

Abstract
The Ah receptor (AhR), a bHLH/PAS transcription factor, mediates dioxin toxicity in the immune system, skin, testis and liver. Toxic phenomena are associated with altered cell proliferation or differentiation, but signaling pathways of AhR in cell cycle regulation are poorly understood. Here we show that AhR induces the p27Kip1 cyclin/cdk inhibitor by altering Kip1 transcription in a direct mode without the need for ongoing protein synthesis or cell proliferation. This is the first example of Kip1 being a direct transcriptional target of a toxic agent that affects cell proliferation. Kip1 causes dioxin-induced suppression of 5L hepatoma cell proliferation because Kip1 antisense-expressing cells are resistant to dioxins. Kip1 is also induced by dioxins in cultures of fetal thymus glands concomitant with inhibition of proliferation and severe reduction of thymocyte recovery. Kip1 expression is likely to mediate these effects as thymic glands of Kip1-deficient mice (Kip1Δ51) are largely, though not completely, resistant.
Keywords: Cell cycle, mRNA induction, dioxins, fetal thymus, cyclin dependent kinase inhibitors, PAS proteins
The Ah receptor (AhR) is a prominent member of the bHLH/PAS transcription factor family characterized by a basic helix–loop–helix (bHLH) DNA-binding domain and a Per/AhR/Arnt/Sim (PAS) homology region for dimerization (Burbach et al. 1992; Ema et al. 1992; Schmidt and Bradfield 1996; Crews 1998). AhR and the hypoxia-inducible factors (HIFs) are the conditionally activated members of the bHLH/PAS family (for review, see Crews 1998). Among the other members, AhRR modulates the activity of AhR (Mimura et al. 1999), Sim (single minded) serves a predominant role in brain midline development, and several other (bHLH/)PAS proteins like Per1, Per2, and Per3, Cycle, BMAL1, and Clock are involved in circadian rhythm regulation (for review, see Dunlap 1998). HIFs allow a cell and the organism to respond specifically to hypoxic conditions. The predominantly studied mode of AhR activation is through binding of nonphysiological compounds, such as heterocyclic food mutagens and aromatic hydrocarbons (Ah). The nonphysiological compounds that have gained the most consideration are the Seveso poison 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and other halogenated aromatic hydrocarbons that are collectively called dioxins and are found ubiquitously generated in the industrialized nations by numerous combustion processes. The AhR in the absence of an activating ligand resides as a complex with chaperoning heat-shock protein 90 (HSP90) in the cytosol. Conditional activation on engagement of a proper ligand such as TCDD involves dissociation of the HSP90 and formation of a nuclear and DNA-binding dimer with the Arnt protein (AhR nuclear translocator) that is another, though nonconditionally regulated, member of the bHLH/PAS transcription factor family. Dimerization occurs through both the amino-terminal bHLH domain as well as a central PAS domain. The AhR/Arnt complex binds to specific DNA-recognition elements, the xenobiotic responsive elements (XREs), which have been mostly mapped in the genes for xenobiotica metabolizing enzymes like cytochromes P450 1A1, 1A2, and 1B1, glutathione S-transferase Ya or UDP–glucuronyltransferase or NADPH/quinone oxidoreductase (for review, see Poland and Knutson 1982; Poellinger et al. 1992; Nebert et al. 1993; Schmidt and Bradfield 1996).
AhR serves a physiological role because targeted inactivation leads to improper proliferation of hepatocyte subsets, liver defects, and delayed population of the peripheral organs of the immune system (for review, see Lahvis and Bradfield 1998). The endogenous mode by which AhR is activated to serve these functions is not known. Also the target gene(s) that have to be regulated by AhR for liver integrity and proper population of lymphatic organs are not known, and it is unlikely that the failure to induce xenobiotica metabolism explains the defects. In addition, inadvertent AhR activation by dioxin poisoning of rodents and humans leads to a wide variety of toxic symptoms, all of which involve changes in cell proliferation or differentiation. They may depend on a uniform initial effect on the cell cycle though tissue specific factors determine the final outcome. Toxic effects are thymus aplasia, reduction in sperm counts, chlorine acne, teratogenicity, and carcinogenicity particularly in the rodent liver. AhR mediates dioxin toxicity (Nebert et al. 1993; Fernandez-Salguero et al. 1996) but none of the symptoms is likely to be a consequence of induced xenobiotica metabolism. Rather, additional AhR dependently regulated genes have to be considered to explain the wide variety of biological consequences of inappropriate gain of AhR activity (Sewall and Lucier 1995). Even the mode of AhR action on these postulated genes cannot be predicted. Detailed studies on a similar family of conditionally regulated transcription factors, for example, the steroid hormone receptors, show that one receptor molecule can serve qualitatively quite distinct functions in activation of gene transcription or cross-talk with other transcription factors (for review, see Göttlicher et al. 1998).
Whereas HIF-1α appears to regulate cell cycle by p53- and p21-dependent pathways (Carmeliet et al. 1998), little is known about AhR target genes that could explain the roles of AhR on cell proliferation. In the present study we searched for an AhR-initiated pathway to the regulation of proliferation using the toxic outcome of inappropriate AhR activation by dioxins as a model. We employed the cell culture system of 5L rat hepatoma cells in which AhR activation severely delays cell cycle progression through G1 (Göttlicher and Wiebel 1991; Weiss et al. 1996). We show that AhR inhibits cell cycle progression in 5L cells by direct induction of the p27Kip1 cyclin/cyclin-dependent kinase (cdk) inhibitor on mRNA and protein levels. Consistent with a primary role of p27Kip1 in the growth arrest response to TCDD, fetal thymus cultures of p27-deficient mice were much less sensitive to TCDD compared to control mice. Resistance was not absolute, consistent with the fact that p27 establishes only one pathway that is partially redundant.
Results
AhR-dependent cell cycle control requires gene transcription
Hyperphosphorylation of the retinoblastoma (Rb) protein characterizes a hallmark in progression through the G1 phase of the cell cycle because Rb is a substrate of the G1 phase cyclin D- and cyclin E-dependent kinase activities and Rb hyperphosphorylation is associated with cells passing the restriction point (Sherr and Roberts 1995; Bartek et al. 1996). The AhR ligand TCDD, much like serum starvation, inhibits G1-phase progression of 5L cells prior to hyperphosphorylation of Rb in asynchronously proliferating cultures (Fig. 1A) or 5L cells synchronously released from a low serum-induced cell cycle arrest (Fig. 1B, cf. lanes 1–3). Two hours of TCDD treatment prior to the expected occurrence of hyperphosphorylated Rb (P-Rb) suffice to substantially reduce Rb phosphorylation. The transcriptional inhibitor actinomycin D is tolerated (lane 4) during the last 2 hr before accumulation of P-Rb and thus could be used to test whether the effect of activated AhR on the cell cycle requires gene transcription. If TCDD was added together with actinomycin D, phosphorylated Rb accumulated at least as efficiently as in untreated cells (cf. lanes 2 and 5), indicating that the effect of dioxin on the cell cycle requires ongoing and presumably induced gene expression. The residual P-Rb in TCDD-treated cells (Fig. 1B, lanes 3 and 5, cf. with Fig. 1A) is most likely due to the short window of treatment, which was minimized to allow the actinomycin D experiment.
Figure 1.



Transcription-dependent delay of cell cycle progression by TCDD. (A) Asynchronously growing 5L cells were starved by serum deprivation or treated with 1 nm TCDD for 48 hr. The degree of Rb phosphorylation was detected by Western blot analysis in which the antibody apparently preferentially recognizes P-Rb. (B) 5L Cells were growth arrested by starvation in serum-free medium for 24 hr and synchronously induced to proliferate by addition of serum. Cell cycle progression was monitored 8 hr later by the phosphorylation status of Rb as in A. Cells were treated 2 hr prior to analysis with 1 nm TCDD, the transcriptional inhibitor actinomycin D (5 μg/ml), or both. (C) Wild-type and mutant AhR (mAhR) were expressed together with GFP in AhR-deficient BP8 cells to reconstitute TCDD-induced cell cycle arrest. A schematic outline of AhR mutations is shown. Proliferation of TCDD-treated cells was determined by flow cytometry after DNA staining with H33258 as the percentage of cells in the G2 and S phases of the cell cycle. Efficiently transfected green fluorescing cells ( ± AhR/mAhR) were compared with the cells that were not efficiently transfected (−AhR) from the same culture dish and FACS analysis. Similar results were obtained in two to six independent experiments.
