

Abstract
On infection, the herpes simplex virus (HSV) virion protein VP16 (Vmw65; αTIF) forms a transcriptional regulatory complex—the VP16-induced complex—with two cellular proteins, HCF and Oct-1, on VP16-responsive cis-regulatory elements in HSV immediate-early promoters called TAATGARAT. Comparison of different HSV VP16 sequences reveals a conserved core region that is sufficient for VP16-induced complex formation. The crystal structure of the VP16 core has been determined at 2.1 Å resolution. The results reveal a novel, seat-like protein structure. Together with the activity of mutant VP16 proteins, the structure of free VP16 suggests that it contains (1) a disordered carboxy-terminal region that associates with HCF, Oct-1, and DNA in the VP16-induced complex, and (2) a structured region involved in virion assembly and possessing a novel DNA-binding surface that differentiates among TAATGARAT VP16-response elements.
Keywords: HSV, protein VP16, crystal structure, DNA recognition
Herpes simplex virus (HSV) is a large double-stranded DNA virus, which is transcribed and replicated in the host cell nucleus. During infection, the viral genes are coordinately expressed in three phases—immediate early (IE) or α, delayed-early or β, and late or γ. This cascade of viral gene expression is initiated by an HSV virion protein called VP16 (also called αTIF, Vmw65, or VF65) (for reviews, see Thompson and McKnight 1992; O’Hare 1993).
VP16 has multiple roles during the HSV infection cycle. It is synthesized late during infection and is incorporated into the tegument of the virion with other viral proteins, including the regulatory virion host shutoff protein vhs (Smibert et al. 1994). On de novo infection, VP16 is released into the cell and associates with two cellular factors, HCF (also called C1, CFF, and VCAF) and Oct-1, on HSV IE promoters at VP16-responsive sites called TAATGARAT. VP16 first binds HCF, a nuclear factor involved in cell proliferation (Goto et al. 1997), and subsequently VP16 and HCF together associate with Oct-1, a POU domain transcription factor, on the TAATGARAT (R = purine) element. Once targeted to HSV IE promoters through association with HCF and Oct-1, VP16 activates IE-gene transcription through a potent carboxy-terminal transcriptional activation domain. Therefore, VP16 is a multifaceted protein that, at different times during the HSV infection cycle, interacts with either viral proteins, as in the virion, or cellular proteins, as in the VP16-induced complex.
The structure and assembly of the VP16-induced complex has been studied extensively. VP16 likely contacts each of the other components of the VP16-induced complex—HCF, Oct-1, and DNA. In HCF, a large protein of >2000 amino acids (Wilson et al. 1993; Kristie et al. 1995), VP16 binds to a stable 380-amino-acid amino-terminal domain (LaBoissière et al. 1997; Wilson et al. 1997). In Oct-1, VP16 interacts with its DNA-binding POU domain (Kristie et al. 1989; Stern et al. 1989). The POU domain is a bipartite DNA-binding domain consisting of two helix–turn–helix DNA-binding structures, a POU-specific (POUS) domain and a POU-homeo (POUH) domain, which are connected by a flexible linker (for review, see Herr and Cleary 1995). VP16 discriminates among the POUH domains of different POU proteins (Stern and Herr 1991; Lai et al. 1992; Pomerantz et al. 1992) and among different TAATGARAT elements present in different HSV genomes (Huang and Herr 1996; Misra et al. 1996).
VP16 is a modular protein of 490 amino acids. The potent transcriptional activation domain is located within the carboxy-terminal 80 amino acids (Sadowski et al. 1988; Triezenberg et al. 1988; Cousens et al. 1989), whereas sequences sufficient for VP16-induced complex assembly encompass much of the remainder of the protein (Greaves and O’Hare 1990). Comparison of the HSV VP16 sequence with those of the related proteins from other herpesviruses reveals that the region sufficient for VP16-induced complex assembly is conserved, whereas the sequence of the transcriptional activation domain is not conserved. Here, we describe the X-ray crystal structure of the free form of the conserved VP16-induced complex forming region. The results reveal a novel, seat-like protein structure. Sequences involved in DNA-sequence recognition and productive virion assembly are structured, but sequences involved in associating with HCF and Oct-1 are disordered, suggesting that VP16 undergoes conformational changes during VP16-induced complex assembly.
Results and Discussion
A conserved VP16 core
The sequence of VP16 homologs has been determined from a number of different herpesviruses (Fig. 1). The length of these VP16 proteins varies from 410 amino acids in varicella zoster virus (VZV) to 504 amino acids in bovine herpesvirus type 1 (BHV-1), but they all contain a conserved core region of ∼350 amino acids (Fig. 1A). We refer to this conserved region of VP16 as the VP16 core. The position of the conserved VP16 core varies from a central location in the HSV type 1 (HSV-1), equine herpesvirus type 1 (EHV-1), and BHV-1 VP16 proteins to a carboxy-terminal location in the VZV and gallus herpesvirus type 1 (GHV-1) VP16 proteins. In HSV-1 VP16, the amino-terminal 48 amino acids and the carboxy-terminal 78 amino acids—the transcriptional activation domain—are not highly conserved. In the studies described here, we focused our structural analysis of VP16 on the VP16 core because (1) deletion analysis of VP16 has shown that this region of VP16 is sufficient for VP16-induced complex assembly (Greaves and O’Hare 1990) and (2) structural studies of the VP16 transcriptional activation domain have indicated that, on its own, its structure is unstable (Donaldson and Capone 1992; O’Hare and Williams 1992; Shen et al. 1996; Uesugi et al. 1997).
Figure 1.

Conserved VP16-core region. (A) Schematic representation of VP16 homologs from five herpesviruses and delineation of the VP16-core protein used for crystallography. The VP16 homologs are derived from HSV-1 (strain 17; Campbell et al. 1984; Dalrymple et al. 1985), VZV (Davison and Scott 1986), GHV-1 (Yanagida et al. 1993), EHV-1 (Telford et al. 1992), and BHV-1 (Carpenter and Misra 1992). A conserved VP16-core region is shown in dark gray and the carboxy-terminal HSV-1 transcriptional activation domain (AD) is shown in light gray. The HSV-1 VP16 core (49–412) protein is shown at the bottom with the structured regions identified by x-ray diffraction shown in black. (B) Electrophoretic mobility retardation assay of GST–VP16 fusion proteins synthesized in E. coli. A labeled (OCTA+)TAATGARAT site from the HSV-1 ICP0 promoter (Stern et al. 1989) was used for electrophoretic mobility retardation as described (Huang and Herr 1996). (Lane 1) Free probe; (lanes 2–6), partially purified HeLa-cell Oct-1 and HCF (the wheat germ agglutinin fraction; Wilson et al. 1993); (lane 3) GST–VP16ΔC; (lane 4) VP16ΔC; (lane 5) GST–VP1649–412 core; (lane 6) VP1649–412 core. The positions of the free probe, Oct-1–DNA (Oct-1) complex, and VP16-induced complex (VIC) are indicated on the left.
Figure 2 shows a sequence alignment of HSV-1 VP16 residues 49–412 and the corresponding residues of the VZV, GHV-1, EHV-1, and BHV-1 VP16 proteins. The sequence alignment reveals sequence conservation throughout the entire region, with invariant or conservatively substituted positions scattered throughout the conserved core. Only three regions of the conserved core have suffered insertions or deletions in the different homologs (around HSV-1 VP16 residues 125, 200, and 347–382) and the size of insertion or deletion is never more than five residues. This sequence comparison suggests that the functions of the conserved VP16 core require extensive sequence conservation and therefore structural conservation.
Figure 2.
