinct phases of transient expression that follow the rostral-to-caudal gradient of normal DRG development: an early phase (Fig. 1I, green line), which is unmasked in the caudal region of E12 ngn1−/− embryos (Fig. 2H, arrow), and a later phase (Fig. 1I, blue line), which is unmasked in the rostral region of E10 ngn2−/− embryos (Fig. 2B, arrow). As there is no neuroD expression in ngn1;ngn2 double mutant embryos (C. Fode and F. Guillemot, unpubl.), the early phase must be NGN2-dependent (Fig. 2A,H, arrows), and conversely the later phase is NGN1-dependent (Fig. 2B,G, arrows).
Figure 2.
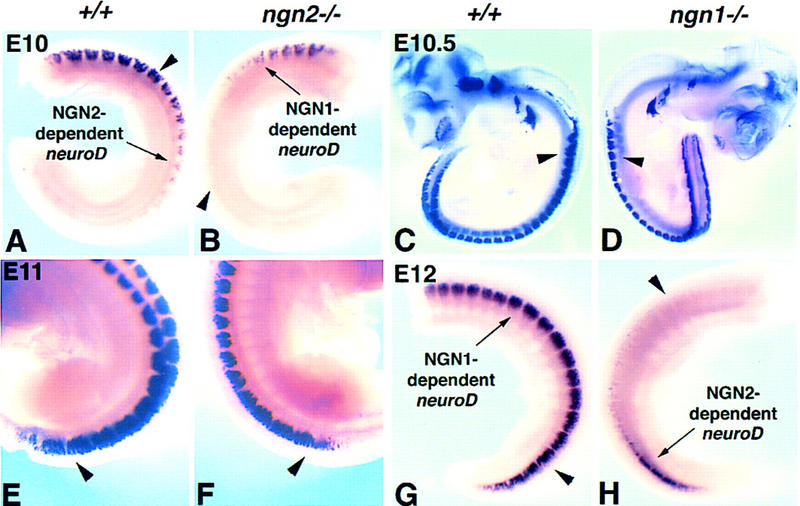
Two transient and overlapping phases of neuroD expression are revealed by comparison of ngn1 and ngn2 single mutants. All panels show whole-mount in situ hybridization with a neuroD probe. Comparison between ngn2 mutant embryos (B,F) and wild-type littermates (A and E, respectively) was performed at E10 (A,B) or E11 (E,F). Comparison between ngn1 mutant embryos (D,H) and wild-type littermates (C,G, respectively) was performed at E10.5 (C,D) or E12 (G,H). Diminished or no expression of neuroD is visible in the caudal region of an E10 ngn2 mutant embryo (B, arrowhead), where earlier-born NGN2-dependent neurons should have been generated (A, arrow), whereas it is detectable in the rostral region where later-born NGN1-dependent neurons are beginning to be generated (B, arrow). Conversely, diminished or no expression of neuroD is visible in the rostral region of an E12 ngn1 mutant embryo (H, arrowhead), where later-born NGN1-dependent neurons would normally be generated at this stage (G, arrow), while the last traces of neuroD expression are visible in caudal regions (H, arrow) where NGN2-dependent neurons are still being generated in these youngest DRG. Note that there is spatial overlap between the NGN2- and NGN1-dependent phases of neuroD expression (cf. intensity of staining in A, arrowhead vs. B; and G, arrowhead, vs. H). Stages E10 and E12 were chosen to reveal most clearly the NGN1-dependent (B, arrow) and NGN2-dependent (H, arrow) phases of neuroD expression, respectively (see also Fig. 1I, NeuroD). In older ngn2−/− embryos (F) and younger ngn1−/− embryos (D), little or no difference in neuroD expression is detected in comparison to wild-type embryos (E and C, respectively). In the case of the older ngn2 mutant, this is presumably because the later wave of NGN1-dependent neuroD expression has already swept into caudal (younger) regions of the embryo (E vs. F, arrowheads). In the case of the younger ngn1 mutant it is presumably because the early wave of NGN2-dependent neuroD expression has not yet been down-regulated in rostral (older) regions of the embryo (C vs. D, arrowheads). Note that comparisons of mutant and wild-type littermates from a given stage are presented at the same magnification, but the magnifications of the different stages are not comparable. In A, B, and G, H the entire trunk region has been dissected away from the rest of the embryo for clarity. In E and F only the caudal region of intact embryos is shown.
The NGN2- and NGN1-dependent phases of neuroD expression were only clearly visible in older (E12) (Fig. 2H) and younger (E10) (Fig. 2B) embryos, respectively. Thus, for example, in ngn1−/− embryos younger than E12 the early NGN2-dependent phase has presumably not yet been down-regulated, and therefore masks the loss of neuroD expression caused by the mutation (Fig. 2C,D, arrowheads). However by E12, down-regulation of NGN2-dependent neuroD expression has occurred already in rostral DRG, unmasking the loss of neuroD expression in this region caused by deletion of ngn1 (Fig. 2H, arrowhead; cf. Fig. 2G, arrow). Conversely, in ngn2−/− embryos older than E10, the later NGN1-dependent wave has evidently already swept into caudal DRG and masks the loss of neuroD expression caused by the mutation (Fig. 2E,F, arrowheads). By contrast, in younger embryos (E10) the later NGN1-dependent phase has not yet been initiated in caudal DRG (Fig. 2B, arrowhead), revealing the loss of neuroD expression caused by the ngn2 mutation (cf. Fig. 2A, arrow vs. 2B). There is clearly some overlap between these two phases, as indicated by the reduced levels of neuroD expression in both the NGN2-dependent and NGN1-dependent regions of mutant embryos in comparison with wild type (Fig. 2, cf. A, arrowhead, and B, arrow; cf. G, arrowhead, and H, arrow). Taken together, these data suggest that neuroD is expressed during DRG development in two sequential and transient, but overlapping, waves that depend on ngn2 and ngn1, respectively.
Loss of the trkA+ neurons and a subset of trkC+ and trkB+ neurons in ngn1 mutants
Neuronal birthdating studies have shown that DRG precursors go through their final divisions in two broad and overlapping successive waves. In the cervical DRG, the first wave, which gives rise to large-diameter neurons, occurs from E9.5 to E11.5, whereas the second wave, which mainly gives rise to small-diameter neurons begins at E10.5 and lasts until E13.5 (Lawson and Biscoe 1979; see Fig. 1I). There is a period of overlap from E10.5 to E11.5 when both sizes of neurons are born concurrently. We noted that the generation of small neurons appears to coincide with the period when only ngn1 is expressed (Fig. 1I). As the small neurons are nociceptive afferents that primarily express trkA, a receptor for nerve growth factor (NGF) (Mu et al. 1993; Wyatt and Davies 1993; Backstrom et al. 1996), we sought to determine if ngn1 is required for the development of the trkA+ subset of sensory neurons. At E15, the cervical DRG of ngn1−/− mutants were much smaller compared with those of wild-type littermates, as indicated by the expression of SCG10, a pan-neuronal marker (Stein et al. 1988) (Fig. 3, cf. B and A). This reduction in size was primarily caused by the complete elimination of trkA+ neurons (Fig. 3, D vs. C), which constitute up to 70% of total ganglionic neurons (Mu et al. 1993). Loss of the NGF-dependent nociceptive population was further confirmed by a lack of expression of two additional markers in newborn ngn1−/− mice: VR1, the capsaicin receptor gene (Fig. 3, F vs. E) (Caterina et al. 1997), and SNS, the tetrodotoxin-resistant voltage-gated sodium channel gene (Fig. 3, H vs. G) (Akopian et al. 1996). In contrast to the complete absence of trkA+ neurons in the ngn1−/− cervical DRG, in more posterior DRG, a few trkA+ neurons could be detected at E14.5 (not shown). Because some trkA+ cells were seen even in cervical DRG at earlier stages (E12.5), and as development of the posterior trunk DRG lags behind that of the cervical ganglia, it is possible that in ngn1−/− embryos some trkA+ neurons or their precursors are transiently generated and then die (see below).
Figure 3.