Further evidence for the need of gene expression was provided by testing AhR mutants for their ability to support TCDD-induced cell cycle delay in a subclone of the 5L cells (BP8AhR−) that lacks AhR mRNA expression (Weiss et al. 1996). Expression vectors for wild-type or mutant AhR were transiently cotransfected together with an expression vector for the green fluorescent protein (GFP) (Heim et al. 1995) to allow identification of efficiently transfected cells by their green fluorescence in a flow cytometric analysis. After transient transfection and culture in the presence of TCDD cell cycle profiles were determined by flow cytometry and analyzed separately for the GFP-expressing cells (+AhR/mAhR) or nonexpressing cells (−AhR). Figure 1C shows that the percentage of TCDD-treated cells in the S and G2 phases is reduced on transfection of wild-type AhR but not of the mutant receptors that either lack their capability to bind to the specific AhR-responsive DNA element (AhRm39, Dong et al. 1996; Gal4-AhR83-805, Weiss et al. 1996) or to transactivate gene expression on DNA binding (AhRΔC). Efficient expression of the receptor mutants was tested in parallel experiments by activation of a Gal4-responsive reporter gene in the case of Gal4–AhR83-805 or dominant-negative effects of the other mutants over wild-type AhR with respect to activation of an XRE-responsive reporter gene. In conclusion the mutational analysis supports the notion that AhR delays cell cycle progression by inducing a specific target gene rather than interfering with pro-mitogenic signaling by other transcription factors or inactivation of the cell cycle machinery by protein–protein interactions.
Induction of the cyclin/cdk inhibitor p27Kip1 by AhR
In a biochemical analysis, the TCDD-induced G1 arrest in 5L cells is characterized by the loss of at least one of the G1-phase-associated cyclin/cdk activities, for example, the cyclin E-dependent histone H1 kinase activity (Fig. 2A). Expression, however, of cyclins as well as the associated cdks (Fig. 2B) required for the G1–S transition in the cell cycle (Lees 1995; Morgan 1995) is not reduced. The apparent induction of cyclins D2 and D3 may be caused by ongoing mitogenic signaling without the cells proceeding to S phase. Inhibition of cyclin/cdk activity without apparent loss of cyclin or cdk proteins together with the finding that AhR apparently has to induce gene expression to inhibit cell cycle progression suggested to us that dioxins induce an inhibitor of cyclin/cdk activity, such as a member of the Ink4 or Cip/Kip protein families that would be able to inhibit both the cyclin D- and cyclin E-dependent kinase activities that contribute to Rb phosphorylation (Sherr and Roberts 1995). By Western blot analysis, p27Kip1 (Polyak et al. 1994; Toyoshima and Hunter 1994) was found at persistently elevated levels after a period of 4–24 hr (Fig. 2C) or even 72 hr (data not shown) of dioxin exposure. This induction is specific because p18Ink and p21Cip1 levels were not changed substantially, and other inhibitors (p57Kip2, p15Ink, p16Ink, p19Ink) could not be detected (data not shown).
Figure 2.







Induction of the p27Kip1 cell cycle inhibitor during TCDD-dependent delay of cell cycle progression. Biochemical properties of G1-phase cyclins and cdks were analyzed from 30%–60% confluent asynchronous 5L cell cultures that, except for those in C and G, had been treated for 24 hr with TCDD or DMSO. Cyclin E-dependent histone H1 kinase activity (A) was measured in an immune complex kinase assay using an anti-cyclin E (lanes 1,2) or a nonspecific antibody (lanes 3,4). Amounts of (B) G1-phase cyclins and cdks, and (C) p27Kip1 were determined by Western blot analysis. (D) Amounts of cdk2 and Kip1 associated with cyclin E were determined by Western blot analysis in immune precipitates similar to those used in A. (E) The amount of free cyclin E not bound in Kip1-containing protein complexes was determined by Western blot analysis of extracts that had been immunodepleted against Kip1 or a matched nonspecific (NS) antigen (N-CAM). (F) Protein levels of p27Kip1 were determined by Western blot analysis of whole-cell extracts from solvent- or TCDD-treated cultures of the AhR-expressing 5L wild-type cells or their AhR-deficient BP8AhR− derivatives. (G) Inducibility of Kip1 protein levels was analyzed in BP8 cells that ectopically expressed AhR (BP8Ahr+). These BP8Ahr+ cells (lanes 3,4) had been generated by transient transfection of expression vectors for AhR and a truncated murine MHC protein. TCDD treatment was from 24 hr to 48 hr after transfection. Subsequently, efficiently transfected cells were isolated for Western blot analysis by magnetic cell sorting directed against the expressed MHC protein. Control cultures (lanes 1,2) received only the MHC selection marker and the empty expression vector.
Increased amounts of p27Kip1 associated with cyclin E after TCDD treatment as shown by immune coprecipitation analysis (Fig. 2D). In the same precipitates we found the expected accumulation of a slower migrating form of cdk2 (Fig. 2D), which presumably lacks phosphorylation at Thr-160 (Polyak et al. 1994). Kip1 in the complex with cyclin E/cdk2 is thought to prevent Thr-160 phosphorylation. The presence of the phosphorylated form of cdk2 in complexes with cyclin E could indicate cyclin E/cdk2 complexes without Kip1. Alternatively, inactive Kip1 complexes with cyclin E/phosphorylated cdk2 could exist such as those found in a previous study (Liu et al. 1997). Cdk2 might be phosphorylated prior to complexation with Kip1. The possibilities were tested by immune depletion directed against Kip1 (Fig. 2E). Already in control cells the αKip antibody precipitated some cyclin E (<30 %). Virtually all cyclin E was depleted after TCDD-treatment of cells by αKip1, but not by an unspecific antibody suggesting that induced Kip1 levels suffice to complex most of the cyclin E/cdk2 complexes. Such complexes are likely to be catalytically inactive irrespective of the state of cdk2 phosphorylation (Sherr and Roberts 1995). A similar analysis addressing Kip1 complexes with D-cyclins was not feasible because even in control cells most of the D-cyclins were found complexed with Kip1, consistent with a role of Kip1 as adaptor or assembly factor for D-cyclins (La Baer et al. 1997).
Induction of p27Kip1 depends on the AhR as it was not found in an AhR-deficient subclone of 5L cells (BP8AhR−, Fig. 2F, lanes 3 and 4). Transient expression of AhR in BP8 cells (Fig. 2G, lanes 3 and 4), but not the transfection of the empty expression vector (lanes 1 and 2), reconstituted TCDD inducibility of the Kip1 protein in a partially purified pool of efficiently transfected cells. Together these data suggest that AhR either directly regulates accumulation of p27Kip1 leading to G1 arrest or arrests cells in G1 allowing p27Kip1 to accumulate. The following experiments focus on clarifying this relationship and the mode of Kip1 induction.
Induction of Kip1 on the mRNA level
Northern blot analysis of 5L cells showed that Kip1 mRNA is induced fourfold by TCDD treatment (Fig. 3A, lanes 1,2). This mRNA induction is direct as it apparently does not involve the induction of intermediary gene products and is observed also in the presence of the translational inhibitor cycloheximide (Fig. 3A, lanes 3,4). The efficiency of cycloheximide treatment is indicated by the lack of p27Kip1 protein accumulation despite the induction of the mRNA (Fig. 3A, third panel). A potential contribution of mRNA stabilization to the observed accumulation of Kip1 mRNA was excluded by following the decay of Kip1 mRNA in control and TCDD-induced 5L cells after inhibition of further transcription by actinomycin D (Fig. 3B,C). Both the basal and the TCDD-induced amounts of Kip1 mRNA are eliminated with similar half-life times of 106 and 112 min, respectively. Hence, a bona fide transcriptional induction appears to prevail. This proposition was confirmed in a nuclear runoff analysis by showing a 2.1 ± 0.4-fold (n = 3) and 5 ± 2-fold (n = 2) increased transcriptional rate after 4 and 24 hr of TCDD treatment, respectively (Fig. 3D).