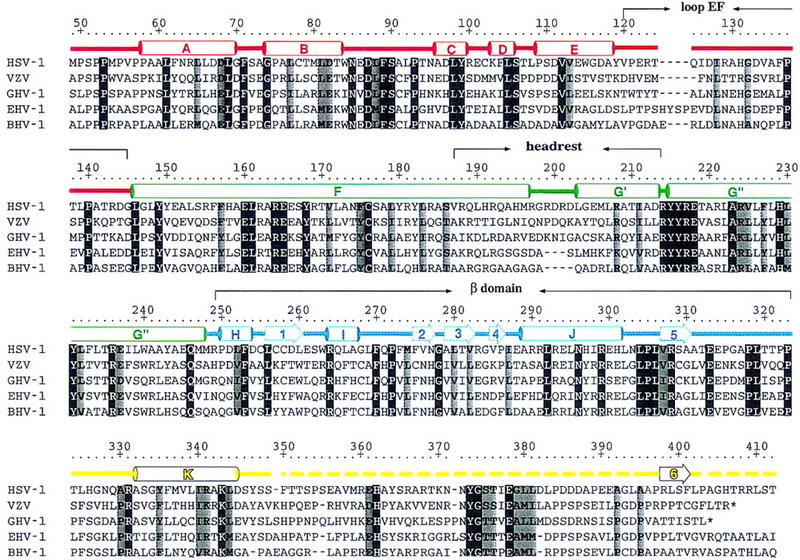
Sequence alignment of the conserved VP16-core region from HSV-1, VZV, GHV-1, EHV-1, and BHV-1 and alignment with structural elements of the VP16-core protein. The HSV-1 residue numbering is shown above the sequence alignment. Amino acids highlighted are either invariant (white against black) among the five proteins or similar (shaded) as defined by the following groupings: V, L, I, and M; F, Y, and W; K and R; E and D; Q and N; S and T; and A, G, and P. The color coding is as described in the text; α helices are labeled A–K (helix αG is subdivided into helices αG′ and αG′′ owing to a discontinuity in this helix); β strands are labeled 1–6; turns and loops (L) are referred to by their flanking secondary structures; broken lines indicate disordered regions. Polar interactions among conserved residues that are seen to exert their effect on the structure of VP16 as a whole include ion pairings (R64–D68, D97–R162, E160–H131, R164–E237, E165–K343, R214–E218, and R308–E313), polar side-chain– side-chain interactions (D87–S90, Y149–Q246, Y99–E165, S106–D111, Y168–H229, Y231–K343), polar side-chain–main-chain interactions (N85–H326, N85–A330, S106–L108, S186–Y182, Y215–F104, Y216–S106, R237–L302, R264–S90, N296–V307, R299–P305, R299–L269), and water-mediated polar interactions (S106-H2O–S348, E160-H2O–E237, Y182-H2O–R214, Y335-H2O–A330).
To prepare the VP16 core for crystallography, we synthesized the HSV-1 VP16 core (residues 49–412) fused at its amino-terminus to glutathione S-transferase (GST–VP16) in Escherichia coli and separated the VP16 sequences from the GST moiety by proteolytic digestion. As shown in Figure 1B, VP16 residues 1–412 fused to GST (GST–VP16ΔC) are active for VP16-induced complex (labeled VIC) assembly in an electrophoretic mobility retardation assay before and after proteolytic digestion (Fig. 1B, cf. lanes 3 and 4). Consistent with previous studies in eukaryotic cells (Greaves and O’Hare 1990), further deletion of VP16 amino-terminal residues 1–48 (labeled Core in Fig. 1B) had no adverse effect on VP16-induced complex formation either as a GST fusion protein (Fig. 1B, lane 5) or as a free protein after proteolysis (Fig. 1B, lane 6). Deletion of the amino-terminal residues, however, offered an important benefit for the crystallography studies described here because synthesis of this protein in E. coli is more efficient than that of the longer VP16 protein. We therefore chose to determine the structure of HSV-1 VP16 residues 49–412.
Structure determination
We calculated electron density maps by isomorphous replacement of mercuric derivatives (Table 1) and were able to build a total of 309 of the 364 residues of the HSV-1 VP16 core protein into a model: residues 49–349 and 395–402. Residues 350–394 (45 residues) and 403–412 (10 residues) are missing and apparently disordered in the crystal (see Fig. 1A, bottom). The model was refined to 2.1 Å resolution with a crystallographic R factor of 0.19 and Rfree value of 0.26 (Table 2).
Table 1.
X-ray data collection and phasing statistics at 3.0 Å resolution
|
|
Native
|
HgCl2
|
|
pCMBS
|
|
|
|
|---|---|---|---|---|---|---|---|
| Crystal | nos. 1 and 2 | no. 3 | no. 3 | no. 4 | no. 5 | no. 6 | no. 6 |
| Cell dimensions (Å) | a = 59.5 | b = 77.1 | c = 84.0 | ||||
| Wavelength (Å) | 1.5418 | 1.01299 | 1.015562 | ||||
| Energy (KeV) | 8.042 | 12.241 | 12.210 | ||||
| I cutoff I/ς(I) | 2 | 2 | 2 | 2 | 2 | 0 | 0 |
| Resolution limit (Å) | 3 | 3 | 3.5 | 3 | 3 | 2.6 | 2.6 |
| Completeness (%)a | 94/86 | 72/71 | 75/76 | 70/38 | 84/65 | 96/94 | 95/91 |
| R linear (%)a | 8.8/20.7 | 10.5/31.9 | 12.9/22.9 | 10.8/22.3 | 11.9/21.2 | 6.1/15.9 | 6.1/16.3 |
| <I/ς(I)>a | 11/5 | 8/3 | 7/3 | 8/3 | 9/3 | 13/6 | 11/5 |
| Observed reflections (no.) | 50575 | 15183 | 10594 | 13886 | 20095 | 37731 | 37130 |
| Unique reflections (no.) | 7498/335 | 5796/277 | 3953/192 | 5746/143 | 6800/255 | 11579/543 | 11751/560 |
| Anomalous pairs (no.) | — | — | — | — | — | 9263 | 8899 |
| Mercury sites (no.) | — | 4 | 4 | 4 | 4 | 5 | 5 |
| Isomorphous/anomalous | — | iso | iso | iso | iso | ano | ano |
| Phasing power | — | 1.95 | 1.75 | 1.27 | 1.47 | 1.28 | 0.95 |
| RCullis | — | 0.537 | 0.505 | 0.633 | 0.533 | — | — |
| RKraut | — | 0.130 | 0.133 | 0.113 | 0.146 | 0.404 | 0.420 |
| Figure of merit | — | 0.374 | 0.363 | 0.301 | 0.326 | 0.277 | 0.234 |
| Overall figure of merit | 0.567 |
Numerical numbers are given for the whole data set/highest resolution bin.
Table 2.
Structural refinement statistics at 2.1 Å resolution
| Crystal | no. 7 |
| Cell dimensions (Å) | 59.5 × 75.2 × 81.1 |
| Temperature (K) | 95 |
| Wavelength | 1.1000 |
| Resolution range (Å) | 20.–2.1/2.14–2.1 (highest resolution bin) |
| Completeness (%) | 96.8/92.9 |
| R linear | 0.068/0.356 |
| <I/ς(I)> | 11.3/2.8 |
| Observed reflections (no.) | 143578/− |
| Unique reflections (no.) | 21163/1008 |
| Protein atoms (no.) | 2460 (the side chain of V225 was modeled in a dual conformation) |
| Water molecules (no.) | 178 |
| Phosphate ions (no.) | 2 |
| Root-mean-square deviation from ideality | |
| bond lengths (Å) | 0.01 |
| bond angles (°) | 1.5 |
| dihedrals (°) | 21.8 |
| improper (°) | 1.4 |
| Average thermal factor (Å2) | |
| main chain | 20.0 ± 1.7 |
| side chain | 22.3 ± 3.3 |
| water | 28.7 |
| Ramachandran plot | |
| most favored regions (%) | 94 |
| additionally allowed regions (%) | 6 |
|
Resolution range (Å)
|
Completeness (%)/Reflections (no.)
|
R value/Rfree
|
|---|---|---|
| 7.00–2.10 | 92.9/19677 | 0.192/0.260 |
| 2.19–2.10 | 86.5/2262 | 0.255/0.320 |
Overall structure
Figure 3 presents two views of the determined VP16-core structure; the location of structural elements with respect to the amino-acid sequence of HSV-1 VP16 is shown in Figure 2. In Figures 2 and 3, the regions of the molecule have been colored, from amino to carboxyl terminus, red (residues 49–145), green (residues 146–247), blue (residues 248–323), and yellow (residues 324–412). In the orientation shown in Figure 3A, the missing amino-terminal segment (residues 1–48) would enter the determined structure from the left side (labeled 49) and the missing carboxy-terminal 78-residue transcriptional activation domain would leave the molecule at the bottom. In the same view, the major disordered region of the determined structure (residues 350–394) lies to the right of the molecule and is indicated by a broken line.