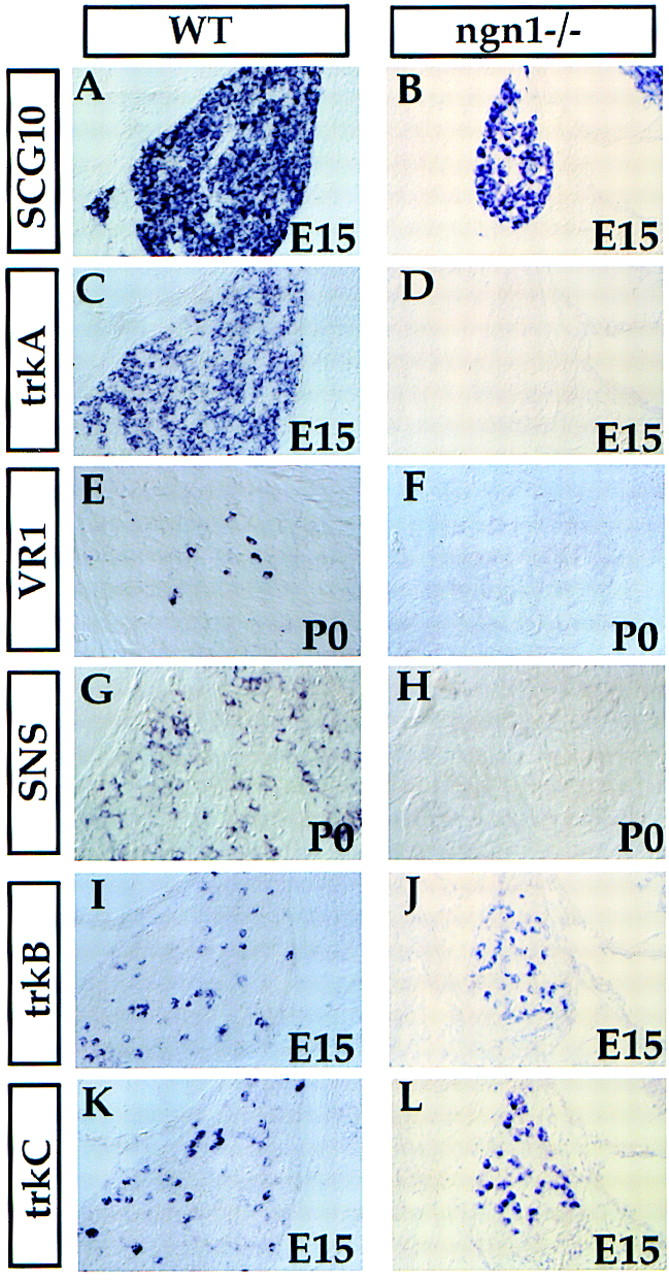
Requirement of NGN1 for the development of the trkA+ neurons. Transverse sections through C2 DRG of wild-type and ngn1 mutant E15 embryos (A–D,I,J) or newborn mice (E–H) are shown. Section in situ hybridization was performed with the indicated probes.
We noted that expression of ngn1 overlaps the period during which large-diameter neurons are born (Fig. 1I). These neurons, which include mechanoceptive and proprioceptive muscle afferents, express trkB and trkC, which are receptors for brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3), respectively (for review, see Chao 1992). Nevertheless, many trkB+ and trkC+ neurons did develop in ngn1 mutant embryos (Fig. 3I–L); in fact their density appears higher because of the loss of intervening trkA+ cells. However, the second cervical DRG (C2) of E16 embryo ngn1 mutants is only about a quarter of the size of the wild-type ganglion, and quantification of these neurons in a series of consecutive sections spanning these ganglia revealed a 35% reduction in the total number of trkC+ neurons, and a 27% reduction in the number of trkB+ neurons, in ngn1 mutants at this stage (Table 1). A 26% reduction in the number of trkC+ neurons in C2 DRG was also observed in ngn1 mutant embryos at E13 (Table 1), arguing against the idea that ngn1 is only required for a later-generated subset of trkC+ neurons (Fariñas et al. 1998). We conclude that ngn1 is required for the development of both a subset of early-generated (trkC+ and trkB+) neurons, as well as for most or all later-generated (trkA+) neurons. TUNEL labeling revealed massive apoptotic cell death in the DRG of ngn1−/− embryos at E11.5 (data not shown), suggesting that the fate of many ngn1-dependent precursors in the mutant is to die.
Table 1.
Numbers of trkB− and trkC+ neurons in wild-type and ngn1−/− embryos
|
|
E16 C2 DRG
|
E13 C2 DRG
|
||||
|---|---|---|---|---|---|---|
|
wild type
|
ngn1−/−
|
percent reduction
|
wild type
|
ngn1−/−
|
percent reduction
|
|
| trkC | 852 ± 56 (4) | 554 ± 28 (6) | 35% (P < 0.0005) | 912 ± 9 (4) | 675 ± 96 (6) | 26% (P < 0.005) |
| trkB | 776 ± 53 (4) | 563 ± 98 (6) | 27% (P < 0.01) | N.D. | N.D. | N.D. |
Number of ganglia counted in parentheses. (N.D.) Not determined.
Development of the trkC+ and trkB+ neurons is normal in ngn2 mutants at E13 and later developmental stages
To determine if the trkC+ and trkB+ neurons spared in ngn1 mutants were, conversely, dependent on ngn2, we examined their development in ngn2 mutants. Surprisingly, the development of the DRG appeared normal in ngn2 mutants at E15 and at P0, as indicated by SCG10 expression (Fig. 4, B vs. A; data not shown). Superficially normal expression was also observed for other markers, including trkA, trkB, trkC, and ER81 (a marker for a subset of the trkC+ neurons) (Fig. 4C–J) (Lin et al. 1998). To determine if the trkC+ neurons in these mutants make proper projections, we placed DiI crystals into the DRG of newborn wild-type or ngn2−/− mutant mice to label the afferents projecting into the spinal cord. The muscle proprioceptive afferents from trkC+ neurons of ngn2 mutants appeared to send normal projections to the ventral spinal cord (Fig. 4, L vs. K, arrows). To determine if there was a loss of a subset of the trkC+ and trkB+ neurons, we counted these neurons in C2 DRG at two embryonic stages: E16 and E13. No significant loss of either subpopulation was found at E16, and the number of trkC+ neurons was normal at E13 as well (Table 2).
Figure 4.
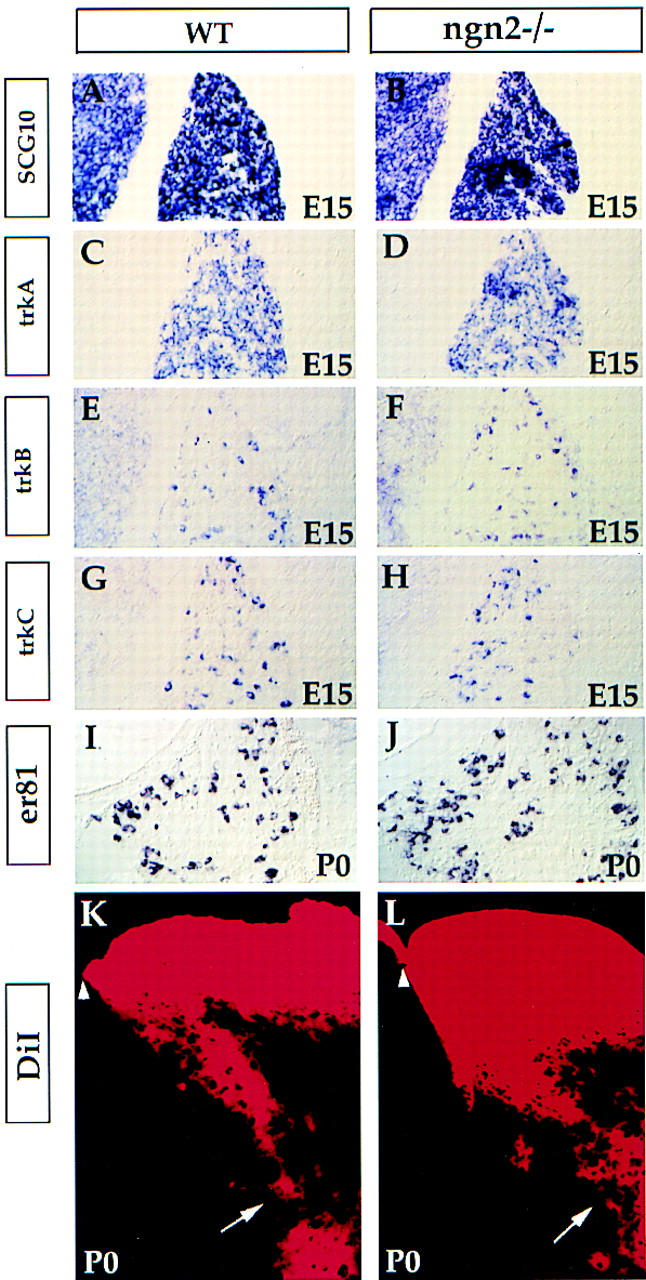
Normal development of the DRG in ngn2 mutants at E15 and P0. Transverse sections through C2 DRG of wild-type and ngn2 mutant E15 embryos (A–H) or newborn (P0) mice (I–L). Section in situ hybridization was performed with the indicated probes. K and L show DiI labeling of the afferents of the DRG neurons projecting to the neural tube. The projection of proprioceptive muscle afferents to the ventral neural tube appears normal (K,L, arrows). Arrowheads indicate the dorsal midline of the spinal cord.
