

Abstract
The identical reaction pathway executed by the spliceosome and self-splicing group II intron ribozymes has prompted the idea that both may be derived from a common molecular ancestor. The minimal sequence and structural similarities between group II introns and the spliceosomal small nuclear RNAs, however, have left this proposal in question. Mechanistic comparisons between group II self-splicing introns and the spliceosome are therefore important in determining whether these two splicing machineries may be related. Here we show that 3′-sulfur substitution at the 5′ splice site of a group II intron causes a metal specificity switch during the first step of splicing. In contrast, 3′-sulfur substitution has no significant effect on the metal specificity of the second step of cis-splicing. Isolation of the second step uncovers a metal specificity switch that is masked during the cis-splicing reaction. These results demonstrate that group II intron ribozymes are metalloenzymes that use a catalytic metal ion for leaving group stabilization during both steps of self-splicing. Furthermore, because 3′-sulfur substitution of a spliceosomal intron has precisely the same effects as were observed during cis-splicing of the group II intron, these results provide striking parallels between the catalytic mechanisms employed by these two systems.
Keywords: Group II intron, spliceosome, ribozyme, metal ion catalysis, 3′-S-phosphorothiolate, phosphotransesterification
Several classes of intervening sequences (introns) exist in eukaryotic genomes, and each class is removed (spliced) post-transcriptionally by a distinct pathway. Most introns are removed by a large RNA–protein complex called the spliceosome (Nilsen 1998; Burge et al. 1999), but some (group I and group II introns) self-splice by virtue of a catalytic activity resident in the intron RNA itself (Cech and Golden 1999). Excision of group II introns occurs by a two-step transesterification pathway involving 5′ splice site cleavage followed by exon ligation; the 2′-hydroxyl group of an adenosine residue within the intron is usually the first-step nucleophile, leading to excision of the intron in the form of a lariat (Michel and Ferat 1995; Pyle 1996). Although spliceosomes are of much greater size and complexity, they catalyze intron removal by the same chemical pathway, leading to speculation that the spliceosome is essentially an RNA catalyst that shares a common molecular ancestor with group II introns (Sharp 1985; Cech 1986).
The discovery of introns immediately prompted many ideas and questions about the roles of introns in the evolution of genomes (e.g., Gilbert 1978; Darnell 1978; Doolittle 1978). The central issue has become one of intron antiquity, that is, whether intron–exon structure predates the divergence of eubacteria, archaebacteria, and eukaryotes. The introns early/late question has been debated vigorously, and most of the evidence has focused on the conservation of intron positions, the correlation between intron positions and protein structure, and the distribution of intron phases for spliceosomal introns from genes present in all three kingdoms (for reviews, see Gilbert et al. 1997; Logsdon 1998). Another line of inquiry concerns the molecular mechanisms of intron removal—if these spliceosomal introns are ancient or are derived from an ancient precursor, then the mechanisms by which they are removed may be ancient as well. Because catalytic RNAs are often thought of as ‘molecular fossils’ from an ancient ‘RNA World’ epoch (Gesteland et al. 1999), understanding whether spliceosomal introns are removed by an RNA-based mechanism is important to the debate over intron antiquity. As a result, the possible evolutionary relationship between the spliceosome and the group II ribozymes has received a great deal of interest.
Evolutionary relationships between macromolecules are often inferred from similarities of sequence and structure. With regard to group II introns and the spliceosomal introns and small nuclear RNAs (U1, U2, U4, U5, and U6), conserved GU dinucleotides at the 5′ splice sites and AGC trinucleotides within presumptive catalytic domains (for reviews, see Pyle 1996; Burge et al. 1999) have been noted, but these similarities are extremely limited and their significance has been questioned (Weiner 1993; Michel and Ferat 1995). As for structure, our understanding of these two systems is still fragmentary, but certain elements of secondary and tertiary structure bear some resemblance to each other. In the spliceosome, the U6/5′ splice site pairing, U2/U6 helix 1 region, U2/branchpoint bulged duplex, and U5/exon interactions have been viewed as analogous (and perhaps homologous) to the ε-ε′ pairing, domain 5, domain 6, and EBS-1/IBS1 and δ-δ′ interactions, respectively, in group II introns (for reviews, see Michel and Ferat 1995; Nilsen 1998). Functional complementation of an EBS1/δ deletion by the U5 conserved loop has made the latter similarity particularly compelling (Hetzer et al. 1997). Although these similarities are tantalizing, some differences exist as well (Michel and Ferat 1995), and the proposed evolutionary kinship between the spliceosome and group II introns remains debatable.
Possible relationships can also be judged by a “mechanistic phylogeny” involving comparisons of reaction pathways and catalytic mechanisms. The only such information obtained for both systems thus far comes from the analysis of chiral phosphorothioates at the splice sites. In both the spliceosome (Maschhoff and Padgett 1993; Moore and Sharp 1993) and group II introns (Padgett et al. 1994), incorporation of an RP phosphorothioate at either splice site blocks the reaction, but incorporation of the SP diastereomer does not; furthermore, reaction at each SP phosphorothioate results in inversion of stereochemistry, indicative of direct in-line SN2 nucleophilic attack. These results provide additional evidence for a common reaction pathway catalyzed by both systems, but whether this commonality extends to the catalytic mechanisms themselves remains unknown.
We reported recently experiments that bear directly on the catalytic mechanisms employed by the spliceosome (Sontheimer et al. 1997). Incorporation of a 3′-S-phosphorothiolate linkage (in which the 3′-oxygen is replaced by sulfur) at the 5′ splice site gives rise to a metal specificity switch for the first step of splicing, providing strong evidence that a catalytic metal ion in the spliceosomal active site stabilizes the leaving group by direct coordination. In contrast, 3′-sulfur substitution at the 3′ splice site has no effect on the metal specificity of the second step of the reaction. This result argues that inner-sphere coordination of the leaving group by a metal ion may not be required for this reaction, although a rate-limiting binding or conformational step may mask an inhibitory effect on the chemical step of 3′ splice site cleavage and exon ligation. Because of the expectation that most catalytic RNAs may use divalent metal ions for catalysis (for review, see Narlikar and Herschlag 1997), the former possibility was viewed as weakening the case for RNA catalysis in the second step of the spliceosome reaction.
We have now extended this analysis to reactions catalyzed by the ai5γ group II intron ribozyme from Saccharomyces cerevisiae mitochondria. We find that 3′-sulfur substitution at the 5′ splice site results in a metal specificity switch for the first reaction step, demonstrating that group II introns, like the spliceosome, use a catalytic metal ion for leaving group stabilization via inner-sphere coordination. The most striking parallel with the spliceosome, however, is that 3′-sulfur substitution at the 3′ splice site has little or no effect on metal ion specificity during cis-splicing. Therefore, spliceosomal and group II introns exhibit the same asymmetric response to 3′-sulfur substitution at the 5′ and 3′ splice sites during cis-splicing. To probe further the effect of 3′-sulfur substitution at the 3′ splice site, we isolated 3′ splice site cleavage and exon ligation from the rest of the cis-splicing pathway using a trans reaction. Under these conditions, a metal specificity switch is uncovered, indicating that a metal ion also stabilizes the leaving group in the second step of splicing and suggesting that a conformational change (Chanfreau and Jacquier 1996) limits the rate of exon ligation during cis-splicing.
Results
3′-S-phosphorothiolate diesters have proven to be useful analogs in the analysis of the catalytic mechanisms used by RNA, protein, and ribonucleoprotein enzymes (Sontheimer 1999). Oxygen and sulfur differ in their abilities to occupy the inner ligand sphere of various metal ions (Sigel et al. 1997 and references therein); for instance, Mg2+ (a “hard” metal) coordinates well to oxygen, but strongly resists coordination to sulfur. In contrast, “soft” metals such as Mn2+, Co2+, Zn2+, and Cd2+ readily accept (and in some cases prefer) sulfur as an inner-sphere ligand. For a divalent-metal-dependent reaction that involves a 3′-oxygen as the leaving group (such as splicing), a change in metal specificity from Mg2+ to a softer metal upon 3′-sulfur substitution implicates a direct metal ion-leaving group interaction.
Group II introns can self-splice by either of two routes—a “branching” or transesterification pathway or a hydrolytic pathway (Fig. 1A). Both can be relevant in vivo (Podar et al. 1998a); in vitro, either pathway can predominate, depending on the ionic conditions (Daniels et al. 1996). The excised intron is stable in vitro and can catalyze hydrolysis at the exon–exon junction of the spliced product. This spliced exons reopening (SER) reaction is mechanistically analogous to the reversal of the second step of splicing (Podar et al. 1995). To test for the presence of catalytic metal ions in the active site(s) of the group II ribozyme, we used a combination of chemical synthesis (Sun et al. 1997) and enzymatic ligation (Moore and Query 1998) to introduce a 3′-S-phosphorothiolate diester at the site of cleavage for the first (Fig. 1B) and second (Fig. 1C) steps of cis-splicing.
Figure 1.
