

Abstract
OBJECTIVES
Some studies have suggested that ursodeoxycholic acid (UDCA) may have a chemopreventive effect on the development of colorectal neoplasia in patients with ulcerative colitis (UC) and primary sclerosing cholangitis (PSC). We examined the effects of high-dose (28–30 mg/kg/day) UDCA on the development of colorectal neoplasia in patients with UC and PSC.
METHODS
Patients with UC and PSC enrolled in a prior, multicenter randomized placebo-controlled trial of high-dose UDCA were evaluated for the development of colorectal neoplasia. Patients with UC and PSC who received UDCA were compared with those who received placebo. We reviewed the pathology and colonoscopy reports for the development of low-grade or high-grade dysplasia or colorectal cancer.
RESULTS
Fifty-six subjects were followed for a total of 235 patient years. Baseline characteristics (including duration of PSC and UC, medications, patient age, family history of colorectal cancer, and smoking status) were similar for both the groups. Patients who received high-dose UDCA had a significantly higher risk of developing colorectal neoplasia (dysplasia and cancer) during the study compared with those who received placebo (hazard ratio: 4.44, 95% confidence interval: 1.30–20.10, P=0.02).
CONCLUSIONS
Long-term use of high-dose UDCA is associated with an increased risk of colorectal neoplasia in patients with UC and PSC.
INTRODUCTION
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease characterized by inflammation and fibrosis of the intrahepatic and extrahepatic bile ducts. Nearly 70% of cases of PSC are associated with inflammatory bowel disease, typically ulcerative colitis (UC). Patients with UC have an increased risk for developing colorectal cancer and dysplasia (1,2). Individuals with concomitant PSC and UC are at a higher risk of developing colorectal cancer than in patients with UC alone (3).
Ursodeoxycholic acid (UDCA) is a synthetic bile acid that is the 7-β-epimer of chenodeoxycholic acid (CDCA). It has been used frequently for therapy in PSC without clear evidence of improved outcomes, though it has been shown to improve liver biochemistries (4–7). Using the Mayo risk score as an end point, a pilot study using 25–30 mg/kg/day of UDCA suggested that such a dose could improve survival (7). This prompted a multicenter, placebo-controlled trial that investigated the use of UDCA 28–30 mg/kg/day in patients with PSC. This study revealed an improvement in liver biochemistries but no improvement in survival and a higher rate in adverse events in the UDCA group. A preliminary analysis reported from this study did not reveal any effect on the development of colonic dysplasia in the initial cohort, including those who did not have UC (4).
Animal and in vitro studies have also suggested that UDCA may have a role as a chemopreventive agent in the prevention of colorectal neoplasia (8–12). Multiple human studies also have investigated the role of UDCA as a chemopreventive agent, and results are conflicting. Two clinical studies suggested that UDCA reduced the incidence of colorectal neoplasia in patients with UC and PSC (13,14); however, a third study indicated that the development of cancer and dysplasia was the same regardless of whether they received UDCA (15). Consequently, it is not clear in clinical practice whether or not patients with PSC and UC should be started on UDCA for the prevention of colonic neoplasia. Because of the limited information, the American Association for the Study of Liver Diseases recommends against the use of UDCA for the chemoprevention of colorectal cancer in patients with PSC and UC (16).
High-dose UDCA has recently been studied as a treatment for PSC (4), and it is not clear whether higher-dose UDCA would have better chemopreventive properties than lower-dose UDCA. This uncertainty prompted us to conduct a retrospective review of a randomized-controlled trial in patients with PSC and UC, to determine the safety and preventive effect of high-dose UDCA on the development of colorectal neoplasia when compared with placebo.
METHODS
We conducted a nested cohort study using data collected during a randomized, double-blind, placebo-controlled multicenter trial of high-dose UDCA (28–30 mg/kg/day). Detailed information on the study design and the main results of this study have been recently published (4). This follow-up study was approved by the Mayo Clinic Institutional Review Board and the initial study was approved by the institutional review boards at each site.
