

Abstract
The etiopathogenesis of many autoimmune disorders has not been identified. The aim of this paper is to focus on the involvement of bacterial exposure in the pathogenesis of primary biliary cirrhosis (PBC) and autoimmune pancreatitis (AIP), both of which are broadly categorized as autoimmune disorders involving hepatobiliary-pancreatic lesions. Avirulent and/or commensal bacteria, which may have important role(s) as initiating factors in the pathogenesis of autoimmune disorders such as PBC and AIP, will be emphasized.
1. Autoimmune Diseases Associated with Microbial Infection
Autoimmune diseases arise from an overactive immune response of the body against tissues normally present in the body. Autoimmune disorders are often described as a condition involving genetic components. A high prevalence of autoimmune diseases is observed within families [1]. However, the fact that the prevalence of autoimmune diseases never reaches 100% among monozygotic twins suggests that both genetic and environmental factors are involved in the etiology of autoimmune diseases.
Several studies have implicated microbes in the environmental etiology of autoimmune disorders based on observations such as the regression of autoimmune thrombocytopenia after the eradication of Helicobacter pylori [2]. In Guillain-Barré syndrome (GBS), amino acid similarities exist between the gangliosides of the nerve system and the lipopolysaccharides (LPSs) of Campylobacter jejuni, suggesting that sensitization by microbes may be based on autoimmunity from molecular mimicry between bacteria and the targeted system of the host [3, 4]. A common recent theory of the cause of autoimmune diseases is that an infectious agent triggers a cycle of events, which leads to the upregulation of the host immune response to self-antigens [5, 6].
2. Innate Immunity Sensitized by Bacteria as an Environmental Factor in the Etiology of PBC
Primary biliary cirrhosis (PBC) is a chronic autoimmune disorder characterized by chronic nonsuppurative destructive cholangitis (CNSDC) of small intrahepatic bile ducts and epithelioid granuloma formation in the portal area, which leads to progressive ductopenia [7, 8]. The pathogenesis of biliary epithelial cell damage in PBC is not clearly understood. However, the relationship between bacterial infection and the pathogenesis of PBC through the mechanism of molecular mimicry has become a focus of attention. Evidence of Propionibacterium acnes DNA has been detected in the epithelioid granulomas of PBC patients [9]. Sera from patients with PBC have been reported to react with both human and Escherichia coli pyruvate dehydrogenase complex E2 (PDC-E2) [10], and such reactivity of antimitochondrial antibodies (AMAs) to both human and bacterial molecules has stimulated speculations that PBC may be induced by exposure to enterobacterial antigens, perhaps by sharing molecular mimicry with mitochondrial antigens in PBC. Other reports indicating bacterial involvement in the etiology of PBC include the reported presence of AMAs, which reacted to Novosphingobium aromaticivorans in sera from PBC patients [11], and cross-reactivity detected between Lactobacillus delbrueckii beta-galactosidase and PDC-E2 with AMA [12]. A link between toll-like receptors (TLRs) and PBC has also been reported. The elevated expression of TLR4 has been observed in intrahepatic bile duct epithelial cells in PBC [13]. Bacterial components, such as LPSs and the TLR9 ligand CpG DNA, trigger peripheral lymphocytes, monocytes, and bile epithelial cells to produce cytokines and AMAs, suggesting that the pathogenesis of PBC is mediated through TLRs [13–17]. Moreover, elevated levels of immunoglobulin M (IgM) in PBC have been reported to result from the CpG DNA stimulation of TLR signalling, leading to the appearance of IgM-positive memory B cells [18].
We previously reported that the Gram-positive bacterial cell wall component lipoteichoic acid (LTA) was detected at the portal tract in the livers of PBC patients with CNSDC and that PBC patients had higher serum IgA class anti-LTA titers, when compared with healthy donors [19]. We analyzed the immunoreactivity against 15 strains from different species of the streptococcal genus using various sera from PBC patients and performed further assays using five strains of the Streptococcus anginosus group, the titers of which were higher than those of the other strains, and found that the PBC patient's sera had the highest IgM class titers to Streptococcus intermedius [20]. Generally, PBC is characterized by a high serum level of IgM [21]. Our observation may indicate that IgM, in part, is involved in the streptococcal-mediated inflammatory response in PBC.
S. intermedius forms part of the commensal bacteria in the oral cavity, attaching to a surface, forming matrix-enclosed biofilms in dental plaques, and thereby exhibiting increased resistance to antimicrobials and to host immune defense mechanisms [22, 23]. The high immunoreactivity of PBC patient sera against oral streptococci and the high familial prevalence of PBC suggest not only a genetic etiology but also a possible environmental transmission during childhood via the mouth of a child's parent. This theory of transmission from parent to child, however, cannot fully explain the dominant occurrence of PBC in women.
Considering the fact that the onset of PBC occurs during the fifth decade of life [7] and that there is no apparent antecedent infection related to PBC, as there is in GBS [3, 4], the environmental etiopathology of bacteria in PBC may possibly suggest the chronic persistence of low-immunogenic or avirulent bacteria, commensally coexisting within the host over the long term, rather than a severe yet transient infection. In this sense, periodontitis isolates and other oral colonizing bacteria satisfy these criteria. How do bacteria in the oral cavity reach the liver? One possibility would be a hematogenous transmission from the oral mucosal layer. Since increased permeability of the stomach and the small intestine is reported in PBC patients [24], transmission from the intestinal tract via the portal vein to the liver should also be an additional possibility.