Figure 3.






Induction of Kip1 mRNA expression in 5L cells by TCDD. (A) Kip1 mRNA levels were determined by Northern blot analysis in 5L cells after 4 hr exposure to TCDD with or without 30 min of pretreatment with the translational inhibitor cycloheximide (50 μg/ml). Cycloheximide treatment was efficient because p27Kip1 protein accumulation was prevented (third panel) whereas levels of a noninducible protein (p38HOG1 kinase) remained unchanged in the same extracts. (B,C) Kip1 mRNA stability in control or TCDD-treated 5L cells was analyzed after induction for 4 hr and disruption of further transcription by actinomycin D (5 μg/ml). The decay of Kip1 mRNA was followed by Northern blot analysis and PhosphorImager quantification in comparison to GAPDH mRNA. mRNA half-life times were determined by regression analysis from the presentation in C. (□) −TCDD; (█) +TCDD. (D) The transcriptional rate of the Kip1 gene was analyzed by a nuclear run-off analysis (Greenberg and Ziff 1984) with nuclei prepared from 5L cells that had been treated for 4 hr or 24 hr with 1 nm TCDD or the DMSO solvent. A representative result (24 hr) is shown. (E) Kip1 promoter reporter gene constructs (Kwon et al. 1996) comprising either 1609, 1382, or 42 bp upstream of the transcriptional start site were stably integrated into 5L cells. Also a Kip1 promoter fragment comprising base pairs −1609 to −1312 in front of a TK81-promoter–luciferase reporter gene (Nordeen 1988) was stably transfected. Inducibility in cell pools (average values of triplicate determinations ± s.d.) was calculated as the ratio of reporter gene activity from cells that had been treated for 24 hr with 1 nm TCDD or the solvent, respectively. (F) Kip1 mRNA expression and inducibility by TCDD were tested in 5L cells that had been serum starved for 24 hr. Cells were treated for additional 4 hr in serum-free medium with 1 nm TCDD or the DMSO solvent prior to Northern blot analysis. Fractions of cells in the S–G2/M segment of the cell cycle were determined from parallel cultures by flow cytometry. (N.D.) Not determined.
Further support for an induction on the level of gene expression was obtained by a reporter gene analysis. A luciferase construct driven by 1609 bp of the upstream regulatory sequences of the murine Kip1 promoter (Kwon et al. 1996) was stably integrated into 5L cells. The reporter gene was found inducible by 2.5-fold after 24 hr of TCDD treatment, whereas control cell pools containing a minimal Kip1 promoter element (42 bp) or a promoter truncated at position −1382 were not inducible (Fig. 3E). A fragment of the Kip1 promoter comprising base pairs −1609 to −1312 was also found twofold inducible by TCDD when cloned into the context of a heterologous promoter (Fig. 3E) indicating that the known Kip1 promoter sequences contain at least one TCDD-responsive element that is located in the −1609 to −1312 fragment. In summary the data identify the AhR as a transcriptional inducer of Kip1 expression that acts directly, presumably in conjunction with other cellular factors, without the need for induced synthesis of intermediary proteins.
Finally, we wanted to exclude the possibility that TCDD arrests 5L cells by a mode independent of Kip1 and that the observed induction of Kip1 expression were the mere consequence of synchronization of cells in G1. Therefore, 5L cells were arrested in the G1 phase by serum starvation and the efficiency of arrest was confirmed by flow cytometric analysis of DNA profiles. The fraction of cells in the S and G2/M phases of the cell cycle was reduced from 29% to 6%. The arrest per se did not suffice to induce Kip1 mRNA (Fig. 3F, lanes 1,3). Moreover, Kip1 mRNA was still inducible in growth-arrested cells that presumably also in the presence of TCDD do not change their status within the cell cycle (Fig. 3F, lanes 3,4). This suggests that the induction of Kip1 mRNA cannot be the consequence of a TCDD-induced arrest in a particular phase of the cell cycle that might be associated with high levels of Kip1 expression. More likely, induced Kip1 expression could cause the inhibition of cell cycle progression by TCDD.
Kip1 is known to be regulated in many cases by post-transcriptional mechanisms (Pagano et al. 1995; Hengst and Reed 1996; Loda et al. 1997). A putative induction of Kip1 protein by TCDD-induced stabilization in addition to the mRNA induction was ruled out by a [35S]methionine pulse-chase experiment. The rate of [35S]methionine incorporation into p27Kip1 was found 2.1-fold induced in the presence of TCDD. The half-life of 35S-labeled Kip1 after addition of nonlabeled methionine was very similar (∼3 hr) in both solvent and TCDD-treated cells (Fig. 4, A and B). The loss of p27Kip1 after cycloheximide treatment suggested a similar protein half-life time of 3 hr irrespective of TCDD treatment (data not shown). Furthermore, Kip1 might be induced by an increased rate of translation of the available mRNA. This was addressed by short time [35S]methionine pulse labeling. In the absence of a transcriptional inhibitor the rate of p27Kip1 synthesis is increased, most likely because of the increased amounts of mRNA (Fig. 4C, left lanes). To prevent an increase in Kip1 mRNA even in the presence of TCDD, the pulse-labeling experiment was performed after pretreatment with the transcriptional inhibitor actinomycin D. Under these conditions, TCDD treatment does not lead to an increased rate of p27Kip1 synthesis. In the most simple case, this argues against a direct effect of TCDD on Kip1 mRNA translation, but it cannot formally be ruled out that TCDD transcriptionally induces an intermediary factor that enhances Kip1 mRNA translation.
Figure 4.



p27Kip1 protein half life and rate of synthesis. (A) Protein half life of p27Kip1 in 5L cells was analyzed by protein pulse labeling with [35S]methionine for 3 hr in the presence of 1 nm TCDD or DMSO solvent followed by a chase for 0, 3, or 6 hr with nonlabeled methionine. The autoradiogram of immune precipitated p27Kip1 is shown (solid arrow) together with a nonspecifically precipitated protein (open arrow). (B) Band intensities in A were quantitated from scanned X-ray films using the calibrated NIH image 1.61 software. (□) −TCDD; (█) +TCDD. (C) The rate of p27Kip1 protein synthesis was determined by [35S]methionine pulse labeling (1 hr) followed by immune precipitation of p27Kip1. Cells had been pretreated for 2 hr 45 min with actinomycin D (5 μg/ml), 2 hr 30 min TCDD (1 nm), both, or the solvent (DMSO) only. One out of two similar experiments, each, are shown.
Causal role of p27Kip1 in the AhR-induced inhibition of proliferation
If Kip1 causes the TCDD-induced inhibition of cell cycle progression and the effects on accumulation of hyperphosphorylated Rb protein (Fig. 1B), it has to be induced in sufficient time, for example, within 2 hr. mRNA and protein are induced as early as 30 min (1.6-fold) and 1 hr (1.5-fold), respectively (Fig. 5). Markedly increased Kip1 levels are found after 2 hr of treatment.
Figure 5.

Time course of Kip1 mRNA and protein induction by TCDD in 5L cells. Expression of Kip1 mRNA and protein in 5L cells after the indicated times of TCDD treatment was determined by Northern blot analysis of poly(A)+ RNA (top) or Western blot analysis of total cell extracts (bottom), respectively.
To provide causal evidence for Kip1 being required for the TCDD-induced delay of cell cycle progression the accumulation of Kip1 was prevented by transient expression of a Kip1 antisense RNA. Coexpression of GFP (Heim et al. 1995) allowed isolation of efficiently transfected cells by fluorescence activated cell sorting (FACS) for the analysis of Kip1 protein expression (Fig. 6A) and incorporation of bromodeoxyuridine (BrdU) into S-phase cells (Fig. 6B). In cells only transfected with GFP and the empty expression vector, TCDD inhibited the proliferation rate by 60% as expected from previous cell cycle analysis (Weiss et al. 1996). After expression of Kip1 antisense RNA no TCDD-induced Kip1 could be detected (Fig. 6A) and TCDD effects on BrdU incorporation were only marginal (Fig. 6B). On microscopic evaluation ∼10% of nontransfected cells were sticking to and copurifying with the sorted fluorescing cells. These cells could well account for the subtle dioxin effects observed in the Kip1-antisense sample. Hence, Kip1 induction is required for the AhR-dependent delay of G1–S phase progression and could sufficiently explain the observed changes in the cell cycle machinery.