Table 2.
Numbers of trkB- and trkC-positive neurons in wild-type and ngn2−/− embryos
|
|
E16 C2 DRG
|
E13 C2 DRG
|
||||
|---|---|---|---|---|---|---|
|
wild type
|
ngn2−/−
|
percent reduction
|
wild type
|
ngn2−/−
|
percent reduction
|
|
| trkC | 1041 ± 91 (6) | 1153 ± 60 (6) | N.S. | 849 ± 62 (6) | 885 ± 86 (6) | N.S. |
| trkB | 654 ± 41 (8) | 642 ± 112 (6) | N.S. | N.D. | N.D. | N.D. |
Number of ganglia counted in parentheses. (N.S.) Not significant. (N.D.) Not determined.
ngn2 and ngn1 together are required for DRG development
One reason for the apparently normal DRG development observed in ngn2 mutants might be functional compensation by ngn1. To examine this possibility, we intercrossed double heterozygous mice to generate ngn1−/−; ngn2−/− double homozygous mutants. DRG were completely absent in E15 double mutant embryos as indicated by the lack of SCG10 expression (Fig. 5, B vs. A). Identical results were obtained at E12.5 (data not shown). Thus, all DRG neurons require ngn1 and/or ngn2. The apparent lack of a defect in ngn2−/− embryos at E13 must reflect redundancy with, or compensation by, ngn1.
Figure 5.
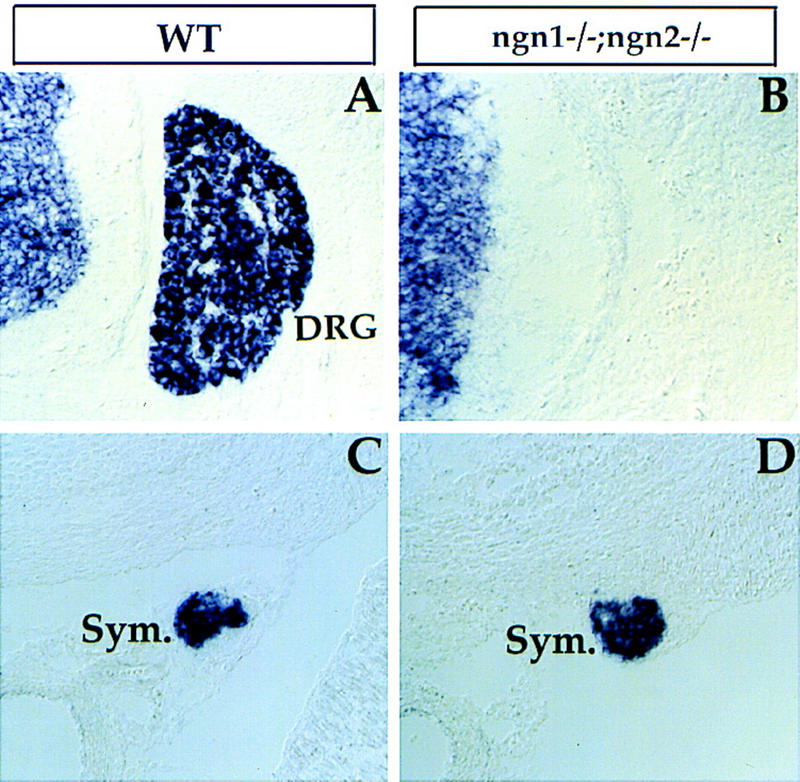
Requirement of NGN1 and NGN2 for DRG development. Transverse sections through an E15 ngn1 and ngn2 double homozygous embryo (B,D) and a wild-type control embryo (A,C) at lumbar level were probed with SCG10, a pan neuronal marker. DRG (A, DRG) were absent in double mutants (B). Expression of SCG10 in sympathetic ganglia (C and D, Sym.) was not affected.
As ngn2 is expressed early in neural crest migration (Fig. 1) and some early migrating neural crest cells have been reported to give rise to both sensory and sympathetic neurons (Fraser and Bronner-Fraser 1991), we examined single and double ngn mutant embryos for effects on autonomic neuron development. The differentiation of neurons in the sympathetic ganglia was not overtly affected in either of the single or in double ngn mutants (Fig. 5C,D; data not shown). Therefore, although ngns are required for the development of all DRG sensory neurons, and although ngn2 is expressed in early migrating neural crest cells, the development of autonomic neurons is unaffected by the lack of these genes.
An early, transient neurogenesis defect in ngn2 mutants
The absence of DRG in double mutants (Fig. 5B) implies that the ∼75% of trkC+ and trkB+ neurons spared in ngn1 mutant embryos (Fig. 3; Table 1) must develop from ngn2-dependent precursors. It was therefore surprising that we did not detect a loss of the trkC+ and trkB+ neurons in our initial analysis of ngn2 single mutants (Fig. 4). These observations suggested that because all trkC+ and trkB+ neurons are lost in ngn1−/−; ngn2−/− double mutants, ngn1 must be compensating in some way for the absence of ngn2. To determine if ngn1 expression was initiated prematurely to substitute for ngn2, we examined ngn1 expression in early ngn2−/− embryos. Rather than observing a precocious induction of ngn1, we found that the earliest detectable phase of ngn1 expression was actually lost in ngn2 mutants (Fig. 6A, arrows). Whereas in wild-type embryos expression of ngn1 in cervical DRG was first detected at E9–E9.25 (20–21 somites), in ngn2 mutants it was delayed until E9.5 (24–25 somites) (Fig. 1I, Δt). These data suggest that the earliest expression of ngn1 in wild-type embryos is NGN2-dependent. Therefore, the apparent compensation by ngn1 in ngn2−/− mutants does not appear to occur by a simple reprogramming of ngn1 expression to substitute for that of ngn2.
Figure 6.