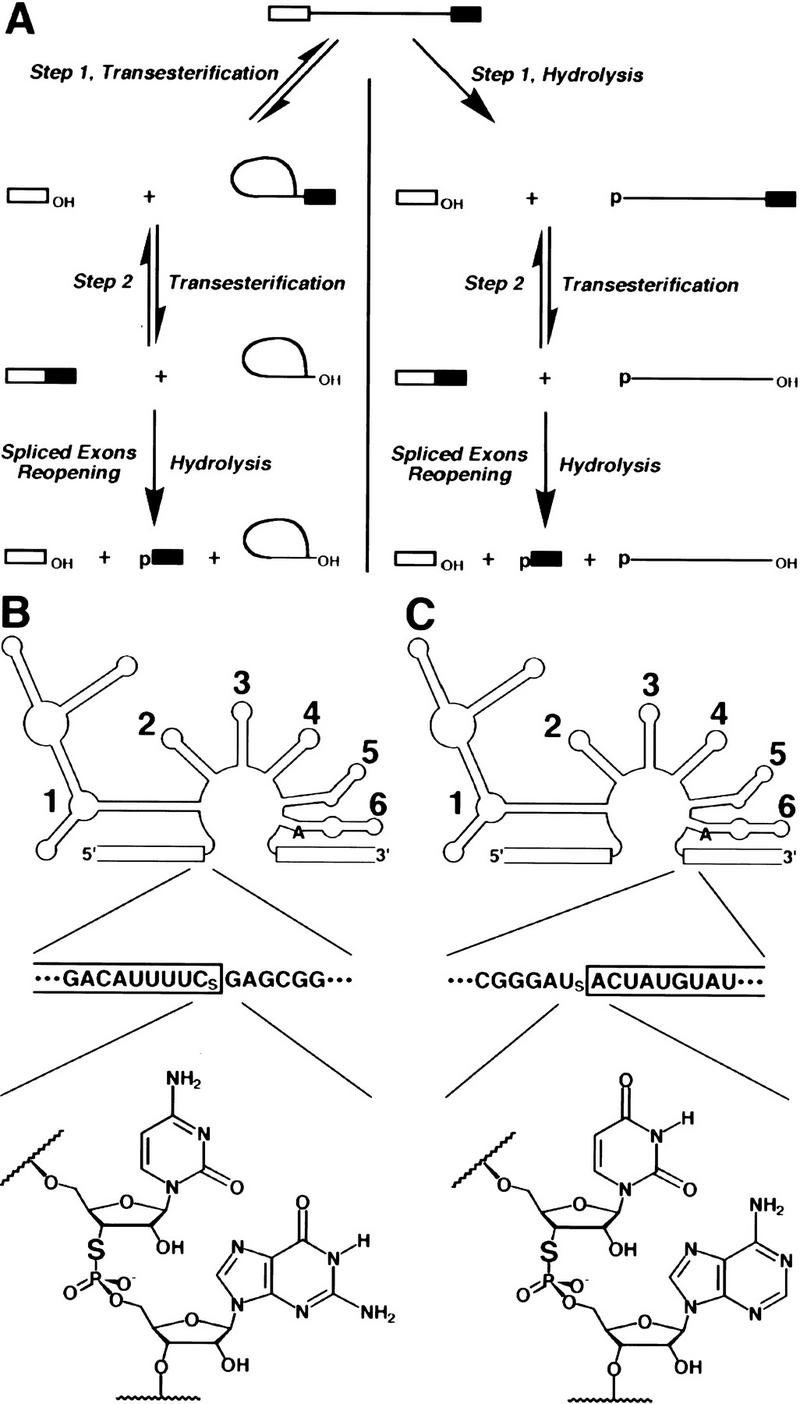
3′-Sulfur substitution at the splice sites of a group II intron. (A) The pathways of group II intron self-splicing. Splicing proceeds by either of two pathways, which differ in the identity of the nucleophile during the first step of the reaction. In the branching or transesterification pathway (left), the 2′-hydroxyl group of an adenosine residue attacks the 5′ splice site, giving rise to the exon 1 and lariat intron/exon 2 intermediates. In the hydrolytic pathway (right), water or hydroxide attacks the 5′ splice site, and the intron/exon 2 intermediate is linear. For both pathways, the second step proceeds by attack of the 3′-hydroxyl group of exon 1 on the 3′ splice site, giving rise to spliced exons and releasing the excised intron. As shown at the bottom, released intron can catalyze a SER hydrolytic reaction. (B) 5′ Splice site substitution. A simplified depiction of the yeast mitochondrial group II intron ai5γ is given at the top; exons are boxed, and each of the six intron domains is indicated. The bulged A residue that acts as a nucleophile in the first reaction step is shown in domain 6. The sequence of the 5′ splice junction is given underneath [(CS) 3′-thiocytidine]. The structure of the 3′-S-phosphorothiolate linkage at the 5′ splice site is given at the bottom. (C) 3′ Splice site substitution. The diagram is as in B, except that the sequence and the 3′-S-phosphorothiolate linkage are for the 3′ splice site [(US) 3′-thiouridine].
3′-Sulfur substitution at the 5′ splice site of a group II intron results in a metal specificity switch
Self-splicing constructs with a 3′-sulfur substitution (Fig. 1B) or a normal 3′-oxygen at the 5′ splice site were constructed and tested for cis-splicing activity in vitro (Fig. 2A) in the presence of 0.5 m (NH4)2SO4 and 100 mm divalent metal ion (Daniels et al. 1996). For the control substrate, no reaction occurred in the absence of divalent metals (Fig. 2A, lane 4), but exon 1 and spliced product were both generated in the presence of 100 mm MgCl2 (Fig. 2A, lane 5). When the reactions included 10 or 20 mm MnCl2, CoCl2, ZnCl2, or CdCl2 (Fig. 2A, lanes 6–13), exon 1 and spliced product were easily detected, indicating that the presence of these metal ions allows efficient splicing (although CoCl2, ZnCl2, and CdCl2 appear to affect the relative rates of the first and second steps, as indicated by the decreased amounts of exon 1 in Fig. 2A, lanes 8–13). For the 3′-sulfur-containing substrate, no reaction was observed in the absence of divalent metals (Fig. 2A, lane 17). Unlike the control substrate, however, no reaction was observed in the presence of 100 mm MgCl2 (Fig. 2A, lane 18). Inclusion of 10 or 20 mm MnCl2, ZnCl2, or CdCl2 restored efficient 5′ splice site cleavage (Fig. 2A, lanes 19, 20, and 23–26), demonstrating a switch in metal specificity for this reaction. CoCl2 (10 or 20 mm) was unable to restore 5′ splice site cleavage (Fig. 2A, lanes 21–22). Although MnCl2, ZnCl2, or CdCl2 rescued the first step of the reaction, no spliced product was generated (Fig. 2A, lanes 19, 20, and 23–26), consistent with the observation that sulfur is a very poor nucleophile at phosphodiester linkages (Pearson 1966; Dantzmann and Kiessling 1996).
Figure 2.
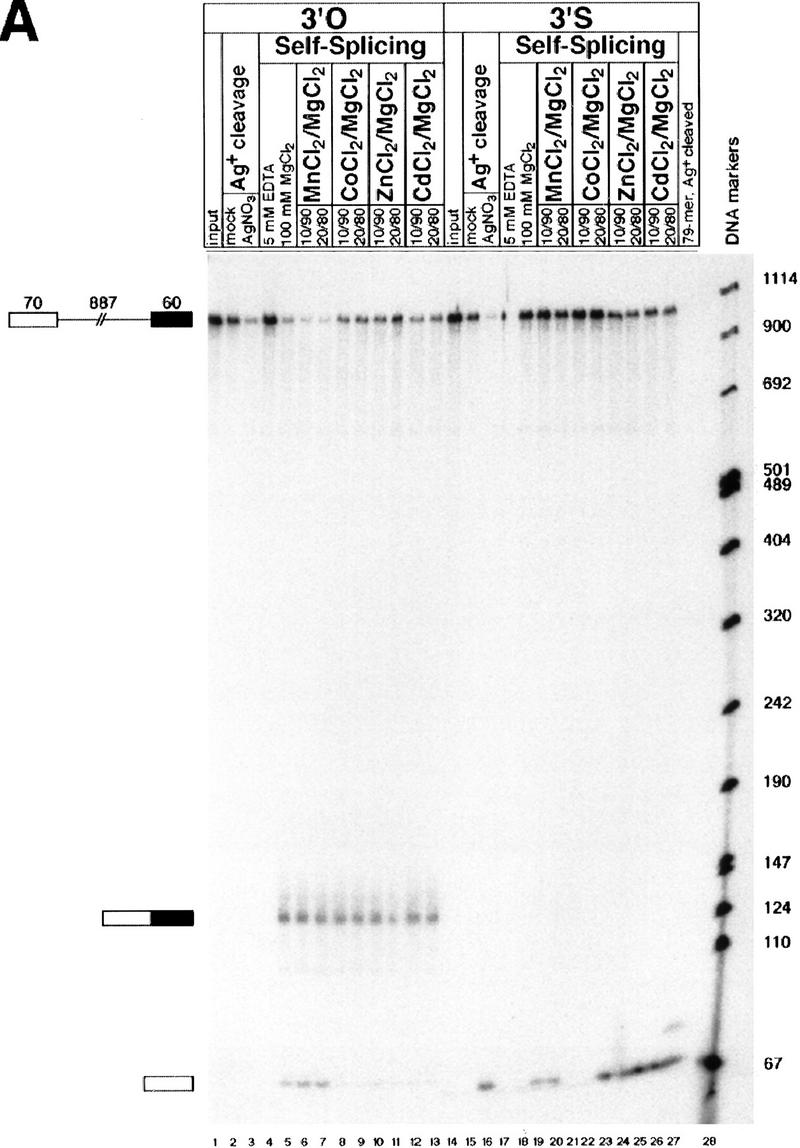

(A) 3′-Sulfur substitution at the 5′ splice site of a group II intron results in a metal specificity switch. Self-splicing reactions were performed with ligated substrates containing a 3′-oxygen (lanes 1–13) or a 3′-sulfur (lanes 14–26) at the 5′ splice site (see Fig. 1B). (Lanes 1,14) Unspliced RNAs; each substrate was also mock-reacted (lanes 2,15) or reacted (lanes 3,16) with silver(I). Self-splicing reactions were carried out for 1 hr and contained 5 mm EDTA (lanes 4, 17), 100 mm MgCl2 (lanes 5,18), or 10 or 20 mm MnCl2, CoCl2, ZnCl2, or CdCl2 (lanes 6–13, 19–26) as indicated at the top of each lane. Reactions in the presence of MnCl2, CoCl2, ZnCl2, or CdCl2 were supplemented with MgCl2 so that the total divalent metal ion concentration was 100 mm. Unspliced precursor (1017 nucleotides), exon 1 intermediate (70 nucleotides), and spliced product (130 nucleotides) are indicated on the left, and the sizes of the DNA markers (in nucleotides) are given on the right. The intron/exon 2 intermediates (lariat and linear) and the corresponding excised intron products contain no radiolabel and are therefore not visible. As an additional standard, lane 27 is a silver cleavage reaction of a 3′-sulfur-containing RNA (79 nucleotides) consisting of exon 1 and only 9 nucleotides of intron sequence. (B) 5′ Splice site cleavage of the 3′-sulfur-substituted substrate occurs accurately in the presence of Mn2+. The strategy for mapping the site of cleavage of the modified RNA is given at the top; the single 32P-labeled phosphate group is indicated with an asterisk. Samples were digested with RNase T1, and half of each sample was also treated with iodoacetamide as indicated at the top of each lane. 3′-oxygen and 3′-sulfur products are indicated on the left and right of the gel, respectively. The 8-nucleotide product with a 3′-terminal thiol (lanes 7,9) runs faster than the same fragment with a 3′-terminal hydroxyl (lanes 3,4) because the sulfur is deprotonated under these electrophoresis conditions, adding an extra negative charge.