Figure 6.


Expression of Kip1 antisense RNA impairs TCDD-induced inhibition of 5L cell proliferation. Proliferation rates in the presence or absence of TCDD were determined in 5L cells that had been transiently cotransfected with expression vectors for a Kip1 antisense RNA and the GFP (anti-Kip1) or the empty expression vector and GFP (control). Green fluorescing cells were collected by FACS and analyzed by either Western blotting for p27Kip1 levels (A) or the percentage of cells that had incorporated BrdU in the presence (solid bars) or absence (open bars) of TCDD (B) (means ± s.d. from three independent experiments in which BrdU staining was evaluated without prior knowledge of sample identity). Significance of differences was tested by Student’s t-test.
The AhR–p27Kip1 pathway in the developing thymus
One symptom of dioxin poisoning in vivo that could be related to alterations in cell proliferation is the atrophy of the thymus. This effect is also found in cultures of fetal thymus glands (FTOC) and involves inhibition of thymocyte proliferation (Lai et al. 1997; Staples et al. 1998). We tested whether TCDD induces Kip1 in the developing thymus and found the Kip1 levels in thymocytes increased by TCDD treatment (Fig. 7A). This coincided with a decrease in thymocyte proliferation as shown by reduced [3H]thymidine incorporation into thymocyte DNA (Fig. 7B) and reduced cell numbers in the mid-S to G2/M phases of the cell cycle (Fig. 7C, 18.9 ± 0.9% vs. 12.7 ± 2.3%). The fraction of cells in S and G2/M was reduced in all thymocyte subpopulations independent of the status of CD4 or CD8 expression. However, effects were more pronounced in the earlier double negative subpopulation. The differential susceptibility of individual subpopulations might also be the reason for the observed slight increase in the representation of an immature CD8+CD4low population (Fig. 7D).
Figure 7.
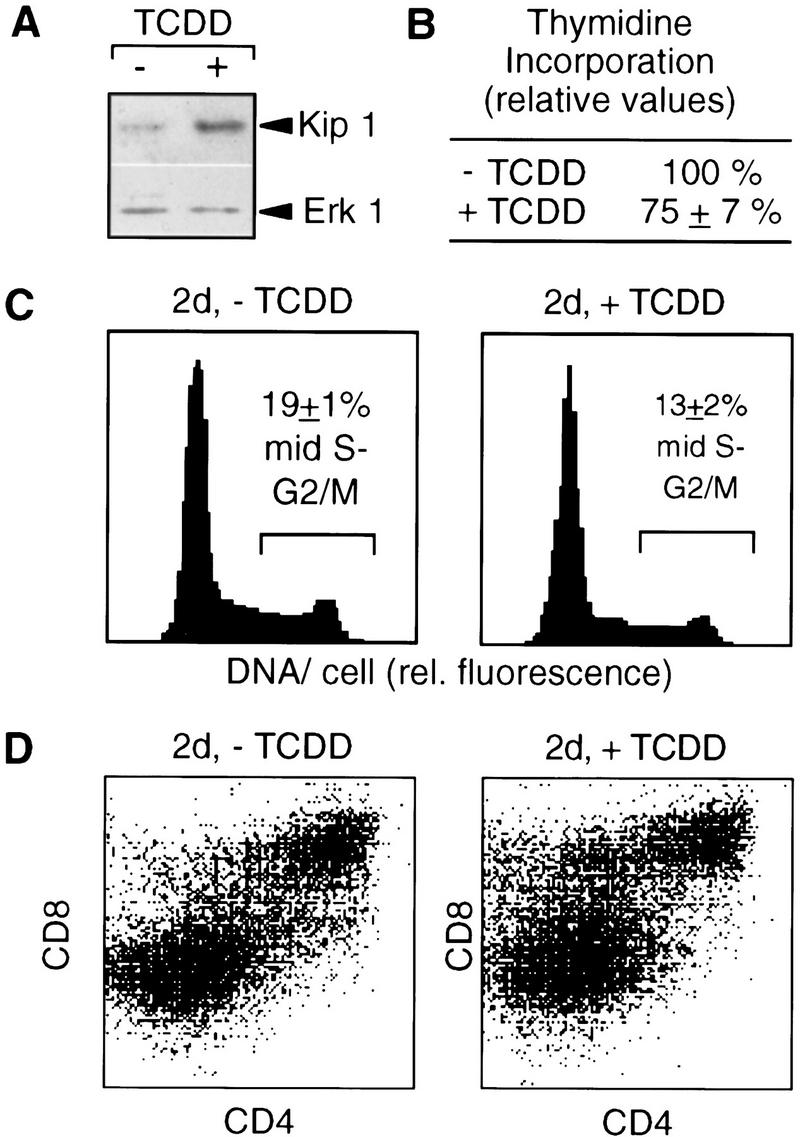
TCDD-induced p27Kip1 protein and cell cycle delay in FTOCs. Fetal thymus glands were cultured for two days in the presence of TCDD or the DMSO solvent before preparation of thymocytes. (A) p27Kip1 protein in thymocytes was determined by Western blot analysis and even loading was confirmed by reprobing with an Erk1 antibody. (B) Incorporation of [3H]thymidine was quantitated per amount of genomic thymocyte DNA after addition of [3H]thymidine for the last 12 hr of culture. (C) The cell cycle distribution of total thymocytes after 2 days of culture was determined by flow cytometry after DNA staining with H33258. (D) Thymocyte subpopulations after 2 days of culture were analyzed by cell surface staining and flow cytometry. One representative out of three experiments is shown; the numbers in B and C are means ± s.d. from three independent experiments (P < 0.01).
Prolonged culture in the presence of TCDD severely reduces the number of recovered thymocytes. Control thymic lobes treated with the solvent for 7 days yielded 8.2 ± 0.6 × 105 thymocytes per lobe, whereas only 9 ± 4 × 104 thymocytes per lobe could be recovered from TCDD-exposed cultures (Table 1). The most mature cells expected, that is, TCRα,β+ (Zuniga-Pflücker and Lenardo 1996), were also found in TCDD-treated cultures suggesting that differentiation per se is not disrupted until this stage (data not shown). The representation of TCRα,β+ cells was for an unknown reason even less efficiently depleted.
Table 1.
TCDD sensitivity of Kipwt/wt FTOCs homozygous (AhRb/b) or heterozygous (AhRb/d) at the AhR gene locus
|
Parameter
|
−TCDD (%)
|
+TCDD (%)a
|
|
|---|---|---|---|
|
AhRb/b
|
AhRb/d
|
||
| Thymocytes in mid-S–G2/M at 2 days of culture | 100 | 68 ± 6 | 66 ± 8 |
| Thymocyte recovery at 7 days | 100 | 11 ± 2 | 11 ± 4 |
Relative values compared to solvent treated cultures, mean ± s.e. (n ≥ 3).
FTOCs from Kip1-deficient mice
To test a causal role of Kip1 in dioxin effects on the fetal thymus cultures were established from wild-type or Kip1 mutant littermate embryos (Fig. 8A). The targeted mutation in the Kip1 gene (Δ51 allele) impairs interaction with the cyclin E/cdk2 complex and hence does not permit inhibition of the kinase activity (Kiyokawa et al. 1996). Embryos were also characterized with respect to AhR expression because a polymorphism for the AhR gene locus has been described and the Kip1-deficient mouse colony still contained some genetic background of the 129/Sv strain from random intercrossing and back-crossing to C57/Bl6 mice. Dominant AhR alleles code for high-affinity receptors (e.g., b1-allele in C57/Bl6 mice) and recessive alleles code for low-affinity receptors (e.g., d allele in 129 mice). Genotyping of the embryos took advantage of the different sizes of the AhR proteins encoded by the b1 allele of C57/Bl6 and the d allele of 129/Sv mice (Fig. 8B, left two lanes). Protein extracts of each embryo used for FTOC were analyzed in a similar way (Fig. 8B, right four lanes). All but one sample showed the presence of an AhR protein encoded by at least one copy of the b1 allele so that most of the embryos could be considered for subsequent analysis. Furthermore, a control experiment confirmed that one copy of the b1 allele confers TCDD sensitivity because FTOCs from heterozygous F1 embryos of a C57/Bl6 × 129/Sv breeding were as sensitive to TCDD as FTOCs from C57/Bl6 mice (Table 1).