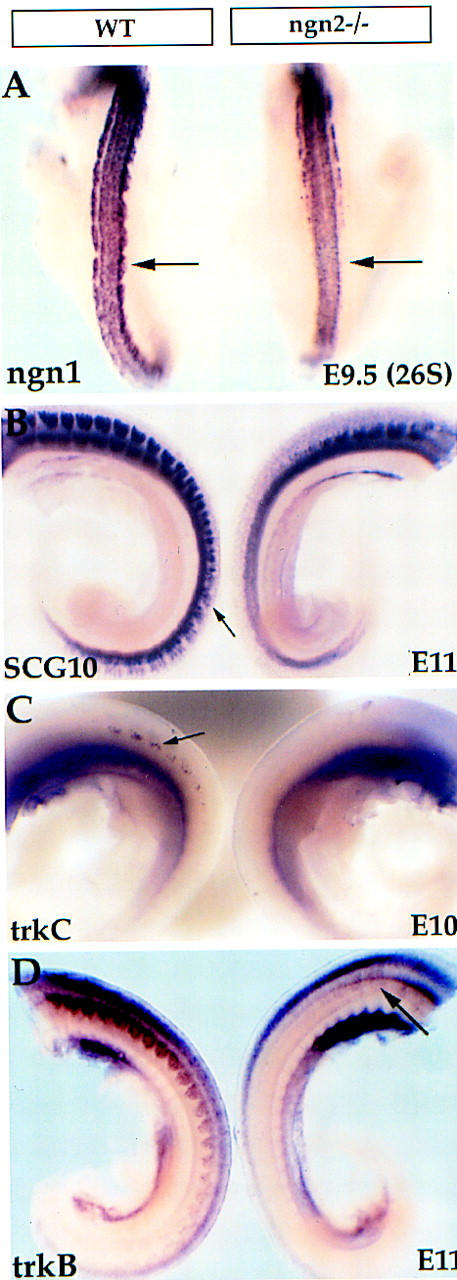
Delayed neurogenesis in ngn2 mutant embryos. (A) E9.5–E9.75 (26S) wild-type and ngn2 mutant embryos were probed with ngn1. Early expression of ngn1 is lost in the ngn2 mutant embryo (arrow, right). (B) Expression of the pan-neuronal marker SCG10 in caudal DRGs is observed in the wild-type embryo (B, left, arrow) but is only detected anteriorly in the ngn2 mutant (B, right), indicating that neurogenesis is delayed (see also Fig. 2B). (C) Initial expression of trkC was detected in wild-type embryos at E10 (left, arrow) but not in ngn2 mutant embryos (right). (D) A delay in the appearance of trkB+ neurons is also detected in ngn2 mutant embryos. In this case, trkB expression is not detected until E11 in the mutant (arrow, right), consistent with the fact that this mRNA is expressed subsequent to trkC mRNA in wild-type embryos as well (not shown).
These data suggested that compensation by ngn1 might involve a delay in neurogenesis in ngn2−/− mutants. Consistent with this, we observed that neuroD expression was initiated substantially later in ngn2 mutants than in wild-type embryos (see above, Fig. 2B). Moreover, overt neuronal differentiation, as marked by expression of SCG10, was also significantly delayed (Fig. 6B). In support of the idea that this delay in neurogenesis reflects the delayed generation of large-diameter neurons, there was a delay in the expression of trkC and trkB in ngn2 mutants as well (Fig. 6C,D). Whereas trkC could be detected as early as E10 in wild-type embryos (Fig. 6C, left, arrow), no such expression was detected in ngn2 mutants at this stage (Fig. 6C, right). Rather, trkC expression was not detected in ngn2−/− embryos until E10.5–E11 (data not shown). The recovery of trkB-expressing neurons occurred even later than that of the trkC-expressing neurons (Fig. 6D, right, arrow), consistent with the sequential expression of these two mRNAs in wild-type embryos (data not shown). By E15, the expression of trkB and trkC in ngn2−/− embryos appeared normal (Fig. 4; Table 2). By contrast, no apparent delay was observed in the generation of trkA+ neurons (not shown). Taken together, these data suggest that the development of trkB+ and trkC+ neurons is initially dependent on ngn2, but that this dependence is compensated at later stages by ngn1.
The delayed neurogenesis in ngn2−/− embryos could mean that ngn2-dependent precursors wait and are later converted into ngn1-expressing cells, which then differentiate into trkC+ and trkB+ neurons. Alternatively, such precursors could die and be replaced by a separate population of ngn1-dependent precursors. We therefore examined programmed cell death in wild-type and ngn2 mutant embryos by the TUNEL method (Gavrieli et al. 1992), at several different stages. Extensive apoptotic cell death was observed at E11 in hindlimb bud-level the DRG of ngn2 mutant embryos, whereas relatively little cell death was observed in wild-type ganglia at similar axial levels (Fig. 7A,B, arrows). Moreover, on adjacent sections ganglia containing such TUNEL+ cells in mutant embryos did not yet exhibit SCG10 expression (Fig. 7, C vs. D, arrows), suggesting that many ngn2-dependent precursors die before undergoing overt neuronal differentiation. TUNEL+ cells were also observed in more anterior DRG of mutant embryos, at axial levels rostral to the forelimb bud (not shown). However, these cells were observed at earlier stages, consistent with the idea that the requirement for ngn2 follows the normal rostral-to-caudal progression of DRG development.
Figure 7.
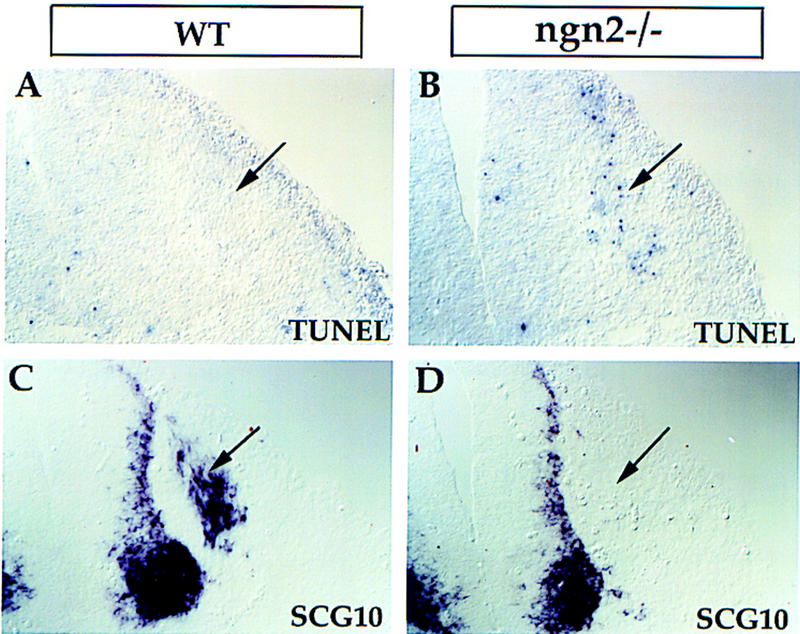
Increased apoptosis precedes overt neurogenesis in ngn2 mutants. Adjacent transverse sections through E11 wild-type (A,C) and ngn2 mutant embryos (B,D) at an axial level close to the hindlimb bud. Dying cells were detected by the TUNEL method. Extensive cell death was detected in mutant DRG (B, arrow) but not in wild-type DRGs (A, arrow) at similar axial levels. No SCG10 expression was detected in an adjacent section through the mutant embryo (D, arrow), whereas in wild-type embryos it was already expressed (C, arrow).
Discussion
The ngns are vertebrate neuronal determination genes that exhibit overlapping expression and apparent functional redundancy in many regions of the CNS. The extent to which this redundancy reflects functional compensation in common or distinct precursors is not clear. We have used the development of the DRG as a model system to address this issue. We find that ngn1 and ngn2 are required during different phases of neurogenesis that generate different classes of sensory neurons: ngn2 is required exclusively during the early phase, whereas ngn1 is required during the early phase and also exclusively during the late phase. The loss of ngn2 function, however, appears compensated by ngn1 after a delay. Many ngn2-dependent cells are fated to die in ngn2−/− embryos, suggesting that the delayed compensation occurs by substitution of separate ngn1-dependent precursors, rather than by reprogramming of ngn1 expression within ngn2-dependent cells.
NGNs and the timing of neurogenesis in the DRG
[3H]Thymidine cell birthdating studies in both mouse (Lawson and Biscoe 1979) and chick (Carr and Simpson 1978) have shown that large- and small-diameter DRG sensory neurons are born in two successive waves. Our observations suggest that these waves may reflect in part the sequential but overlapping utilization of ngn2 and ngn1, respectively (Fig. 8D). This correspondence is not absolute, however, as ngn1 and ngn2 are coexpressed during much of the early phase and are both required for the generation of trkB+ and trkC+ sensory neurons. However ngn1 is exclusively expressed during the later phase and is required for most or all of the trkA+ neurons.
Figure 8.