Figure 3.

Ribbon (Carson 1991) diagrams of the VP16-core protein, colored red (residues 49–145), green (146–247), light blue (248–323), and yellow (324–412). (A) Front view; (B) right-side view.
The overall structure of the VP16 core, as viewed in Figure 3B, resembles a seat with the back of the seat at the right. We use this seat analogy to describe the molecule and refer to the view shown in Figure 3A as the front view and the view shown in Figure 3B as the right-side view (the point of view of an observer from the right side of Fig. 3A). To describe the structure, we divide it into three parts: the back (green), bottom (blue), and the top surface (red and yellow) of the seat. With respect to the primary sequence, the seat back and bottom each consist of contiguous sequences, whereas the surface of the seat is composed of elements from the amino-terminal region (helix αB in red) and the carboxy-terminal region (helix αK in yellow).
Structured segments
Viewed from the front (Fig. 3A), the structured regions of VP16 display considerable internal structural symmetry. There are two principal elements to this symmetry. The first element is the seat surface, consisting of helices αB (red) and αK (yellow), which form a V-like structure that links the amino- and carboxy-terminal regions of VP16 together. The second element represents the most dominating feature of the VP16 structure: a two-stranded antiparallel coiled–coil formed by two long α helices—helix αF of 51 amino acids and helix αG of 45 amino acids—which form the seat back (colored green; note that in Fig. 2 helix αG is labeled G′/G′′ owing to a kink in this helix that is described below). These two long helices are connected by a short six-amino-acid loop (L-FG) at the top of the seat back, forming the center of what we refer to as a headrest (see Fig. 2). The two pairs of symmetrical helices—αB and αK on the seat surface, and αF and αG at the seat back—lie nearly perpendicular to one another and are largely responsible for the seat-like appearance of VP16.
Two other overall structural elements of the VP16 structure are the amino-terminal region shown in red and the seat bottom shown in blue (Fig. 3). The amino-terminal region (red) is critical to the structural integrity of the molecule; it wraps around the middle of the molecule. Helix αE is positioned approximately perpendicular to the axis of the coiled–coil structure forming a support for the headrest, followed by the most pronounced loop of the VP16-core structure: the 27-amino-acid-long L-EF loop (residues 119–145); this loop extends along nearly the entire length of the back of the molecule (see Fig. 3B) and connects to helix αF of the coiled–coil structure.
The seat bottom contains all of the strands (β1–β6) present in the VP16 core. The strand β6 shown in yellow is near the carboxyl terminus of the VP16-core protein. The strands form two β sheets: a two-stranded parallel β sheet (strands β5 and β2) and a four-stranded mixed antiparallel β sheet (strands β4, β3, β1, and β6). Strands β2 and β3 are connected by a sharp turn at the invariant glycine G278 (see Fig. 2), causing a nearly perpendicular orientation of the two β sheets (see Fig. 3B).
Segment involved in HCF/Oct-1/DNA interactions is disordered
The dashed yellow lines in Figures 2 and 3 identify the two segments of the crystallized VP16-core protein that are not represented in the determined structure: residues 350–394 and carboxy-terminal residues 403–412. The disorder of the carboxy-terminal residues (403–412) may indicate the beginning of a disordered transcriptional activation domain (Donaldson and Capone 1992; O’Hare and Williams 1992; Shen et al. 1996). The other, larger disordered segment (residues 350–394) is critical for VP16-induced complex assembly. Numerous insertion, deletion, and point mutation analyses (Ace et al. 1988; Werstuck and Capone 1989; Greaves and O’Hare 1990; Stern and Herr 1991; Walker et al. 1994; Shaw et al. 1995; Lai and Herr 1997), and peptide studies (Haigh et al. 1990; Stern and Herr 1991; Hayes and O’Hare 1993; Wu et al. 1994; Simmen et al. 1997) have identified this segment as important for VP16-induced complex formation and interaction with HCF, Oct-1, and DNA. The activity of individual point mutations in VP16 and synthetic peptides has shown that residues E361, H362, and Y364 are involved in contacting HCF (Freiman and Herr 1997; Lai and Herr 1997; Simmen et al. 1997; Lu et al. 1998); residues Y373, G374, and S375 are involved in recognizing Oct-1 (Walker et al. 1994; Lai and Herr 1997); and residues R360, R366, R368, and K370 are involved in binding DNA (Shaw et al. 1995; Lai and Herr 1997). We hypothesize that this HCF/Oct-1/DNA-interaction segment of VP16 is unstructured in its native state, but adopts structure on binding to HCF, Oct-1, and DNA in the VP16-induced complex. Consistent with this hypothesis, VP16 residues 365–370 are susceptible to proteolytic digestion in the free VP16 molecule but are protected from digestion in the VP16-induced complex, suggesting that these residues represent a major surface-exposed site that is covered in the VP16-induced complex (Hayes and O’Hare 1993).
There is an excellent precedent for the hypothesis that a disordered segment of VP16 adopts a stable structure on formation of VP16-induced complex. Structural studies have shown that a disordered segment of the yeast transcriptional regulator MATα2 adopts a stable α helical structure on complex formation with the homeodomain of the MATα2-coregulator MATa1 (Li et al. 1995). Mutational analyses of the Oct-1 POUH domain (Pomerantz et al. 1992) and VP16 (Stern and Herr 1991; Walker et al. 1994; Lai and Herr 1997), and sequence similarity between VP16 (372-NYGSTIEGLL-381) and MATα2 (59-TIAPELADLL-68) suggested that VP16 contacts the Oct-1 POUH domain in the same manner as MATα2 contacts the MATa1 homeodomain (Li et al. 1995). Our finding that, as in MATα2, the homeodomain interaction region of free VP16 is disordered is consistent with this hypothesis and suggests that this region of VP16 undergoes a conformational change on binding to Oct-1. These results extend the remarkable similarity between a yeast transcriptional coregulator interaction and a human viral and cellular coregulator interaction.
Between the disordered HCF/Oct-1/DNA-interaction segment of VP16 and the disordered carboxy-terminal segment leading to the transcriptional activation domain is a short structured segment of eight amino acids that contains strand β6 (yellow) and forms part of the seat bottom (Fig. 3). This segment is of considerable interest because it tethers the HCF/Oct-1/DNA-interaction region to the body of VP16 and positions the transcriptional activation domain at the bottom of the structure (see Fig. 3). Furthermore, the position of strand β6 as the outermost strand of the four-stranded sheet of the seat bottom suggests that it could be detached from the body of VP16 without disrupting the structured region of the free VP16 core. Perhaps, during VP16-induced complex assembly or transcriptional activation, strand β6 becomes detached from the remainder of VP16 providing a mechanism for conformational changes in VP16 during regulation of transcription.
Virion assembly surface
Ace et al. (1988) described the first mutational analysis of VP16 and the results of this study are easily interpreted with respect to the VP16-core structure presented here. In that study, four-amino-acid insertions were made throughout the VP16 molecule and the corresponding mutants were assayed for virion assembly, VP16-induced complex formation, and transcriptional activation. Seven of these insertions were within the VP16-core region crystallized here, between residues G74/P75, G145/L146, L172/A173, S177/A178, R249/P250, E288/A289, and G379/L380. The position of five of the seven mutants is shown in the rear and left-side views of VP16 shown in Figure 4; one of the seven mutants, G379/L380, lies within the disordered region, and another, S177/A178, is buried in the VP16 structure and is therefore not visible in the molecular surface representation shown in Figure 4.
Figure 4.