Figure 8.
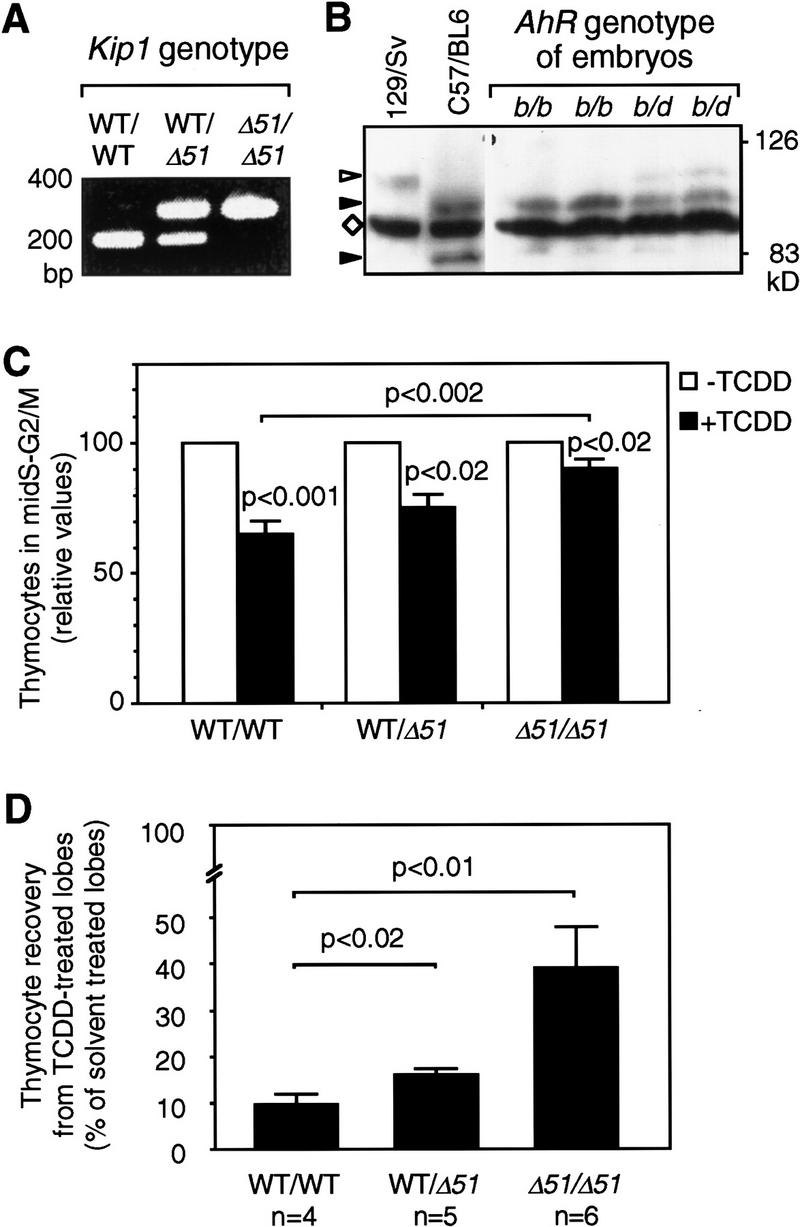
FTOC from wild-type or Kip1 mutant mice. FTOCs were prepared from wild-type (WT), Kip1 mutant (Δ51/Δ51), or heterozygous littermate embryos. A representative PCR-based genotyping for Kip1 is shown in A. Genotyping for AhR alleles (B) was performed by Western blot analysis of the encoded proteins. Liver cytosolic extracts from C57/Bl6 (AhRb1 allele) and 129/Sv (AhRdallele) mice were loaded as reference together with extracts from AhRb/b or AhRb/d embryos. (Solid arrowhead) The size of AhRb1 (95 kD) and a putative degradation product (82 kD) that is specific for the C57Bl6 mouse; (open arrowhead) AhRd; (⋄) a prominent unspecific band. (C) The effect of TCDD on cell cycle distribution depending on the Kip1 genotype in FTOCs from embryos that carry at least one allele coding for a high-affinity AhR (b1 allele). The fraction of cells in mid-S–G2/M in each of the solvent-treated control thymic lobes was set to 100% and the pair-matched TCDD-treated lobe was evaluated relative to that. Data from six litters were grouped according to genotyping of the embryos (Means ± s.e.: nWT = 10; nKip1 WT/Δ51 = 4; nKip1 Δ51/Δ51 = 5). (Solid bars) +TCDD; (open bars) −TCDD. TCDD effects in each group and differences between groups were statistically evaluated by Student’s t-test. (D) Thymocyte recovery after 7 days of FTOCs in the absence or presence of TCDD was determined by hemocytometer counting. For each pair of thymic lobes isolated from an individual embryo the cell number recovered from the solvent (0.1% DMSO) treated lobe was set to 100% and the recovery from the TCDD-treated lobe (1 nm) was calculated as relative value. Data from three litters were grouped according to Kip1 genotyping and confirmation of the presence of at least one high-affinity AhR allele. Values are means ± s.e. (nwt = 4, nhet = 5, nKip1 Δ51/Δ51 = 6) and significance levels were calculated by Student’s t-test.
Kip1-deficient embryos together with wild-type or heterozygous littermate embryos were obtained from heterozygous breeding. Analysis of cell cycle distribution in non-TCDD-treated 2-day-old FTOCs from Kip1 mutant mice showed a slight but only poorly significant increase in the percentage of cells in the mid-S–G2/M segment of the cycle (18.6 ± 2.7% (Kip1 wild type) to 22.2 ± 3.0% (Kip1-deficient), P < 0.05, data not shown). To assess TCDD effects on thymocytes depending on the Kip1 genotype the lobes of each thymus were cultured separately in the absence or presence of TCDD. The fraction of cells in the mid-S–G2/M segment of the cell cycle was determined in each individual lobe as an indicator of the proliferation rate. Values of solvent-treated lobes were set to 100% and matched TCDD-treated lobes were evaluated relative to the control lobe. Data were grouped according to Kip1 genotype after confirmation that at least one copy of the AhR allele coding for the high-affinity AhR had been present (Fig. 8C). TCDD treatment for two days reduces the proliferation rate, that is, percentage of cells in S–G2/M, in FTOCs from Kip1 wild-type embryos by 35% (Fig. 8C, cf. also Fig. 7C). In FTOCs established from Kip1 mutant embryos, the effect of TCDD was severely impaired, for example, the proliferation rate is reduced only by 10% (Fig. 8C). This decrease in TCDD responsiveness of FTOCs from Kip1-deficient embryos is significant compared to wild-type littermates (P < 0.002). The remaining reduction in proliferation rate, though minor, is still significant (P < 0.02). After 7 days of culture, the recovery of thymocytes is reduced to 10% in cultures from wild-type embryos (Fig. 8D). In Kip1-deficient thymic lobes the reduction of thymocyte recovery is still detectable but is much less pronounced as almost 40% of thymocytes are recovered from TCDD, compared with solvent-treated lobes (Fig. 8D).
Discussion
TCDD induces the p27Kip1 cell cycle inhibitor in an AhR-dependent way and this induction occurs by increased expression of the Kip1 mRNA. In contrast to other stimuli, there is no indication that TCDD induces p27Kip1 protein stability or rate of mRNA translation. The induction of Kip1 in 5L cells is, as in other experimental systems, associated with reduced proliferation and the biochemical data could plausibly explain this effect. The evident question is whether increased Kip1 levels inhibit proliferation or whether elevated Kip1 levels are found as a consequence of cell cycle arrest by a putative alternate mechanism. Successful application of Kip1 antisense RNA or antisense oligonucleotides has been reported a few times and here the loss of TCDD sensitivity in Kip1 antisense RNA-expressing 5L cells strongly indicates that Kip1 induction is cause rather than consequence of the TCDD-induced delay of cell cycle progression.