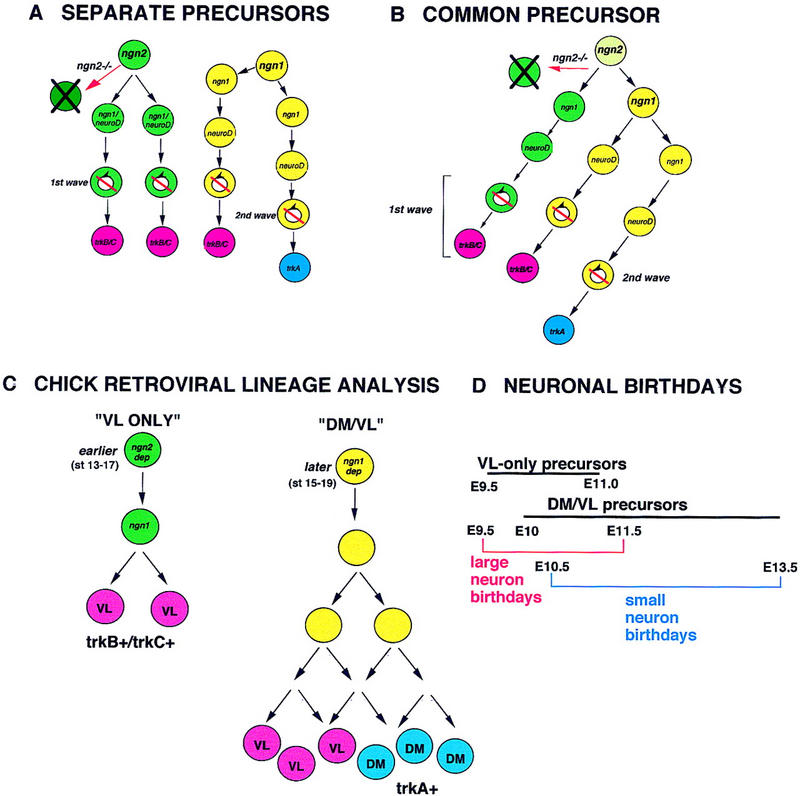
Genetic and lineage relationships between sensory precursors in the DRG. (A,B) Alternative lineage relationships between ngn2- and ngn1-dependent precursors. (A) ngn2-dependent precursors are distinct from ngn1-dependent precursors in the DRG. For simplicity, the expression of ngn1 in the ngn2-dependent lineage (see B) is shown together with that of NeuroD. (B) A common precursor within the DRG that initially expresses ngn2 generates both ngn2-dependent and ngn1-dependent precursors. As the early expression of ngn1 and neuroD is lost in ngn2−/− mutants (Figs. 2B, 6A), this model requires that such mutant precursors divide to generate some daughters that fail to express ngn1 or neuroD and die (X-ed out cell), and/or other daughters that survive and do express ngn1 and neuroD. Although the model illustrates an asymmetric division of an individual ngn2-dependent cell, it is possible that identical cells divide symmetrically to generate either two daughters that survive, or two daughters that die. Slashed circular arrows indicate cell cycle withdrawal. (C) Correlation of genetic dependence on ngn2 or ngn1 with different sensory neuron precursor populations identified by retroviral lineage tracing in chick (Frank and Sanes 1991; and unpubl.). (Earlier; later) The embryonic stages (Ham-burger and Hamilton 1951) at which such precursors are observed. VL neurons (red circles) located in the ventrolateral region of the DRG and correspond to large-diameter trkB+ and/or trkC+ neurons; DM neurons (blue circles) are located in the dorsomedial region of the DRG and correspond to small-diameter trkA+ neurons. (D) Time line emphasizing that the birthdays of large-diameter neurons in the DRG (Lawson and Biscoe 1979) likely are initiated by VL-only (ngn2-dependent) precursors but extend into the period when DM/VL (ngn1-dependent) precursors are also giving rise to trkB+ and/or trkC+ (VL) neurons. We cannot exclude that some VL-only precursors are ngn1-dependent. Small-diameter neurons are produced primarily or exclusively by DM/VL precursors, which are ngn1-dependent.
The mechanisms that control the timing of the two waves of neurogenesis are unknown. Despite the absence of ngn2-dependent precursors, the generation of ngn1-dependent neurons in ngn2−/− embryos was still delayed in comparison with wild-type embryos (Fig. 6). This argues against the idea that the delayed differentiation of ngn1-dependent precursors is normally controlled by a negative-feedback signal from the earlier-generated ngn2-dependent neurons. We cannot exclude the possibility that there is nevertheless some acceleration in the differentiation of ngn1-dependent neurons in ngn2 mutants. However, it seems more likely that the timing of neurogenesis is controlled by the timing of independent inducing mechanisms for ngn2 and ngn1.
The correlation between the utilization of different ngns and the generation of different sensory neuron subtypes should not be taken to imply that the NGNs autonomously specify these subtypes, although they could contribute to this specification. In fact, the compensation by ngn1 observed in the ngn2−/− mutant makes it more likely that the NGNs control essentially equivalent programs of sensory neurogenesis. Indeed, we observed a small number of trkA+ neurons in more posterior DRG of ngn1 mutants, suggesting that some ngn2-dependent precursors may contribute to this class of neurons in wild-type animals. If so, then different sensory neuron subtypes are likely determined by factors other than the ngns. The action of such factors may nevertheless require NEUROGENIN function. In that case, the sequential utilization of ngn2 and ngn1 could simply control the timing of neurogenesis, with the action of subtype-determining factors dependent on this timing (Tanabe 1998; Edlund 1999).
Do NGN2-dependent cells constitute a distinct subpopulation of sensory precursors?
The fact that different subclasses of sensory neurons are born on different schedules raises the possibility that they may be generated from distinct subsets of neural crest-derived precursors (Fig. 8A,B). Alternatively, a common precursor within the DRG could divide asymmetrically to sequentially generate early- and late-born neurons (Fig. 8A,B). Our data favor (but do not prove) the former possibility. In ngn2−/− embryos, extensive apoptosis is observed in ganglia that have not yet expressed SCG10 (Fig. 7B,D), and therefore likely reflects the death of ngn2-dependent precursor cells. However, these ganglia already exhibit neuroD expression on adjacent sections (Q. Ma, unpubl.). As neuroD expression is initially prevented in ngn2−/− embryos (Fig. 2B), the subsequent expression of neuroD in the face of extensive cell death must occur in surviving precursors that have already expressed ngn1. For such surviving cells and those killed by the ngn2 mutation to derive from the same progenitor, it is necessary to postulate that this progenitor divides to generate both a daughter cell that fails to express ngn1 (or neuroD) and dies (Fig. 8B, ngn2−/−), and a daughter that survives to express ngn1 and neuroD and that differentiates. Although such an explanation is formally possible, it seems simpler to think that ngn2-dependent and ngn1-dependent precursors represent distinct lineages in the DRG (Fig. 8A).
Interestingly, evidence for the existence of two types of sensory precursors has been provided by a retroviral lineage analysis of avian DRG development (Frank and Sanes 1991). In the chick, large-diameter trkC+ (and some trkB+) neurons tend to be located in the ventrolateral (VL) region of the DRG, whereas small-diameter trkA+ neurons tend to be located in the dorsomedial (DM) region (Carr and Simpson 1978; Kahane and Kalcheim 1994; Wright and Snider 1995; Backstrom et al. 1996). Two types of retrovirally marked clones containing neurons were obtained in this experiment. One, which is small (3 cells/clone), contained exclusively large diameter VL neurons; the other, which is large (∼35 cells/clone), contained both small diameter DM neurons and larger VL neurons (Fig. 8C). Interestingly, VL-only clones were encountered only when viral infection was performed at earlier stages (Hamilton and Hamburger 1951; stage 13–16); whereas DM/VL clones were more frequent when infection was carried out later (stage 17) and were rare for infections made before stage 15 (E. Frank, pers. comm.). This observation is opposite to what one would expect if VL-only clones simply represented more restricted progeny of DM/VL precursors. Furthermore, DM/VL clones were large even when obtained with earlier (before stage 17) injections, whereas VL-only clones in the same embryos were small. As retroviral infection occurred within the neural tube, prior to emigration, these data strongly suggest the existence of a distinct, early precursor population with limited proliferative capacity that generates only large-diameter neurons. The sequential but overlapping waves of neuronal birthdays observed in thymidine-labeling experiments (Lawson and Biscoe 1979) would then reflect the fact that VL-only precursors are generated first, followed by a period of overlap with DM/VL precursors, followed by a period when only DM/VL precursors are observed (Frank and Sanes 1991; E. Frank, pers. comm.; see Fig. 8C,D).