Inclusion criteria
The diagnosis of PSC was based on the following criteria: (i) chronic cholestatic disease for at least 6 months; (ii) serum alkaline phosphatase at least 1.5 times the upper limits of normal; (iii) retrograde, operative, magnetic resonance, or percutaneous cholangiography, revealing intrahepatic and/or extrahepatic biliary duct obstruction, beading, or narrowing within 1 year of study entry; and (iv) liver biopsy within the past year that was available for review and compatible with PSC. The diagnosis of UC was made by the typical clinical, endoscopic, and histologic criteria (17) and only those patients with a diagnosis of UC at the time of study entry were included.
Exclusion criteria
Patients were excluded in the following circumstances: (i) colectomy before study entry; (ii) prior history of colorectal dysplasia or cancer; (iii) comorbidities that would limit life expectancy to <2 years; (iv) inability to give consent; (v) treatment with UDCA, pentoxifylline, corticosteroids, cyclosporin, colchicine, azathioprine, methotrexate, D-penicillamine, budesonide, nicotine, pirfenidone, or tacrolimus in the 3 months before study entry; (vi) if they did not undergo a screening colonoscopy after randomization; (vii) if they had received therapy for UC in the past 3 months other than maintenance therapy with 5-ASA compounds (individuals on non-5-ASA therapies before the trial were not asked to stop such medications, rather they were not enrolled into the study); (viii) end-stage liver disease determined by clinical or laboratory parameters with or without an anticipated need for a liver transplant within the next 2 years; (ix) pregnancy or lactation; (x) age <18 years or >75 years; (xi) prior intraductal stones or biliary operations other than cholecystectomy; (xii) presence of other chronic liver disease; and (xiii) recurrent cholangitis requiring hospitalization more than two times per year.
Randomization and study drug
All eligible patients were randomized by computer to study drug or placebo stratified by histologic stage of PSC, Mayo risk score, and presence of varices. Patients received UDCA 28–30 mg/kg/day (Axcan Pharma, Mont-St Hilare, Canada) or an identical placebo in divided doses given with meals and a bedtime snack. In July 2008, participants were asked to stop taking the study drug or placebo secondary to an increase in adverse events in the UDCA group.
Data abstraction
The data were abstracted on a standardized template. The database from the previous UDCA trial and clinical records, when available, were reviewed to obtain patient demographics, date of randomization and event (neoplasia, liver transplant, colectomy, death), age at diagnosis of PSC, duration of PSC and histologic stage (I–IV) at study entry, duration of UC, severity of UC, colonoscopy and pathology reports, smoking status, family history of colorectal cancer, medication use, presence of pancolitis, and the number of surveillance biopsies per colonoscopy. The study protocol included colonoscopy and surveillance biopsies within 1 year of entry and annually thereafter in individuals with colitis. If colorectal neoplasia was present on the colonoscopy within 1 year of entry or at another time point before the study, they were excluded. During the initial study, if colorectal neoplasia was detected, study personnel entered this into standard abstraction and adverse event forms. If colonoscopies were available at other time points, they were included in our review. For example, the initial prospective high-dose UDCA trial ended in July 2008. Participants were then asked to continue ongoing surveillance per the initial protocol, which included surveillance colonoscopies with biopsies. The number of surveillance biopsies was determined per institutional protocol. Patients who did not undergo a surveillance colonoscopy after randomization were excluded.
Pathology, colonoscopy, and adverse event reports were examined to identify cases of colorectal neoplasia. Cases of colorectal neoplasia were determined by expert gastrointestinal pathologists at each study site who were blinded to the treatment group. Th e standard grading and classification of dysplasia was applied, except lesions interpreted as indefinite for dysplasia were not subdivided into categories of probably positive or negative (18). Indefinite lesions were not included as an end point. All cases were reviewed by two coauthors and adjudicated. All cases of colorectal neoplasia were included in the primary analysis. In the secondary analysis, cases of dysplasia were excluded if they may have occurred in an adenoma-like lesion (19,20).