Furthermore, we show for the first time a primary and direct mode of Kip1 mRNA induction that is not secondary to a cell cycle arrest by a putative Kip1-independent mechanism because Kip1 mRNA is well inducible in serum-starved cell cycle-arrested cells. Kip1 mRNA induction by TCDD and the AhR is also direct, that is, resistant to the translational inhibitor cycloheximide, in the sense that no intermediary protein synthesis is required. This still leaves several possibilities to explain how AhR could regulate Kip1 expression.
The reporter gene analysis indicates that a fragment of 1609 bp of murine Kip1 promoter upstream sequence contains at least one of the required cis-regulatory elements although the reporter gene is only twofold inducible as compared to a fivefold inducibility of the endogenous gene. This may depend on the often-observed lower inducibility of randomly integrated reporter genes as compared to the properly arranged endogenous gene but it may also indicate that there are additional TCDD-inducible elements outside the cloned fragment. A potential recognition sequence for an AhR/Arnt heterodimer (CACGCTA) is found 1120 bp upstream of the transcriptional start site, though orientation is opposite to the consensus element (Poellinger et al. 1992; Swanson et al. 1995). AhR could together with Arnt induce Kip1 expression through this site in a similar way as other target genes such as CYP450 1A1 are regulated. This is, however, not likely because overexpression of Arnt in an analysis similar to that shown in Figure 1C enhances as expected the inducibility of a CYP450 1A1-derived reporter gene but diminishes the effect of TCDD on the cell cycle. Overexpression of a dominantly negative-acting DNA-binding deficient mutant of Arnt impairs inducibility of the CYP450 1A1-derived reporter gene but does not affect the TCDD effects on cell cycle progression (data not shown). These findings suggest that induction of Kip1 by the AhR may occur independent of Arnt. Furthermore, deletional analysis of the Kip1 promoter suggests that TCDD induces the Kip1 promoter through a fragment from −1609 to −1312 bp that does not contain a consensus binding site for the AhR/Arnt heterodimer.
Steroid hormone and retinoid receptors that are functionally but not structurally similar to AhR serve as the precedent that one receptor can act in several qualitatively distinct ways. Modes of action involve the binding to the appropriate recognition sequences (Beato et al. 1995; Mangelsdorf and Evans 1995) as homo- or heterodimers. A second type of receptor action depends on interaction with other transcription factors (for review, see Göttlicher et al. 1998) and cellular signaling pathways (Migliaccio et al. 1998). There is evidence for several types of AhR action, for example, by a rapid activation of the Src tyrosine kinase (Enan and Matsumura 1995), and also dimer formation with partner proteins distinct from Arnt could be assumed. The TCDD responsive fragment of the Kip1 promoter (base pairs −1609 to −1312) contains elements at positions −1427, −1350, −1314 and elements in reverse orientation at positions −1562, −1531, −1434, and −1351 that resemble recognition half-sites of AhR, for example, TYGC (Swanson et al. 1995), but differ in the adjacent base pairs that should bind Arnt (GTG). A detailed future analysis of the Kip1 promoter may by the identification of the cis-regulatory element(s) and the trans-acting factor(s) lead to an understanding of the exact mode of AhR action.
Kip1 is the first target gene of the AhR that has been shown to mediate TCDD toxicity. A number of other candidate genes such as the genes for interleukin-1β, tissue plasminogen activator inhibitor 2 (Sutter et al. 1991), down-regulation of the receptors for epidermal growth factor and estrogens as well as induction of the AP-1 proteins c-Jun and c-Fos (for review, see Nebert et al. 1993) had been identified before but a clear causal link to a toxic dioxin action had never been established. Also a direct interaction of AhR with the Rb protein has been proposed (Ge and Elferink 1998) to delay cell cycle progression in 5L cells. However, this interaction should, in contrast to our findings with respect to inhibition of proliferation by TCDD, not depend on the induction of Kip1.
In addition to inhibition of proliferation of cultured cell lines like the 5L cells, certain symptoms of dioxin toxicity in vivo are associated with reduced cell proliferation and could possibly be caused by the induction of p27Kip1. Examples are the antimitotic effects during liver regeneration (Bauman et al. 1995) and in primary hepatocytes (Hushka and Greenlee 1995), a severe delay in body weight gain, and possibly the dioxin-induced wasting syndrome of the adult, as well as reduced sperm count and thymic weight (for review, see Poland and Knutson 1982; Sewall and Lucier 1995). A role of Kip1 in at least some of these processes is likely since they are largely the opposite of the phenotype of Kip1-deficient mice generated by germ-line mutagenesis that results in increased overall body weight and multiple organ hyperplasia including hyperplastic thymus and testis (Fero et al. 1996; Kiyokawa et al. 1996; Nakayama et al. 1996).
The analysis of FTOCs supports the proposition of a major role of Kip1 in dioxin toxicity because Kip1 is induced by TCDD and Kip1-deficient thymic glands are much less sensitive than glands from wild-type embryos. Thymocyte development comprises phases of differentiation, TCR-gene rearrangement, and proliferation. At least one of these transitions, for example, after completion of the TCRβ-chain rearrangement, is associated with a drop in Kip1 expression (Hoffman et al. 1996). TCDD treatment could induce or maintain inappropriately high levels of Kip1 protein that impede proliferation when a rapid expansion of a thymocyte population is required. Even a relatively moderate decrease in the proliferation rate by 35% as that found in our analysis could account for a 10-fold drop in cell number considering the hypothetical and simplified calculation that the number of cell cycles in the observed time period of 7 days drops, for example, from nine to less than six.
Kip1 accounts for a major part of the dioxin effect but a minor activity is still found in Kip1-deficient glands. Theoretically, this residual activity of TCDD would only require the loss of approximately one cell doubling over the observation time. The TCDD effects in Kip1-deficient thymic glands may be mediated by alternate pathways in addition to Kip1 or may even depend on residual activities in the Δ51 mutant Kip1 protein, such as a weak interaction capacity with cdk2 (Kwon and Nordin 1998) or hitherto poorly defined functions in the carboxyl terminus (Uren et al. 1997). It is, however, remarkable that the mutation of Kip1 affects the sensitivity to TCDD at all because other agents that, like serum starvation, high cell density, and treatment with rapamycin or TGFβ, were thought to affect proliferation in a Kip1-dependent manner are mostly functional in Kip1-deficient primary cells (Nakayama et al. 1996; Coats et al. 1999), and there is only one report that supports an important, though not unique, role of Kip1 in response to rapamycin (Luo et al. 1996). Furthermore, it is well accepted in a similar case that p53-dependent induction of the p21Cip1/Waf cyclin/cdk inhibitor mediates cell cycle arrest by DNA damage. Yet, p21-deficient cells are only partially impaired in their capacity to arrest proliferation on DNA damage (Brugarolas et al. 1995; Deng et al. 1995) presumably because of alternate pathways that allow even p21-deficient cells to inhibit proliferation on damage.
Other effects of dioxins like the carcinogenicity and tumor-promoting activity in rodents (Kociba et al. 1978; Pitot et al. 1980) and human (for review, see Huff et al. 1994) are not likely to be explained by the induction of a cell cycle inhibitor. Kip1 rather appears as a tumor suppressor gene considering the spontaneous occurrence of pituitary tumors in Kip1-deficient mice (Fero et al. 1996; Kiyokawa et al. 1996; Nakayama et al. 1996) and the enhanced susceptibility to cancer at many different sites even of Kip1 hemizygous mice (Fero et al. 1998). Furthermore, reduced levels of Kip1 protein have been found associated with poor prognosis cancer in human (Steeg and Abrams 1997). This tumor suppressor activity of Kip1 could be relevant for the anticarcinogenic activity of TCDD found with respect to the development of mammary tumors in rodents (Kociba et al. 1978; Holcomb and Safe 1994). One might even imagine exploiting the inducibility of Kip1 by ligands to the AhR as part of a cytostatic anticancer therapy, particularly if it were feasible to separate different activities of the AhR by appropriate design of the ligand much as it is possible to specifically address different activities of the glucocorticoid receptor by the choice of the ligand (Göttlicher et al. 1998).
In conclusion, we show the first example of directly inducible control of Kip1 mRNA expression by a low-molecular-weight ligand to the AhR. This pathway mediates part of the toxicity of dioxins and one might even envision states of disease where this pathway could be therapeutically exploited by the use of appropriately designed ligands to the AhR.