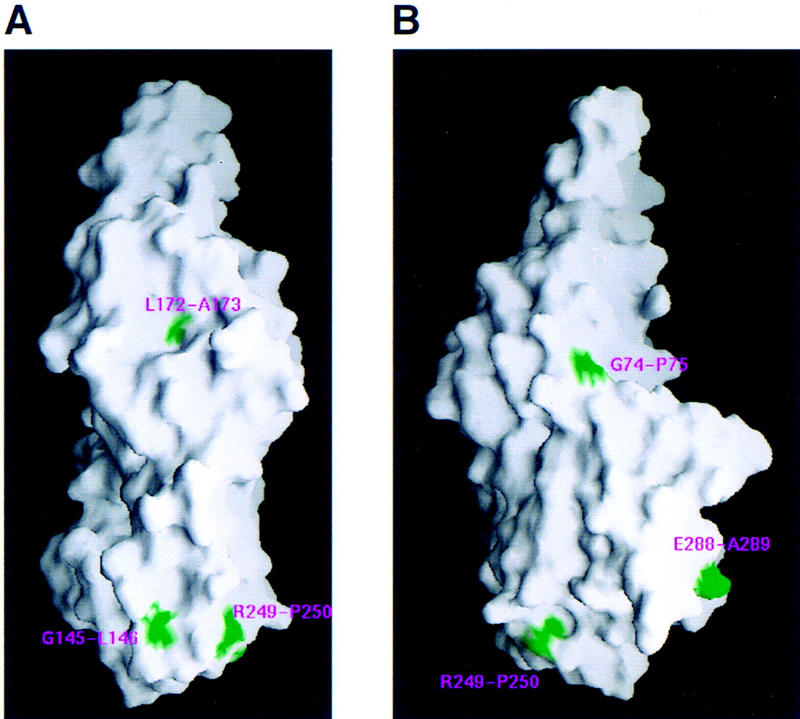
Position of insertion mutations (green) prepared by Ace et al. (1988) on a molecular surface representation of VP16 (Nicholls et al. 1991). (A) Rear view; (B) Left-side view.
The seven insertion mutants fall into four different classes: (1) wild-type activity (G145/L146); (2) active for virion assembly but not VP16-induced complex formation (G379/L380); (3) active for VP16-induced complex formation or transactivation but not virion assembly (G74/P75, R249/P250, and E288/A289); and (4) inactive for both VP16-induced complex formation and virion assembly (L172/A173 and S177/A178) (Ace et al. 1988). Consistent with these results, the inactive mutants L172/A173 and S177/A178 map within the center of the protein in the coiled–coil helix αF (see Fig. 4A)—these mutations probably disrupt the overall structure of VP16—whereas the mutant with wild-type activity, G145/L146, lies on the surface of VP16 (at the end of loop L-EF). The mutation that is active for virion assembly but not VP16-induced complex formation, G379/L380, lies within the disordered HCF/Oct-1/DNA-interaction segment, and is now known to affect interaction with Oct-1 (Stern and Herr 1991). Interestingly, the three mutations that are active for VP16-induced complex formation or transactivation but not virion assembly all map on the side of the molecule opposite to the HCF/Oct-1/DNA-interaction segment (Fig. 4B). Therefore, the activities of these VP16 insertion mutants suggest that different surfaces of VP16 are used for different functions—the right side (Fig. 3B) of VP16 is intimately involved in the control of gene expression by promoting VP16-induced complex assembly and activation of HSV immediate-early gene expression, and the left side (Fig. 4B, below) is intimately involved in virion assembly. Perhaps, the virion-assembly surface is involved in association with the virion tegument protein vhs (Smibert et al. 1994).
Conserved residues
As noted above, highly conserved residues of VP16 are scattered throughout the conserved core region (see Fig. 2). The structure of the VP16 core offers a rationale for their conservation: The large majority of the conserved amino acids is involved in structural packing and intramolecular interactions. For example, conserved hydrophobic side chains intercalate with each other at the interfaces of the α helices to form the hydrophobic core of the molecule. Unexpectedly, however, many of the invariant and conserved residues are polar or charged, and are involved in intramolecular interactions that likely confer stability to the molecule (see legend to Fig. 2).
Figure 5 illustrates three of these interactions in detail. Figure 5A shows a network of polar interactions involving the invariant residue K343. In a previous study, substitution of K343 for alanine resulted in complete inactivation of VP16 and poor VP16 synthesis in E. coli suggesting a major disruption of the VP16 structure (Lai and Herr 1997). Consistent with this hypothesis, K343 is buried in the center of the structure and is critical for stabilizing a network of polar interactions involving the amino-terminal (red), coiled–coil (green), and carboxy-terminal (yellow) segments of the VP16 core. This network of interactions involves (1) a main-chain carbonyl group of the amino-terminal residue P93 interacting with Y231 of helix αG of the coiled–coil; (2) Y231 interaction with K343 of carboxy-terminal helix αK; (3) K343 interaction with E165 of helix αF also of the coiled–coil; (4) E165 interaction with amino-terminal residues Y99 and N95; (5) N95 interaction with R162 of helix αF; and (6) R162 interaction with amino-terminal residue D97.
Figure 5.
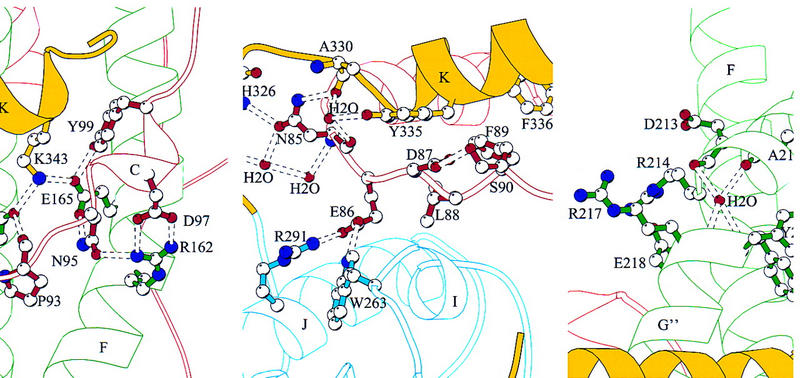
Views of defined elements in the VP16 structure. (A) Network of internal charged interactions centered on residue K343. (B) Loop L-BC (red) between helix αK (yellow) and the seat bottom (blue). (C) Solvent-exposed and invariant arginines 214 and 217 and the discontinuity point of helix αG. [Produced by MOLSCRIPT (Kraulis 1991)].
Emphasizing the importance of these interactions is the observation that residues P93, Y99, E165, Y231, and K343 are invariant in the five VP16 homologs shown in Figure 2. Furthermore, in the case of the three other residues, N95 varies in only one of the VP16 homologs—this position is a histidine in EHV-1, a conservative change—and residues D97 and R162 also only differ in GHV-1 and here there is covariation: in GHV-1 residues corresponding to D97 and R162 are histidine and glutamic acid, respectively. Therefore, at the center of the VP16 core lies an extensive network of stabilizing polar interactions.
Figure 5B shows a highly conserved stretch of six amino acids, of which three are charged or polar (residues 85–90; see Fig. 2), located in the amino-terminal region of the VP16 core. These residues pass through the molecule between the carboxy-terminal helix αK (yellow) and the seat bottom (blue), performing an apparently critical function of stabilizing the association of the seat bottom with the surface of the seat. The invariant residue N85 is stabilized by a network of water molecules as well as interactions with the main chain. The following residue, E86, forms hydrogen bonds with two residues in the seat bottom, W263 in loop L-1I and R291 in helix αJ. Although the set of three residues in the R291–E86–W263 interaction (R–E–W) is not present in any of the other VP16 homologs, there is covariation that suggests a conserved interaction: The corresponding positions in VZV are A–E–R, in EHV-1, D–E–R, and in BHV-1, E–E–R. Therefore, in these proteins an arginine is present at the position corresponding to 263 rather than 291, probably still maintaining the ion-pair interaction with the E86 position. GHV-1 (with residues E–V–E) is the exception in which neither E86 nor an arginine are conserved; these residues may form an alternate interaction.
Although most of the conserved residues in VP16 are involved in intramolecular interactions, the most highly conserved segment—the five consecutive invariant residues RYYRE (residues 214–218)—involves residues that are exposed on the surface of the molecule (Fig. 5C). The tyrosine doublet Y215 and Y216 is buried, making main-chain contacts with residues F104 and S106, respectively. R217, however, is solvent exposed and R214 and E218 form an ion pair on the solvent-exposed surface of the helix. Just above the RYYRE segment, there is a discontinuity in helix αG where a water molecule is situated in the kink by hydrogen bonding to the main-chain oxygen atoms of A212 and D213 and to the main-chain nitrogen atoms of Y216 and R217 (Fig. 5C). As described below, we hypothesize that the conserved arginines 214 and 217 are involved in DNA binding; perhaps, the discontinuity in helix αG can be altered to cause a conformational change in the headrest upon interaction with DNA or some other molecule.