Treatment of the 3′-sulfur-substituted substrate with silver(I), which induces the specific hydrolysis of the sulfur–phosphorus bond of a 3′-S-phosphorothiolate linkage (Cosstick and Vyle 1990), gave rise to a product of the same size (Fig. 2A, lane 16), confirming the presence of the 3′-sulfur modification in the substrate and suggesting that 5′ splice site cleavage occurred accurately. Although the 70-nucleotide exon 1 intermediates and the silver-cleaved product comigrated in this 5% polyacrylamide gel, it is possible that the resolution was not sufficient to detect very small size differences (1–2 nucleotides). To confirm the accuracy of 5′ splice site cleavage of the modified substrate, unspliced precursor and the purified products of silver cleavage and Mn2+-rescued self-splicing were digested with RNase T1, which cleaves after guanosine residues. Unmodified precursor and exon 1 intermediate were digested in parallel for comparison. We also treated portions of each sample with iodoacetamide (which reacts with thiols but not hydroxyls) to test for the presence of the free 3′-thiol (Weinstein et al. 1996). All samples were then subjected to electrophoresis in a 20% polyacrylamide gel, which provides sufficient resolution to detect single-nucleotide and even single-functional-group differences. As shown in Figure 2B, the RNase T1-digested products of silver cleavage (lane 7) and self-splicing (lane 9) comigrated precisely. Furthermore, both fragments reacted quantitatively with iodoacetamide, which decreased their mobilities by exactly the same extent (Fig. 2B, cf. lanes 8 and 10). Iodoacetamide did not react with the fragments derived from unspliced precursors or unmodified exon 1 intermediate (Fig. 2B, cf. lanes 1, 3, and 5 with lanes 2, 4, and 6, respectively), confirming the specificity of the modification reaction. These results identify the cleavage site as the sulfur-phosphorus bond of the 3′-S-phosphorothiolate linkage. Similar analyses demonstrated the accuracy of 5′ splice site cleavage of the modified substrate in the presence of Zn2+ and Cd2+ (data not shown).
To confirm that group II ribozyme activity was required for the observed cleavage in the presence of thiophilic metal ions, we took advantage of a trans reaction characterized by Pyle and coworkers (Fig. 3, top panel). An RNA consisting of exon 1 and intron domains 1, 2, and 3 (ExD123) has no 5′ splice site cleavage activity on its own, but addition of a separate domain 5 RNA (D5) causes specific 5′ splice site cleavage (Pyle and Green 1994). This reaction appears to be a faithful mimic of the first step of self-splicing (Pyle and Green 1994; Peebles et al. 1995; Podar et al. 1995). We incorporated a 3′-S-phosphorothiolate linkage into the 5′ splice site of an ExD123 RNA and tested the ability of saturating levels of D5 (Pyle and Green 1994) to catalyze the hydrolysis of the sulfur–phosphorus bond. 3′-S-Phosphorothiolate linkages in RNA undergo base-catalyzed breakdown two to three orders of magnitude faster than unmodified phosphodiesters, giving rise to cleavage products with 2′-O,3′-S-cyclic phosphorothiolate and 5′-hydroxyl termini (Liu and Reese 1996; Weinstein et al. 1996). Therefore, the 70-nucleotide exon 1 resulting from either enzymatic hydrolysis or background cleavage differ only in the presence or absence of a 3′-terminal cyclic phosphorothiolate, and cannot be resolved reliably by gel electrophoresis (data not shown). Because the enzymatic reaction is relatively slow (Pyle and Green 1994), the levels of background cleavage are prohibitively high to assay D5-catalyzed hydrolysis directly. Therefore, we treated reaction mixtures with iodoacetamide and RNase T1 to generate fragments that could be resolved from those derived from unreacted or background-cleaved molecules. This assay has the additional advantage of confirming the site of D5-catalyzed 5′ splice site hydrolysis with single-nucleotide accuracy. The reactions are shown in the lower panel of Figure 3. For the 3′-oxygen control substrate, accurate D5-dependent hydrolysis was observed in 100 mm MgCl2 (Fig. 3, lane 3), and inclusion of 10 mm MnCl2 (Fig. 3, lane 5) or 10 mm CdCl2 (Fig. 3, lane 7) did not impair the reaction. [10 mm ZnCl2 is insoluble and causes RNA degradation under the high-KCl conditions of this assay (Pyle and Green 1994) , and therefore could not be tested.] Substitution of the 3′-oxygen leaving group with sulfur blocked the hydrolysis reaction when Mg2+ was the sole divalent metal ion present (Fig. 3, cf. lanes 3 and 11). Inclusion of 10 mm MnCl2 (Fig. 3, lane 13) or CdCl2 (Fig. 3, lane 15) relieved this negative effect. The Mn2+- and Cd2+-rescued reactions were D5-dependent (Fig. 3, lanes 12,14) and accurate, as judged by the comigration with a silver-cleaved, iodoacetamide-modified standard (Fig. 3, lane 9). Therefore, ribozyme activity is required for 5′ splice site cleavage in the presence of these thiophilic divalent metals.
Figure 3.
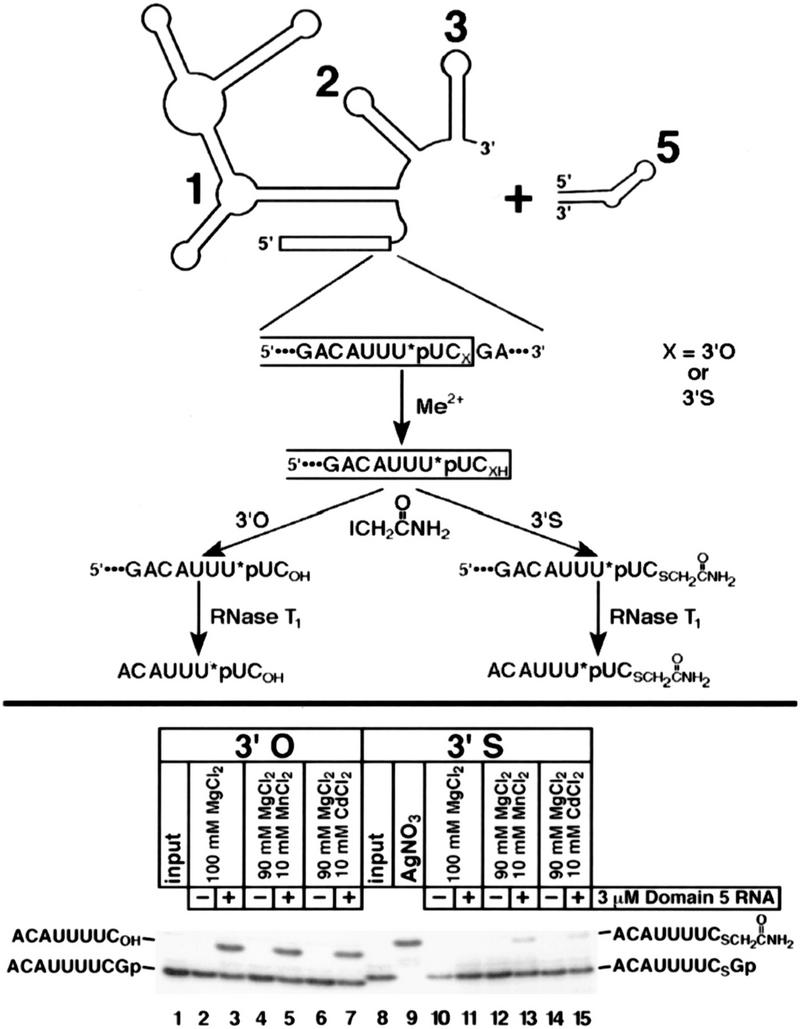
Domain 5 of the group II intron acts in trans to catalyze hydrolysis of the 3′-sulfur-substituted 5′ splice site in a Mn2+- or Cd2+-dependent manner. The substrate (ExD123) and enzyme (D5) are diagrammed at the top (the single 32P-labeled phosphate is denoted as *p). To resolve the product of accurate cleavage from that of background cleavage (see text), all reaction mixtures were treated with iodoacetamide, digested with RNase T1, and subjected to electrophoresis (bottom) as in Fig. 2B. RNase T1-digested background cleavage product was run off the bottom of the gel. (Lanes 1,8) Unreacted RNAs; (lane 9) a size standard generated by silver cleavage of the 3′-sulfur-containing substrate. Reactions contained 3 μm D5 RNA (lanes 3,5,7,11,13,15) and 100 mm MgCl2, 90 mm MgCl2/10 mm MnCl2, or 90 mm MgCl2/10 mm CdCl2 as shown at the top of each lane. Parallel reactions in the absence of D5 RNA (lanes 2,4,6,10,12,14) were done as controls. 3′-Oxygen and 3′-sulfur products are indicated on the left and right of the gel, respectively.