3. Infection-Induced Mouse Model of PBC
Whereas strong evidence exists that bacterial infection may trigger PBC, subsequent implications have been difficult to elucidate. Oral streptococcal isolates, namely, S. intermedius, S. sanguinis, and Streptococcus mitis as well as Micrococcus luteus and E. coli, were repeatedly used to inoculate BALB/c mice, mimicking chronic bacterial exposure, and pathological alterations in the liver and the production of autoantibodies were investigated. The livers of mice at 8 weeks after inoculation with S. intermedius, S. sanguinis, or S. mitis showed CNSDC-like inflammatory cellular infiltrates in the portal area [25]. When BALB/c mice were left inoculation-free for an additional 20 months after the completion of an initial 8 week-S. intermedius inoculation, CNSDC-like portal inflammation was still observed [25]. LTA immunoreactivity was detectable in not all but some of the cytoplasm of polymorphic inflammatory cells around biliary epithelial cells and connective tissues around bile ducts, and CD3-positive cells were predominantly observed in the cellular infiltrates around the bile ducts. In the portal area, LTA immunoreactivity was detectable in not all but some of the cytoplasm of polymorphic inflammatory cells around the biliary epithelial cells and connective tissues around the bile ducts, and CD3-positive cells were predominantly observed in the cellular infiltrates around the bile ducts. Other tissue damages that are occasionally associated with PBC were also detected. The salivary glands of live S. intermedius-inoculated mice exhibited Sjögren's syndrome-like periductal lymphocytic infiltration [25].
An investigation of PBC-like autoantibodies in our model detected a high frequency of the production of anti-gp210 antibodies, which are commonly detected in PBC patients, especially those with a poor prognosis [26]. We previously reported that IgM class antibody titers against histone-like DNA-binding protein (HLP) of S. intermedius were significantly high in the sera of PBC patients and that immunoreactivity to anti-S. intermedius-HLP was detected in the cytoplasm of biliary epithelial cells and inflammatory cells in the portal area of the livers of PBC patients [20]. In S. intermedius-inoculated BALB/c mice, consistent with observations in human patients, immunoreactivity to S. intermedius-HLP was detected around the sites of CNSDC in the portal area of the livers both soon after and 8 weeks after S. intermedius inoculation. Furthermore, the affinity of anti-S. intermedius-HLP antibody to the c-terminus synthetic peptide of gp210 was detected in a dose-dependent and specific manner, suggesting cross-reactivity between gp210 and S. intermedius-HLP [25]. These results suggested that chronic exposure to S. intermedius could trigger PBC-like pathological alterations in BALB/c mice resembling the pathology of PBC in humans.
4. Autoimmune Pancreatitis and IgG4-Related Diseases
Autoimmune pancreatitis (AIP) is another putative autoimmune disease of the hepatobiliary-pancreatic system and is a chronically progressing inflammatory disease of the pancreas [27, 28]. The morphological characteristics of AIP include diffuse or localized enlargement of the pancreas and irregular narrowing of the main pancreatic duct. Histologically, the disease is also associated with progressive lymphoplasmacytic infiltration, predominantly localized to the ductal structures, and varying degrees of parenchymal and acinar destruction [29].
There are two types of AIP that differ in their clinical features, such as the gender ratio, mean age, and associated immune-related diseases. Type 1 AIP is associated with the histological finding of lymphoplasmacytic sclerosing pancreatitis (LPSP). Its serological hallmark is an elevation in the serum levels of the IgG4 subclass of IgG [27]. Type 1 AIP appears to be the pancreatic manifestation of a systemic disease called IgG4-associated systemic disease (ISD), affecting not only the pancreas but also other organs including the bile duct, retroperitoneum, kidney, lymph nodes, and salivary glands [30]. Type 2 AIP is a form of idiopathic chronic pancreatitis, histologically associated with granulocyte-epithelial lesions [31]. The pathogenesis of AIP remains unknown. Genetic associations between susceptibility to the disease and the human leukocyte antigen (HLA) DRB1*0405-DQB1*0401 haplotype [32–34], Fc receptor-like gene 3 (FCRL3) [35], and the CTLA4 gene [36] have been suggested.
An outstanding finding in type 1 AIP is hypergammaglobulinemia and the existence of a high serum concentration for IgG4, which has been documented in 90% of patients [37]. This occurs in parallel to an abundant IgG4-positive plasma cell infiltration in the pancreatic tissue [38]. The fibroinflammatory process characterizing AIP occurs at the pancreatic basement membranes where IgG4/IgG/complement-immune complexes are deposited [39]. AIP is occasionally associated with elevated circulating immune complex levels, which are significantly linked to increased serum IgG1 and complement activation via the classical pathway [33]. IgG4 is unable to activate the classical pathway of complements but binds IgG1, 2, and 3 and forms an Fc–Fc interaction immune complex in patients with AIP [40]. Although the role of IgG4 in the immune response and autoimmunity has not yet been fully elucidated, it may be hypothesized that IgG4 blocks the Fc-mediated effector functions of IgG1 and dampens the inflammatory response to an as-yet-unidentified primary trigger of the inflammatory process in AIP [40, 41].