Materials and methods
Cell culture and biochemical analysis
Cell culture and gene transfer protocols have been described (Weiss et al. 1996). For Western blot analysis normally whole cell extracts were prepared in denaturing SDS sample buffer. Immune complex kinase assays and precipitations were performed as published (Matsushime et al. 1994). Antibodies (from Santa Cruz, if not stated otherwise) were rabbit anti-Rb (C-15), rabbit anti-cyclin D1 (H-295), mouse anti-cyclin D2 (mAb, Neomarkers), rat anti-cyclin D3 (mAb 18B6), rabbit anti-cyclin E (M-20), goat anti-CDK2 (M-2), goat anti-CDK4 (H-22), goat (for immune depletion) or rabbit anti-Kip1 (C-19), rabbit anti-Erk (K-23), and mouse anti-AhR (Affinity Bioreagents). Northern blots were usually performed on 10 μg of total RNA with a rat probe generated by RT-PCR using primers corresponding to nucleotides 28–54 and nucleotides 548–577 of the murine Kip1 cDNA (Polyak et al. 1994). Equal loading of protein gels was confirmed by staining parts of the gels, and RNA loading was controlled by hybridization with a GAPDH probe. For 35S-labeling experiments appropriately pretreated cells were deprived of methionine and serum for 30 min prior to addition of 3.7 MBq of [35S]methionine/ml of medium. Labeled proteins were immunoprecipitated in standard RIPA buffer with a rabbit α-Kip1 (C19) antibody.
Transient transfection studies and flow cytometric analysis
Expression vectors for wild-type and mutant AhR have been described (Weiss et al. 1996). The point mutation R39A was introduced by PCR. The Kip1 antisense RNA expression vector was generated by cloning the PCR fragment used for Northern hybridization into pCMV5 (Weiss et al. 1996) to generate pCMV5–anti-Kip1. pCMV–hGFP contains a XhoI–HindIII open reading frame for GFP (Heim et al. 1995) in pCMV5 prepared by digestion with SalI and HindIII. Cells were transfected by electroporation with 1.5 μg of pCMV–hGFP and 10 μg of AhR expression vectors, pCMV5–anti-Kip1, or the empty expression vector, respectively. For analysis of AhR mutants, cells were treated with 1 nm TCDD or DMSO, harvested 45 hr later using trypsin, fixed in 4% formaldehyde, permeabilized in 0.05% Tween 20, and stained for their DNA content with bisbenzimide H33258. Three color analysis was performed (FACS-Star Plus, Becton Dickinson) using an FITC filter for detection of GFP, a PE filter for compensation of cellular autofluorescence, and UV excitation for analysis of DNA content.
For expression of Kip1 antisense RNA, cells were left untreated for 32 hr prior to exposure to 1 nM of TCDD or DMSO for additional 18 hr of which during the last 5 hr BrdU was present in the culture medium. Cells were harvested using trypsin and green fluorescing cells were sorted to a purity >80% without prior fixation. For Western blot analysis 2 × 104 cells were lysed in sample buffer and comparable loading of lanes within a factor of 2 was confirmed by reprobing the blot against ERK1 and ERK2. Remaining cells were spun onto glass slides for immunocytochemical detection (Kit by Boehinger, Mannheim) of cells having incorporated BrdU.
Enrichment of transfected cells by ferromagnetic beads
Cells were transfected by electroporation with 2.5 μg of pMACSKK (Miltenyi, Bergisch-Gladbach, Germany), 2.5 μg of pCMV–GFP, and 10 μg of pCMV–AhR or pCMV5, respectively. Twenty-four hours later, cells were treated for additional 24 hr with 1 nm TCDD or DMSO. Cells were collected using 0.25% trypsin and 10 μg/ml DNase I. Attachment of anti-KK-coated ferromagnetic beads and enrichment for efficiently transfected cells to a purity of ∼50% was performed on V+MACS-columns as described by the manufacturer.
FTOC
Fetal thymus glands were prepared at day 14.5 of pregnancy and cultured on 3-μm pore grids (Costar). Culture medium was Dulbecco’s modified Eagle medium supplemented with 20% fetal bovine serum, 2 mm glutamine, Dulbecco’s nonessential amino acids, 2-mercaptoethanol, and penicillin/streptomycin with addition of 1 nm TCDD or 0.1% of the DMSO solvent. Thymocytes were prepared after 2 or 7 days of culture by passing through a 70-μm mesh and either lysed for Western blot analysis (105 cells) or stained with fluorochrome-conjugated antibodies against CD4, CD8, or TCRα,β. Fixation and DNA staining for FACS analysis was as described above.
Genotyping of Kip1-deficient embryos used for FTOC
PCR genotyping of the targeted Kip1 gene locus was performed using a common 5′-primer (5′-agcccgagcctggagcggatggacgcc) and two 3′-primers specific either for the wild-type (5′-ctctccacctcctgccattcgtatctgccc) or the mutant allele (5′-ggacatagcgttggctacccgtgatattgctga) in a single-tube PCR reaction of 35 cycles at 94°C, 63°C, and 72°C for 1 min each. Products of 220 and 290 bp were indicative of the wild-type or mutant allele, respectively. Genotyping for the AhR was performed by Western blot analysis of sonicated embryonal head and trunk tissue exploiting the difference in molecular mass of the AhR expressed in C57Bl6 mice (95 kD) and 129/Sv mice (104 kD).
Acknowledgments
We thank Peter Herrlich, Jonathan P. Sleeman, and Timothy J. Soos for critical and constructive discussions, Norma Howells for help with the animal work, Jürgen Moll for introduction to FACS analysis, and Anke Pelzer, Elke Martin, and Margarethe Litfin for excellent technical support. C.A. Bradfield is gratefully acknowledged for providing AhR and Arnt cDNAs. This work was supported by the Deutsche Forschungsgemeinschaft (GO-473/3 and GO-473/5) and a fellowship by the Forschungszentrum Karlsruhe (S.K.K.). A.K. is a Pew Scholar in Biomedical Science and is supported by the Ine T. Hirschl foundation, the Frederek R. Adler chair, and the National Institutes of Health.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL martin.goettlicher@igen.fzk.de; FAX 49-7247-82-3354.