These results are remarkably congruent with our observation that ngn2 is required only for the development of early-generated trkC+ and trkB+ neurons, whereas ngn1 is required for most or all later-generated trkA+ neurons (as well as for a subset of trkC+ and trkB+ neurons). This correlation suggests that most VL-only precursors likely correspond to ngn2-dependent precursors (and perhaps to a subset of ngn1-dependent precursors), whereas most DM/VL precursors likely correspond to ngn1-dependent cells. We emphasize that this correspondence is based on the differential genetic requirement for either ngn2 or ngn1, not on mere expression of these genes that likely can occur in both lineages. Thus, for example, the initial phase of ngn1 expression is ngn2-dependent, and likely occurs in VL-only precursors (Fig. 8C, VL only), whereas the later, ngn2-independent phase of ngn1 expression likely reflects expression in DM/VL precursors. This interpretation is supported by the observation that within developing cranial sensory ganglia, NGN2 activates ngn1 expression in epibranchial placode-derived neuronal precursors, whereas in trigeminal placode-derived precursors, ngn1 expression is independent of NGN2, but conversely activates ngn2 expression (Fode et al. 1998; Ma et al. 1998). These data argue that expression of the ngns can be both independently regulated in distinct sensory lineages, as well as cross-regulated within a given lineage. Consistent with this, preliminary analysis of ngn2–lacZ knock-in mice suggests that most or all sensory neurons express ngn2 at some point in their developmental history (Q. Ma, C. Fode, F. Guillemot, and D. Anderson, unpubl.). Whether this reflects an early, transient expression of ngn2 in DM/VL precursors, or cross-regulation of ngn2 by ngn1 at a later stage, is currently being investigated. Whatever the case, the results emphasize that expression patterns of the ngns alone do not predict lineage relationships in the DRG.
The association of ngn1- and ngn2-dependency with distinct types of sensory precursors would also be consistent with the observation that VL-only clones are smaller than DM/VL clones (Frank and Sanes 1991; Fig. 8C). Expression of ngns leads to expression of neuroD (Ma et al. 1996), which in turn may promote cell-cycle withdrawal (Morrow et al. 1999). Cells that already express ngn2 early in crest migration (Fig. 1) might therefore undergo relatively few divisions after arriving at the DRG. This would explain why VL-only clones are small. Conversely, as most ngn1 expression occurs after ganglion condensation, ngn1-dependent precursors would have more time to divide between the time they emigrated from the neural tube and the time they expressed neuroD. Consequently, DM/VL clones would be relatively larger, as observed in the chick studies.
Why have two separate lineages of sensory precursors? The fact that ngn2 is expressed early in neural crest migration suggests that the VL-only precursors that require its function may be specified for a sensory fate shortly after they emigrate from the neural tube. Consistent with this idea, forced expression of ngn2 in premigratory chick neural crest cells can bias them to a sensory fate (Perez et al. 1999). Furthermore, ngn2-expressing precursors in neural crest explant cultures appear committed to a sensory neuron fate in the presence of the autonomic-inducing signal, BMP2 (Greenwood and Anderson 1999). If VL-only precursors are determined for a sensory fate, they could provide a population of pioneer neurons to seed the developing DRG, and form a cellular scaffold around which to assemble the other neuronal subtypes that develop in these ganglia. Analagous pioneer precursors have been described in muscle development (Kahane et al. 1998), and in this respect it is interesting that the sensory pioneer neurons are those that innervate muscle.
Possible compensation mechanisms in ngn2 mutants
If the ngn1-dependent (DM/VL) precursor population has the capacity to generate a larger number of trkC+ and trkB+ neurons than it is normally required to, then in wild-type embryos ngn2-dependent (VL-only) precursors (or their progeny) must limit the number of such neurons generated from the former ngn1-dependent precursor pool. There are several possible mechanisms by which this could occur. One is by competition for limiting amounts of neurotrophic factors. Perhaps surplus neurons generated from the ngn1-dependent precursors that normally die are rescued to provide a full complement of neurons in the ngn2 mutant. Alternatively, production of trkB+ and trkC+ neurons may ultimately be limited by a negative-feedback signal, threshhold levels of which are normally reached before ngn1-dependent precursors are able to generate >25%–30% of all these neurons. In ngn2 mutants release from such feedback inhibition would allow the production of extra trkB+ and trkC+ neurons from ngn1-dependent precursors. It is important to emphasize that this negative feedback is postulated to control the number of trkB+ and trkC+ neurons that are generated, not the time at which they are produced (see above). There is precedent for such feedback inhibition of neuronal subtype generation in other systems (Reh 1986; Belliveau 1999; Belliveau and Cepko 1999).
Cellular mechanisms underlying apparent genetic redundancy
Our observations for ngn1 and ngn2 show remarkable parallels to those made previously for the myogenic determination factors myf5 and MyoD. During somitic development, myf5 is expressed initially by the earliest myogenic precursors at the DM edge of the myotome that give rise to epaxial (deep back) muscle, whereas MyoD is expressed later-differentiating hypaxial precursors located in the VL myotome (for reviews, see Molkentin and Olson 1996; Yun and Wold 1996; Tajbakhsh and Cossu 1997). The idea that myf5 and MyoD are expressed initially by distinct muscle lineages is also supported by in vitro studies (Braun and Arnold 1996). These results provide a precedent for the idea that ngn2 and ngn1 may define distinct populations of sensory precursors in the DRG, although this remains to be formally proven. Later in development, cross-regulation by the MRFs occurs in both myogenic sublineages (for review, see Tajbakhsh and Cossu 1997). Similarly, cross-regulation by the ngns can occur in distinct sensory sublineages (Fode et al. 1998; Ma et al. 1998); a subtle difference is that in the DRG, cross-regulation of ngn1 by NGN2 appears to occur in the VL-only lineage before ngn1 is activated independently in the DM/VL lineage.
Striking parallels are also seen in the compensation phenomena observed in single mutants. In myf5 null mice, the development of epaxial muscle is initially blocked, but is recovered after a delay in a MyoD-dependent manner (Braun et al. 1992, 1994; Tajbakhsh et al. 1996). This is remarkably analogous to the delayed recovery of trkB+ and trkC+ neurons in ngn2−/− mice. In the case of muscle, controversy still exists regarding the question of whether such compensation reflects the substitution of MyoD-dependent precursors for myf5-dependent precursors (Patapoutian et al. 1995; Braun and Arnold 1996; Molkentin and Olson 1996; Yun and Wold 1996), or rather a delayed activation of MyoD expression in the normally myf5-dependent lineage (Tajbakhsh and Cossu 1997; Tajbakhsh et al. 1998). As discussed earlier, our apoptosis data support the idea of cellular compensation in ngn2−/− mice. However, we cannot exclude the possibility completely that only a subset of ngn2-dependent precursors die in ngn2−/− mice, and that the rest wait and then go on to express ngn1. An apparent difference between the muscle and neural systems is that no obvious muscle defects are seen in either myf5 or MyoD single mutants, whereas in ngn1 single mutants the trkA+ population is almost completely eliminated. As a few trkA+ cells are found in ngn1−/− embryos and therefore presumably can be produced by precursors that express ngn2, we think that the inability of the ngn2-dependent VL-only lineage to compensate for the loss of trkA+ neurons in ngn1 mutants most likely reflects its limited proliferative capacity (Frank and Sanes 1991), rather than an inherent functional difference between the NGNs.
The initial expression of myf5 and MyoD in epaxial and hypaxial myoblasts is induced by distinct signals produced by different tissues: the neural tube and dorsal ectoderm, respectively (for reviews, see Cossu et al. 1996; Tajbakhsh and Cossu 1997). Although little is known about the nature of the signals that control the initial expression of ngn2 and ngn1, by analogy to muscle it seems likely that they are induced by distinct factors, or by related factors produced by different tissues at different times. Moreover, the fact that the DRG develop in close physical association with the somites, taken together with the striking parallels in the expression and function of myogenic and neurogenic bHLH factors, raises the possibility that the induction of the ngns is controlled by analogous, if not homologous, signaling mechanisms. The identification of these signals should help clarify the mechanisms that control the timing of neurogenesis in the DRG.