5. Autoantibodies in AIP
To which antigens the immunoglobulins in the deposits along the basal membranes of the pancreatic ducts and acini react remains unclear. Circulating antibodies in AIP include autoantibodies against carbonic anhydrase- (CA-) II [62], CA-IV [63], lactoferrin (LF) [62], pancreatic secretary trypsin inhibitor (PSTI) [64], and heat shock protein- (HSP-) 10 [65]. Thymectomized mice immunized with CA-II or LF develop a pathology that closely resembles AIP under a regulatory T cell- (Treg-) depleted background [54]. However, T cells specific for CA-II and LF were unable to induce pancreatitis in the adoptive transfer of an amylase-specific rat model [52], implying that autoantibodies against these enzymes in AIP represent a late consequence of tissue destruction and perhaps not a fundamental pathogenic mechanism. Additionally, these potential antigens reside in the cytoplasm of pancreatic cells and are not associated with the basement membranes targeted for the fibroinflammatory process in AIP [39]. Moreover, the major subclasses of these autoantibodies in the sera of patients with AIP are classified as the IgG1 subtype, and not IgG4 [33, 40, 64]. This implies that these antigens may be secondarily involved in the AIP immune process, as it is directed toward still-unknown antigens in pancreatic basal membranes.
6. Innate Immunity Sensitized by Infectious Agents in the Etiology of AIP
Although the initial events triggering AIP are not known, the pancreatic cells may become targets of immune-mediated processes through viral or other infectious agents. Guarneri et al. showed a significant homology between human CA-II and α-CA of H. pylori. Moreover, the homologous segments contained the binding motif of DRB1*0405 [66]. Notably, the possession of the HLA DRB1*0405-DQB1*0401 genotype confers a risk for AIP development [32]. Frulloni et al. reported that 94% of AIP patients, but only 5% of pancreatic cancer patients, exhibit IgG antibodies to a plasminogen-binding protein (PBP) that is homologous to the human protein ubiquitin-protein ligase E3 component n-recognin 2 (UBR2), which is expressed in pancreatic acinar cells and is also homologous to the PBP of H. pylori [67]. These data suggest that H. pylori infection may trigger AIP in genetically predisposed subjects through autoimmune responses triggered by molecular mimicry.
Several experimental models of AIP have been described (Table 1). Virus-induced AIP models, such as C57BL/6 mice infected with the murine leukemia retrovirus LP-BM5, developed histological findings similar to human AIP [59, 60]. The spontaneous development of pancreatitis via an autoimmune mechanism in MRL/Mp mice is accelerated by the administration of polyinosinic : polycytidylic acid (poly I:C), a synthetic double-stranded RNA and TLR3 ligand [55–58]. Sensitization occurs with not only viral components, such as double-stranded RNA poly I:C, but also bacterial LPS-induced pancreatitis in interleukin- (IL-) 10-deficient mice [55]. TLRs play important roles in innate immunity and initiate intracellular signaling to macrophages and dendritic cells after stimulation with various antigens. The majority of known TLRs mediate the development of Th1 cell-inducing dendritic cells [68]. Hence, pattern-recognition receptors (PRRs) that bind pathogen-associated molecular patterns (PAMPs) may trigger an autoimmune response.
Table 1.
Experimental animal models of autoimmune pancreatitis.
| Animal | Organs with lesions | Induction | Target antigen | Effector cells | References |
|---|---|---|---|---|---|
| MRL/lpr mice | Pancreas | Spontaneous | ? | T cells | [42] |
| Pancreas, salivary gland | [43] | ||||
| aly/aly mice | Pancreas, salivary gland, lung | Spontaneous | ? | CD4+ T cells | [44] |
| Pancreas | [45, 46] | ||||
| WBN/Kob rats, male | Pancreas, salivary gland, bile duct | Spontaneous | ? | CD8+ T cells | [47] |
| MHC-II−/− mice | Pancreas | Spontaneous | ? | CD8+ T cells | [48] |
| T-cell+ HLADR*0405Ab0 NOD mice | Pancreas, lung | Spontaneous | ? | T cells | [49] |
| Tgfbr2(fspKO) mice | Pancreas, salivary gland | Spontaneous | ? | T cells | [50] |
| NOD.CD28KO mice | Pancreas | Spontaneous | Amylase | CD4+ T cells | [51] |
| DA(RP) rats Lewis rats |
? Pancreas |
Amylase-specific T cell | ? | CD4+ and CD8+ T cells | [52] |
| PL/J mice (H-2s, H-2u) | Pancreas, salivary gland, kidney | CA-II | CA-II | ? | [53] |
| nTx-BALBc mice | Pancreas, salivary gland, bile duct, kidney | CA-II | CA-II | CD4+ Th1 cells | [54] |
| nTx-BALBc mice | Pancreas, salivary gland, bile duct, kidney | LF | LF | CD4+ Th1 cells | [54] |
| MRL/Mp, MRL/lpr mice | Pancreas, salivary gland, bile duct, kidney | ? | CD4+ T cells | [57] | |
| MRL/Mp mice | Pancreas, salivary gland, liver | Poly I:C | PSTI | ? | [58] |
| Pancreas | ? | ? | [55] | ||
| IL-10KO mice | Pancreas | Poly I:C, LPS | ? | ? | [55] |
| C57BL/6 mice | Pancreas, salivary gland, liver, kidney, lung | LP-BM5 | ? | CD4+ T cells | [59, 60] |
| C57BL/6 mice | Pancreas, salivary gland | E. coli | ? | T cells | [61] |
7. Infection-Induced Mouse Model of AIP
We previously reported that when C57BL/6 mice were inoculated intraperitoneally (i.p.) with heat-killed E. coli weekly for 8 weeks, marked cellular infiltration with fibrosis was observed in the exocrine pancreas accompanied by a high serum gamma-globulin level and the production of autoantibodies against CA-II and LF. Bacterial infection apparently triggered autoimmune pancreatitis-like pathological alterations in mice that strikingly resembled AIP in humans [61].