Mg2+ supports the second reaction step of cis-splicing of a group II intron containing a 3′-sulfur substitution at the 3′ splice site
To determine whether the second reaction step of self-splicing requires direct coordination of a metal ion to the 3′-oxygen leaving group, we synthesized a self-splicing construct with a 3′-sulfur substitution (Fig. 1C) or a normal 3′-oxygen at the 3′ splice site, and tested them for cis-splicing activity in vitro in the presence of 0.5 m (NH4)2SO4 and 100 mm divalent metal ion (Fig. 4A). Separate aliquots of the same reaction were subjected to electrophoresis in 5% (Fig. 4A, top) and 20% (Fig. 4A, bottom) polyacrylamide gels. The latter was necessary to visualize the released 10-nucleotide exon 2, generated by the hydrolysis of the exon–exon junction of the spliced product (spliced exons reopening; see Fig. 1A). For the control substrate, no reaction occurred in the absence of divalent metals (Fig. 4A, lanes 4–7), but intron–exon 2 intermediates (both linear and lariat), spliced product, and released exon 2 were all generated in the presence of 100 mm MgCl2 (Fig. 4A, lanes 8–11). For the 3′-sulfur-containing substrate, no reaction was observed in the absence of divalent metals (Fig. 4A, lanes 19–22). In striking contrast to the results obtained with the 5′ splice site, however, 100 mm MgCl2 supported both steps of cis-splicing (Fig. 4A, lanes 23–26), as judged by the appearance of lariat intron–exon 2 intermediates, spliced product, and released exon 2. The addition of 10 mm MnCl2 had no significant effect on the reaction rate (data not shown). This asymmetry in the response to 3′-sulfur substitution at the 5′ and 3′ splice sites is exactly what we observed in the spliceosome (Sontheimer et al. 1997). To diminish the unlikely possibility that the second reaction step was supported by contaminating traces of thiophilic metals, we carried out reactions with 110 mm MgCl2 and 10 mm EDTA (Fig. 4A, lanes 12–15 and 27–30). Because EDTA chelates most thiophilic divalent metal ions five to eight orders of magnitude more tightly than it chelates Mg2+ (Anderegg 1987), its inclusion would be expected to abolish the ability of trace contaminants to support the reaction. The added EDTA, however, had no effect on the second reaction step with the 3′-sulfur-substituted substrate (Fig. 4A, cf. lanes 23–26 with lanes 27–30), arguing against this possibility. Treatment of the 3′-sulfur-substituted substrate with silver(I) gave rise to the 10-nucleotide exon 2 fragment (Fig. 4A, lane 18), confirming the presence of the 3′-sulfur modification in the substrate. Although the high salt present in the self-splicing reactions distorted the electrophoretic mobilities of the exon 2 fragments (Fig. 4A, lanes 8–15, 23–30), they appeared to comigrate with the silver-cleaved exon 2, providing a preliminary indication that 3′ splice site cleavage occurred accurately.
Figure 4.

(A) Mg2+ supports 3′ splice site cleavage and exon ligation of the 3′-sulfur-substituted substrate. Self-splicing reactions were performed with ligated 3′-end-labeled substrates containing a 3′-oxygen (lanes 1–15) or a 3′-sulfur (lanes 16–30) at the 3′ splice site (see Fig. 1C). Separate aliquots of each sample were subjected to electrophoresis in a 5% polyacrylamide (top) or 20% polyacrylamide (bottom) denaturing gel. (Lanes 1,16) Unspliced RNAs; each substrate was also mock-reacted (lanes 2,17) or reacted (lanes 3,18) with silver(I). Self-splicing reactions were carried out for 1 hr and contained 10 mm EDTA (lanes 4–7, 19–22), 100 mm MgCl2 (lanes 8–11, 23–26), or 10 mm EDTA mixed with 110 mm MgCl2 (lanes 27–30). Samples were incubated for the times (in min) indicated at the top of each lane. Unspliced precursor (967 nucleotides), lariat and linear intron/exon 2 intermediates (897 nucleotides), spliced product (80 nucleotides), and released exon 2 (10 nucleotides) are indicated on the left. The sizes of the DNA markers (in nucleotides) are given on the right. The exon 1 intermediate and the excised intron products (lariat and linear) contain no radiolabel and are therefore not visible. (B) 3′ Splice site cleavage of the 3′-sulfur-substituted substrate occurs accurately in the presence of Mg2+. The strategy for mapping the site of cleavage of the modified RNA is given at the top, and is similar to that described in Figure 2B. The single 32P-labeled phosphate in the intron is denoted with an asterisk. Samples were digested with RNase T1, and half of each sample was treated with iodoacetamide as indicated at the top of each lane. The products are described on the right; the A*pUSH dinucleotide (lanes 3,5,7) and its acetamide-modified derivative (lanes 4,6,8) are indicated by dots.
To obtain further evidence for the accuracy of 3′ splice site cleavage and exon ligation, we mapped the 3′-terminus of the excised intron RNA directly. The mapping strategy (Fig. 4B, top) involved incorporation of a single 32P-labeled phosphate adjacent to the 3′ splice site, RNase T1 digestion, iodoacetamide modification, and comparison with the identically treated product of silver cleavage. If the correct 3′ splice site was used, then RNase T1 digestion should yield the dinucleotide A*pUSH as the only radiolabeled product, and this dinucleotide should be modifiable with iodoacetamide. The data are shown at the bottom of Figure 4B. Silver cleavage generated the A*pUSH standard (Fig. 4B, lane 3), which on reaction with iodoacetamide yielded the slower-migrating product A*pUSCH2C(O)NH2. In the RNase T1 digestions of total RNA from self-splicing reactions in either 100 mm MgCl2 (Fig. 4B, lane 5) or 90 mm MgCl2/10 mm MnCl2 (Fig. 4B, lane 7), the A*pUSH dinucleotide is largely obscured by background; however, treatment with iodoacetamide clearly generates the identical A*pUSCH2C(O)NH2 modified dinucleotide in both cases (Fig. 4B, lanes 6, 8). This product is absent from the RNA derived from unspliced precursor (Fig. 4B, lane 2). We conclude that the sulfur-phosphorus bond of the 3′ splice site 3′-S-phosphorothiolate linkage is cleaved during self-splicing in the presence of Mg2+. Furthermore, the efficiency of accurate 3′ splice site cleavage is not altered by the inclusion of the thiophilic metal Mn2+ (Fig. 4B, cf. lanes 6 and 8), providing further evidence against a metal specificity switch during the second step of cis-splicing.
Isolation of the second step of self-splicing uncovers a metal specificity switch
As with the spliceosome (Sontheimer et al. 1997), the ability of Mg2+ alone to support exon ligation with the 3′-splice-site-substituted substrate could indicate that inner-sphere coordination of the leaving group by a metal ion is not required for the reaction. An alternative possibility, however, is that 3′-sulfur substitution does reduce the rate of the chemical step of exon ligation in the presence of Mg2+ alone, but this effect is masked by a rate-limiting conformational step (Sontheimer et al. 1997). To distinguish between these possibilities, we assayed the exon ligation reaction in isolation. We took advantage of a recently developed tripartite reaction (A. Bar-Shalom and M. Moore, pers. comm.) in which a 3′ splice site oligonucleotide is added separately to an exon 1 oligonucleotide and a ribozyme containing all but the six 3′-terminal nucleotides of the intron (Fig. 5A). Unlike other group II exon ligation systems (Podar et al. 1998b; Deme et al. 1999), this reaction circumvents the requirement for the inefficient enzymatic ligation step in the construction of the 3′-splice-site-containing substrate. We synthesized and 3′-end-labeled substrate oligonucleotides containing either a 3′-oxygen or a 3′-sulfur at the scissile phosphate and tested them in tripartite exon ligation reactions with an exon 1 oligonucleotide containing a 3′-terminal 2′-deoxycytidine (E1dC) (Fig. 5B). Although these experiments were done with subsaturating levels of 3′ splice site oligonucleotide (i.e., kcat/KM conditions), the rate of the reaction was log-linear with pH (slope ∼1 between pH 5.0 and 6.5), which is consistent with the possibility that the rate is sensitive to the chemical step of the reaction (P.M. Gordon, E.J. Sontheimer, and J.A. Piccirilli, in prep.).
Figure 5.
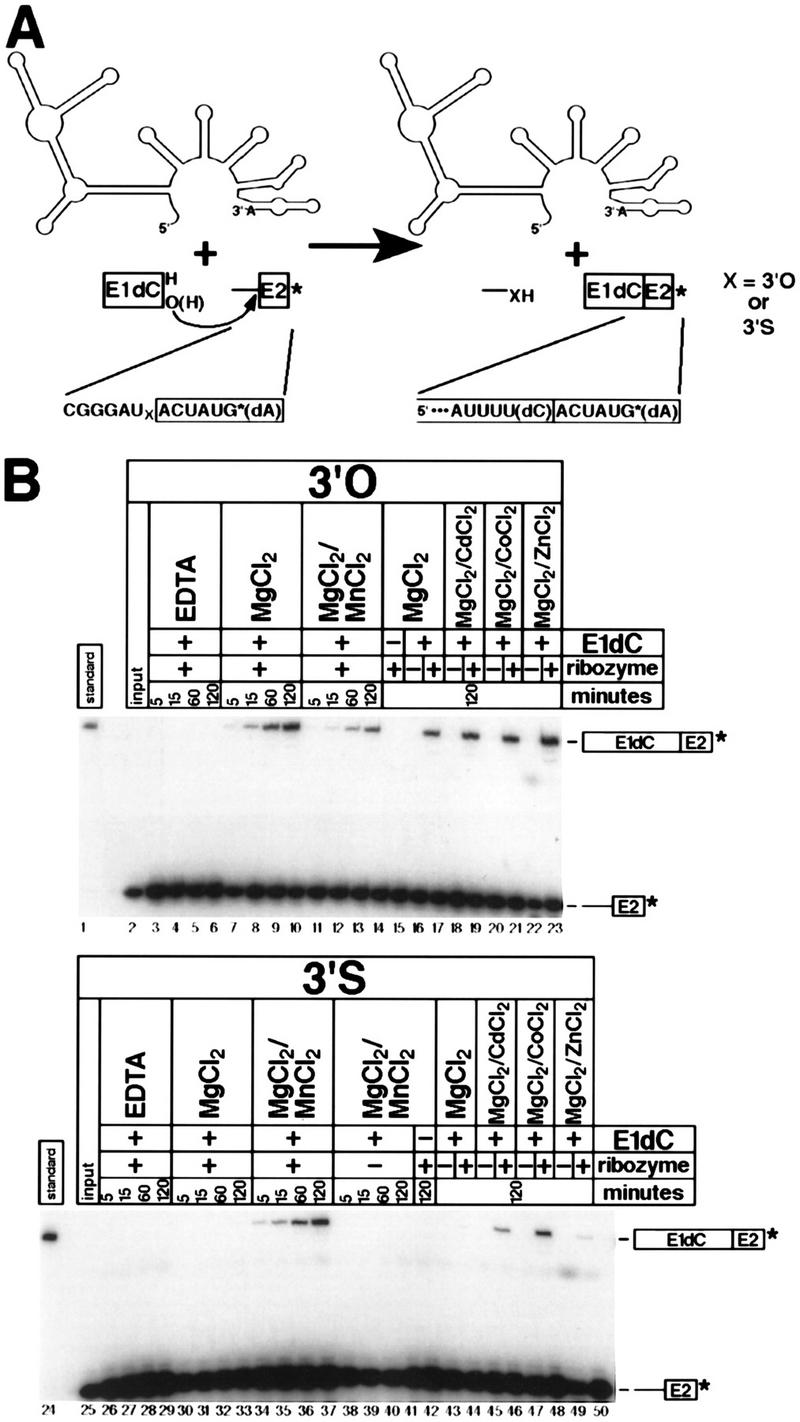
(A) The tripartite step 2 reaction involves attack of an 18-nucleotide exon 1 RNA with a 3′-terminal 2′-deoxycytidine residue (E1dC) on a 13-nucleotide 3′ splice site oligoribonucleotide, catalyzed by an ai5γ ribozyme. The 3′ splice site oligonucleotides contained either a 3′-oxygen or a 3′-sulfur at the scissile phosphate. The products are the 25-nucleotide spliced exons (labeled) and the 6-nucleotide intron fragment (unlabeled). (B) Isolation of the second step of group II self-splicing uncovers a metal specificity switch. Reactions with the 3′-oxygen (lanes 2–23) and 3′-sulfur (lanes 25–50) substrates are shown in the top and bottom panels, respectively. Reactions were incubated for the times (in min) given at the top of each lane. Parallel reactions in the absence of ribozyme (lanes 16,18,20,22,38–41,43,45,47,49) or the absence of E1dC (lanes 15,42) were done as controls. Reactions contained 10 mm EDTA (lanes 3–6, 26–29), 100 mm MgCl2 (lanes 7–10, 15–17, 30–33, 43–44), 90 mm MgCl2/10 mm MnCl2 (lanes 11–14, 34–42), 90 mm MgCl2/10 mm CdCl2 (lanes 18,19,45,46), 90 mm MgCl2/10 mm CoCl2 (lanes 20,21,47,48), or 90 mm MgCl2/10 mm ZnCl2 (lanes 22,23,49,50).