We also reviewed data regarding serum bile acid composition in these patients, when available, before and after treatment with high-dose UDCA. Serum samples were taken at entry to the study and at the end of treatment with UDCA. We determined the change in lithocholic acid (LCA), deoxycholic acid (DCA), UDCA, CDCA, CA (cholic acid), and total bile acid levels before and after treatment. More details on the changes in bile composition from the original study have been previously published (21).
Statistical analysis
Continuous data were expressed as the mean ± s.d. or medians and ranges. Categorical data were expressed as the number of subjects (and percentage). Differences in continuous variables between the study groups were assessed using two-sided t -tests and non-parametric Wilcoxon tests. Follow-up was calculated from the time of enrollment to the time of an end point (neoplasia) or censoring event (death, liver transplant, or colectomy), whichever was earliest, and if neither occurred then the last colonoscopy with biopsy. Comparison of categorical data between the study groups was performed using κ2 or Fisher's exact method where appropriate. Overall survival free of neoplasia curves were plotted for the groups of interest using the Kaplan-Meier method. Multivariate analysis was performed using a Cox-proportional hazard regression, with P≤0.10 in univariate analysis required for entry into the model. All tests were two-sided, and the chosen level of significance was P < 0.05. Analysis was performed using JMP software (version 8.0; SAS Institute, Cary, NC).
RESULTS
We identified 91 patients with PSC and concomitant UC at the time of enrollment into the original study. For the purposes of this study, 35 patients were excluded: 26 had a colectomy or prior history of colorectal neoplasia before enrollment and 9 patients did not undergo a colonoscopy after randomization. Therefore, 65 patients could have been eligible but 9 (14%) were excluded due to the lack of a colonoscopy. Of the 56 subjects included in our study, 25 were randomized to UDCA and 31 to placebo. Of the eight cases of dysplasia reported in the initial high-dose UDCA trial (4), four individuals (placebo) were excluded secondary having Crohn's disease or a prior history of colorectal neoplasia.
Table 1 summarizes the patient characteristics. The duration of follow-up was 235 patient years, with a mean duration of follow-up and drug use of 4.0 ± 1.54 years in the UDCA group and 4.35 ± 2.02 years in placebo group (P =0.48). During follow-up, the number of surveillance colonoscopies in both groups was similar (median, 4, range 1–8, in the UDCA group vs. 4, 1–7, in the placebo group). The time between colonoscopies after randomization was also similar (median, 377 days, range 98–1,482 days, in the UDCA group vs. 389 days, 21–1,386 days, in the placebo group). Endoscopic surveillance was balanced between the two groups and further adjustments were not made. After the initial trial was stopped, participants were asked to continue the same surveillance in a prospective follow-up study. This included annual colonoscopies with surveillance biopsies. Eleven (four UDCA, seven placebo) out of 37 people who would have been eligible for ongoing surveillance (did not reach an end point or were censored previously) did not consent for ongoing participation or were lost to follow-up after the initial high-dose UDCA trial was stopped in July 2008. The proportion of those who did not have ongoing surveillance colonoscopies was similar between groups (UDCA 4/12 (33%); placebo 7/25 (28%)). The remaining 26 (70%) people (8 UDCA and 18 placebo) had at least one colonoscopy with biopsies after the initial study ended that was reviewed. No new cases of colorectal neoplasia were detected in these 26 individuals after cessation of the initial study.
Table 1.
