

Abstract
Gastrulation in mice is associated with the start of extreme proliferation and differentiation. The potential cost to the embryo of a very rapid proliferation rate is a high production of damaged cells. We demonstrate a novel surveillance mechanism for the elimination of cells damaged by ionizing radiation during mouse gastrulation. During this restricted developmental window, the embryo becomes hypersensitive to DNA damage induced by low dose irradiation (<0.5 Gy) and undergoes apoptosis without cell cycle arrest. Intriguingly, embryonic cells, including germ cell progenitors, but not extraembryonic cells, become hypersensitive to genotoxic stress and undergo Atm- and p53-dependent apoptosis. Thus, hypersensitivity to apoptosis in the early mouse embryo is a cell fate-dependent mechanism to ensure genomic integrity during a period of extreme proliferation and differentiation.
Keywords: DNA damage, apoptosis, p53, Atm, irradiation, primordial germ cells, gastrulation, embryo
Early postimplantation development in mammals is associated with a dramatic increase in the proliferation rate of undifferentiated stem cells that form the epiblast and with the start of differentiation within the embryo (Solter et al. 1971; Snow 1977; MacAuley et al. 1993). During this period of rapid proliferation, the cell cycle is shorter than for adult cells, ranging from 3 to 7.5 hr (MacAuley et al. 1993). The embryonic and extraembryonic lineages are separated first. Then, the embryonic cells are separated into three distinct populations. These populations of cells interact with each other to define the primitive pattern and generate the tissue primordials. During development, as during an animal's entire life span, cells are constantly subjected to environmental and metabolic conditions that may cause damage to genomic DNA. If left unrepaired, these modifications cause mutations that could result in loss of viability (Lim and Hasty 1996; Ludwig et al. 1998). Mutations incurred in early embryogenesis could be transmitted to large populations of cells as embryogenesis proceeds. These mutated cells might contribute to teratogenesis and might be incorporated into the germ line. Therefore, to ensure survival, cells are equipped with mechanisms to repair these DNA modifications (for review, see Evan and Littlewood 1998; Wang 1998). In addition, proliferating cells can also delay cell cycle progression to avoid replication or segregation of damaged DNA. Finally, as a safeguard, cells of multicellular organisms have the option of undergoing apoptosis in response to DNA damage.
Which of these survival strategies is used by mammalian cells during early development in response to DNA damage is unknown. Although embryos do show some DNA repair capacity, the extent of DNA damage that embryonic cells can tolerate during early gastrulation is unclear (Lim and Hasty 1996; Ludwig et al. 1998). The level of damage to which embryonic cells might respond is unknown. To understand the response of early embryonic cells to the lowest levels of genotoxic stress, we examined the response of mouse embryos exposed to low doses of X-rays from the preimplantation phase through gastrulation to the beginning of neurulation.
Results
The cell cycle does not arrest after low dose irradiation of early mouse embryos
The most common response of cells to DNA damage is perturbation of progression through the cell cycle. To study the effects of DNA damage on cell proliferation during early gastrulation, a period of extreme proliferation and growth, we injected pregnant females with 5-bromo-2′-deoxyuridine (BrdU), immediately irradiated them with 0.5 Gy of X-rays, and sacrificed them 1 hr later. We chose a dose of X-rays that would produce minimal damage (∼20 double strand breaks per cell; Ward 1988). We calculated a proliferative index on the basis of the ratio of proliferating cells (BrdU-positive nuclei) to total cell number. We analyzed the embryonic and extraembryonic regions in sections of non-irradiated and irradiated embryos (Fig. 1). The number and the percentage of BrdU-labeled cells was approximately the same in the irradiated embryos and in the controls at both embryonic day 6.5 (E6.5) and E7.5 (Fig. 1B). In addition, there was no difference between the number of mitoses observed in control and irradiated embryos in either the embryonic or the extraembryonic regions (data not shown). These results show that DNA damage induced by low dose irradiation does not perturb the cell cycle during early gastrulation.
Figure 1.


Cellular proliferation is not impaired in irradiated embryos. (A) BrdU labeling of E6.5 and E7.5 embryos after low dose irradiation (0.5 Gy). Strongly BrdU-positive nuclei (brown) can be seen throughout the control embryos and irradiated embryos. The boundary between the extraembryonic (ex) and embryonic (e) regions is indicated. (B) Percentage of total number of cells that are BrdU labeled. At E6.5, 70.3 ± 2.9% and 67 ± 1.3% of cells were strongly BrdU positive for the controls (n = 5) and irradiated embryos (n = 4), respectively. At E7.5, 73.25 ± 4.4% and 72.8 ± 5.6% of the nuclei were labeled in the controls (n = 4) and irradiated embryos (n = 7), respectively.
Low dose irradiation induces apoptosis in the embryonic, but not in the extraembryonic, region of the early mouse embryo
Because embryos did not show cell cycle arrest after irradiation, we asked whether low levels of DNA damage induces apoptosis. Remarkably, we found that irradiation of the mouse embryo between E6.5 and E7.5 with 0.5 Gy, a dose that has little effect on somatic cells and later stage embryos, led to a dramatic elevation of apoptosis in the embryonic, but not in the extraembryonic, region. Apoptosis induced by irradiation was confined to the embryonic portion of the egg cylinder exclusively (Fig. 2,3). Less than 5% of the embryonic cells underwent spontaneous apoptosis in control embryos. However, 40.4% ± 4.4% of the embryonic cells underwent apoptosis 6 hr after low dose irradiation. In >200 embryos analyzed, there was no evidence of the induction of apoptosis in the extraembryonic tissue by irradiation at any dose tested (up to 5 Gy) or any time point examined (up to 48 hr post-irradiation; data not shown). In the embryo, the localization of the apoptotic cells was restricted mostly to the embryonic ectoderm (Fig. 2), but a few apoptotic cells were also detected in the embryonic mesoderm (Fig. 2). These results suggest first, that the early embryo has a very low threshold for DNA damage during gastrulation and second, that the developmental fate determines whether a cell activates apoptosis in response to the lowest levels of DNA damage: Only embryonic cells activate apoptosis in response to extremely low doses of X-rays during early gastrulation.
Figure 2.