For the 3′-oxygen control substrate, the reaction in the presence of 100 mm MgCl2 yielded spliced product (Fig. 5B, lanes 7–10) that comigrated with an independently synthesized and purified standard (Fig. 5B, lane 1) under conditions where single-nucleotide differences are easily detected. The reaction is dependent on the presence of divalent metal ions (Fig. 5B, lanes 3–6), E1dC (Fig. 5B, lane 15), and intron ribozyme (Fig. 5B, lane 16). Reaction also occurred in the presence of 10 mm MnCl2 (Fig. 5B, lanes 11–14), although the rate was reduced by approximately twofold. Inclusion of 10 mm CdCl2 (Fig. 5B, lanes 18,19), CoCl2 (Fig. 5B, lanes 20,21), or ZnCl2 (Fig. 5B, lanes 22,23) also allowed efficient exon ligation in a ribozyme-dependent manner. For the 3′-sulfur-substituted substrate, no reaction was observed in the absence of divalent metal ions (Fig. 5B, lanes 26–29), ribozyme (Fig. 5B, lanes 38–41), or E1dC (Fig. 5B, lane 42). In the presence of all reaction components, however, the metal ion dependence was very different from that observed in the context of cis-splicing—reaction in the presence of MgCl2 alone (Fig. 5B, lanes 30–33) resulted in a rate reduction of 100-fold relative to the 3′-oxygen substrate. Inclusion of 10 mm MnCl2 (Fig. 5B, lanes 34–37) restored the rate to within three- or fourfold of that of the 3′-oxygen substrate under the same conditions (Fig. 5B, lanes 11–14). Furthermore, 10 mm CdCl2 (Fig. 5B, lanes 45,46) or CoCl2 (Fig. 5B, lanes 47,48) also provided strong ribozyme-dependent rate enhancements; 10 mm ZnCl2 had only a modest effect (Fig. 5B, lanes 49,50). Subsequent experiments with saturating amounts of ribozyme showed a similar inhibition in Mg2+ and rescue in Mn2+ (data not shown). Therefore, isolation of the second step of self-splicing allowed us to detect a metal ion-leaving group interaction that is obscured during canonical cis-splicing (Fig. 4A).
Reopening of spliced exons is blocked by 3′-sulfur substitution
Two metal ions have been proposed to catalyze phosphoryl transfer in each step of group II intron self-splicing: one that facilitates deprotonation and activation of the incoming 2′- or 3′-hydroxyl nucleophile, and one that stabilizes the developing negative charge on the oxyanion leaving group (Steitz and Steitz 1993). Although we have provided strong evidence for the latter in both steps of splicing (see above), there is currently no evidence regarding the former. Replacement of an oxygen nucleophile with sulfur is not optimal for the detection of metal-nucleophile interactions because of sulfur’s weak nucleophilicity at phosphate diesters (Dantzmann and Kiessling 1996; Pearson 1966). The principle of microscopic reversibility (which states that forward and reverse reactions must proceed through the same transition state) dictates that a metal specificity switch in the reverse reaction is evidence for a metal ion-nucleophile interaction in the forward reaction. Because the SER hydrolytic reaction (Fig. 1A) is mechanistically analogous to the reverse of the second step of splicing (Podar et al. 1995), 3′-sulfur substitution at the exon–exon junction of the spliced product allows a direct test of the presence of the second metal ion postulated to exist in the group II intron second-step active site (Steitz and Steitz 1993).
We constructed 80-nucleotide spliced exons RNAs with a 3′-sulfur substitution or a normal 3′-oxygen at the exon–exon junction. In the presence of the linear intron ribozyme, MgCl2 supported miscleavage at the two unmodified phosphodiester bonds flanking the exon/exon junction, but no accurately cleaved exon 2 was detected (data not shown). Therefore, substitution of the 3′-oxygen leaving group with sulfur blocks the ability of Mg2+ to support the accurate SER reaction. We were unable to rescue accurate exon–exon junction hydrolysis, however, despite testing multiple concentrations of many different divalent metal ions (data not shown). Although the loss of activity in Mg2+ is consistent with the possibility of a direct metal ion interaction, sulfur differs from oxygen in other ways besides metal ion specificity. Accordingly, the absence of rescue by a thiophilic metal means we cannot confidently ascribe the inhibition to the disruption of a metal ion-leaving group interaction, and the proposal for a metal ion-nucleophile interaction in the second step of group II intron self-splicing (Steitz and Steitz 1993) remains tentative.
Discussion
We have demonstrated that 3′-sulfur substitution at the 5′ splice site causes a metal specificity switch for the first step of group II intron self-splicing, and that this switch is also evident in a first-step intermolecular reaction. Furthermore, we have shown that 3′-sulfur substitution at the 3′ splice site has no significant effect on the metal specificity of the second step of cis-splicing, as observed in the spliceosome (Sontheimer et al. 1997). When the second step is assayed in a tripartite reaction that bypasses the first step, however, a metal specificity switch becomes evident. These results have significant implications for the mechanism of group II intron self-splicing, and for the possible relationship between group II introns and the spliceosome.
Metal ion catalysis by group II intron ribozymes
The inhibition of the first step of splicing in Mg2+ by 3′-sulfur substitution at the 5′ splice site, and the ability of Mn2+, Zn2+, and Cd2+ to relieve this inhibition, provide very strong evidence for a metal ion-leaving group interaction that is essential for 5′ splice site cleavage (Figs. 2 and 3). Although we have not determined whether this interaction occurs in the ground state or the transition state, the latter possibility is more likely (Fig. 6A). Because the bridging oxygen of a phosphoester linkage is electropositive (Bourne and Williams 1980), interaction with a divalent cation in the ground state is expected to be weak or even repulsive. This interaction should provide a stabilizing effect only when the leaving group develops negative charge during bond breaking in the transition state. This effect has been documented in a group I intron ribozyme (Piccirilli et al. 1993; Narlikar et al. 1995) and may be true for other catalysts that employ a divalent metal for leaving group stabilization during phosphotransesterification.
Figure 6.