C57BL/6 mice inoculated weekly with E. coli for 8 weeks were utilized as donors, and the spleens were intravenously transferred to RAG2−/− mice. The pancreas in the recipient RAG2−/− mice showed cellular infiltration in the exocrine pancreas, especially around the pancreatic ducts, indicating that the E. coli-inoculated mouse spleen cells possess the ability to reproduce pathological alterations in the pancreas of naïve mice. Similarly, when the spleen cells of donor S. intermedius-inoculated mice were transferred to RAG2−/− mice, CNSDC-like cholangitis in the liver was induced, similar to that seen in the donor [61].
The AIP-like inflammatory region in the pancreases of recipient mice with spleen cells transferred from E. coli-inoculated mice [61], as well as the CNSDC-like inflammatory region in the livers of recipient mice with spleen cells transferred from S. intermedius-inoculated mice [25], both showed that most of the cellular infiltrates in the target organs were CD3 positive, indicating that these cells in both models originated from the donor mice. The findings observed indicated that our animal models of PBC and AIP are of autoimmune etiology.
The criteria for determining whether a condition may be considered to be autoimmune, according to Witebsky's postulates with modern revision by Rose and Bona [69], include (i) indirect evidence based on the reproduction of the autoimmune disease in experimental animals, (ii) direct evidence of the transfer of pathogenic antibodies, or (iii) pathogenic T cells and indirect evidence of the isolation of autoantibodies or autoreactive T cells. Several lines of evidence have established our animal models to be of autoimmune origin. The current approach in our research is to search for a possible supply of antigenic stimulant that is similar to or that will cross-react with autoantigens in vivo; so far we have narrowed our search down to bacterial species possessing a candidate epitope.
8. Conclusions
We propose a hypothetical pathogenetic mechanism for bacteria-induced PBC and AIP. During the initiation phase, weak but silently infiltrating PAMPs and/or antigen(s), such as avirulent bacteria, trigger and upregulate the innate immune system. Second, the progressive phase features the persistence of this PAMP attack or stimulation by molecular mimicry and/or exposure or stimulation from commensal flora possessing the same antigenic epitope that the initial pathogen and/or PAMP possessed, thereby upregulating the host immune response to the target antigen. These slowly progressive steps eventually lead to the development of autoimmune diseases (Figure 1).
Figure 1.
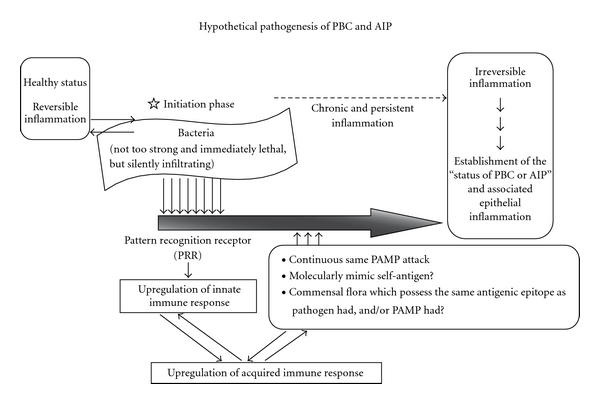
Hypothetical pathogenesis of PBC and AIP. During the initiation phase, weak but silently infiltrating PAMPs and/or antigen(s), such as avirulent bacteria, trigger and upregulate the innate immune system. Second, the progressive phase features the persistence of this PAMP attack or stimulation by molecular mimicry and/or exposure or stimulation from commensal flora possessing the same antigenic epitope that the initial pathogen and/or PAMP possessed, thereby upregulating the host immune response to the target antigen. These slowly progressive steps eventually lead to the development of autoimmune diseases. Modified from previous papers [25, 61].
Recently, NOD.c3c4 [70], TGFb receptor II dominant-negative [71], IL-2 receptor α knockout [72], and Ae2a,b-deficient mice [73] have been reported as spontaneous PBC animal models. The spontaneous development of AIP in T-cell-competent HLA-DR*0405 transgenic Ab0 NOD mice [49], Treg-deficient backgrounds in neonatally thymectomized mice [54], NOD.CD28 knockout mice [51], and Tgfbr2fsp knockout mice [50] have demonstrated genetic polymorphisms of the effector cells in the etiologies of AIP. However, as these mice are genetically engineered, they may not completely reflect the onset of human diseases. Our models of PBC and AIP developed in genetically normal BALB/c and C57BL/6 mice, respectively, by the inoculation of human commensal bacteria are more clinically relevant, as they reflect the conditions of human patients, and should be useful for advancing our understanding of the pathological mechanism(s) underlying PBC and AIP.
Under normal conditions, commensal bacteria are not pathogenic and in fact may inhabit the host from early life. Commensal bacteria in the gastrointestinal tract have been recognized to interact with the innate immune system and to drive regulatory T-cell differentiation [74, 75]. The fact that commensal bacteria can induce autoimmune diseases associated with genetic polymorphisms, immune susceptibility, and other environmental factors may expand our concept of the pathoetiology of autoimmune diseases.
Conflict of Interests
The authors declare no conflicts of financial interest.