Time and dose dependence of apoptosis in response to low dose irradiation. (A) E6.5 embryos were exposed in utero to 0.5 Gy and collected at different time points from 1 to 24 hr post-irradiation as indicated. (Top) Embryos probed for apoptotic cells (green); (bottom) embryos stained with Hoechst dye (nuclei detection). (Arrows) Apoptotic embryonic cells. (ac) Amniotic cavity; (am) amnion; (ec) embryonic ectoderm; (exo) exocoelom; (mes) mesoderm, (ve) visceral endoderm. (B) E6.5 embryos were exposed to increasing doses of X-ray from 0 to 1.5 Gy as indicated and collected 6 hr post-irradiation. (Top) Embryos probed for apoptotic cells (green); (bottom) embryos stained with Hoechst dye. (Horizontal white bars) Boundary between the extraembryonic (ex) and embryonic (e) regions.
Figure 3.

Apoptosis in response to low dose irradiation (0.5 Gy) is timed with the onset of gastrulation. Embryos were exposed in utero to 0.5 Gy at different time points during development (from E3.5 to E8.5) and collected 6 hr post-irradiation. For each time point, at least 20 embryos were analyzed. The staining in the center of E8.5 embryo is background. The embryos were subsequently probed for apoptotic cells. (Arrows) Apoptotic cells. (Bottom) Graphic representation of the change in hypersensitivity/apoptosis in response to low dose irradiation during early mouse development.
Induction of apoptosis in response to X-rays was detected by 1 hr post-irradiation with TUNEL analysis (Fig. 2A) and DNA laddering (data not shown). The number of apoptotic cells increased until 6 hr following irradiation and decreased subsequently (Fig. 2A) reaching a basal level 12 to 24 hr post-irradiation. Doses as low as 0.05 Gy induced apoptosis in the early mouse embryo (Fig. 2B). The number of apoptotic cells slightly increased with doses up to 1.5 Gy (Fig. 2B), but did not increase further at higher doses (data not shown).
The apoptotic response to low dose irradiation is regulated in parallel with the onset of gastrulation
Endogenous apoptosis occurs in the mouse embryo starting shortly after implantation and persists throughout the rest of development at a low level (Coucouvanis and Martin 1995; Sanders et al. 1997) indicating that apoptosis plays a role in many developmental processes. We found that induced apoptosis in response to the lowest levels of DNA damage (caused by low dose irradiation) is temporally restricted to the short period of time of gastrulation (E6.5 to E7.5). Whole embryos were irradiated between the preimplantation phase and neurulation and were analyzed by the TUNEL assay. In contrast to earlier studies (Russell and Russell 1954), which were conducted with 2 Gy, our results show that low dose irradiation up to 0.5 Gy during the preimplantation phase did not lead to apoptosis or increased prenatal death (Fig. 3). After implantation, but shortly before gastrulation at E5.5 to E6.0, low dose irradiation also did not increase the number of apoptotic cells (Fig. 3). However, with the onset of gastrulation by E6.5, low dose irradiation of mouse embryos led to a dramatic elevation in the number of apoptotic cells in embryonic, but not in extraembryonic, tissue (Fig. 3). This increased apoptosis in response to the lowest levels of DNA damage persisted through gastrulation (Fig. 3). By E8.5, after gastrulation, low dose irradiation no longer induced apoptosis in the embryo (Fig. 3). These results show that the apoptotic response to low dose irradiation is restricted to the time period corresponding to gastrulation.
Cultured blastocysts acquire the ability to undergo apoptosis in response to low dose irradiation concomitant with gastrulation
Our in vivo analysis indicates that the embryo becomes susceptible to the induction of apoptosis in response to DNA damage by low dose irradiation during early gastrulation. Next, we asked whether the environment in culture, where the rates of proliferation and differentiation are slower, is sufficient to allow differentiation and acquisition of hypersensitivity to low dose irradiation. We compared apoptosis in response to depletion of growth factors such as EGF and in response to low dose irradiation in blastocysts in culture for 2 and 4 days. E3.5 blastocysts that were placed in culture attached after one day and developed differentiated trophoblast giant cells and an egg cylinder that underwent gastrulation with a frequency of about 65% after 3 days (Fig. 4A; Behrendtsen and Werb 1997). Blastocyst outgrowths expressed Brachyury, an early mesodermal/gastrulation marker (Herrmann 1991) after 4 days of outgrowth, when gastrulation was morphologically evident (Fig. 4B). We observed that the initial 2 days of growth and differentiation of the blastocyst in culture required exogenous EGF. The blastocysts cultured in medium without EGF showed a relatively high rate of endogenous apoptosis at 2 days (Fig. 4C). However, by 4 days in culture, basal apoptosis in blastocyst outgrowths became independent of exogenous EGF (Fig. 4C).
Figure 4.
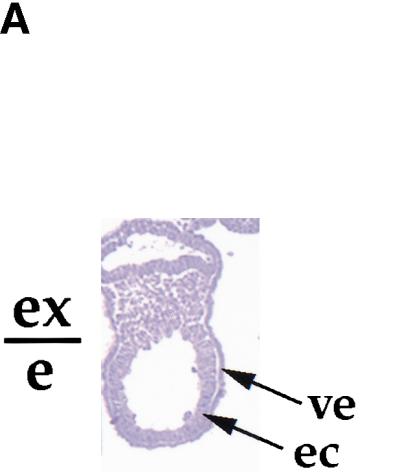



Gastrulating embryos derived from blastocyst outgrowths in culture acquire hypersensitivity to low dose irradiation. (A) Sagittal section of a 4-day outgrowth. The boundary between the extraembryonic (ex) and embryonic (e) regions is indicated. (B) Brachyury expression in blastocyst outgrowths after 2 and 4 days in culture. RT–PCR was performed with Brachyury-specific primers. GAPDH was amplified as a control. (C) Blastocysts were collected at E3.5 and individually cultured with or without EGF. (Top) Phase contrast image; (bottom) apoptotic cells (white). (Arrows) Apoptotic cells. In 2-day outgrowths, apoptosis is induced in the absence of EGF. In 4-day outgrowths, apoptosis is not induced in the absence of EGF. (D) Blastocysts were collected at E3.5 and individually cultured in the presence of EGF; half were irradiated at day 2 or 4 of outgrowth with 0.5 Gy. Irradiation with 0.5 Gy induced apoptosis in 4-day outgrowths but not in 2-day outgrowths. Addition of ZVAD-FMK reduced basal apoptosis in 4-day outgrowths, but did not have an effect on apoptosis induced by X-irradiation. (Open bars) Controls; (black bars) 0.5 Gy irradiation; (gray bars) ZVAD-FMK added. (ec) Embryonic ectoderm; (GC) giant cell; (outg) outgrowth; (ve) visceral endoderm.