Clinical features of the study patients according to randomization groups
| UDCA (25) | Placebo (31) | N | P value | |
|---|---|---|---|---|
| Age at study entry | 45 (21–77) | 45 (18–64) | 56 | NS |
| Gender | 56 | |||
| Male | 64% (16/25) | 58% (18/31) | NS | |
| Female | 36% (9/25) | 42% (13/31) | NS | |
| BMI | 26 (13–35) | 26 (19–44) | 54 | NS |
| Smoking history | 11% (2/18) | 13% (3/23) | 41 | NS |
| Family history colon cancer | 0% (0/18) | 4% (1/23) | 41 | NS |
| 5-ASA use | 78% (14/18) | 93% (25/27) | 45 | NS |
| Immunomodulator usea | 0% (0/13) | 5% (1/18) | 31 | NS |
| Steroid use | 0% (0/13) | 6% (1/18) | 31 | NS |
| NSAID use | 29% (4/14) | 22% (4/18) | 32 | NS |
| Non-dietary folic acid use | 0% (0/14) | 11% (2/18) | 32 | NS |
| Age at diagnosis of PSC | 38 (20 – 71) | 39 (17 – 64) | 56 | NS |
| PSC duration (months) | 75 (1 – 299) | 49 (1 – 603) | 56 | NS |
| Histologic stage | 56 | |||
| I | 36% (9/25) | 26% (8/31) | NS | |
| II | 28% (7/25) | 42% (13/31) | NS | |
| III | 24% (6/25) | 16% (5/31) | NS | |
| IV | 12% (3/25) | 16% (5/31) | NS | |
| UC duration (years) | 11 (0 – 34) | 8 (0 – 38) | 56 | NS |
| Severity of colitis | 53 | |||
| Inactive | 13% (3/23) | 10% (3/30) | NS | |
| Mild | 65% (15/23) | 53% (16/30) | NS | |
| Moderate | 17% (4/23) | 37% (11/30) | NS | |
| Severe | 4% (1/23) | 0% (0/30) | NS | |
| Pancolitis | 90% (19/21) | 90% (26/29) | 50 | NS |
| Surveillance biopsiesb | 30±6.8 | 31±5.6 | 41 | NS |
BMI, body mass index; N, number of patients where data were available for review; NS, not significant; NSAID, non-steroidal anti-inflammatory drug; PSC, primary sclerosing cholangitis; UC, ulcerative colitis; UDCA, ursodeoxycholic acid; 5-ASA, 5-aminosalicalate.
lmmunomodulator consisted of azathioprine or 6-mercaptopurine.
Surveillance biopsy expressed as mean value are obtained per colonoscopy±s.d. Forty-one patients had surveillance biopsy data available for review (17 in UDCA and 24 in placebo group).
Variables are expressed in percentages (numbers) or medians (ranges).
In our primary analysis, a total of 12 patients developed colorectal neoplasia during follow-up. Nine patients had received high-dose UDCA (one colon cancer, one high-grade dysplasia, and seven low-grade dysplasia), and three patients had received placebo (one colon cancer, one high-grade dysplasia, and one low-grade dysplasia). Eleven of the 12 cases of neoplasia occurred during the initial high-dose UDCA trial. One new case of dysplasia was detected 4 months after cessation of high-dose UDCA. This patient ultimately underwent a colectomy for an unresectable lesion. Of the nine patients in the UDCA group with colorectal neoplasia, six had lesions proximal to the splenic flexure. In the three patients who received placebo, one patient had two low-grade lesions (one proximal and one distal to the splenic flexure). For the remaining two patients, one had a lesion proximal to the splenic flexure and the other patient had a lesion distal to the splenic flexure. In the UDCA group, the majority (78%) of patients developed colorectal neoplasia after 2 years of use. Survival free of dysplasia is illustrated in Figure 1. The use of high-dose UDCA was associated with a higher incidence of colorectal neoplasia compared with placebo (hazard ratio (HR): 4.44, 95% confidence interval (CI): 1.30–20.10, P =0.02).
Figure 1.
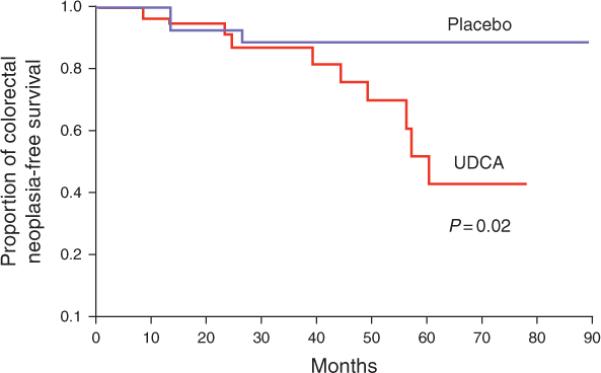
Kaplan–Meier estimates of proportion of patients free of colorectal neoplasia according to randomization group. UDCA, ursodeoxycholic acid.