Acknowledgments
This work was financially supported in part by the Takako Satake Scientific Award (to N. Yanagisawa) and the 28th Itoe Okamoto Scientific Award (to N. Yanagisawa) from Shiseikai, Tokyo Women's Medical University, and Grants-in-Aid for Scientific Research (C21590496 to J. Yagi and C23590522 to I. Haruta) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Abbreviations
- AIP:
Autoimmune pancreatitis
- AMA:
Antimitochondrial antibody
- ANA:
Antinuclear antibody
- CA-II:
Carbonic anhydrase II
- CTLA4:
Cytotoxic T-lymphocyte antigen 4
- FCRL3:
Fc receptor-like gene 3
- GEL:
Granulocyte epithelial lesions
- GBS;
Guillain-Barré syndrome
- HLA:
Human leukocyte antigen
- HSP:
Heat shock protein
- HLP:
Histone-like DNA-binding protein
- IDCP:
Idiopathic duct-centric pancreatitis
- LF:
Lactoferrin
- LPS:
Lipopolysaccharide
- LPSP:
Lymphoplasmacytic sclerosing pancreatitis
- LTA:
Lipoteichoic acid
- PAMP:
Pathogen-associated molecular pattern
- PRR:
Pattern recognition receptor
- poly I:C:
Polyinosinic : polycytidylic acid
- PBC:
Primary biliary cirrhosis
- PBP:
Plasminogen-binding protein
- Treg:
Regulatory T cell
- TLR:
Toll-like receptor
- TGF:
Transforming growth factor
- UBR2:
Ubiquitin-protein ligase E3 component N-recognin 2.
References
- 1.Selmi C, Mayo MJ, Bach N, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127(2):485–492. doi: 10.1053/j.gastro.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Gasbarrini A, Franceschi F, Tartaglione R, Landolfi R, Pola P, Gasbarrini G. Regression of autoimmune thrombocytopenia after eradication of Helicobacter pylori . The Lancet. 1998;352(9131):p. 878. doi: 10.1016/S0140-6736(05)60004-9. [DOI] [PubMed] [Google Scholar]
- 3.Yuki N, Susuki K, Koga M, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barré syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(31):11404–11409. doi: 10.1073/pnas.0402391101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Houliston RS, Vinogradov E, Dzieciatkowska M, et al. Lipooligosaccharide of Campylobacter jejuni: similarity with multiple types of mammalian glycans beyond gangliosides. The Journal of Biological Chemistry. 2011;286(14):12361–12370. doi: 10.1074/jbc.M110.181750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tlaskalová-Hogenova H, Štĕpánková R, Hudcovic T, et al. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunology Letters. 2004;93(2-3):97–108. doi: 10.1016/j.imlet.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Aoki S. Rheumatoid arthritis and enteric bacteria. Japanese Journal of Rheumatology. 1999;9(4):325–352. [Google Scholar]
- 7.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. The New England Journal of Medicine. 2005;353(12):1261–1273. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 8.Selmi C, MacKay IR, Gershwin ME. The autoimmunity of primary biliary cirrhosis and the clonal selection theory. Immunology and Cell Biology. 2011;89(1):70–80. doi: 10.1038/icb.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harada K, Tsuneyama K, Sudo Y, Masuda S, Nakanuma Y. Molecular identification of bacterial 16S ribosomal RNA gene in liver tissue of primary biliary cirrhosis: is Propionibacterium acnes involved in granuloma formation? Hepatology. 2001;33(3):530–536. doi: 10.1053/jhep.2001.22653. [DOI] [PubMed] [Google Scholar]
- 10.Bogdanos DP, Baum H, Grasso A, et al. Microbial mimics are major targets of crossreactivity with human pyruvate dehydrogenase in primary biliary cirrhosis. Journal of Hepatology. 2004;40(1):31–39. doi: 10.1016/s0168-8278(03)00501-4. [DOI] [PubMed] [Google Scholar]
- 11.Olafsson S, Gudjonsson H, Selmi C, et al. Antimitochondrial antibodies and reactivity to N. aromaticivorans proteins in icelandic patients with primary biliary cirrhosis and their relatives. The American Journal of Gastroenterology. 2004;99(11):2143–2146. doi: 10.1111/j.1572-0241.2004.40397.x. [DOI] [PubMed] [Google Scholar]
- 12.Bogdanos D, Pusl T, Rust C, Vergani D, Beuers U. Primary biliary cirrhosis following lactobacillus vaccination for recurrent vaginitis. Journal of Hepatology. 2008;49(3):466–473. doi: 10.1016/j.jhep.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 13.Wang AIP, Migita K, Ito M, et al. Hepatic expression of toll-like receptor 4 in primary biliary cirrhosis. Journal of Autoimmunity. 2005;25(1):85–91. doi: 10.1016/j.jaut.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama T, Komori A, Nakamura M, et al. Human intrahepatic biliary epithelial cells function in innate immunity by producing IL-6 and IL-8 via the TLR4-NF-κB and -MAPK signaling pathways. Liver International. 2006;26(4):467–476. doi: 10.1111/j.1478-3231.2006.01254.x. [DOI] [PubMed] [Google Scholar]
- 15.Moritoki Y, Lian ZX, Wulff H, et al. AMA production in primary biliary cirrhosis is promoted by the TLR9 ligand CpG and suppressed by potassium channel blockers. Hepatology. 2007;45(2):314–322. doi: 10.1002/hep.21522. [DOI] [PubMed] [Google Scholar]