Next, we asked whether there are any differences in apoptotic response induced by a deficiency of growth and survival factors as compared with the apoptotic response induced by low dose irradiation. Blastocyst outgrowths were cultured in the presence of EGF to inhibit apoptosis in response to growth factor deficiency during the first phase of outgrowth. After 2 or 4 days of outgrowth, blastocysts were exposed to low dose irradiation (0.5 Gy). In contrast with apoptosis in response to deficiency of growth and survival factors, blastocyst outgrowths after 2 days in culture were resistant to the induction of apoptosis in response to low dose irradiation (Fig. 4D). However, after 4 days of blastocyst outgrowth, when gastrulation was evident, low dose irradiation induced apoptosis significantly (Fig. 4D). The apoptosis induced by low dose irradiation at 4 days of outgrowth could not be inhibited by addition of ZVAD-FMK (Fig. 4D). These data suggest that, as the embryos move to the next developmental stage, they become relatively independent of exogenous EGF, either because they produce sufficient endogenous growth factors or because they no longer require EGF as a survival factor. But, at the same time, embryonic cells acquire the ability to respond to the lowest levels of DNA damage by inducing the apoptotic pathway. The inability to inhibit apoptosis with ZVAD-FMK distinguishes the apoptotic response to low dose irradiation from that of growth factor withdrawal by its lack of independence on small domain caspases.
Irradiation impairs formation of primordial germ cells
A high cost of DNA damage to the embryo would be mutations in the germ line. Thus, we determined whether primordial germ cell (PGC) progenitors respond to low dose irradiation by undergoing apoptosis during early embryogenesis. At E6, PGC progenitors are distributed all around the proximal epiblast. By late E6, they are found closer to the side where the primitive streak has formed (Lawson and Hage 1994). The cells then move through the streak and into the extraembryonic region. Germ cell specification takes place in the extraembryonic region at around E7.2 (for review, see McLaren 1999).
We noted that the distribution of apoptotic cells resembles the distribution of PGC progenitors in the epiblast at E6 to E6.5 (Fig. 3). To determine whether the formation of PGCs is impaired by DNA damage, we irradiated embryos at E6.25 and E7.25 and examined them for the formation of PGCs by staining for the alkaline phosphatase marker in whole embryos on E8 (before turning). The number of stained PGCs in controls and irradiated embryos was determined in whole embryos and on sections. Irradiation between E6.25 and E7.25 with 0.5 Gy reduced the number of PGCs to half in the irradiated embryos (n = 45) in comparison with the control embryos analyzed on E8 (Fig. 5B–D). Thus, the formation of PGCs is also sensitive to low dose irradiation during early gastrulation.
Figure 5.

Irradiation of embryos reduces the number of PGCs. (A) Schematic representation of the localization of PGCs. At E6.5, epiblast cells that become directed to the PGC fate are located in the proximal region of the embryo close to the embryonic/extraembryonic boundary (shaded area). At E8.5, PGCs are located at the base of the allantois around the hindgut pocket (Arrow shows the view for B–D). (B –D). Embryos were stained with alkaline phosphatase (blue) to mark PGCs. (B) Control; (C,D) embryos irradiated with 0.5 Gy at E6.25 or E7.25, respectively, and collected at E8.5. (Arrows) Stained PGCs. (e) Embryonic region; (ex) extraembryonic region; (D) distal; (Pr) proximal.
Next, we asked whether low dose irradiation during early gastrulation affects further development. The irradiation of 51 embryos between E6.25 and E7.25 led to resorption of 36.3% and malformation in 13.7% of the embryos observed at E11.5, and to a 50% reduction in the number of pups born, in comparison with the controls. However, all pups born were healthy and fertile and did not show any abnormalities by 10 months of age. These results suggest that the lowest levels of DNA damage during early gastrulation perturb normal embryonic development in an all-or-nothing fashion.
p53 and Atm are required for embryonic response to low dose irradiation
Next, we sought to determine the molecular mechanisms required for acquisition of this regulated embryonic response to low dose irradiation. p53 is one of the key regulators in the DNA damage pathway to cell cycle arrest and apoptosis. Inactivation of p53 in response to DNA damage attenuates both of these cellular responses (for review, see Ko and Prives 1996; Levine 1997; Giaccia and Kastan 1998; Sherr 1998). p53 null mice are mostly developmentally viable, yet show high-frequency developmental abnormalities and develop tumors very rapidly (Donehower et al. 1992; Jacks et al. 1994; Armstrong et al. 1995). Recent work indicates that p53 is involved in the response of embryonic stem cells to DNA-damaging agents such as UV irradiation and X irradiation of higher doses (>4 Gy; Sabapathy et al. 1997; Aladjem et al. 1998; Corbet et al. 1999). To investigate whether p53 also plays a role in the embryonic response to the lowest levels of damage, we analyzed the effects of low doses of irradiation on wild-type and p53 heterozygous and homozygous null embryos in vivo. We mated p53 heterozygous animals and irradiated the embryos with 0.5 Gy between E6.5 and E7.5 in utero, fixed them 6 hr later, and then analyzed them for apoptotic cells. Whereas irradiation increased apoptosis in wild-type embryos, there was no change in apoptosis in p53 homozygous null embryos after irradiation (Fig. 6). p53 heterozygous null embryos showed an intermediate level of apoptosis in response to low dose irradiation (Fig. 6E). The apoptotic response to DNA damage in the p53 null embryos was absent, not delayed. Apoptosis did not increase at any time point up to 48 hr after irradiation in the p53-null embryos (data not shown). The requirement for p53 was specific. We noted no differences from wild type when we tested E6.5 and E7.5 embryos deficient in two other genes involved in cell cycle regulation and DNA damage response: Embryos deficient in p19ARF (Serrano et al. 1996) or DNA-PKScid/Scid (Bosma and Carroll 1991) had a wild-type apoptotic response to low dose irradiation (data not shown). These data clearly indicate that p53 is involved in the embryonic response to DNA damage during the early gastrulation.
Figure 6.