DNA-binding specificity determinants of VP16
VP16 can bind DNA on its own (Kristie and Sharp 1990; Stern and Herr 1991) and homologs of VP16 from different herpesviruses can display different DNA-binding specificities. For example, HSV-1 and BHV-1 VP16 proteins recognize different TAATGARAT VP16-response elements (Huang and Herr 1996; Misra et al. 1996). The DNA sequences responsible for this difference in DNA-binding specificity are a 3-bp determinant (D) element that lies 3′ of the core TAATGARAT consensus sequence TAATGAG/AAT. HSV-1 VP16 recognizes the D-element sequence CTT, but not GGC, in the TAATGARAT-element sequence ATGCTAATGAGATTCTT (the D element is underlined), whereas BHV-1 VP16 recognizes the D-element sequence GGC, but not CTT (Huang and Herr 1996). The region of VP16 responsible for D-element recognition is not known. Because, in addition to associating with HCF and Oct-1, the disordered region of the HSV-1 VP16 core interacts with DNA (Shaw et al. 1995; Lai and Herr 1997), we asked whether substituting the corresponding region of BHV-1 VP16 with the disordered region of HSV-1 VP16 would alter the DNA-binding specificity of BHV-1 VP16 (Fig. 6).
Figure 6.

The structured region of the conserved VP16 core is involved in DNA-sequence recognition. (A) Electrophoretic mobility retardation assay comparing the TAATGARAT-element recognition properties of HSV-1 VP16 (H-VP16) lacking the transcriptional activation domain (residues 1–412), full-length BHV-1 VP16 (B-VP16), and a recombinant VP16 protein (B/H-VP16) containing BHV-1 VP16 residues 1–356 and HSV-1 VP16 residues 353–412. All samples contained partially purified HeLa-cell Oct-1 and HCF (the wheat germ agglutinin fraction; Wilson et al. 1993). Odd-numbered lanes contained the TAATGARAT DNA probe H-TAATP; even-numbered lanes contained the TAATGARAT DNA probe B-TAATP described previously (Huang and Herr 1996). (Lanes 1,2) No VP16; (lanes 3,4) H-VP16; (lanes 5,6) B/H-VP16; (lanes 7,8) B-VP16. Only the Oct-1-DNA (Oct-1) and VP16-induced complexes (VIC) are shown. (B) Diagrams of the H-VP16, B-VP16, and recombinant B/H-VP16 proteins. The dark (H-VP16) and light (B-VP16) shaded segments represent the conserved VP16-core sequences from HSV-1 and BHV-1, respectively.
We created a BHV-1/HSV-1 VP16 (B/H-VP16) recombinant carrying BHV-1 VP16 (B-VP16) sequences covering the amino-terminal structured region of VP16 and HSV-1 VP16 (H-VP16) sequences representing the carboxy-terminal disordered region of VP16 (see Fig. 6B). As shown in Figure 6A, we compared the ability of H-VP16, B-VP16, and the recombinant B/H-VP16 proteins to form VP16-induced complexes in an electrophoretic mobility retardation assay on two related TAATGARAT elements. One element, called H-TAAT, contains the HSV-1 VP16-responsive D-element sequence CTT, whereas the other element, called B-TAAT, contains the BHV-1 VP16-responsive D-element sequence GGC. Consistent with previous results (Huang and Herr 1996), H-VP16 recognized the H-TAAT element (Fig. 6A, lane 3), but not the B-TAAT element (Fig. 6A, lane 4), whereas B-VP16 recognized the B-TAAT element (Fig. 6A, lane 8) but not the H-TAAT element (Fig 6A, lane 7). To our surprise, the B/H-VP16 recombinant displayed the same DNA-recognition properties as B-VP16: It recognized the B-TAAT element (Fig. 6A, lane 6) but not the H-TAAT element (Fig. 6A, lane 5). These results indicate that the disordered HCF/Oct-1/DNA-interaction region of native VP16 is not responsible for the difference in DNA-sequence recognition by the HSV-1 and BHV-1 VP16 proteins, and suggest instead that the structured region of the conserved VP16 core is responsible for D-element recognition.
An alternative hypothesis is that the nonconserved amino-terminal region of VP16 (residues 1–48 in HSV-1 VP16), which was retained in the constructs assayed in this experiment, is responsible for this difference in VP16 DNA-binding specificity. We do not favor this hypothesis because this nonconserved region is not required for DNA binding (see Fig. 1B) or D-element recognition (R. Babb and W. Herr, unpubl.) by VP16. We therefore examined the VP16-core structure for a possible DNA-binding surface.
A potential structured VP16 DNA-binding surface
To identify a DNA-binding surface on VP16, we displayed a molecular surface representation of the VP16 core showing the surface charge distribution. Figure 7 shows three views of such a representation of the VP16 core. Acidic (red) and basic (blue) surfaces are not evenly distributed over VP16. For example, there are regions on the right side of the VP16 core that are enriched in acidic residues (Fig. 7C). In contrast, in the left-side view shown in Figure 7A, two prominent basic surfaces can be seen: the convex surface on the back of the headrest consisting of residues R181, R184, R188, and R192, and the concave surface of the seat consisting of residues R214, R217, R221, R224, R331, and R341. The basic region on the concave surface can also be seen in the front view shown in Figure 7B. We suggest that the basic concave surface represents a DNA-binding surface because (1) substitution of arginines 331 and 341 with alanine impairs VP16-induced complex formation by disrupting the ability of VP16 to bind DNA (Lai and Herr 1997), and (2) the solvent exposed residues R214 and R217 (see Fig. 5C) are invariant among the VP16 homologs shown in Figure 2, suggesting that they are involved in a conserved function such as DNA contacts. In contrast, the arginines on the back of the convex headrest are not highly conserved among VP16 homologs (see Fig. 2).
Figure 7.
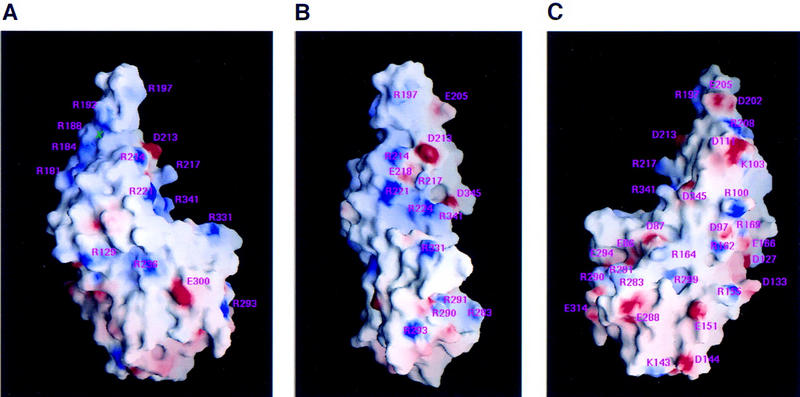
Molecular surface representation (Nicholls et al. 1991) of the electrostatic potential of the VP16 core. The surface charge distribution was computed at neutral pH and displayed at the level of the solvent-accessible surface, assuming a probe radius of 1.4 Å to represent a water molecule, and dielectric constants of 80 and 2 for the solvent and protein, respectively. The surface is colored: blue for positive (20 kBT), red for negative (−20 kBT), and white for neutral, where kB is the Boltzmann’s constant and T is the temperature. Charged side chains are labeled. (A) Left-side view. (B) front view; (C) right-side view. A green cross indicates the location of a phosphate ion.
A hypothetical model of a VP16/Oct-1 DNA complex
Encouraged by the identification of a potential DNA-binding surface on VP16, we attempted to create a model of VP16 bound to a TAATGARAT element with the Oct-1 POU domain. To prepare the model, we used the coordinates for the cocrystal structure of the Oct-1 POU domain bound to an octamer element (Klemm et al. 1994) and extended the DNA with B-helical DNA to include the D element of a complete TAATGARAT element. We then positioned the putative concave DNA-binding surface of VP16 over the 3-bp D element, in an orientation such that the unstructured region of VP16 that interacts with the Oct-1 POUH domain faces the Oct-1 molecule (Fig. 8). In this representation the orientation of VP16 is the same as the front view shown in Figures 3A and 7B.
Figure 8.