- 16.Shimoda S, Harada K, Niiro H, et al. Biliary epithelial cells and primary biliary cirrhosis: the role of liver-infiltrating mononuclear cells. Hepatology. 2008;47(3):958–965. doi: 10.1002/hep.22102. [DOI] [PubMed] [Google Scholar]
- 17.Mao TK, Lian ZX, Selmi C, et al. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology. 2005;42(4):802–808. doi: 10.1002/hep.20859. [DOI] [PubMed] [Google Scholar]
- 18.Kikuchi K, Lian ZX, Yang GX, et al. Bacterial CpG induces hyper-IgM production in CD27+ memory B cells in primary biliary cirrhosis. Gastroenterology. 2005;128(2):304–312. doi: 10.1053/j.gastro.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 19.Haruta I, Hashimoto E, Shibata N, Kato Y, Kobayashi M, Shiratori K. Lipoteichoic acid may affect the pathogenesis of PBC-like bile duct damage and might be involved in systemic multifocal epithelial inflammations in chronic colitis-harboring TCRα-/- × AIM-/- mice. Autoimmunity. 2007;40(5):372–379. doi: 10.1080/08916930701402392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haruta I, Kikuchi K, Hashimoto E, et al. A possible role of histone-like DNA-binding protein of Streptococcus intermedius in the pathogenesis of bile duct damage in primary biliary cirrhosis. Clinical Immunology. 2008;127(2):245–251. doi: 10.1016/j.clim.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 21.Selmi C, Diana A, Cocchi CA, Zuin M, Gershwin ME. Environmental factors and the induction of autoimmunity in primary biliary cirrhosis. Expert Review of Clinical Immunology. 2008;4(2):239–245. doi: 10.1586/1744666X.4.2.239. [DOI] [PubMed] [Google Scholar]
- 22.Whiley RA, Beighton D, Winstanley TG, Fraser HY, Hardie JM. Streptococcus intermedius, Streptococcus constellatus, and Streptococcus anginosus (the Streptococcus milleri group): association with different body sites and clinical infections. Journal of Clinical Microbiology. 1992;30(1):243–244. doi: 10.1128/jcm.30.1.243-244.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petersen FC, Pecharki D, Scheie AA. Biofilm mode of growth of Streptococcus intermedius favored by a competence-stimulating signaling peptide. The Journal of Bacteriology. 2004;186(18):6327–6331. doi: 10.1128/JB.186.18.6327-6331.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feld JJ, Meddings J, Heathcote EJ. Abnormal intestinal permeability in primary biliary cirrhosis. Digestive Diseases and Sciences. 2006;51(9):1607–1613. doi: 10.1007/s10620-006-9544-z. [DOI] [PubMed] [Google Scholar]
- 25.Haruta I, Kikuchi K, Hashimoto E, et al. Long-term bacterial exposure can trigger nonsuppurative destructive cholangitis associated with multifocal epithelial inflammation. Laboratory Investigation. 2010;90(4):577–588. doi: 10.1038/labinvest.2010.40. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura M, Kondo H, Mori T, et al. Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis. Hepatology. 2007;45(1):118–127. doi: 10.1002/hep.21472. [DOI] [PubMed] [Google Scholar]
- 27.Okazaki K, Uchida K, Miyoshi H, Ikeura T, Takaoka M, Nishio A. Recent concepts of autoimmune pancreatitis and IgG4-related disease. doi: 10.1007/s12016-010-8214-2. Clinical Reviews in Allergy and Immunology. In press. [DOI] [PubMed] [Google Scholar]
- 28.Park DH, Kim MH, Chari ST. Recent advances in autoimmune pancreatitis. Gut. 2009;58(12):1680–1689. doi: 10.1136/gut.2008.155853. [DOI] [PubMed] [Google Scholar]
- 29.Okazaki K, Uchida K, Fukui T. Recent advances in autoimmune pancreatitis: concept, diagnosis, and pathogenesis. Journal of Gastroenterology. 2008;43(6):409–418. doi: 10.1007/s00535-008-2190-9. [DOI] [PubMed] [Google Scholar]
- 30.Okazaki K, Uchida K, Koyabu M, Miyoshi H, Takaoka M. Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease. Journal of Gastroenterology. 2011;46(3):277–288. doi: 10.1007/s00535-011-0386-x. [DOI] [PubMed] [Google Scholar]
- 31.Shimosegawa T, Kanno A. Autoimmune pancreatitis in Japan: overview and perspective. Journal of Gastroenterology. 2009;44(6):503–517. doi: 10.1007/s00535-009-0054-6. [DOI] [PubMed] [Google Scholar]
- 32.Ota M, Katsuyama Y, Hamano H, et al. Two critical genes (HLA-DRB1 and ABCF1) in the HLA region are associated with the susceptibility to autoimmune pancreatitis. Immunogenetics. 2007;59(1):45–52. doi: 10.1007/s00251-006-0178-2. [DOI] [PubMed] [Google Scholar]
- 33.Muraki T, Hamano H, Ochi Y, et al. Autoimmune pancreatitis and complement activation system. Pancreas. 2006;32(1):16–21. doi: 10.1097/01.mpa.0000188308.75043.e4. [DOI] [PubMed] [Google Scholar]
- 34.Kawa S, Ota M, Yoshizawa K, et al. HLA DRB10405-DQB10401 haplotype is associated with autoimmune pancreatitis in the Japanese population. Gastroenterology. 2002;122(5):1264–1269. doi: 10.1053/gast.2002.33022. [DOI] [PubMed] [Google Scholar]
- 35.Kojima M, Sipos B, Klapper W, et al. Autoimmune pancreatitis: frequency, IgG4 expression, and clonality of T and B cells. The American Journal of Surgical Pathology. 2007;31(4):521–528. doi: 10.1097/01.pas.0000213390.55536.47. [DOI] [PubMed] [Google Scholar]