p53 is a key regulator in the embryonic response to low dose irradiation. (A) E6.5 p53+/+ control; (B) E6.5 p53+/+, 0.5 Gy irradiated; (C) E6.5 p53−/− control; (D) E6.5 p53−/−, 0.5 Gy irradiated. In p53-null mutants the endogenous apoptosis is reduced. In response to low dose irradiation (0.5 Gy), fewer apoptotic cells are seen in the p53-null embryo (D) than in the wild-type littermate (B). (E) The number of apoptotic cells in embryos from p53 heterozygous matings. (Open bars) Controls; (black bars) 0.5 Gy irradiated. The pooled data show that the number of apoptotic cells in response to low dose irradiation is reduced in the absence of p53 (P < 0.001).
Because p53 is involved in the embryonic response to DNA damage, we asked which molecules might function upstream of p53 in this embryonic apoptosis pathway. A candidate gene is Atm, encoding the protein deficient in the human neurodegenerative and cancer predisposition condition ataxia telangiectasia. Atm has been proposed to be a DNA damage sensor (for review, see Jeggo et al. 1998; Wang 1998; Smith and Jackson 1999). The induction of p53 is impaired in cells lacking Atm, indicating that Atm acts upstream of p53 in the signal transduction pathway (Kastan et al. 1992). Interestingly, by RT–PCR, we found that the expression of Atm and p53 was upregulated in both the embryonic and extraembryonic region within 1 hr post-irradiation in wild-type embryos (Fig. 7A). However, only in the embryonic region did this up-regulation of Atm and p53 expression lead to an induction of apoptosis.
Figure 7.


Atm mediates apoptosis in response to low dose irradiation in the early embryo. (A) Atm and p53 expression in embryonic and extraembryonic regions at E7 after low dose irradiation (0.5 Gy). RT–PCR was performed with Atm- and p53-specific primers. (B) p53 expression in Atm heterozygous and homozygous null embryos at E7 after low dose irradiation (0.5 Gy). RT–PCR was performed with p53-specific primers. GAPDH was amplified as control in A and B. (C) E6.5 Atm+/− control. (D) E6.5 Atm+/−, 0.5 Gy irradiated. (E) E6.5 Atm−/− control. (F) E6.5 Atm −/−, 0.5 Gy irradiated. In response to low dose irradiation, fewer apoptotic cells are seen in the Atm -null embryo (F) than in the heterozygous littermate (D).
To determine whether Atm functions in the embryonic apoptosis pathway through early gastrulation, embryos (E6.5 to E7.5) from Atm heterozygous matings were irradiated with 0.5 Gy in utero and analyzed for apoptotic cells (Fig. 7C–F). As expected, apoptosis was increased in wild-type and Atm heterozygous null embryos after irradiation in four litters. However, apoptosis did not increase after low dose irradiation in Atm homozygous null embryos. Interestingly, by RT–PCR, we found that the expression of p53 was impaired in Atm homozygous null embryos after low dose irradiation with 0.5 Gy (Fig. 7B). These data indicate that Atm functions upstream of p53 in the embryonic apoptotic response to low dose irradiation during early gastrulation.
Next, we determined the biological consequences of the lack of Atm- and p53-dependent apoptosis in response to low dose irradiation during gastrulation (E6.5 to E7.5). Embryos of heterozygous matings (3 litters each) were irradiated with 0.5 Gy at E6.5. Only 25 embryos from the p53+/− matings (75% heterozygous and 25% wild-type pups) and 5 embryos from the Atm+/− matings (40% heterozygous and 60% wild type) survived to birth. None of the Atm or p53 homozygous null embryos survived to birth, indicating the importance of the Atm- and p53-dependent safeguard apoptosis pathway to embryonic development.
Discussion
In this study, we have demonstrated that the early mouse embryo has a safeguard mechanism to protect genomic integrity through early gastrulation, a phase of extreme proliferation, growth, and differentiation. We show that, by the onset of gastrulation, the sensitivity threshold of the mouse embryo to DNA damage is extremely low, such hypersensitive response has not been reported previously for any other developmental stage or cell types. We demonstrate further that damaged embryonic cells are eliminated by an Atm- and p53-dependent apoptotic pathway.
The onset of gastrulation is associated with the start of differentiation and a dramatic increase in the rate of proliferation and growth. Through this developmental period, the structure of the cell cycle differs from that of adult cells: The cell cycle is extremely shortened (MacAuley et al. 1993). In general, two major pathways could be activated in response to DNA damage: cell cycle arrest or apoptosis. However, at early gastrulation, the cell cycle is not perturbed in response to low dose irradiation, as shown by the same pattern of BrdU incorporation in control and irradiated embryos. Because of the extremely short cell cycle, cell cycle arrest and accurate repair of DNA damage may not be feasible. Instead, damaged cells are eliminated by apoptosis.
We observed that, with the onset of gastrulation, the embryo regulates the response to induced DNA damage by low dose irradiation by achieving a greater sensitivity to DNA damage. Doses as low as 0.05 Gy (which is equivalent to two double strand breaks per cell; Ward 1988) led to an elevation of apoptosis in the embryo. Interestingly, in our study, irradiation with up to 0.5 Gy during the preimplantation phase did not cause a high incidence of prenatal mortality as has been reported for irradiation with 2 Gy (Russell and Russell 1954). Increased sensitivity of tissues and organs to irradiation correlates with their proliferation and differentiation status (Streffer 1997). Each organ is most sensitive in the period during which differentiation starts. However, the unique feature of embryonic cells at the start of gastrulation is that this increased sensitivity has a much lower threshold and is associated with immediate apoptosis of the damaged cells.
Apoptosis is specific to the embryonic cell lineage
Several lines of evidence suggest that spatial and microenvironmental cues regulate the hyper-responsiveness of embryos to genotoxic stress, such as low dose irradiation. First, irradiation during early gastrulation leads to apoptosis of cells in the embryonic region, but not in the extraembryonic region. Both lineages detect DNA damage and induce expression of Atm and p53 within 1 hr post-irradiation. Our results indicate clearly that developmental fate determines whether the cell undergoes apoptosis in response to DNA damage or not. What are the differences between the embryonic and extraembryonic lineages that lead to a difference in the ability to undergo apoptosis in response to DNA damage? There are a number of genes that have distinct expression patterns in the embryonic versus extraembryonic regions during the peri-gastrulation period, for example, Evx1 and Fgf5 versus Bmp4 and H19, respectively (Haub and Goldfarb 1991; Hébert et al. 1991; Poirier et al. 1991; Dush and Martin 1992; Lawson and Hage 1994). However, whether any of these genes are responsible remains to be determined. The extraembryonic cells contribute to tissues that enable the embryo to survive within the maternal uterus. But these extraembryonic cells are transient and do not contribute to the embryo. Therefore, DNA damage might be tolerated or be repaired in this cell type. In the embryo itself, however, the cells undergo complex morphogenetic movements, proliferation, and differentiation. In addition, all embryonic cells give rise to critical lineages. Therefore, it may be safer for damaged embryonic cells to undergo apoptosis than to delay the cell cycle or repair DNA damage through this complex phase.