Materials and methods
Animals
The generation of ngn1 and ngn2 mutant mice has been described previously (Fode et al. 1998; Ma et al. 1998). Single heterozygous or double heterozygous mice were intercrossed to generate homozygous embryos. The morning that vaginal plugs were observed has been considered as E0.5. Embryos at E10, E11, and E12 were collected in PBS, fixed in 4% paraformaldehyde overnight, and stored in methanol for whole-mount in situ hybridization. Embryos at E13, E15, and E16 were fixed overnight in 4% paraformaldehyde, sunk in 30% sucrose, and embedded in OCT (Tissue-Tek) for cryostat sectioning. Newborn mice were perfused with PBS followed by 4% paraformaldehyde, immersed overnight in 4% paraformaldehyde, sunk in 15% sucrose, and embedded in OCT for sectioning.
In situ hybridization
Section and whole-mount in situ hybridization were performed as described previously (Ma et al. 1998). Detailed protocols are available upon request. Probes used in this study include ngn1 (Ma et al. 1998), ngn2 (Fode et al. 1998), rat SCG10, probe SCG10-8 (Stein et al. 1988), mouse neuroD (Lee et al. 1995), trkA and trkB (Birren et al. 1993), trkC (kinase-domain-containing probe, from Mariano Barbacid), er81 (Lin et al. 1998), and Hfh2 (Labosky and Kaestner 1998).
TUNEL staining on cryostat sections
Frozen sections were collected on Super-Frost/plus microscope slides (Fisher), dried at 37°C for 20 min, fixed in 4% paraformaldehyde for 30 min at room temperature, and washed twice with PBS for 10 min each. Slides were then incubated in permeabiliation solution (0.1% Triton X-100 plus 0.1% sodium citrate) for 10 min on ice, rinsed twice with PBS, incubated with 200 μl of 3% H2O2 with coverslips for 15 min, preincubated with 100 μl TDT (terminal deoxynucleotide transferase, Boehringer Mannheim) buffer (30 mm Tris at pH 7.2, 140 mm sodium cacodylate, 1 mm CoCl2, 0.5 mm DTT) with coverslips for several minutes, incubated with a 100 μl of reaction mixture (30 mm Tris at pH 7.2, 140 mm sodium cacodylate, 1 mm CoCl2, 0.5 mm DTT, 10 μm biotin–dUTP, 0.3 U/μl TDT) for 1 hr at 37°C in a humidified chamber, and washed three times with PBS for 5 min each. The Vectastain ABC kit was used to detect the biotin-labeled DNA.
Cell counts
Sections from E13 and E16 embryos were used. Three sets of 10-μm transverse sections through C2 DRG were collected and probed with trkC, trkB, and trkA, respectively. Nuclear-containing positive cells were counted bilaterally. Numbers were not corrected.
DiI tracing
Newborn mice were fixed by perfusion with 4% paraformaldehyde. DiI crystals were inserted into lumbar/thoracic DRG. Mice were then incubated in 4% paraformaldehyde for 2 weeks at 37°C, sunk in 15% sucrose overnight, and embedded in OCT. Transverse sections (30 μm) were collected and viewed using a Bio-Rad MRC600 confocal microscope.
Acknowledgments
We thank Shirley Pease and the staff of the Transgenic Animal Facility at Caltech for assistance with mouse breeding and care, Lan Dinh for genotyping, Gaby Mosconi for lab management, P. Labosky for the gift of the Hfh2 probe, Sandra Rebelo for advice on DiI labeling, and Eric Frank and Josh Sanes for sharing unpublished data. We also thank S. Perez, S. Gerety, A. Greenwood, B. Wold, E. Frank, and J. Sanes for their helpful comments on the manuscript. Q.M. is an Associate and D.J.A. an Investigator of the Howard Hughes Medical Institute. This work was supported in part by a grant from the National Institutes of Health to the Silvio Conte Center for Neuroscience Research (H. Lester, P.I.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL c/o mancusog@cco.caltech.edu; FAX (626) 395-8374.
Reference
- Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium-channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- Alonso MC, Cabrera CV. The achaete-scute complex of Drosophila melanogaster comprises four homologous genes. EMBO J. 1988;7:2585–2591. doi: 10.1002/j.1460-2075.1988.tb03108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ, Jan YN. The determination of the neuronal phenotype. In: Cowan WM, editor. Molecular and cellular approaches to neural development. New York, NY: Oxford University Press; 1997. pp. 26–63. [Google Scholar]
- Backstrom A, Soderstrom S, Kylberg A, Ebendal T. Molecular cloning of the chicken trkA and its expression in early peripheral ganglia. J Neurosci Res. 1996;46:67–81. doi: 10.1002/(SICI)1097-4547(19961001)46:1<67::AID-JNR9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Birren SJ, Lo LC, Anderson DJ. Sympathetic neurons undergo a developmental switch in trophic dependence. Development. 1993;119:597–610. doi: 10.1242/dev.119.3.597. [DOI] [PubMed] [Google Scholar]
- Braun T, Arnold H. myf5 and myoD genes are activated in distinct mesenchymal stem cells and determine different skeletal muscle cell lineages. EMBO J. 1996;15:310–318. [PMC free article] [PubMed] [Google Scholar]
- Braun T, Rudnicki MA, Arnold HH, Jaenisch R. Targeted inactivation of the muscle regulatory gene Myf-5 results in abnormal rib development and perinatal death. Cell. 1992;71:369–382. doi: 10.1016/0092-8674(92)90507-9. [DOI] [PubMed] [Google Scholar]
- Braun T, Bober E, Rudnicki MA, Jaenisch R, Arnold HH. MyoD expression marks the onset of skeletal myogenesis in Myf-5 mutant mice. Development. 1994;120:3083–3092. doi: 10.1242/dev.120.11.3083. [DOI] [PubMed] [Google Scholar]
- Campuzano S, Modolell J. Patterning of the Drosophila nervous system: The achaete–scute gene complex. Trends Genet. 1992;8:202–208. doi: 10.1016/0168-9525(92)90234-u. [DOI] [PubMed] [Google Scholar]
- Carr VM, Simpson SB. Proliferative and degenerative events in the early development of chick dorsal root ganglia. J Comp Neurol. 1978;182:727–740. doi: 10.1002/cne.901820410. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophin receptors: A window into neuronal differentiation. Neuron. 1992;9:583–593. doi: 10.1016/0896-6273(92)90023-7. [DOI] [PubMed] [Google Scholar]
- Cossu G, Tajbakhsh S, Buckingham M. How is myogenesis initiated in the embryo? Trends Genet. 1996;12:218–223. doi: 10.1016/0168-9525(96)10025-1. [DOI] [PubMed] [Google Scholar]
- Fariñas I, Wilkinson GA, Backus C, Reichardt LF, Patapoutian A. Characterization of neurotrophin and trk receptor functions in developing sensory ganglia: Direct NT-3 activation of trkB neurons in vivo. Neuron. 1998;21:325–334. doi: 10.1016/s0896-6273(00)80542-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fode C, Gradwohl G, Morin X, Dierich A, LeMeur M, Goridis C, Guillemot F. The bHLH protein NEUROGENIN 2 is a determination factor for epibranchial placode-derived sensory neurons. Neuron. 1998;20:483–494. doi: 10.1016/s0896-6273(00)80989-7. [DOI] [PubMed] [Google Scholar]
- Frank E, Sanes JR. Lineage of neurons and glia in chick dorsal root ganglia: Analysis in vivo with a recombinant retrovirus. Development. 1991;111:895–908. doi: 10.1242/dev.111.4.895. [DOI] [PubMed] [Google Scholar]
- Fraser SE, Bronner-Fraser ME. Migrating neural crest cells in the trunk of the avian embryo are multipotent. Development. 1991;112:913–920. doi: 10.1242/dev.112.4.913. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradwohl G, Fode C, Guillemot F. Restricted expression of a novel murine atonal-related bHLH protein in undifferentiated neural precursors. Dev Biol. 1996;180:227–241. doi: 10.1006/dbio.1996.0297. [DOI] [PubMed] [Google Scholar]
- Greenwood, A.L., E.E. Turner, and D.J. Anderson. 1999. Identification of dividing, determined sensory neuron precursors in the mammalian neural crest. Development (in press). [DOI] [PubMed]