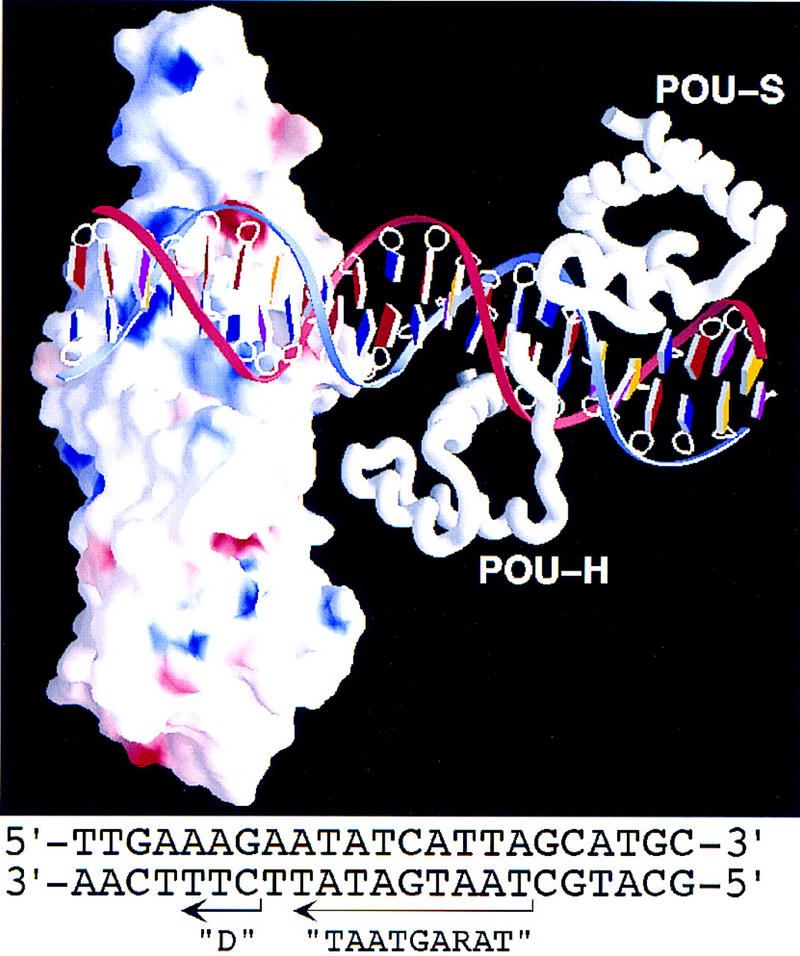
Model of VP16 bound to a TAATGARAT element with the Oct-1 POU domain. The presentation of VP16 is similar to that shown in Fig. 7B. The nucleotide sequence of the DNA in the model is shown at the bottom and originates from the HSV ICP0 gene promoter. In the graphic, the bases are color coded as follows: A (red), T (blue), G (yellow), and C (purple). The blue DNA strand represents the top strand; the red strand represents the bottom strand of the sequence shown below. The Oct-1 POUS (POU-S) and POUH (POU-H) domain structures and the Oct-1 DNA-binding site structure are from Klemm et al. (1994).
Because the unstructured region of VP16 is also involved in binding DNA (Shaw et al. 1995; Lai and Herr 1997), we imagine that on association with Oct-1 the unstructured region adopts a stable conformation that includes contacts with the DNA. The GARAT sequence of the TAATGARAT element lies between the D element VP16-binding site and the TAAT POUH domain-binding site in the model, and therefore the GARAT sequence is an attractive site for interaction with the unstructured region of VP16. We do not know how the other member of the VP16-induced complex, HCF, may bind in the complex. Because the sequences in VP16 bound by HCF lie adjacent to the sequences involved in interaction with the DNA and the Oct-1 POUH domain, we imagine that HCF could bind VP16 near the Oct-1 POUH domain and perhaps stabilize the structure of VP16 that contacts the Oct-1 POUH domain.
The model shown in Figure 8 may explain how the Oct-1 POUS domain can display flexibility in its interaction with DNA in the VP16-induced complex. UV-induced protein-to-DNA crosslinking has shown that, in a VP16-induced complex, the Oct-1 POUS domain can lie to the right of the POUH domain as shown in Figure 8 or on the opposite side of the POUH domain over the GARAT element (Cleary et al. 1997). As can be seen from the center of the model in Figure 8, there is space for the Oct-1 POUS domain to adopt such a configuration in the major groove of the DNA between the Oct-1 POUH domain and VP16.
Conclusions
We have described the crystallographic structure of the conserved region of HSV-1 VP16. This region of VP16 is responsible for formation of the VP16-induced complex with Oct-1, HCF, and DNA. Although the majority of the conserved VP16 core is highly structured with many internal networks of ionic interactions, a carboxy-terminal region that interacts with Oct-1, HCF, and DNA is disordered in the crystal structure. By analogy to the yeast MATα2 protein (Li et al. 1995), we suggest that this region of VP16 becomes structured on VP16-induced complex formation. The results of a mutagenesis study (Ace et al. 1988) suggest that one surface of VP16 is involved in virion assembly and the other in VP16-induced complex formation. Surprisingly, the structured region of the VP16 core is involved in DNA recognition in the VP16-induced complex. Examination of the electrostatic surfaces of the VP16 core suggest that a concave surface—the seat—forms a DNA-binding surface. In this view, VP16 represents a throne on which the DNA sits.
Materials and methods
Overexpression and purification of HSV-1 VP16 core
The glutathione S-transferase (GST)–VP16 fusion expression construct pET11c–GST–VP16ΔC (containing HSV-1 residues 1–412) was used as a template for oligonucleotide-directed mutagenesis to remove the sequences encoding VP16 residues 1–48. The resulting fusion protein, GST–VP1649–412 was purified on a glutathione–agarose column (Smith and Johnson 1988), and the VP1649–412 core moiety bound to the column was cleaved from the GST moiety by thrombin digestion, resulting in the final VP1649–412 core protein and three additional amino-terminal GST fusion related amino acids (Gly–Ser–Arg). The VP1649–412 core protein was separated from thrombin by Q–Sepharose column chromatography and further purified by Mono-S chromatography. Typically, frozen E. coli from 2 liters of culture were thawed and resuspended in 40 ml of lysis buffer (50 mm Tris-HCl at pH 8.0, 100 mm NaCl, 10% glycerol, 2 mm EDTA, 0.1% 2-mercaptoethanol, 2 mm PMSF, and 1 mg/ml of lysozyme), ruptured by two passages through a French press and sonication, and centrifuged to yield a crude supernatant. Nucleic acids were precipitated from the crude extract by the slow addition of one-half volume of a 10 mg/ml stock solution of protamine sulfate and removed by centrifugation. The cleared supernatant was loaded onto a glutathione–agarose column (20 ml bed volume) equilibrated with the lysis buffer. The flowthrough was collected and loaded onto the column again. The column was then washed twice with five bed volumes of buffer A (50 mm Tris-HCl at pH 8.3, 10% glycerol, 2.5 mm Ca2Cl) with first 300 mm NaCl and then 100 mm NaCl. One-bed volume of buffer A with 25 Units of thrombin (Calbiochem) was loaded onto the column and digestion was performed for 1 hr at room temperature. The protein was eluted from the column by addition of a second one-bed volume of buffer A with thrombin and the digestion and collection repeated. Thrombin was removed by chromatography over a 20-ml Q–Sepharose column in which the VP1649–412 core protein passed through the column. Remaining contaminants were removed by chromatography over a Pharmacia Mono-S (10/10) FPLC column. The protein was eluted in buffer A with a linear NaCl gradient; the VP1649–412 core protein eluted at ∼300 mm NaCl.
Crystallization and X-ray data collection
Crystals were observed under the condition of 20% (wt/vol) polyethylene glycol 8000, 50 mm K phosphate (pH 4.0), of a sparse matrix screening (Hampton Research; Jancarik and Kim 1991) at 16 °C in a 6 μl hanging drop containing equal volumes of protein solution (10 mg/ml or higher) and the mother liquor. Rod-like crystals with an average dimension of 0.2 × 0.2 × 1.0 mm3 were only reproduced by using the microseeding technique. Crystals were of the orthorhombic space group P212121, determined based on the systematic absences of reflections along the three axes. There was one molecule per asymmetric unit. The crystals diffracted from 3.5–3 Å to 2.1 Å resolutions (Tables 1 and 2), by X-rays produced by an Elliott GX-21 rotating anode generator and the synchrotron radiation at the National Synchrotron Light Source. The diffraction limits varied from crystal to crystal depending largely on the success of microseeding. The best data collected was 2.1-Å resolution from crystal number 7, which had slightly smaller cell dimensions in b and c axes (Table 2). X-ray data sets collected from crystal number 1 to 5 were measured at approximately 16°C by an R-AXIS II imaging plate detector (λ = 1.54 Å). Data sets collected from crystal number 6 to 8 were measured by a MarResearch imaging plate detector on beam line X12-C under cryogenic condition. Crystals were quenched into liquid nitrogen and then maintained at 95 K in a stream of liquid nitrogen gas. The resulting images were processed using the program HKL (Otwinowski and Minor 1997).