Second, cultured embryos also undergo apoptosis in response to low dose irradiation around the time of gastrulation. This observation indicates that development in culture is sufficient for embryonic cells to achieve the ability to undergo apoptosis in response to the lowest levels of genotoxic stress, even though the rates of proliferation and development are significantly slower than in vivo. Thus, it can not be the cell cycle rate, per se, that triggers the hyper-responsiveness. Embryonic stem (ES) cells, which can contribute to all embryonic lineages, and their differentiated progeny (embryoid bodies) do not develop the apoptotic response to low dose irradiation in culture, but can when placed back into embryos (B. Heyer and Z. Werb, unpubl.). These observations indicate that signaling from the early embryonic environment is necessary for embryonic cells to acquire the ability to undergo apoptosis in response to low dose irradiation. It will be interesting to elucidate the cellular and molecular differences between ES cells and early embryonic cells that regulate the difference in hypersensitivity and the ability to undergo apoptosis in response to the lowest doses of irradiation, and how ES cells acquire sensitivity in vivo after implantation into an embryo.
In addition to being a response to DNA damage, apoptosis is also a response to other fundamental decisions during early mouse development (Coucouvanis and Martin 1995). Embryonic cells are also most sensitive to the abundance of growth factors before gastrulation. Our study and others (Manova et al. 1998) have shown that the early streak embryo displays increased cell death in the embryonic region, but not in the extraembryonic region, when the embryos are cultured in media with reduced levels of growth factors. Therefore, growth factor sensitivity may reflect a mechanism for regulating the number of cells in the embryo. Excess cells that do not receive sufficient growth factors are removed by apoptosis. However, the apoptotic response to growth factor withdrawal and DNA damage are regulated independently during early development.
Damaged cells eliminated by apoptosis ensure not only correct cell fate determination, but also genomic integrity. Progenitors of PGCs also undergo apoptosis in response to DNA damage. These cells are located through early gastrulation in the proximal epiblast and migrate from there into the extraembryonic region. Low dose irradiation reduced the number of PGCs. This reduction is most likely due to apoptosis of embryonic epiblast cells that become committed to PGC fate, but are not replaced.
p53 and Atm are key regulators of the embryonic apoptosis pathway
We found that p53 is a key regulator in maintaining genomic integrity during early gastrulation. By activating the embryonic apoptotic response, the early mouse embryo has the ability to activate the p53-dependent apoptosis pathway. Embryonic cells of p53-null embryos do not undergo apoptosis in response to low dose irradiation, indicating that p53 is a key regulator in response to low dose irradiation in the early mouse embryo. We showed that low dose irradiation during early gastrulation led to a complete ablation of surviving p53 homozygous null embryos. In addition, although p53 homozygous null mice do survive to birth, Armstrong et al. (1995) and Jacks et al. (1994) demonstrated a link between p53 deficiency and developmental abnormalities. Our data and theirs demonstrate clearly the importance of p53 in development. Early mouse embryo cells that exhibit the strongest p53 response are rapidly proliferating, similar to p53-dependent cell populations in the adult mouse or older embryo (Gottlieb et al. 1997). In this context, it is interesting that undifferentiated ES cells express high levels of functional p53 in a wild-type conformation; upon in vitro differentiation ES cells show reduced p53 levels and alteration to the mutant conformation (Sabapathy et al. 1997). p53 may be an important regulator in an early proliferation and differentiation process during gastrulation, controlling the balance between differentiating cells and cells undergoing apoptosis. Although, the conformational status of p53 may be the key for the regulation of the sensitivity threshold during early development, the levels of p53 in gastrulating embryos was below the level of detection and lower than in ES cells (data not shown).
Mice carrying null mutations in genes encoding proteins involved in DNA double strand repair (Brca1, Brca2, Rad51) die in the same developmental period in which we observed p53-dependent apoptosis in response to low dose irradiation (Hakem et al. 1996; Lim and Hasty 1996; Ludwig et al. 1997; Suzuki et al. 1997). As in the case of the response to low dose irradiation, the embryonic part of the mutant embryos is much more sensitive to apoptotic death. Genetic analysis demonstrates that their development is shifted to a more advanced stage when p53 is absent, but complete rescue of the mutations does not occur (Lim and Hasty 1996; Ludwig et al. 1997). Because these null mutants die of a lack of proliferation/growth, and the absence of p53 does not rescue their mutant phenotype completely, it is unlikely that Brca1, Brca2, and Rad51 are components of the p53-mediated apoptosis pathway through early gastrulation.
Recently, it has been proposed that DNA-PK is an essential activator of p53 in response to DNA damage (Woo et al. 1998; for review, see Prives 1998), but the evidence for its involvement in this pathway is still controversial (Woo et al. 1998; Jimenez et al. 1999; Smith and Jackson 1999). DNA-PK activation in response to low dose irradiation could induce apoptosis or cell cycle arrest. However, we did not detect any differences between the embryos deficient in DNA-PK and control embryos in the response to low dose irradiation. Therefore, we conclude that DNA-PK is not a component of the p53-mediated apoptosis pathway through early gastrulation.