Six individuals eventually underwent a liver transplant (four in the UDCA group and two in placebo group). Of those six, only one (assigned to UDCA group) developed colorectal neoplasia and the transplant occurred after they reached our primary end point. One patient in the placebo group died of liver failure after they reached an end point. One person in the UDCA group died secondary to a gastrointestinal hemorrhage. After randomization, seven had a colectomy (four in UDCA and three in placebo group). In the UDCA group, all four individuals with a colectomy had a history of colorectal neoplasia and two of the three people in the placebo group who received a colectomy had a history of dysplasia or colon cancer. Of the five people with dysplasia in the UDCA group who did not undergo a colectomy, four had recurrent or persistent dysplasia detected on subsequent colonoscopies while they were still taking high-dose UDCA. Four of those five individuals had at least one colonoscopy after cessation of high-dose UDCA. After high-dose UDCA was stopped, three of those four patients did not have dysplasia detected on a subsequent colonoscopy.
We evaluated the association of multiple covariates with the development of colorectal neoplasia using univariate analyses and Cox-proportional hazards regression, as shown in Table 2. In univariate analysis, only smoking history and UC duration met our threshold for inclusion in the multivariate analysis. After adjusting for smoking, and UC duration, the use of high-dose UDCA was still associated with a higher incidence (HR: 5.97, 95% CI: 1.39–41.44, P =0.02)of colorectalneoplasia.
Table 2.
The role of covarlates in the development of cancer or dysplasia
| Variable | Univariate | Multivariate | ||
|---|---|---|---|---|
| HR (95% Cl) | P value | HR (95% Cl) | P value | |
| Age at study entry | 1.03(0.99–1.08) | 0.16 | — | — |
| Gender female vs. male | 0.89 (0.24–2.85) | 0.86 | — | — |
| BMI | 0.98(0.85–1.10) | 0.72 | — | — |
| Smoking history | 0(−)b | 0.10 | 0(−)b | 0.10 |
| Family history of colon cancer | 0(−)b | 0.42 | — | — |
| 5-ASA use | 1.24 (0.22–23.20) | 0.84 | — | — |
| Immunomodulator usea | 0(−)b | 0.50 | — | — |
| Steroid use | 0(−)b | 0.50 | — | — |
| NSAID use | 1.96 (0.27 – 10.06) | 0.46 | — | — |
| Non-dietary folic acid use | 0(−)b | 0.29 | — | — |
| UDCA | 4.44 (1.30 – 20.10) | 0.02 | 5.97(1.39 – 41.44) | 0.02 |
| Age at diagnosis of PSC | 1.02(0.97 – 1.07) | 0.40 | — | — |
| PSC duration (months) | 1.00(0.99 – 1.00) | 0.75 | — | — |
| Histologic stage (I –IV) | 1.06(0.61 – 1.81) | 0.83 | — | — |
| UC duration (years) | 1.04(0.99 – 1.09) | 0.10 | 1.05(0.98 – 1.13) | 0.16 |
| UC severity (inactive-severe) | 0.68(0.28 – 1.68) | 0.40 | — | — |
| Pancolitis | 0.39 (0.10 – 2.55) | 0.28 | — | — |
BMI, body mass index; Cl, confidence interval; HR, hazard ratio; NSAID, non-steroidal anti-inflammatory drug; PSC, primary sclerosing cholangitis; UC, ulcerative colitis; UDCA, ursodeoxycholic acid; –, not performed; 5-ASA, 5-aminosalicalate.
lmmunomodulator consisted of azathioprine or 6-mercaptopurine.