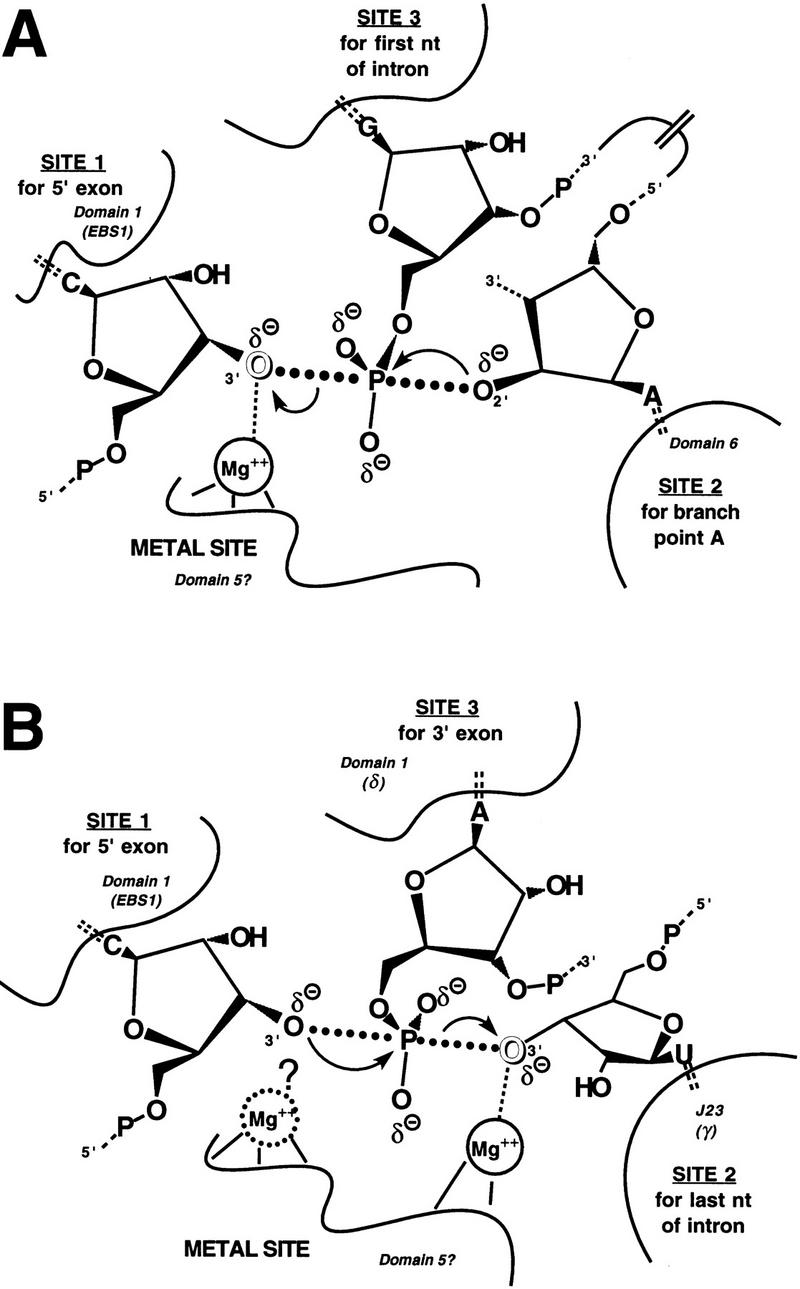
A catalytic metal ion is present in the active site of a group II intron during the first (A) and second (B) steps of self-splicing. The three reactive nucleotides in each step of self-splicing are shown bound to sites 1, 2, and 3 within the group II active sites, as proposed (Steitz and Steitz 1993). Cataytic Mg2+ ions (inferred from this work) directly coordinate the 3′-oxyanion leaving groups (outlined) in the proposed transition states. In the second step (B), an additional catalytic metal ion may coordinate directly to the incoming 3′-oxyanion nucleophile, based on the inhibition of the SER reaction in Mg2+ following 3′-sulfur substitution (see text), but the inability to recoveractivity in the prsence of thiophilic divalent metal ions makes this proposal tentative. Adapted from Steitz and Steitz (1993).
It has been shown recently that Mn2+ has a marked stimulatory effect on the chemical step of a model reverse-branching reaction with this ribozyme (Deme et al. 1999). Therefore, the rescue of the 3′-thio inhibition by Mn2+ could be attributable to a general stimulatory effect on the reaction rather than a specific rescue of the metal ion/leaving group interaction. However, 10 mm Cd2+ or Zn2+ (which also rescue the 3′-thio inhibition) do not stimulate the rate of this model reaction (A. Nolte and A. Jacquier, pers. comm.). Furthermore, Mn2+ does not generally stimulate the rate of the D5-catalyzed 5′ splice site hydrolysis (Deme et al. 1999), despite the fact that it rescues the hydrolysis of the sulfur-substituted substrate (Fig. 3). We conclude that the relief of 3′-thio inhibition by Mn2+, Zn2+, and Cd2+ is not attributable to a general stimulatory effect of these metals on the first chemical step of self-splicing.
Because ribozymes appear to be poorly suited for acid-base catalysis (for review, see Narlikar and Herschlag 1997), it has been thought that they usually rely on divalent metal ions for efficient catalysis. The observation that the second step of self-splicing proceeds accurately in the presence of Mg2+ alone even after 3′-sulfur substitution at the 3′ splice site (Fig. 4) was therefore surprising. The appearance of a metal specificity switch on isolation of the second reaction step (Fig. 5), however, provides a rationale for this observation: a different step (presumably conformational) that precedes 3′ splice site cleavage and exon ligation is likely to limit the rate of the overall reaction, obscuring the metal specificity switch in the subsequent (presumably chemical) phase of the reaction. An interaction (η–η′) that intervenes between the two catalytic steps of splicing has been described by Chanfreau and Jacquier (1996) and likely has a role in such a conformational change. In the tripartite second-step reaction, a different step likely becomes rate-limiting, and the metal specificity switch is revealed. This provides strong evidence for a metal ion-leaving group interaction that is important for 3′ splice site cleavage (Fig. 6B). The observation that 3′-sulfur substitution slows the rate of exon ligation at least 100-fold in Mg2+ without significantly changing the overall rate of cis-splicing suggests that the conformational step that occurs during cis-splicing is at least 100-fold slower than the chemical step of exon ligation.
The effects of Mn2+, Co2+, Zn2+, and Cd2+ were tested on both steps of splicing, and we found that different metal ions rescue cleavage of the 5′ and 3′ modified substrates to different extents. Although Mn2+ and Cd2+ relieve the inhibition at both sites, Zn2+ rescues the 5′ splice site far more efficiently than the 3′ splice site, and the reverse is true for Co2+, indicating that rescue may be conferred by distinct metal binding sites with unique coordination environments. This would not be expected if the sulfur substitution caused the recruitment of thiophilic metals from solution, leading to non-native cleavage activity.
The possibility that the group II second-step active site contains a second metal ion that activates the 3′-hydroxyl nucleophile (Steitz and Steitz 1993; Fig. 6B) remains viable, given the strong inhibition of the SER reaction by 3′-sulfur substitution at the exon–exon junction. The inability of thiophilic metals to rescue activity, however, means that the inhibition cannot yet be attributed to the loss of metal ion-leaving group coordination (Fig. 6B). The inability to rescue this inhibitory effect raises questions about the commonality of the active sites for the two steps of splicing. The original two-metal-ion model (Steitz and Steitz 1993) depicted the two steps of group II self-splicing as forward and reverse reactions in a single active site, as in group I introns. The effects of chiral phosphorothioates at the splice sites call this model into question (Padgett et al. 1994) and indicate that the two reaction steps are mechanistically distinguishable (Podar et al. 1998b). In the original model (Steitz and Steitz 1993), the same metal ion interacts with the leaving group in the first step and the nucleophile in the second step. Our experiments document that Mn2+, Zn2+, and Cd2+ can serve the first-step function in the context of a 3′-sulfur substitution (Figs. 2 and 3) but cannot serve the second-step function. Therefore, if a single metal ion serves both functions, the ligand environment that positions this metal ion is likely to be different for the two steps. Alternatively, there could be a single active site that carries out the two steps as parallel rather than reverse reactions; that is, the same metal ion stabilizes the leaving group in both steps. Although a metal ion-leaving group interaction is important for both steps (Fig. 6), different metals rescue each step to different extents (Figs. 2 and 5), again arguing for nonidentical ligand environments in the first-step and second-step active sites. Detailed thermodynamic and structural analysis of metal ion function will be required to settle this issue definitively, and the results described herein provide a starting point for these analyses.
Parallels with the spliceosome
The results with a group II intron have a very significant effect on how we view the results obtained previously with the spliceosome (Sontheimer et al. 1997). The identical asymmetric responses of splice site 3′-sulfur substitution during cis-splicing in the spliceosome and group II introns—a clear metal specificity switch during the first reaction step, and no apparent metal specificity switch in the second reaction step—is striking, and provides a fundamental parallel in the actual chemical mechanisms of these two systems. Furthermore, it is unnecessary to invoke a protein catalyst to explain the lack of a metal specificity switch in the second step of nuclear pre-mRNA splicing, because the same effect has now been observed in a true ribozyme that catalyzes the same chemistry. Two explanations for the asymmetric effect were noted for the spliceosome: either a rate-limiting conformational step masks a metal specificity switch during 3′ splice site cleavage and exon ligation, or there is no essential metal ion-leaving group interaction during the reaction (Sontheimer et al. 1997). Initially, both explanations were possible for the group II effects as well. We were able to distinguish between the two possible explanations by examining exon ligation in isolation, and we uncovered a metal specificity switch (Fig. 5). The lack of any effect (stimulatory or inhibitory) of 3′-sulfur substitution at the 3′ splice site during nuclear pre-mRNA splicing (Sontheimer et al. 1997) strongly suggests that it too is limited by a conformational change. Because it is clear that conformational rearrangements intervene between the two steps of pre-mRNA splicing (Umen and Guthrie 1995), it will now be important to determine whether a switch can be uncovered for the second step in the spliceosome.
It has been argued that in general, catalytic mechanisms will be among the features of enzymes that are most tightly constrained from drifting during evolution (Benner and Ellington 1988). Therefore, it is significant for the debate over a common molecular ancestry, and therefore intron antiquity, that we observe mechanistic parallels between the two systems. Other biochemical similarities continue to accumulate. In addition to the identical reaction pathways, stereochemical requirements, and 3′-S-phosphorothiolate effects on cis-splicing noted above, similar asymmetric responses to 2′-deoxynucleoside substitution at the splice sites have also been observed in both systems (Moore and Sharp 1992; Griffin et al. 1995; Podar et al. 1998b; A. Bar-Shalom and M. Moore, pers. comm.; E.J. Sontheimer et al., unpubl.). Although it is possible that the parallels between group II introns and the spliceosome could have arisen by convergent evolution from two independent lineages, as in mammalian and bacterial serine proteases (Fersht 1985), an increasingly large number of coincidences would have to be invoked to explain the cumulative similarities in reaction pathways, secondary structural motifs, and now catalytic mechanisms.