Atm-deficient mice are viable, but infertile, growth retarded, and hypersensitive to radiation at higher doses (>4 Gy; Barlow et al. 1996; Xu et al. 1996). In contrast, our studies show that Atm-deficient embryos, similar to p53-deficient embryos, do not undergo apoptosis in response to low dose irradiation. This implies that Atm mediates apoptosis in the early mouse embryo in response to low dose irradiation. Because Atm can activate p53 by direct phosphorylation (Banin et al. 1998), it may act upstream of p53. Atm acts as a key regulator in response to DNA damage by activating different responses such as cell cycle checkpoints, DNA repair, and apoptosis. We showed that, in the embryo during early gastrulation, Atm is required for apoptosis, most likely by a p53-mediated mechanism. In addition, we demonstrated that lack of p53- and Atm-dependent apoptosis through gastrulation interferes with survival, demonstrating the importance of this new pathway to embryonic development. Interestingly, Atm and p53 are up-regulated within 1 hr post-irradiation in both the embryonic and extraembryonic regions after low dose irradiation. But this up-regulation leads to apoptosis only in the embryonic region. Atm is also necessary for the apoptotic response to irradiation in specific regions of the developing central nervous system (Herzog et al. 1998). This Atm-mediated apoptosis is specific to particular areas of the central nervous system and is not seen in other regions of the central nervous system or in the thymus, which show normal response to irradiation (Xu et al. 1996; Barlow et al. 1997; Herzog et al. 1998). Taken together with our data, these data suggest that Atm may function in a cell-lineage-specific manner to sense DNA damage and activate the apoptosis pathway in response to irradiation.
We have found that the early mouse embryo has a specific surveillance mechanism to remove damaged cells from the embryonic region and, therefore, prevent such defective cells from contributing to critical lineages later in development. In the highly proliferating cells of the gastrulating embryo, cells undergo Atm- and p53-mediated apoptosis in response to even lowest levels of DNA damage. A major challenge for the future will be to understand what specific genes have to be expressed before the surveillance mechanism can be turned on, why it shuts down by the end of gastrulation, and how molecular differences between the embryonic and the extraembryonic cells are achieved.
Perspective
The Atm- and p53-dependent embryonic apoptosis pathway in response to the lowest levels of DNA damage may be a general feature of early embryogenesis in higher eukaryotes to ensure genomic integrity. Like mouse embryos, early embryos of Drosophila, Caenorhabditis, and Xenopus are sensitive to any DNA damage during early embryogenesis (Bownes and Sunnell 1977; Ishii and Suzuki 1990; Fernandez et al. 1993), and damaged cells may be eliminated by apoptosis. It is possible that the Drosophila Atm homolog Mei-41, which is essential for the midblastula transition during early Drosophila embryogenesis (Sibon et al. 1999), may also play a role in the embryonic apoptosis pathway. These model organisms may prove useful for dissecting the interactions needed for regulating this important pathway.
Materials and methods
Embryo dissection and culture
Wild-type CD-1 females were obtained from Charles River. p53 heterozygous animals (Stock 28899, FVBN/NJrp53tmlTyl) and Atm heterozygous animals (Stock, JR2753, 129/SvEvAtm tml Awb) were obtained from the Jackson Laboratories. Colonies were kept under clean conditions.
Blastocysts were flushed of the uteri of E3.5 pregnant females. Embryos were dissected from pregnant females on the day of interest, fixed overnight in 4% paraformaldehyde, and processed for detection of apoptotic cells. Blastocysts for outgrowth were cultured as described earlier (Behrendtsen and Werb 1997). In brief, blastocysts were cultured overnight in 2× TAAT medium supplemented with 20% fetal calf serum (outgrowth medium) at 37°C in 5% CO2. On the next day, blastocysts were coated for 2 hr with vitronection (20 μg/ml), subsequently plated in individual 5-μl drops of outgrowth medium plus EGF (20 ng/ml), and cultured for 2–4 days. Outgrowths were fixed in acetone/methanol 1:1 and probed for apoptotic cells (Apotag kit, Oncor). Three experiments, with at least 50 embryos per group, were performed.
PCR analysis of p53 and Atm genotypes
Genomic DNA was isolated from neonate tails, embryos, or outgrowths and used for PCR amplification. Tissues were incubated overnight at 55°C in lysis buffer (100 mm Tris at pH 8.5, 5 mm EDTA, 200 mm NaCl, 0.2% SDS) and 100 μg/ml proteinase K. The DNA was phenol extracted and ethanol precipitated. For PCR amplification, 0.2 μg genomic DNA was used. For p53 genotyping, primers p53-3 (5′-AGAGCAAGAATAAGTCAGAAGCCG-3′) and p53-5 (5′-GTCCGCGCCATGGCCATCTA-3′), as well as neo-3 (5′-GCTCTTCAGCAATATCACGG-3′) and neo-5 (5′-GGAGAGGCTATTCGGCTATG-3′) specific for the intact p53 gene and the neomycin resistance cassette, respectively, were used to detect the recombinant allele. Temperature cycling conditions were one initial cycle at 94°C for 5 min, followed by 30 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 45 sec. For Atm genotyping, primers Atm-1 (5′-GCTGCCATACTTGATCCATG-3′) and Atm-2 (5′-TCCGAATTTGCAGGAGTTG-3′), as well as Atm-3 (5′-CTTGGGTGGAGAGGCTATTC-3′) and Atm-4 (5′-AGGTGAGATGACAGGAGATC-3′) specific for the intact Atm gene and the neomycin resistance cassette, respectively, were used to detect the recombinant allele. Temperature cycling conditions were one initial cycle at 94°C for 5 min, followed by 30 cycles of 94°C for 30 sec, 58°C for 30 sec and 72°C for 30 sec. Half of each reaction mixture was subjected to electrophoresis on a 1.5% agarose gel and stained with ethidium bromide.
RNA isolation and RT–PCR
Embryos were irradiated with 0.5 Gy in utero and collected 1 hr post-irradiation. Polyadenylated RNA from pools of 15 embryonic or extraembryonic regions of control and irradiated embryos or 10 outgrowths was prepared by use of the Oligotex direct mRNA kit (Qiagen). For the RT–PCR in Figure 7B, Atm embryos were first genotyped by use of the extraembryonic region, and then polyadenylated RNA from pools of three embryos with the same genotype was prepared with the Oligotex direct mRNA kit (Qiagen). RT reactions were performed for 1 hr at 42°C, 50 mm Tris-HCl (pH 8.3), 75 mm KCl, 3 mm MgCl2, 2 mm dNTPs, 10 mm DTT and 200 units of reverse transcriptase (Superscript, GIBCO). The samples were treated with 1 unit of Rnase H (Boehringer) for 10 min at room temperature and heated at 95°C for 5 min prior to PCR or storage. The PCR was performed as following: One-sixth of the RT-reaction was mixed with PCR-buffer (10 mm Tris-HCl at pH 8.3, 3.45 mm MgCl2, 0.2 mm dNTPs), 1 unit of Taq polymerase (Perkin Elmer Cetus) and 2 pmoles each of the 5′ and 3′ primers. The samples were denatured for 5 min at 94°C before cycling at 94°C for 30 sec, 58°C for 30 sec, and 72°C for 30 sec for 25 to 40 cycles. The primer sets used were GAPDH-1 (5′-TGATGACATCAAGAAGGTGGTGAAG-3′), GAPDH-2 (5′-TTCTTGGAGGCCATGTAGGCCAT-3′), Atm-1 (5′- ATCGCAGAGCGCCTCCATGTC-3′), Atm-2 (5′-GAAGAACATGATCTGTGGGTG-3′), p53-1 (5′-GGATAGCAAAGAGCACAGAGC-3′), p53-2 (5′-CCAGTCTTCGGACAAGCGTGAC-3′), Brachyury-1 (5′-TCCAGGTGCTATATATTGCC-3′), Brachyury-2 (5′-TGCTGCCTGTGAGTCATAAC-3′). At least three experiments with pools of RNA isolated from different embryos/outgrowths were performed.