References
- Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway and the restriction point. Curr Opin Cell Biol. 1996;8:805–814. doi: 10.1016/s0955-0674(96)80081-0. [DOI] [PubMed] [Google Scholar]
- Bauman JW, Goldsworthy TL, Dunn CS, Fox TR. Inhibitory effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on rat hepatocyte proliferation induced by 2/3 partial hepatectomy. Cell Prolif. 1995;28:437–451. doi: 10.1111/j.1365-2184.1995.tb00084.x. [DOI] [PubMed] [Google Scholar]
- Beato M, Herrlich P, Schütz G. Steroid hormone receptors: Many actors in search of a plot. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshet E. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- Coats S, Whyte P, Fero ML, Lacy S, Chung G, Randel E, Firpo E, Roberts JM. A new pathway for mitogen-dependent cdk2 regulation uncovered in p27(Kip1)-deficient cells. Curr Biol. 1999;9:163–173. doi: 10.1016/s0960-9822(99)80086-4. [DOI] [PubMed] [Google Scholar]
- Crews ST. Control of cell lineage-specific development and transcription by bHLH-PAS proteins. Genes & Dev. 1998;12:607–620. doi: 10.1101/gad.12.5.607. [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Dong L, Ma Q, Whitlock JP., Jr DNA binding by the heterodimeric Ah receptor. Relationship to dioxin-induced CYP1A1 transcription in vivo. J Biol Chem. 1996;271:7942–7948. doi: 10.1074/jbc.271.14.7942. [DOI] [PubMed] [Google Scholar]
- Dunlap JC. Common threads in eukaryotic circadian systems. Curr Opin Genet Dev. 1998;8:400–406. doi: 10.1016/s0959-437x(98)80109-3. [DOI] [PubMed] [Google Scholar]
- Ema M, Sogawa K, Watanabe N, Chujoh Y, Matsushita N, Gotoh O, Funae Y, Fujii-Kuriyama Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem Biophys Res Commun. 1992;184:246–253. doi: 10.1016/0006-291x(92)91185-s. [DOI] [PubMed] [Google Scholar]
- Enan E, Matsumura F. Evidence for a second pathway in the action mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Significance of Ah-receptor mediated activation of protein kinase under cell-free conditions. Biochem Pharmacol. 1995;49:249–261. doi: 10.1016/s0006-2952(94)00430-7. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge NL, Elferink CJ. A direct interaction between the aryl hydrocarbon receptor and retinoblastoma protein. Linking dioxin signaling to the cell cycle. J Biol Chem. 1998;273:22708–22713. doi: 10.1074/jbc.273.35.22708. [DOI] [PubMed] [Google Scholar]
- Göttlicher M, Wiebel FJ. 2,3,7,8-Tetrachlorodibenzo-p-dioxin causes unbalanced growth in 5L rat hepatoma cells. Toxicol Appl Pharmacol. 1991;111:496–503. doi: 10.1016/0041-008x(91)90253-b. [DOI] [PubMed] [Google Scholar]
- Göttlicher M, Heck S, Herrlich P. Transcriptional cross-talk, the second mode of steroid hormone receptor action. J Mol Med. 1998;76:480–489. doi: 10.1007/s001090050242. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Ziff EB. Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature. 1984;311:433–438. doi: 10.1038/311433a0. [DOI] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271:1861–1864. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- Hoffman ES, Passoni L, Crompton T, Leu TM, Schatz DG, Koff A, Owen MJ, Hayday AC. Productive T-cell receptor β-chain gene rearrangement: Coincident regulation of cell cycle and clonality during development in vivo. Genes & Dev. 1996;10:948–962. doi: 10.1101/gad.10.8.948. [DOI] [PubMed] [Google Scholar]
- Holcomb M, Safe S. Inhibition of 7,12-dimethylbenzanthracene-induced rat mammary tumor growth by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cancer Lett. 1994;82:43–47. doi: 10.1016/0304-3835(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Huff J, Lucier G, Tritscher A. Carcinogenicity of TCDD: Experimental, mechanistic, and epidemiologic evidence. Annu Rev Pharmacol Toxicol. 1994;34:343–372. doi: 10.1146/annurev.pa.34.040194.002015. [DOI] [PubMed] [Google Scholar]
- Hushka DR, Greenlee WF. 2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibits DNA synthesis in rat primary hepatocytes. Mutat Res. 1995;333:89–99. doi: 10.1016/0027-5107(95)00135-2. [DOI] [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- Kociba RJ, Keyes DG, Beyer JE, Carreon RM, Wade CE, Dittenber DA, Kalnins RP, Frauson LE, Park CN, Barnard SD, Hummel RA, Humiston CG. Results of a two-year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Toxicol Appl Pharmacol. 1978;46:279–303. doi: 10.1016/0041-008x(78)90075-3. [DOI] [PubMed] [Google Scholar]
- Kwon TK, Nordin AA. Identification of cdk2 binding sites on the p27Kip1 cyclin-dependent kinase inhibitor. Oncogene. 1998;16:755–762. doi: 10.1038/sj.onc.1201586. [DOI] [PubMed] [Google Scholar]
- Kwon TK, Nagel JE, Buchholz MA, Nordin AA. Characterization of the murine cyclin-dependent kinase inhibitor gene p27Kip1. Gene. 1996;180:113–120. doi: 10.1016/s0378-1119(96)00416-7. [DOI] [PubMed] [Google Scholar]
- La Baer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes & Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Lahvis GP, Bradfield CA. Ahr null alleles: Distinctive or different? Biochem Pharmacol. 1998;56:781–787. doi: 10.1016/s0006-2952(98)00134-8. [DOI] [PubMed] [Google Scholar]
- Lai ZW, Hundeiker C, Gleichmann E, Esser C. Cytokine gene expression during ontogeny in murine thymus on activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol Pharmacol. 1997;52:30–37. doi: 10.1124/mol.52.1.30. [DOI] [PubMed] [Google Scholar]
- Lees E. Cyclin dependent kinase regulation. Curr Opin Cell Biol. 1995;7:773–780. doi: 10.1016/0955-0674(95)80060-3. [DOI] [PubMed] [Google Scholar]
- Liu J, Flanagan WM, Drazba JA, Estes ML, Barnett GH, Haqqi T, Kondo S, Barna BP. The CDK inhibitor, p27Kip1, is required for IL-4 regulation of astrocyte proliferation. J Immunol. 1997;159:812–819. [PubMed] [Google Scholar]
- Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med. 1997;3:231–234. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- Luo Y, Marx SO, Kiyokawa H, Koff A, Massagué J, Marks AR. Rapamycin resistance tied to defective regulation of p27Kip1. Mol Cell Biol. 1996;16:6744–6751. doi: 10.1128/mcb.16.12.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio A, Piccolo D, Castoria G, Di Domenico M, Bilancio A, Lombardi M, Gong W, Beato M, Auricchio F. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 1998;17:2008–2018. doi: 10.1093/emboj/17.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura J, Ema M, Sogawa K, Fujii-Kuriyama Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes & Dev. 1999;13:20–25. doi: 10.1101/gad.13.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Puga A, Vasiliou V. Role of the Ah receptor and the dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal transduction. Ann NY Acad Sci. 1993;685:624–640. doi: 10.1111/j.1749-6632.1993.tb35928.x. [DOI] [PubMed] [Google Scholar]
- Nordeen SK. Luciferase reporter gene vectors for analysis of promoters and enhancers. Biotechniques. 1988;6:454–458. [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Pitot HC, Goldsworthy T, Campbell HA, Poland A. Quantitative evaluation of the promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from diethylnitrosamine. Cancer Res. 1980;40:3616–3620. [PubMed] [Google Scholar]
- Poellinger L, Göttlicher M, Gustafsson JA. The dioxin and peroxisome proliferator-activated receptors: Nuclear receptors in search of endogenous ligands. Trends Pharmacol Sci. 1992;13:241–245. doi: 10.1016/0165-6147(92)90076-i. [DOI] [PubMed] [Google Scholar]
- Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Ann Rev Cell Dev Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- Sewall CH, Lucier GW. Receptor-mediated events and the evaluation of the Environmental Protection Agency (EPA) of dioxin risks. Mutat Res. 1995;333:111–122. doi: 10.1016/0027-5107(95)00137-9. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes & Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Staples JE, Murante FG, Fiore NC, Gasiewicz TA, Silverstone AE. Thymic alterations induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are strictly dependent on aryl hydrocarbon receptor activation in hemopoietic cells. J Immunol. 1998;160:3844–3854. [PubMed] [Google Scholar]
- Steeg PS, Abrams JS. Cancer prognostics: Past, present and p27. Nat Med. 1997;3:152–154. doi: 10.1038/nm0297-152. [DOI] [PubMed] [Google Scholar]
- Sutter TR, Guzman K, Dold KM, Greenlee WF. Targets for dioxin: Genes for plasminogen activator inhibitor-2 and interleukin-1b. Science. 1991;254:415–418. doi: 10.1126/science.1925598. [DOI] [PubMed] [Google Scholar]
- Swanson HI, Chan WK, Bradfield CA. DNA binding specificities and pairing rules of the Ah receptor, ARNT, and SIM proteins. J Biol Chem. 1995;270:26292–26302. doi: 10.1074/jbc.270.44.26292. [DOI] [PubMed] [Google Scholar]
- Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- Uren A, Jakus J, de Mora JF, Yeudall A, Santos E, Gutkind S, Heidaran MA. Carboxyl-terminal domain of p27Kip1 activates CDC2. J Biol Chem. 1997;272:21669–21672. doi: 10.1074/jbc.272.35.21669. [DOI] [PubMed] [Google Scholar]
- Weiss C, Kolluri SK, Kiefer F, Göttlicher M. Complementation of Ah receptor deficiency in hepatoma cells: Negative feedback regulation and cell cycle control by the Ah receptor. Exp Cell Res. 1996;226:154–163. doi: 10.1006/excr.1996.0214. [DOI] [PubMed] [Google Scholar]
- Zuniga-Pflücker JC, Lenardo MJ. Regulation of thymocyte development from immature progenitors. Curr Opin Immunol. 1996;8:215–224. doi: 10.1016/s0952-7915(96)80060-4. [DOI] [PubMed] [Google Scholar]