Materials and methods
Plasmids and transcription
All constructs were based on the ai5γ group II intron and flanking exons from the mitochondrial COX1 gene from S. cerevisiae. Linear intron ribozyme (D1-6) was transcribed from EcoRV-digested pKC.D1-6 (a gift from K. Chin and A. Pyle, Columbia University, New York, NY), and domain 5 RNA was transcribed from HpaII-digested pJDI5′-75 (Jarrell et al. 1988). All constructs for RNA ligation reactions (except for G2.5+10D123 and pG2.1-881/FokI; see below) were derivatives of the pJD20 plasmid (Jarrell et al. 1988). The following plasmids were constructed, which when linearized and transcribed with T7 RNA polymerase generate RNAs as indicated: pG2.5+10D1-6Ex (HincII; starting 10 nucleotides downstream of the 5′ splice site and ending 60 nucleotides downstream of the 3′ splice site), pG2.3-2ExLD1-6 (NlaIV; starting 293 nucleotides upstream of the 5′ splice site and ending with the second nucleotide upstream of the 3′ splice site), pG2.3-2ExSD1-6 (NlaIV; starting 70 nucleotides upstream of the 5′ splice site and ending with the second nucleotide upstream of the 3′ splice site), and pG2.3-3D1-6 (HaeIII; starting with the first intron nucleotide and ending with the third nucleotide upstream of the 3′ splice site). pG2.5+10D123 was derived from the pJDI3′-673 plasmid (Jarrell et al. 1988); HindIII digestion and T7 transcription generates an RNA starting 10 nucleotides downstream of the 5′ splice site and ending 673 nucleotides downstream of the 5′ splice site, plus an additional 37 nucleotides of 3′-terminal polylinker sequence. All of these plasmids except pG2.3-2ExLD1-6 were made by PCR amplification. PCR products were cloned into pCR2.1 (Invitrogen), sequenced in their entirety, and subcloned into pSP64 poly(A) (Promega). pG2.3-2ExLD1-6 was made by subcloning a fragment of pG2.3-2ExSD1-6 into pJD20. To generate linear intron ribozyme (D1-6/1-881) containing all but the six 3′-terminal nucleotides of the intron, we made pG2.1-881/FokI by cloning annealed synthetic oligonucleotides into the 3′ splice site of pG2.3-3D1-6; digestion with FokI and transcription with T7 RNA polymerase generates the ribozyme. Finally, 5-3ExS RNA (68 nucleotides), starting 70 nucleotides upstream of the 5′ splice site and ending with the third nucleotide upstream of the 5′ splice site, was transcribed directly from annealed synthetic oligonucleotides incorporating a T7 promoter. Transcription of the 5+10D123 and 5+10D1-6Ex RNAs were done in 100-μl reactions (37°C, 3–5 hr) containing 40 mm Tris-HCl (pH 8.0), 2 mm each NTP, 10 mm GMP, 10 mm DTT, 20 mm MgCl2, 2 mm spermidine, 0.6 U/μl RNase inhibitor (Promega), 40 ng/μl linearized plasmid DNA, and 0.16 μg/μl T7 RNA polymerase. The fivefold excess of GMP over GTP was included to generate RNAs with a 5′-monophosphate to serve as substrates in the ligation reactions.
Oligonucleotide synthesis
All oligoribonucleotides were synthesized on a Millipore solid-phase DNA/RNA synthesizer. Coupling of unmodified RNA phosphoramidites (Glen Research) followed standard protocols; 3′-S-phosphoramidites were synthesized and coupled as described by Sun et al. (1997). All oligoribonucleotides were deprotected following standard techniques and purified by anion exchange HPLC, except for G2.18/6, which was purified by denaturing polyacrylamide gel electrophoresis. The following oligoribonucleotides were used in this study (subscript S refers to a 3′-S-phosphorothiolate linkage and d refers to a 2′-deoxynucleoside): G2.5R (5′-UCGAGCGGUCU-3′), G2.5S (5′-UCSGAGCGGUCU-3′), G2.3R (5′-UACUAUGUAU-3′), G2.3S (5′-USACUAUGUAU-3′), G2.SER (5′-UCACUAUGUAU-3′), G2.SES (5′-UCSACUAUGUAU-3′), G2.3RTP (5′-CGGGAUACUAUG-3′), G2.3STP (5′-CGGGAUSACUAUG-3′), E1dC (5′-ACGUGGUGGGACAUUUU(dC)-3′), and G2.18/6 (5′- ACGUGGUGGGACAUUUU(dC)ACUAUG-3′).
Construction of substrate RNAs
Full-length RNAs were generated by ligation of synthetic oligoribonucleotides to flanking RNAs, using a bridging oligonucleotide and T4 DNA ligase (Moore and Query 1988). The 5′ splice site bridging oligonucleotide was complementary to the last 22 nucleotides of exon 1 and the first 28 nucleotides of the intron, the 3′ splice site bridging oligonucleotide was complementary to the last 22 nucleotides of the intron and the first 28 nucleotides of exon 2, and the spliced exons bridging oligonucleotide was complementary to the last 20 nucleotides of exon 1 and the first 10 nucleotides of exon 2. For 5′ splice site ligations, the 5′-32P-phosphorylated G2.5R or G2.5S oligoribonucleotides, bridging oligonucleotide, and 5-3ExS RNA were annealed and ligated as described (Query et al. 1994), and purified by denaturing polyacrylamide gel electrophoresis. The recovered 79-nucleotide RNA was then ligated to the 5+10D123 or 5+10D1-6Ex RNAs as described (Podar et al. 1995) and gel-purified. 3′ splice site ligations were done essentially as described (Podar et al. 1995), except that the G2.3R and G2.3S oligoribonucleotides were 3′-end-labeled with α-32P-labeled 3′-deoxyadenosine triphosphate (New England Nuclear) and yeast poly(A) polymerase (Amersham Pharmacia), and ligation reactions contained a ‘disrupter’ oligonucleotide (3 μm) complementary to domain 5 (nucleotides 813–848 of the intron). For RNase T1 mapping of the 3′ splice site, the G2.3S oligoribonucleotide was 5′-32P-phosphorylated, and the 3′-end-labeling was omitted. For unknown reasons, yields of 3′ splice site ligations were always very low (<1%) and often gave no detectable ligation products, despite extensive efforts to optimize the reactions. For the SER assay, spliced exons were generated with G2.SER and G2.SES oligoribonucleotides and 5-3ExS RNAs as described above for the 5′ splice site ligations, except that the oligonucleotides were 3′-end-labeled with α-32P-labeled 3′-deoxyadenosine triphosphate and 5′-phosphorylated with nonradioactive ATP.
Ribozyme reactions
Cis-splicing assays containing trace radiolabeled substrate, 0.5 m (NH4)2SO4, 40 mm MOPS (pH 7.5), and EDTA or metal ion chlorides as indicated in Figures 2 and 4 were preincubated and reacted at 42°C as described (Daniels et al. 1996). Domain 5-catalyzed hydrolysis reactions of ExD123 containing trace radiolabeled substrate, 3 μm D5 RNA, 0.5 m KCl, 40 mm MOPS (pH 7.0), and metal ion chlorides as indicated in Figure 3 were preincubated and reacted for 2 hr at 45°C as described (Pyle and Green 1994). The reactions were then treated with 40 mm iodoacetamide and 50 mm HEPES (pH 8.0) for 1 hr at room temperature, precipitated with ethanol, and digested with RNase T1 as described below. Electrophoresis was as described below for 5′ splice site mapping, except that DTT and the long prerun were omitted. SER reactions containing trace radiolabeled substrate, 0.5 μm D1-6 RNA, 1.0 m KCl, 40 mm MOPS (pH 7.5), and 100 mm MgCl2 were preincubated and reacted at 45°C as described (Podar et al. 1995). Tripartite step 2 reactions containing trace 3′-end-labeled G2.3RTP or G2.3STP substrate, 0.5 μm E1dC oligonucleotide, 0.2 μm D1-6/1-881 RNA, 0.5 m (NH4)2SO4, 40 mm MOPS (pH 7.5), and EDTA or metal ion chlorides as indicated in Figure 5 were done at 42°C (A. Bar-Shalom and M.J. Moore, pers. comm.). Before reaction, all components except substrate and thiophilic metal ions were preincubated at 75°C for 2 min and then at 42°C for 90 min. All ribozyme reactions containing EDTA, MgCl2, and/or MnCl2 were supplemented with 10 mm DTT to avoid 3′-thiol oxidation; for reactions containing CoCl2, ZnCl2, or CdCl2, tricarboxyethylphosphine (Strem Chemicals) was used instead to avoid precipitation of metal/DTT complexes. KCl, (NH4)2SO4, and divalent metal ion chlorides (Aldrich) were of the highest purity available (>99.99%).
Splice site mapping
For mapping the position of 5′ splice site cleavage, substrates were subjected to cis-splicing in 90 mm MgCl2 and 10 mm MnCl2 for 1 hr as described above. A separate 3′-sulfur-substituted sample was cleaved with silver(I) as described (Sontheimer 1999). The exon 1 intermediates and Ag+-cleaved product were purified by electrophoresis in an 8% polyacrylamide:bis (19:1)/0.5× TBE/10 mm DTT gel. Half of each sample (as well as the corresponding unspliced precursor) was kept frozen in 10 mm DTT, while the other half was treated with iodoacetamide/HEPES as described above. All samples were then digested to completion with RNase T1 as described (Sontheimer et al. 1997), and subjected to electrophoresis in a 20% polyacrylamide:bis (29:1)/0.5× TBE/10 mm DTT gel that had been prerun at low wattage (12 W) overnight. The bromophenol blue tracking dye (which runs just ahead of the 8- to 9-nucleotide RNase T1 fragments) was run to the bottom of a 40-cm gel.
To map the position of 3′ splice site cleavage, a 3′-sulfur-substituted substrate with a single 32P label in the adjacent intron phosphate was prepared as described above. Samples were subjected to Ag+-cleavage or cis-splicing in 100 mm MgCl2 or 90 mm MgCl2/10 mm MnCl2 for 1 hr as described above; half of each sample (as well as unspliced precursor) was reacted with iodoacetamide, digested with RNase T1, and subjected to electrophoresis as described above for 5′ splice site mapping.