Detection of BrdU-labeled and apoptotic cells
BrdU (100 μg/gram of body weight) was injected intraperitoneally into pregnant females at E6.5 and E7.5, and the females were sacrificed 1 hr after the injection. The decidua were dissected, fixed in 4% paraformaldehyde, and processed for immunohistochemistry. Serial sagittal sections of each embryo were incubated with an anti-BrdU antibody by use of a kit from Zymed. BrdU-positive cells were counted and the number compared to the total number of cells.
Apoptotic cells were identified by use of the TUNEL method with fluorescence-conjugated or alkaline phosphatase-conjugated antibodies and a kit from Oncor or Boehringer. Cells on sections were counterstained with Hoechst dye. Apoptotic cells were counted on serial sections and the number compared to the total number of cells. For the experiments described in Figures 2 and 3, at least three experiments with a minimum of 20 embryos per time point/dose group were performed.
Detection and count of PGCs
Embryos (45) irradiated at E6.25 or E7.25 and controls were dissected at E8.5 in PBS (before turning, ∼10 somite stage), and fixed in 70% ethanol at least for 2 hr at 4°C. The intact embryos were split in the anterior and posterior region. The posterior regions were washed three times in distilled water. Subsequently, they were stained with α-naphthyl phosphate/fast red (Ginsburg et al. 1990) for 10 min at room temperature. They were rinsed in water and cleared in 50% glycerol. In the stained region from the posterior region, PGCs were identified and counted by use of a stereomicroscope. Stained regions were refixed in 4 % paraformaldehyde and embedded in plastic resin (JB-4 catalyzed resin, Polysciences) according to the manufacturer's protocol. Sections of 6 μm were cut, and the number of PGCs was counted again.
Irradiation
All irradiation procedures were completed using a Phillips RT250 X-ray machine (250-kV peak, 15 mA, half-value layer 1.0 mm copper) at a dose rate of 1 Gy/min. The pregnant females were placed in a rotating plexiglas wheel. The wheel rotated within the irradiation field to assure homogeneity of exposure. If not otherwise specified, embryos and cells were collected 6 hr post-irradiation.
Acknowledgments
We would like to thank Dr. W. Morgan for discussions and critical comments on the manuscript and Dr. D. Beach for the p19 ARF-deficient mice. We are also grateful to Drs. C. Kenyon and D. Toczyski, and our laboratory colleagues for critical reading of the manuscript. This work was supported by a grant of the National Cancer Institute (CA75072). B.S.H. is grateful to the Alexander von Humboldt-Foundation for support (Feodor-Lynen-Program).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Babette@itsa.ucsf.edu; FAX (415) 476-4565.
References
- Aladjem MI, Spike BT, Rodewald LW, Hope TJ, Klemm M, Jaenisch R, Wahl GM. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol. 1998;8:145–155. doi: 10.1016/s0960-9822(98)70061-2. [DOI] [PubMed] [Google Scholar]
- Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995;5:931–936. doi: 10.1016/s0960-9822(95)00183-7. [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Barlow C, Brown KD, Deng CX, Tagle DA, Wynshaw-Boris A. Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat Genet. 1997;17:453–456. doi: 10.1038/ng1297-453. [DOI] [PubMed] [Google Scholar]
- Behrendtsen O, Werb Z. Metalloproteinases regulate parietal endoderm differentiating and migrating in cultured mouse embryos. Dev Dyn. 1997;208:255–265. doi: 10.1002/(SICI)1097-0177(199702)208:2<255::AID-AJA12>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Bosma MJ, Carroll AM. The SCID mouse mutant: Definition, characterization, and potential uses. Annu Re Immunol. 1991;9:323–350. doi: 10.1146/annurev.iy.09.040191.001543. [DOI] [PubMed] [Google Scholar]
- Bownes M, Sunnell LA. Developmental effects of X-irradiation of early Drosophila embryos. J Embryol Exp Morphol. 1977;39:253–259. [PubMed] [Google Scholar]
- Corbet SW, Clarke AR, Gledhill S, Wyllie AH. p53-dependent and -independent links between DNA-damage, apoptosis and mutation frequency in ES cells. Oncogene. 1999;18:1537–1544. doi: 10.1038/sj.onc.1202436. [DOI] [PubMed] [Google Scholar]
- Coucouvanis E, Martin GR. Signals for death and survival: A two-step mechanism for cavitation in the vertebrate embryo. Cell. 1995;83:279–287. doi: 10.1016/0092-8674(95)90169-8. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Dush MK, Martin GR. Analysis of mouse Evx genes: Evx-1 displays graded expression in the primitive streak. Dev Biol. 1992;151:273–287. doi: 10.1016/0012-1606(92)90232-6. [DOI] [PubMed] [Google Scholar]
- Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- Fernandez M, L'Haridon J, Gauthier L, Zoll-Moreux C. Amphibian micronucleus test(s): A simple and reliable method for evaluating in vivo genotoxic effects of freshwater pollutants and radiations. Mutat Res. 1993;292:83–99. doi: 10.1016/0165-1161(93)90010-w. [DOI] [PubMed] [Google Scholar]
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes & Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- Ginsburg M, Snow MH, McLaren A. Primordial germ cells in the mouse embryo during gastrulation. Development. 1990;110:521–528. doi: 10.1242/dev.110.2.521. [DOI] [PubMed] [Google Scholar]
- Gottlieb E, Haffner R, King A, Asher G, Gruss P, Lonai P, Oren M. Transgenic mouse model for studying the transcriptional activity of the p53 protein: Age- and tissue-dependent changes in radiation-induced activation during embryogenesis. EMBO J. 1997;16:1381–1390. doi: 10.1093/emboj/16.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Firpo F, et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- Haub O, Goldfarb M. Expression of the fibroblast growth factor-5 gene in the mouse embryo. Development. 1991;112:397–406. doi: 10.1242/dev.112.2.397. [DOI] [PubMed] [Google Scholar]