- 36.Umemura T, Ota M, Hamano H, et al. Association of autoimmune pancreatitis with cytotoxic T-lymphocyte antigen 4 gene polymorphisms in Japanese patients. The American Journal of Gastroenterology. 2008;103(3):588–594. doi: 10.1111/j.1572-0241.2007.01750.x. [DOI] [PubMed] [Google Scholar]
- 37.Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. The New England Journal of Medicine. 2001;344(10):732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 38.Aoki S, Nakazawa T, Ohara H, et al. Immunohistochemical study of autoimmune pancreatitis using anti-IgG4 antibody and patients’ sera. Histopathology. 2005;47(2):147–158. doi: 10.1111/j.1365-2559.2005.02204.x. [DOI] [PubMed] [Google Scholar]
- 39.Detlefsen S, Bräsen JH, Zamboni G, Capelli P, Klöppel G. Deposition of complement C3c, immunoglobulin (Ig)G4 and IgG at the basement membrane of pancreatic ducts and acini in autoimmune pancreatitis. Histopathology. 2010;57(6):825–835. doi: 10.1111/j.1365-2559.2010.03717.x. [DOI] [PubMed] [Google Scholar]
- 40.Kawa S, Kitahara K, Hamano H, et al. A novel immunoglobulin-immunoglobulin interaction in autoimmunity. PLoS One. 2008;3(2, article e1637) doi: 10.1371/journal.pone.0001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito T, Kitahara K, Umemura T, et al. A novel heterophilic antibody interaction involves igG4. Scandinavian Journal of Immunology. 2010;71(2):109–114. doi: 10.1111/j.1365-3083.2009.02353.x. [DOI] [PubMed] [Google Scholar]
- 42.Kanno H, Nose M, Itoh J, Taniguchi Y, Kyogoku M. Spontaneous development of pancreatitis in the MRL/Mp strain of mice in autoimmune mechanism. Clinical and Experimental Immunology. 1992;89(1):68–73. doi: 10.1111/j.1365-2249.1992.tb06879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hosaka N, Nose M, Kyogoku M, et al. Thymus transplantation, a critical factor for correction of autoimmune disease in aging MRL/+ mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(16):8558–8562. doi: 10.1073/pnas.93.16.8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsubata R, Tsubata T, Hiai H, et al. Autoimmune disease of exocrine organs in immunodeficient alymphoplasia mice: a spontaneous model for Sjogren’s syndrome. European Journal of Immunology. 1996;26(11):2742–2748. doi: 10.1002/eji.1830261129. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura Y, Yi SQ, Terayama H, et al. Sequential histopathology of pancreatic tissues in aly/aly mice. Cells Tissues Organs. 2007;186(3):204–209. doi: 10.1159/000105675. [DOI] [PubMed] [Google Scholar]
- 46.Wang HX, Yi SQ, Li J, et al. Effects of splenectomy on spontaneously chronic pancreatitis in aly/aly mice. Clinical and Developmental Immunology. 2010;2010:8 pages. doi: 10.1155/2010/614890. Article ID 614890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakaguchi Y, Inaba M, Tsuda M, et al. The Wistar Bonn Kobori rat, a unique animal model for autoimmune pancreatitis with extrapancreatic exocrinopathy. Clinical and Experimental Immunology. 2008;152(1):1–12. doi: 10.1111/j.1365-2249.2008.03588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vallance BA, Hewlett BR, Snider DP, Collins SM. T cell-mediated exocrine pancreatic damage in major histocompatibility complex class II-deficient mice. Gastroenterology. 1998;115(4):978–987. doi: 10.1016/s0016-5085(98)70270-7. [DOI] [PubMed] [Google Scholar]
- 49.Freitag TL, Cham C, Sung HH, et al. Human risk Allele HLA-DRB1∗0405 predisposes class II transgenic Ab0 NOD mice to autoimmune pancreatitis. Gastroenterology. 2010;139(1):281–291. doi: 10.1053/j.gastro.2010.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boomershine CS, Chamberlain A, Kendall P, et al. Autoimmune pancreatitis results from loss of TGFβ signalling in S100A4-positive dendritic cells. Gut. 2009;58(9):1267–1274. doi: 10.1136/gut.2008.170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meagher C, Tang Q, Fife BT, et al. Spontaneous development of a pancreatic exocrine disease in CD28-deficient NOD mice. The Journal of Immunology. 2008;180(12):7793–7803. doi: 10.4049/jimmunol.180.12.7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davidson TS, Longnecker DS, Hickey WF. An experimental model of autoimmune pancreatitis in the rat. The American Journal of Pathology. 2005;166(3):729–736. doi: 10.1016/S0002-9440(10)62294-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishimori I, Bratanova T, Toshkov I, et al. Induction of experimental autoimmune sialoadenitis by immunization of PL/J mice with carbonic anhydrase II. The Journal of Immunology. 1995;154(9):4865–4873. [PubMed] [Google Scholar]
- 54.Uchida K, Okazaki K, Nishi T, et al. Experimental immune-mediated pancreatitis in neonatally thymectomized mice immunized with carbonic anhydrase II and lactoferrin. Laboratory Investigation. 2002;82(4):411–424. doi: 10.1038/labinvest.3780435. [DOI] [PubMed] [Google Scholar]