Acknowledgments
We thank Michelle Hamm and Cecilia Cortez for oligonucleotide synthesis; Barbara Golden, Manyuan Long, Nipam Patel, and Thomas Tuschl for comments on the manuscript; and members of the Piccirilli laboratory for advice, discussions, and comments on the manuscript. We are grateful to Barbara Golden for T4 DNA ligase; Richard Padgett for advice on ligation reactions; and Melissa Moore, Anna Marie Pyle, and Alain Jacquier for plasmids, discussions, and communication of unpublished results. E.J.S. was supported in part as a research associate of the Howard Hughes Medical Institute. J.A.P. is an assistant investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Anderegg G. Complexones. In: Wilkinson G, Gillard RD, McCleverty JA, editors. Comprehensive coordination chemistry:The synthesis, reactions, properties & applications of coordination compounds. Oxford, UK: Pergamon Press; 1987. pp. 777–792. [Google Scholar]
- Benner S, Ellington AD. Interpreting the behavior of enzymes: Purpose or pedigree? CRC Crit Rev Biochem. 1988;23:369–426. doi: 10.3109/10409238809082549. [DOI] [PubMed] [Google Scholar]
- Bourne N, Williams A. Effective charge on oxygen in phosphoryl (-PO32-) group transfer from an oxygen donor. J Org Chem. 1980;49:1200–1204. [Google Scholar]
- Burge CB, Tuschl T, Sharp PA. Splicing of precursors to mRNAs by the spliceosomes. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA world. second edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. pp. 525–560. [Google Scholar]
- Cech TR. The generality of self-splicing RNA: Relationship to nuclear RNA splicing. Cell. 1986;44:207–210. doi: 10.1016/0092-8674(86)90751-8. [DOI] [PubMed] [Google Scholar]
- Cech TR, Golden BL. Building a catalytic active site using only RNA. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA world. second edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. pp. 321–349. [Google Scholar]
- Chanfreau G, Jacquier A. An RNA conformational change between the two chemical steps of group II self-splicing. EMBO J. 1996;15:3466–3476. [PMC free article] [PubMed] [Google Scholar]
- Cosstick R, Vyle JS. Synthesis and properties of dithymidine phosphate analogues containing 3′-thiothymidine. Nucleic Acids Res. 1990;18:829–835. doi: 10.1093/nar/18.4.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DL, Michels WJ, Jr, Pyle AM. Two competing pathways for self-splicing by group II introns: A quantitative analysis of in vitro reaction rates and products. J Mol Biol. 1996;256:31–49. doi: 10.1006/jmbi.1996.0066. [DOI] [PubMed] [Google Scholar]
- Dantzmann CL, Kiessling LL. Reactivity of a 2′-thio nucleotide analog. J Am Chem Soc. 1996;118:11715–11719. [Google Scholar]
- Darnell J. Implications of RNA-RNA splicing in evolution of eukaryotic cells. Science. 1978;202:1257–1260. doi: 10.1126/science.364651. [DOI] [PubMed] [Google Scholar]
- Deme E, Nolte A, Jacquier A. Unexpected metal ion requirements specific for catalysis of the branching reaction in a group II intron. Biochemistry. 1999;38:3157–3167. doi: 10.1021/bi982462j. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. Genes in pieces: Were they ever together? Nature. 1978;272:581–582. [Google Scholar]
- Fersht A. Enzyme structure and mechanism. second edition. New York, NY: W.H. Freeman & Co.; 1985. pp. 17–23. [Google Scholar]
- Gesteland RF, Cech TR, Atkins JF, editors. The RNA world. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. [Google Scholar]
- Gilbert W. Why genes in pieces? Nature. 1978;271:501. doi: 10.1038/271501a0. [DOI] [PubMed] [Google Scholar]
- Gilbert W, de Souza SJ, Long M. Origin of genes. Proc Natl Acad Sci. 1997;94:7698–7703. doi: 10.1073/pnas.94.15.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin EA, Jr, Qin Z, Michels WJ, Jr, Pyle AM. Group II intron ribozymes that cleave DNA and RNA linkages with similar efficiency, and lack contacts with substrate 2′-hydroxyl groups. ChemBiol. 1995;2:761–770. doi: 10.1016/1074-5521(95)90104-3. [DOI] [PubMed] [Google Scholar]
- Hetzer M, Würzer G, Schweyen RJ, Müller MW. Trans-activation of group II intron splicing by nuclear U5 snRNA. Nature. 1997;386:417–420. doi: 10.1038/386417a0. [DOI] [PubMed] [Google Scholar]
- Jarrell KA, Dietrich RC, Perlman PS. Group II intron domain 5 facilitates a trans-splicing reaction. Mol Cell Biol. 1988;8:2361–2366. doi: 10.1128/mcb.8.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Reese CB. 3′-Thiouridylyl-(3′ → 5′)-uridine. Tetrahedron Lett. 1996;37:925–928. [Google Scholar]
- Logsdon JM., Jr The recent origins of spliceosomal introns revisited. Curr Opin Genet Dev. 1998;8:637–648. doi: 10.1016/s0959-437x(98)80031-2. [DOI] [PubMed] [Google Scholar]
- Maschhoff KL, Padgett RA. The stereochemical course of the first step of pre-mRNA splicing. Nucleic Acids Res. 1993;21:5456–5462. doi: 10.1093/nar/21.23.5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Ferat J-L. Structure and activities of group II introns. Annu Rev Biochem. 1995;64:435–461. doi: 10.1146/annurev.bi.64.070195.002251. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Query CC. Uses of site-specifically modified RNAs constructed by RNA ligation. In: Smith C, editor. RNA-protein interactions: A practical approach. London, UK: Oxford University Press; 1998. pp. 75–108. [Google Scholar]
- Moore MJ, Sharp PA. Site-specific modification of pre-mRNA: The 2′-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- ————— Evidence for two active sites in the spliceosome provided by stereochemistry of pre-mRNA splicing. Nature. 1993;365:364–368. doi: 10.1038/365364a0. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Herschlag D. Mechanistic aspects of enzymatic catalysis: Lessons from comparison of RNA and protein enzymes. Annu Rev Biochem. 1997;66:19–59. doi: 10.1146/annurev.biochem.66.1.19. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Gopalakrishnan V, McConnell TS, Usman N, Herschlag D. Use of binding energy by an RNA enzyme for catalysis by positioning and substrate destabilization. Proc Natl Acad Sci. 1995;92:3668–3672. doi: 10.1073/pnas.92.9.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW. RNA-RNA interactions in nuclear pre-mRNA splicing. In: Simons RW, Grunberg-Manago M, editors. RNA structure and function. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1998. pp. 279–307. [Google Scholar]
- Padgett RA, Podar M, Boulanger SC, Perlman PS. The stereochemical course of group II intron self-splicing. Science. 1994;266:1685–1688. doi: 10.1126/science.7527587. [DOI] [PubMed] [Google Scholar]
- Pearson RG. Acids and Bases. Science. 1966;151:172–177. doi: 10.1126/science.151.3707.172. [DOI] [PubMed] [Google Scholar]
- Peebles CL, Zhang M, Perlman PS, Franzen JS. Catalytically critical nucleotides in domain 5 of a group II intron. Proc Natl Acad Sci. 1995;92:4422–4426. doi: 10.1073/pnas.92.10.4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirilli JA, Vyle JS, Caruthers MH, Cech TR. Metal ion catalysis in the Tetrahymena ribozyme reaction. Nature. 1993;361:85–88. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- Podar M, Perlman PS, Padgett RA. Stereochemical selectivity of group II intron splicing, reverse splicing, and hydrolysis reactions. Mol Cell Biol. 1995;15:4466–4478. doi: 10.1128/mcb.15.8.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podar M, Chu VT, Pyle AM, Perlman PS. Group II intron splicing in vivo by first-step hydrolysis. Nature. 1998a;391:915–918. doi: 10.1038/36142. [DOI] [PubMed] [Google Scholar]
- Podar M, Perlman PS, Padgett RA. The two steps of group II intron self-splicing are mechanistically distinguishable. RNA. 1998b;4:890–900. doi: 10.1017/s1355838298971643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle AM. Catalytic reaction mechanisms and structural features of group II intron ribozymes. In: Eckstein F, Lilley DMJ, editors. Catalytic RNA. Berlin, Germany: Springer-Verlag; 1996. pp. 75–107. [Google Scholar]
- Pyle AM, Green JB. Building a kinetic framework for group II intron ribozyme activity: Quantitation of interdomain binding and reaction rate. Biochemistry. 1994;33:2716–2725. doi: 10.1021/bi00175a047. [DOI] [PubMed] [Google Scholar]
- Query CC, Moore MJ, Sharp PA. Branch nucleophile selection in pre-mRNA splicing: Evidence for the bulged duplex model. Genes & Dev. 1994;8:587–597. doi: 10.1101/gad.8.5.587. [DOI] [PubMed] [Google Scholar]
- Sharp PA. On the origin of RNA splicing and introns. Cell. 1985;42:397–400. doi: 10.1016/0092-8674(85)90092-3. [DOI] [PubMed] [Google Scholar]
- Sigel RKO, Song B, Sigel H. Stabilities and structures of metal ion complexes of adenosine-5′-O-thiomonophosphate (AMPS2-) in comparison with those of its parent nucleotide (AMP2-) in aqueous solution. J Am Chem Soc. 1997;119:744–755. [Google Scholar]
- Sontheimer EJ. Bridging sulfur substitutions in the analysis of pre-mRNA splicing. Methods. 1999;18:29–37. doi: 10.1006/meth.1999.0754. [DOI] [PubMed] [Google Scholar]
- Sontheimer EJ, Sun S, Piccirilli JA. Metal ion catalysis during splicing of premessenger RNA. Nature. 1997;388:801–805. doi: 10.1038/42068. [DOI] [PubMed] [Google Scholar]
- Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Yoshida A, Piccirilli JA. Synthesis of 3′-thioribonucleosides and their incorporation into oligoribonucleotides via phosphoramidite chemistry. RNA. 1997;3:1352–1363. [PMC free article] [PubMed] [Google Scholar]
- Umen JG, Guthrie C. The second catalytic step of pre-mRNA splicing. RNA. 1995;1:869–885. [PMC free article] [PubMed] [Google Scholar]
- Weiner AM. mRNA splicing and autocatalytic introns: Distant cousins or the products of chemical determinism? Cell. 1993;72:161–164. doi: 10.1016/0092-8674(93)90654-9. [DOI] [PubMed] [Google Scholar]
- Weinstein LB, Earnshaw DJ, Cosstick R, Cech TR. Synthesis and characterization of an RNA dinucleotide containing a 3′-S-phosphorothiolate linkage. J Am Chem Soc. 1996;118:10341–10350. [Google Scholar]