Phasing, model building, and refinement
Two useful heavy atom derivatives, a smaller mercurial compound (HgCl2) and a larger one (p-chloromercuriphenyl sulfonate; pCMBS) were obtained by soaking the crystals in 0.25 mm HgCl2 for ∼8 hr and 3.0 mm pCMBS for ∼1–3 days. The mercury atom had reacted with the sulfur atom of five (C78, C176, C255, C257, and C258) out of six cysteines. The positions of the mercury atoms were first determined from difference Patterson syntheses and confirmed by difference Fourier methods. The multiple isomorphous replacement method was used to generate initial protein phases, using the program package PHASES (Furey and Swaminathan 1997). The resulting initial map at 3 Å was of sufficient quality to place several helices, particularly the long helix αF, in recognizable densities using the molecular modeling program O (Jones et al. 1991). The position of one of the mercury atoms immediately identified the side chain of the conserved C176 in helix αF. The surface loops connecting the secondary structures, however, were undetectable in the Wang’s (1985) solvent flatten MIR map.
A pCMBS-soaked crystal (number 6) was then used for data collected at two distinct wavelengths. These two wavelengths correspond to the peak (maximum Δf′′, λ = 1.01299 Å) and the inflection point (minimum Δf′, λ = 1.015562 Å) of the Hg-containing crystal absorption spectrum. The multiwavelength anomalous diffraction data at the absorption K-edge of mercury were treated as if they were from a multiple isomorphous replacement experiment (Ramakrishnan and Biou 1997). In our case, we combined data from a conventional rotating-anode generator and data from the synchrotron source as well (Xu et al. 1997). With these data we combined up to as many as six different sources of phase information: four isomorphous sets from crystal number 3 to 5, and two anomalous scattering sets from λ1 and λ2 of crystal number 6. By systematically trying various combinations/modifications of phases, with the indications from the positions of mercury atoms, we placed amino acids 48–349 and 395–402 of VP16 into the density. Parallel analysis of selenomethionine VP16 core (49–412) protein was used to confirm the location of methionines (Hendrikson et al. 1990). The resultant model was refined against a higher resolution data set (up to 2.1 Å) collected from crystal number 7 (Table 2) in the resolution range of 7.0–2.1 Å by simulated annealing and least-square minimization using the X-PLOR program suite (Brünger 1992). The pdb coordinates code is 16VP.
Acknowledgments
We thank K. McCloy for initial protein purification and crystallization; J.R. Horton for help with SeMet-containing protein production; R. Babb for unpublished results; N. Hernandez for critically reading the manuscript; and R.M. Sweet for help with X-ray data collection at Brookhaven National Laboratory in the Biology Department single-crystal diffraction facility at beamline X12-C in the National Synchrotron Light Source. This facility is supported by the U.S. Department of Energy, Office of Health and Environmental Research, and by the National Science Foundation. These studies were funded by W.M. Keck Foundation and Georgia Research Alliance to X.C. and U.S. Public Health Service grant CA-13106 from the National Cancer Institute to W.H.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL xcheng@emory.edu; FAX (404) 727-3746.
References
- Ace CI, Dalrymple MA, Ramsay FH, Preston VG, Preston CM. Mutational analysis of the herpes simplex virus type 1 trans-inducing factor, Vmw65. J Gen Virol. 1988;69:2595–2605. doi: 10.1099/0022-1317-69-10-2595. [DOI] [PubMed] [Google Scholar]
- Brünger AT. X-PLOR. A system for X-ray crystallography and NMR, version 3.1. New Haven, CT: Yale University; 1992. [Google Scholar]
- Campbell ME, Palfreman JW, Preston CM. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J Mol Biol. 1984;180:1–19. doi: 10.1016/0022-2836(84)90427-3. [DOI] [PubMed] [Google Scholar]
- Carpenter DE, Misra V. Sequences of the bovine herpesvirus 1 homologue of herpes simplex virus α-trans-inducing factor (UL48) Gene. 1992;119:259–263. doi: 10.1016/0378-1119(92)90280-3. [DOI] [PubMed] [Google Scholar]
- Carson M. Ribbons 2.0. J Appl Crystallogr. 1991;24:958–961. [Google Scholar]
- Cleary MA, Pendergrast PS, Herr W. Structural flexibility in transcription complex formation revealed by protein-DNA photocrosslinking. Proc Natl Acad Sci. 1997;94:8450–8455. doi: 10.1073/pnas.94.16.8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousens DJ, Greaves R, Goding CR, O’Hare P. The C-terminal 79 amino acids of the herpes simplex virus regulatory protein, Vmw65, efficiently activate transcription in yeast and mammalian cells in chimeric DNA-binding proteins. EMBO J. 1989;8:2337–2342. doi: 10.1002/j.1460-2075.1989.tb08361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalrymple MA, McGeoch DJ, Davison AJ, Preston CM. DNA sequence of the herpes simplex virus type 1 gene whose product is responsible for transcriptional activation of immediate early promoters. Nucleic Acids Res. 1985;13:7865–7879. doi: 10.1093/nar/13.21.7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison AJ, Scott JE. The complete DNA sequence of varicella-zoster virus. J Gen Virol. 1986;67:1759–1816. doi: 10.1099/0022-1317-67-9-1759. [DOI] [PubMed] [Google Scholar]
- Donaldson L, Capone JP. Purification and characterization of the carboxyl-terminal transactivation domain of Vmw65 from herpes simplex virus type I. J Biol Chem. 1992;267:1411–1414. [PubMed] [Google Scholar]
- Freiman RN, Herr W. Viral mimicry: Common mode of association with HCF by VP16 and the cellular protein LZIP. Genes & Dev. 1997;11:3122–3127. doi: 10.1101/gad.11.23.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey W, Swaminathan S. PHASES-95: A program package for the processing and analysis of diffraction data from macromolecules. Methods Enzymol. 1997;277:590–620. doi: 10.1016/s0076-6879(97)77033-2. [DOI] [PubMed] [Google Scholar]
- Goto H, Motomura S, Wilson AC, Freiman RN, Nakabeppu Y, Fukushima K, Fujishima M, Herr W, Nishimoto T. A single-point mutation in HCF causes temperature-sensitive cell-cycle arrest and disrupts VP16 function. Genes & Dev. 1997;11:726–737. doi: 10.1101/gad.11.6.726. [DOI] [PubMed] [Google Scholar]
- Greaves RF, O’Hare P. Structural requirements in the herpes simplex virus type 1 transactivator Vmw65 for interaction with the cellular octamer-binding proteins and target TAATGARAT sequences. J Virol. 1990;64:2716–2724. doi: 10.1128/jvi.64.6.2716-2724.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigh A, Greaves RF, O’Hare P. Interference with the assembly of a virus-host transcription complex by peptide competition. Nature. 1990;344:257–259. doi: 10.1038/344257a0. [DOI] [PubMed] [Google Scholar]
- Hayes S, O’Hare P. Mapping of a major surface-exposed site in herpes simplex virus protein Vmw65 to a region of direct interaction in a transcription complex assembly. J Virol. 1993;67:852–862. doi: 10.1128/jvi.67.2.852-862.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson WA, Horton JR, LeMaster DM. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. EMBO J. 1990;9:1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr W, Cleary MA. The POU domain: Versatility in transcriptional regulation by a flexible two-in-one DNA binding domain. Genes & Dev. 1995;9:1679–1693. doi: 10.1101/gad.9.14.1679. [DOI] [PubMed] [Google Scholar]