- Hébert JM, Boyle M, Martin GR. mRNA localization studies suggest that murine FGF-5 plays a role in gastrulation. Development. 1991;112:407–415. doi: 10.1242/dev.112.2.407. [DOI] [PubMed] [Google Scholar]
- Herrmann BG. Expression pattern of Brachyury gene in whole-mount Twis/Twis mutant embryos. Development. 1991;113:913–917. doi: 10.1242/dev.113.3.913. [DOI] [PubMed] [Google Scholar]
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280:1089–1091. doi: 10.1126/science.280.5366.1089. [DOI] [PubMed] [Google Scholar]
- Ishii N, Suzuki K. X-ray inactivation of Caenorhabditis elegans embryos or larvae. Int J Radiat Biol. 1990;58:827–833. doi: 10.1080/09553009014552201. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Jeggo PA, Carr AM, Lehmann AR. Splitting the ATM: Distinct repair and checkpoint defects in ataxia-telangiectasia. Trends Genet. 1998;14:312–316. doi: 10.1016/s0168-9525(98)01511-x. [DOI] [PubMed] [Google Scholar]
- Jimenez GS, Bryntesson F, Torres-Arzayus MI, Priestley A, Beeche M, Saito S, Sakaguchi K, Appella E, Jeggo PA, Taccioli GE, et al. DNA-dependent protein kinase is not required for the p53-dependent response to DNA damage. Nature. 1999;400:81–83. doi: 10.1038/21913. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Ko LJ, Prives C. p53: Puzzle and paradigm. Genes & Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Lawson K, Hage W. Clonal analysis of the origin of primordial germ cells in the mouse. Ciba Found Symp. 1994;182:68–84. doi: 10.1002/9780470514573.ch5. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A. Targeted mutations of breast cancer susceptibility gene homologs in mice: Lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes & Dev. 1997;11:1226–1241. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, Meneses J, Pedersen RA, Chen DJ. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat Res. 1998;409:17–29. doi: 10.1016/s0921-8777(98)00039-1. [DOI] [PubMed] [Google Scholar]
- MacAuley A, Werb Z, Mirkes PE. Characterization of the unusually rapid cell cycles during rat gastrulation. Development. 1993;117:873–883. doi: 10.1242/dev.117.3.873. [DOI] [PubMed] [Google Scholar]
- Manova K, Tomihara-Newberger C, Wang S, Godelman A, Kalantry S, Witty-Blease K, De Leon V, Chen WS, Lacy E, Bachvarova RF. Apoptosis in mouse embryos: Elevated levels in pregastrulae and in the distal anterior region of gastrulae of normal and mutant mice. Dev Dyn. 1998;213:293–308. doi: 10.1002/(SICI)1097-0177(199811)213:3<293::AID-AJA6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- McLaren A. Signaling for germ cells. Genes & Dev. 1999;13:373–376. doi: 10.1101/gad.13.4.373. [DOI] [PubMed] [Google Scholar]
- Prives C. Signaling to p53: Breaking the MDM2–p53 circuit. Cell. 1998;95:5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- Poirier F, Chan CT, Timmons PM, Robertson EJ, Evans MJ, Rigby PW. The murine H19 gene is activated during embryonic stem cell differentiation in vitro and at the time of implantation in the developing embryo. Development. 1991;113:1105–1114. doi: 10.1242/dev.113.4.1105. [DOI] [PubMed] [Google Scholar]
- Russell L, Russell WL. An analysis of the changing radiation response of the developing mouse embryo. Cell Comp Physiol. 1954;43:103–120. doi: 10.1002/jcp.1030430407. [DOI] [PubMed] [Google Scholar]
- Sabapathy K, Klemm M, Jaenisch R, Wagner EF. Regulation of ES cell differentiation by functional and conformational modulation of p53. EMBO J. 1997;16:6217–6229. doi: 10.1093/emboj/16.20.6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders EJ, Torkkeli PH, French AS. Patterns of cell death during gastrulation in chick and mouse embryos. Anat Embryol. 1997;195:147–154. doi: 10.1007/s004290050033. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes & Dev. 1998;12:2984–2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- Sibon OCM, Laurencon A, Hawley RS, Theurkauf WE. The Drosophila Atm homologue Mei-41 has an essential checkpoint function at midblastula transition. Curr Biol. 1999;9:302–312. doi: 10.1016/s0960-9822(99)80138-9. [DOI] [PubMed] [Google Scholar]
- Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes & Dev. 1999;13:916–934. doi: 10.1101/gad.13.8.916. [DOI] [PubMed] [Google Scholar]
- Snow MHL. Gastrulation in the mouse: Growth and regionalisation of the epiblast. J Embryol Exp Morphol. 1977;42:293–303. [Google Scholar]
- Solter D, Skreb N, Damjanov I. Cell cycle analysis in the mouse egg-cylinder. Exp Cell Res. 1971;64:331–334. doi: 10.1016/0014-4827(71)90084-x. [DOI] [PubMed] [Google Scholar]
- Streffer C. Biological effects of prenatal irradiation. Ciba Found Symp. 1997;203:155–166. [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Hakem R, Elia A, Yoshida R, Mo R, Nishina H, Chuang T, Wakeham A, Itie A, et al. Brca2 is required for embryonic cellular proliferation in the mouse. Genes & Dev. 1997;11:1242–1252. doi: 10.1101/gad.11.10.1242. [DOI] [PubMed] [Google Scholar]
- Wang JY. Cellular responses to DNA damage. Curr Opin Cell Biol. 1998;10:240–247. doi: 10.1016/s0955-0674(98)80146-4. [DOI] [PubMed] [Google Scholar]
- Ward JF. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol. 1988;35:95–125. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- Woo RA, McLure KG, Lees-Miller SP, Rancourt DE, Lee PW. DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature. 1998;394:700–704. doi: 10.1038/29343. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes & Dev. 1996;10:2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]