- Hamburger V, Hamilton H. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Jan YN, Jan LY. HLH proteins, fly neurogenesis and vertebrate myogenesis. Cell. 1993;75:827–830. doi: 10.1016/0092-8674(93)90525-u. [DOI] [PubMed] [Google Scholar]
- Jarman AP, Grau Y, Jan LY, Jan Y-N. atonal is a proneural gene that directs chordotonal organ formation in the Drosophila peripheral nervous system. Cell. 1993;73:1307–1321. doi: 10.1016/0092-8674(93)90358-w. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Birren SJ, Anderson DJ. Two rat homologues of Drosophila achaete–scute specifically expressed in neuronal precursors. Nature. 1990;346:858–861. doi: 10.1038/346858a0. [DOI] [PubMed] [Google Scholar]
- Kageyama R, Nakanishi S. Helix-loop-helix factors in growth and differentiation of the vertebrate nervous system. Curr Opin Gen Dev. 1997;7:659–665. doi: 10.1016/s0959-437x(97)80014-7. [DOI] [PubMed] [Google Scholar]
- Kahane N, Kalcheim C. Expression of trkC receptor mRNA during development of the avian nervous system. J Neurobiol. 1994;25:571–584. doi: 10.1002/neu.480250509. [DOI] [PubMed] [Google Scholar]
- Kahane N, Cinnamon Y, Kalcheim C. The origin and fate of pioneer myotomal cells in the avian embryo. Mech Dev. 1998;74:59–73. doi: 10.1016/s0925-4773(98)00066-5. [DOI] [PubMed] [Google Scholar]
- Labosky PA, Kaestner KH. The winged helix transcription factor Hfh2 is expressed in neural crest and spinal cord during mouse development. Mech Dev. 1998;76:185–190. doi: 10.1016/s0925-4773(98)00105-1. [DOI] [PubMed] [Google Scholar]
- Lawson SN, Biscoe TJ. Development of mouse dorsal root ganglia: An autoradiographic and quantitative study. J Neurocytol. 1979;8:265–274. doi: 10.1007/BF01236122. [DOI] [PubMed] [Google Scholar]
- Lee JE. Basic helix-loop-helix genes in neural development. Curr Opin Neurobiol. 1997;7:13–20. doi: 10.1016/s0959-4388(97)80115-8. [DOI] [PubMed] [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- Lin JH, Saito T, Anderson DJ, Lance-Jones C, Jessell TM, Arber S. Functionally-related motor neuron pool and muscle sensory afferent subtypes defined by coordinate ETS gene expression. Cell. 1998;95:393–407. doi: 10.1016/s0092-8674(00)81770-5. [DOI] [PubMed] [Google Scholar]
- Ma Q, Kintner C, Anderson DJ. Identification of neurogenin, a vertebrate neuronal determination gene. Cell. 1996;87:43–52. doi: 10.1016/s0092-8674(00)81321-5. [DOI] [PubMed] [Google Scholar]
- Ma Q, Sommer L, Cserjesi P, Anderson DJ. Mash1 and neurogenin1 expression patterns define complementary domains of neuroepithelium in the developing CNS and are correlated with regions expressing Notch ligands. J Neurosci. 1997;17:3644–3652. doi: 10.1523/JNEUROSCI.17-10-03644.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Chen ZF, Barrantes IB, de la Pompa JL, Anderson DJ. Neurogenin 1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron. 1998;20:469–482. doi: 10.1016/s0896-6273(00)80988-5. [DOI] [PubMed] [Google Scholar]
- McCormick MB, Tamimi RM, Snider L, Asakura A, Bergstrom D, Tapscott SJ. neuroD2 and neuroD3: Distinct expression patterns and transcriptional activation potentials within the neuroD gene family. Mol Cell Biol. 1996;16:5792–5800. doi: 10.1128/mcb.16.10.5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson AM, Abmayr SM, Bate M, Arias AM, Maniatis T. Expression of a MyoD family member prefigures muscle pattern in Drosophila embryos. Genes & Dev. 1990;4:2086–2097. doi: 10.1101/gad.4.12a.2086. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Olson EN. Defining the regulatory networks for muscle development. Curr Opin Gen Dev. 1996;6:445–453. doi: 10.1016/s0959-437x(96)80066-9. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Furukawa T, Lee JE, Cepko CL. NeuroD regulates multiple functions in the developing neural retina in rodent. Development. 1999;126:23–36. doi: 10.1242/dev.126.1.23. [DOI] [PubMed] [Google Scholar]
- Mu X, Silos-Santiago I, Carrol SL, Snider WD. Neurotrophin receptor genes are expressed in distinct patterns in developing dorsal root ganglia. J Neurosci. 1993;13:4029–4041. doi: 10.1523/JNEUROSCI.13-09-04029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapoutian A, Yoon JK, Miner JH, Wang S, Stark K, Wold B. Disruption of the mouse MRF4 gene identifies multiple waves of myogenesis in the myotome. Development. 1995;121:3347–3358. doi: 10.1242/dev.121.10.3347. [DOI] [PubMed] [Google Scholar]
- Perez SE, Rebelo S, Anderson DJ. Early specification of sensory neuron fate revealed by expression and function of neurogenins in the chick embryo. Development. 1999;126:1715–1728. doi: 10.1242/dev.126.8.1715. [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Braun T, Hinuma S, Jaenisch R. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Schnegelsberg PNJ, Stead RH, Braun T, Arnold HH, Jaenisch R. MyoD or Myf5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- Snider WD. Functions of the neurotrophins during nervous system development—what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Snider WD, Wright DE. Neurotrophins cause a new sensaton. Neuron. 1996;16:229–232. doi: 10.1016/s0896-6273(00)80039-2. [DOI] [PubMed] [Google Scholar]
- Sommer L, Ma Q, Anderson DJ. neurogenins, a novel family of atonal-related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Mol Cell Neurosci. 1996;8:221–241. doi: 10.1006/mcne.1996.0060. [DOI] [PubMed] [Google Scholar]
- Stein R, Mori N, Matthews K, Lo L-C, Anderson DJ. The NGF-inducible SCG10 mRNA encodes a novel membrane-bound protein present in growth cones and abundant in developing neurons. Neuron. 1988;1:463–476. doi: 10.1016/0896-6273(88)90177-8. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S, Cossu G. Establishing myogenic identity during somitogenesis. Curr Opin Genet Dev. 1997;7:634–641. doi: 10.1016/s0959-437x(97)80011-1. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S, Rocancourt D, Buckingham M. Muscle progenitor cells failing to repond to positional cues adopt non-myogenic fates in Myf5 null mice. Nature. 1996;384:266–270. doi: 10.1038/384266a0. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S, Borello U, Vivarelli E, Kelly R, Papkoff J, Duprez D, Buckingham M, Cossu G. Differential activation of Myf5 and MyoD by different Wnts in explants of mouse paraxial mesoderm and the later activation of myogenesis in the absence of Myf5. Development. 1998;125:4155–4162. doi: 10.1242/dev.125.21.4155. [DOI] [PubMed] [Google Scholar]
- Weintraub H. The MyoD family and myogenesis: Redundancy, networks and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- Weintraub H, Davis R, Tapscott S, Thayer M, Krause M, Benezra R, Blackwell TK, Turner D, Rupp R, Hollenberg S, Zhuang Y, Lassar A. The myoD gene family: Nodal point during specification of the muscle cell lineage. Science. 1991;251:761–766. doi: 10.1126/science.1846704. [DOI] [PubMed] [Google Scholar]
- Wright DE, Snider WD. Neurotrophin receptor mRNA expression defines distinct populations of neurons in rat dorsal root ganglia. J Comp Neurol. 1995;351:329–338. doi: 10.1002/cne.903510302. [DOI] [PubMed] [Google Scholar]
- Wyatt S, Davies AM. Regulation of expression of mRNAs encoding the nerve growth factor receptors p75 and trkA in developing sensory neurons. Development. 1993;119:635–647. doi: 10.1242/dev.119.3.635. [DOI] [PubMed] [Google Scholar]
- Yun KS, Wold B. Skeletal muscle determination and differentiation—story of a core regulatory network and its context. Curr Opin Cell Biol. 1996;8:877–889. doi: 10.1016/s0955-0674(96)80091-3. [DOI] [PubMed] [Google Scholar]