- Huang CC, Herr W. Differential control of transcription by homologous homeodomain coregulators. Mol Cell Biol. 1996;16:2967–2976. doi: 10.1128/mcb.16.6.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancarik J, Kim SH. Sparse matrix sampling: A screening method for crystallization of proteins. J Appl Crystallogr. 1991;24:409–411. [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Klemm JD, Rould MA, Aurora R, Herr W, Pabo CO. Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA-binding modules. Cell. 1994;77:21–32. doi: 10.1016/0092-8674(94)90231-3. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- Kristie TM, LeBowowitz JH, Sharp PA. The octamer-binding proteins form multi-protein-DNA complexes with the HSV α-TIF regulatory protein. EMBO J. 1989;8:4229–4238. doi: 10.1002/j.1460-2075.1989.tb08608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, Sharp PA. Interactions of the Oct-1 POU subdomains with specific DNA sequences and the HSV α-trans-activator protein. Genes & Dev. 1990;4:2383–2396. doi: 10.1101/gad.4.12b.2383. [DOI] [PubMed] [Google Scholar]
- Kristie TM, Pomerantz JL, Twomey TC, Parent SA, Sharp PA. The cellular C1 factor of the herpes simplex virus enhancer complex is a family of polypeptides. J Biol Chem. 1995;270:4387–4394. doi: 10.1074/jbc.270.9.4387. [DOI] [PubMed] [Google Scholar]
- LaBoissière S, Walker S, O’Hare P. Concerted activity of host cell factor subregions in promoting stable VP16 complex assembly and preventing interference by the acidic activation domain. Mol Cell Biol. 1997;17:7108–7118. doi: 10.1128/mcb.17.12.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J-S, Herr W. Interdigitated residues within a small region of VP16 interact with Oct-1, HCF, and DNA. Mol Cell Biol. 1997;17:3937–3946. doi: 10.1128/mcb.17.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J-S, Cleary MA, Herr W. A single amino acid exchange transfers VP16-induced positive control from the Oct-1 to the Oct-2 homeo domain. Genes & Dev. 1992;6:2058–2065. doi: 10.1101/gad.6.11.2058. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Li T, Stark MR, Johnson AD, Wolberger C. Crystal structure of the MATa1/α2 homeodomain heterodimer bound to DNA. Science. 1995;270:262–269. doi: 10.1126/science.270.5234.262. [DOI] [PubMed] [Google Scholar]
- Lu R, Yang P, Padmakumar S, Misra V. The herpesvirus transactivator VP16 mimics a human basic domain leucine zipper protein, luman, in its interaction with HCF. J Virol. 1998;72:6291–6297. doi: 10.1128/jvi.72.8.6291-6297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra V, Walker S, Yang P, Hayes S, O’Hare P. Conformational alteration of Oct-1 upon DNA binding dictates selectivity in differential interactions with related transcriptional coactivator. Mol Cell Biol. 1996;16:4404–4413. doi: 10.1128/mcb.16.8.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Protein Struct Funct Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- O’Hare P. The virion transactivator of herpes simplex virus. Semin Virol. 1993;4:145–155. [Google Scholar]
- O’Hare PO, Williams G. Structural studies of the acidic transactivation domain of the Vmw65 protein of herpes simplex virus using 1H NMR. Biochemistry. 1992;31:4150–4156. doi: 10.1021/bi00131a035. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz JL, Kristie TM, Sharp PA. Recognition of the surface of a homeo domain protein. Genes & Dev. 1992;6:2047–2057. doi: 10.1101/gad.6.11.2047. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan V, Biou V. Treatment of MAD data as a specific case of MIR. Methods Enzymol. 1997;276:538–557. [PubMed] [Google Scholar]
- Sadowski I, Ma J, Triezenberg SJ, Ptashne M. GAL4-VP16: An unusual potent transcriptional activator. Nature. 1988;335:551–557. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- Shaw P, Knez J, Capone JP. Amino acid substitutions in the herpes simplex virus transactivator VP16 uncouple direct protein-protein interaction and DNA binding from complex assembly and transactivation. J Biol Chem. 1995;270:29030–29037. doi: 10.1074/jbc.270.48.29030. [DOI] [PubMed] [Google Scholar]
- Shen F, Triezenberg SJ, Hensley P, Porter D, Knutson JR. Transcriptional activation domain of the herpesvirus protein VP16 becomes conformationally constrained upon interaction with basal transcription factors. J Biol Chem. 1996;271:4827–4837. doi: 10.1074/jbc.271.9.4827. [DOI] [PubMed] [Google Scholar]
- Simmen KA, Newell A, Robinson M, Mills JS, Canning G, Handa R, Parkes K, Borkakoti N, Jupp R. Protein interactions in the herpes simplex virus type 1 VP16-induced complex: VP16 peptide inhibition and mutational analysis of host cell factor requirements. J Virol. 1997;71:3886–3894. doi: 10.1128/jvi.71.5.3886-3894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smibert CA, Popova B, Xiao P, Capone JA, Smiley JR. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J Virol. 1994;68:2339–2346. doi: 10.1128/jvi.68.4.2339-2346.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DO, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione-S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Stern S, Tanaka M, Herr W. The Oct-1 homeodomain directs formation of a multiprotein-DNA complex with the HSV transactivator VP16. Nature. 1989;341:624–630. doi: 10.1038/341624a0. [DOI] [PubMed] [Google Scholar]
- Stern S, Herr W. The herpes simplex virus trans-activator VP16 recognizes the Oct-1 homeo domain: Evidence for a homeo domain recognition subdomain. Genes & Dev. 1991;5:2555–2566. doi: 10.1101/gad.5.12b.2555. [DOI] [PubMed] [Google Scholar]
- Telford EA, Watson MS, McBride K, Davison AJ. The DNA sequence of equine herpesvirus-1. Virology. 1992;189:304–316. doi: 10.1016/0042-6822(92)90706-u. [DOI] [PubMed] [Google Scholar]
- Thompson CC, McKnight SL. Anatomy of an enhancer. Trends Genet. 1992;8:232–236. [Google Scholar]
- Triezenberg SJ, Kingsbury RG, McKnight SL. Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes & Dev. 1988;2:718–729. doi: 10.1101/gad.2.6.718. [DOI] [PubMed] [Google Scholar]
- Uesugi M, Nyanguile O, Lu H, Levine AJ, Verdine GL. Induced α helix in the VP16 activation domain upon binding to a human TAF. Science. 1997;277:1310–1313. doi: 10.1126/science.277.5330.1310. [DOI] [PubMed] [Google Scholar]
- Walker S, Hayes S, O’Hare P. Site-specific conformational alteration of the Oct-1 POU domain-DNA complex as the basis for differential recognition by Vmw65 (VP16) Cell. 1994;79:841–852. doi: 10.1016/0092-8674(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Wang B-C. Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 1985;115:90–112. doi: 10.1016/0076-6879(85)15009-3. [DOI] [PubMed] [Google Scholar]
- Werstuck G, Capone JP. Mutational analysis of the herpes simplex virus trans-inducing factor Vmw65. Gene. 1989;75:213–224. doi: 10.1016/0378-1119(89)90267-9. [DOI] [PubMed] [Google Scholar]
- Wilson AC, LaMarco K, Peterson MG, Herr W. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell. 1993;74:115–125. doi: 10.1016/0092-8674(93)90299-6. [DOI] [PubMed] [Google Scholar]
- Wilson AC, Freiman RN, Goto H, Nishimoto T, Herr W. VP16 targets an amino-terminal domain of HCF involved in cell cycle progression. Mol Cell Biol. 1997;17:6139–6146. doi: 10.1128/mcb.17.10.6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T-J, Monokian G, Mark D, Wobbe CR. Transcriptional activation by herpes simplex virus type 1 VP16 in vitro and its inhibition by oligopeptide. Mol Cell Biol. 1994;14:3484–3493. doi: 10.1128/mcb.14.5.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R-M, Koch C, Liu Y, Horton JR, Knapp D, Nasmyth K, Cheng X. Crystal structure of the DNA-binding domain of Mbp1, a transcription factor important in cell-cycle control of DNA synthesis. Structure. 1997;5:349–358. doi: 10.1016/s0969-2126(97)00192-5. [DOI] [PubMed] [Google Scholar]
- Yanagida N, Yoshida S, Nazerian K, Lee LF. Nucleotide and predicted amino acid sequence of Marek’s disease virus homologues of herpes simplex virus major tegument proteins. J Gen Virol. 1993;74:1837–1845. doi: 10.1099/0022-1317-74-9-1837. [DOI] [PubMed] [Google Scholar]