Hazard ratio is zero.
Proportion of patients with neoplasia vs. no neoplasia taking the following medications: 5/5 vs. 34/40 5-ASA; 1/5 vs. 0/26 immunomodulator; 1/5 vs. 0/26 steroid; 2/6 vs. 6/26 NSAID; 0/6 vs. 2/26 folic acid.
Serum bile composition before and after treatment was available for review in 14 patients who received high-dose UDCA. Of those, four developed colorectal neoplasia and the other 10 did not. The change in bile acid composition between those who did and did not develop colorectal neoplasia was not statistically significant (Table 3). However, there was a trend toward a greater increase in LCA (0.6 ± 0.6 vs. 0.2 ± 0.2, P=0.28) and CDCA (0.6±0.8 vs. 0.1±0.3, P=0.22) in those who developed colorectal neoplasia compared with those who did not.
Table 3.
Change in serum bile acid composition before and after therapy with high-dose UDCA
| LCA (μmol/l) | DCA (μmol/l) | UDCA (μmol/l) | CDCA (μmol/l) | CA (μmol/l) | Total BA (μmol/l) | |
|---|---|---|---|---|---|---|
| Patients on UDCA with colorectal neoplasia (n=4) | 0.6±0.6 | 0.1±0.1 | 21.9±34.8 | 0.6±0.8 | 0.1±0.4 | 23.2±36.8 |
| Patients on UDCA without colorectal neoplasia (n=10) | 0.2±0.2 | 0.1±0.2 | 16.9±35.9 | 0.1±0.3 | 0.01±0.7 | 17.1±36.1 |
| P value | 0.28 | 0.72 | 0.43 | 0.22 | 0.88 | 0.28 |
CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; total BA, total bile acids; UDCA, ursodeoxycholic acid.
Data represent change from baseline and are expressed as means±s.d.
In a secondary analysis, four patients with a lesion possibly consistent with an adenoma-like dysplastic lesion were censored at the time of liver transplant, death, or last colonoscopy with biopsy, but not counted as having reached a dysplasia end point. Of the remaining eight patients, six received UDCA and two received placebo. The use of high-dose UDCA trended toward an increase in colorectal neoplasia in this analysis, although this difference was not statistically significant (HR: 4.00, 95% CI: 0.92–27.39, P = 0.07). Multivariate analysis was not performed after excluding these four cases, as none of the covariates met our threshold for inclusion.
DISCUSSION
Our study revealed that high-dose UDCA administration in patients with PSC and UC was associated with a higher rate of colorectal neoplasia compared with placebo. This association remained statistically significant after adjusting for smoking history and UC duration. Other important covariates such as age at study entry were not associated with the development of colorectal neoplasia. The majority of patients developed colorectal neoplasia after ≥2 years of UDCA use. After excluding lesions possibly consistent with an adenoma (19,20), the HR for the association with colorectal neoplasia did not change significantly, but due to the small number of cases, the CI for this association was wide, and the use of high-dose UDCA was not associated with a statistically significant risk of colorectal neoplasia.
Our results are in contrast with several prior studies. In vitro and animal studies suggested that UDCA may have a role as a chemopreventive agent. Several mechanisms for which UDCA may act to prevent colorectal cancer have been proposed, including downregulation of cyclooxygenase-2 expression, prevention of carcinogen-induced changes in protein c, inhibiting cell proliferation by suppressing epidermal growth factor receptor, which is typically activated by DCA, and altering the bile acid milieu to reduce secondary bile acid levels (8–12). Lower doses of UDCA may have a protective role as it may have an antiapoptotic effect. For example, exposing human colon cancer cell lines to UDCA can decrease DCA-induced apoptosis (22).