- 55.Nishio A, Asada M, Uchida K, Fukui T, Chiba T, Okazaki K. The role of innate immunity in the pathogenesis of experimental autoimmune pancreatitis in mice. Pancreas. 2011;40(1):95–102. doi: 10.1097/MPA.0b013e3181f3a5d4. [DOI] [PubMed] [Google Scholar]
- 56.Soga Y, Komori H, Miyazaki T, et al. Toll-like receptor 3 signaling induces chronic pancreatitis through the Fas/Fas ligand-mediated cytotoxicity. Tohoku Journal of Experimental Medicine. 2009;217(3):175–184. doi: 10.1620/tjem.217.175. [DOI] [PubMed] [Google Scholar]
- 57.Qu WM, Miyazaki T, Terada M, et al. A novel autoimmune pancreatitis model in MRL mice treated with polyinosinic:polycytidylic acid. Clinical and Experimental Immunology. 2002;129(1):27–34. doi: 10.1046/j.1365-2249.2002.01881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asada M, Nishio A, Akamatsu T, et al. Analysis of humoral immune response in experimental autoimmune pancreatitis in mice. Pancreas. 2010;39(2):224–231. doi: 10.1097/MPA.0b013e3181bab5e2. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe S, Suzuki K, Kawauchi Y, et al. Kinetic analysis of the development of pancreatic lesions in mice infected with a murine retrovirus. Clinical Immunology. 2003;109(2):212–223. doi: 10.1016/s1521-6616(03)00197-9. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki K, Makino M, Okada Y, et al. Exocrinopathy resembling Sjogren’s syndrome induced by a murine retrovirus. Laboratory Investigation. 1993;69(4):430–435. [PubMed] [Google Scholar]
- 61.Haruta I, Yanagisawa N, Kawamura S, et al. A mouse model of autoimmune pancreatitis with salivary gland involvement triggered by innate immunity via persistent exposure to avirulent bacteria. Laboratory Investigation. 2010;90(12):1757–1769. doi: 10.1038/labinvest.2010.153. [DOI] [PubMed] [Google Scholar]
- 62.Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology. 2000;118(3):573–581. doi: 10.1016/s0016-5085(00)70264-2. [DOI] [PubMed] [Google Scholar]
- 63.Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut. 2005;54(2):274–281. doi: 10.1136/gut.2004.049064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Asada M, Nishio A, Uchida K, et al. Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas. 2006;33(1):20–26. doi: 10.1097/01.mpa.0000226881.48204.fd. [DOI] [PubMed] [Google Scholar]
- 65.Takizawa S, Endo T, Wanjia X, Tanaka S, Takahashi M, Kobayashi T. HSP 10 is a new autoantigen in both autoimmune pancreatitis and fulminant type 1 diabetes. Biochemical and Biophysical Research Communications. 2009;386(1):192–196. doi: 10.1016/j.bbrc.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 66.Guarneri F, Guarneri C, Benvenga S. Helicobacter pylori and autoimmune pancreatitis: role of carbonic anhydrase via molecular mimicry? Journal of Cellular and Molecular Medicine. 2005;9(3):741–744. doi: 10.1111/j.1582-4934.2005.tb00506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. The New England Journal of Medicine. 2009;361(22):2135–2142. doi: 10.1056/NEJMoa0903068. [DOI] [PubMed] [Google Scholar]
- 68.Li M, Zhou Y, Feng G, Su SB. The critical role of toll-like receptor signaling pathways in the induction and progression of autoimmune disease. Current Molecular Medicine. 2009;9(3):365–374. doi: 10.2174/156652409787847137. [DOI] [PubMed] [Google Scholar]
- 69.Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky’s postulates revisited) Immunology Today. 1993;14(9):426–430. doi: 10.1016/0167-5699(93)90244-F. [DOI] [PubMed] [Google Scholar]
- 70.Irie J, Wu Y, Wicker LS, et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. Journal of Experimental Medicine. 2006;203(5):1209–1219. doi: 10.1084/jem.20051911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oertelt S, Lian ZX, Cheng CM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-βreceptor II dominant-negative mice. The Journal of Immunology. 2006;177(3):1655–1660. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 72.Wakabayashi K, Lian ZX, Moritoki Y, et al. IL-2 receptor α mice and the development of primary biliary cirrhosis. Hepatology. 2006;44(5):1240–1249. doi: 10.1002/hep.21385. [DOI] [PubMed] [Google Scholar]
- 73.Salas JT, Banales JM, Sarvide S, et al. Ae2-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology. 2008;134(5):1482–1493. doi: 10.1053/j.gastro.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 74.de Moreno de LeBlanc A, del Carmen S, Zurita-Turk M, et al. Importance of IL-10 modulation by probiotic microorganisms in gastrointestinal inflammatory diseases. ISRN Gastroenterology. 2011;2011:11 pages. doi: 10.5402/2011/892971. Article ID 892971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McLoughlin RM, Mills KHG. Influence of gastrointestinal commensal bacteria on the immune responses that mediate allergy and asthma. Journal of Allergy and Clinical Immunology. 2011;127(5):1097–1107. doi: 10.1016/j.jaci.2011.02.012. [DOI] [PubMed] [Google Scholar]