There is limited data regarding the role of UDCA in colorectal neoplasia prevention in PSC patients with UC. Two studies have suggested that UDCA may have a preventive role in UC patients. Tung et al. (13) performed a retrospective review of 59 patients and found that UDCA was associated with a significant reduction in the odds ratio for colonic dysplasia development. However, after excluding cases of indefinite dysplasia, multivariate analysis did not reveal a statistically significant association with UDCA. A secondary analysis revealed a significant association between UDCA use and development of high-grade dysplasia only. Furthermore, in this study, the control group had a high proportion of dysplasia (72%), and 50% of those who received UDCA went on to develop dysplasia. Compared with those who did not receive UDCA, the UDCA-treated group had a shorter duration of colitis and were older at the diagnosis of colitis. It is unknown to what extent this may have impacted the results. Although the mean duration of UDCA was similar to our study, our dose was nearly three times higher.
Our group performed a previous study that examined a lower dose of UDCA at 13–15 mg/kg/day. The present study and the one conducted by Pardi et al. (14) were similar in that they are both retrospective reviews of randomized-controlled trials. In addition, the proportion of patients that could have been appropriate for inclusion but had to be excluded were also similar. With the exception for censoring at the time of transplant, both studies had similar censoring events. In contrast to the primary analysis presented in this study, Pardi et al. excluded cases of dysplasia occurring in a typical adenoma for the primary end point after confirmation of dysplasia by a single blinded pathologist and found that a relative risk for developing dysplasia was 0.26 (95% CI: 0.06–0.92). However, when we excluded cases of dysplasia that may have occurred in a typical adenoma, the HR for the increased risk did not change significantly, although the association no longer reached statistical significance. Therefore, it is unlikely that methodologic differences between the studies would be sufficient enough to explain the differing results. Another distinction is that the initial study reviewed by Pardi et al. did not exclude patients on therapy with non-5-ASA compounds within 3 months before enrollment. This is in contrast with the present study and that may have been selected for a different population of UC patients despite a small proportion of these individuals requiring the initiation of non-5-ASA agents after enrollment. Furthermore, individuals in the present study tended to have more intensive endoscopic surveillance in terms of the median number of colonoscopies, less time between colonoscopies, and number of surveillance biopsies. Though the duration of UC and follow-up time were longer compared with the current study, the duration of UDCA use was slightly shorter in that study. In the present study, the duration of PSC was shorter and we had fewer men. In addition, the age in the UDCA group was older.
Wolf et al. (15) conducted a retrospective cohort study over 120 patients (92 controls and 28 cases) over a median period of 21 and 22 years for the UDCA and control groups, respectively. The mean duration of UDCA use was 3.4 years, which is slightly less than our study. This study did not find a reduction in colorectal cancer or dysplasia in the UDCA group nor did they find that increasing the dose provided any benefit. It is unclear what the mean dose was but it isunlikely that they utilized doses as high as 28–30 mg/kg/day. Moreover, it may be possible that this study was underpowered.
It has been postulated that bile acids may have a role in colon carcinogenesis, particularly the secondary bile acids (LCA and DCA) (23–25). In vitro studies have suggested that certain bile acids can induce DNA damage. For example, LCA has been shown to cause DNA damage in colonic cells (26,27). In addition, hydrophobic bile acids may induce oxidative stress in gastrointestinal cancer cells (28). Therefore, it has been suggested that bile acids may have a role in gastrointestinal cancers via a prolonged exposure to high bile acid concentrations leading to oxidative stress, selection for cells resistant to apoptosis, and replication with unrepaired DNA damage (25,29). Thus, one possible biologic mechanism behind our surprising results could be an increase in supraphysiologic levels of hydrophobic bile acids after a prolonged exposure to high doses of UDCA. A potential reason why this effect has not been observed in lower doses of UDCA is high-dose UDCA (28–30 mg/kg/day) has resulted greater increases in the serum UDCA levels than at lower doses (21). We reported that there was a statistically significant increase in the serum UDCA and LCA levels in the treatment group compared with placebo and an expansion of the total serum bile acid pool without significant changes in CA, DCA, or CDCA levels (21). It is unknown if prolonged exposure to high-dose UDCA leads to increased colonic levels of LCA, DCA, or other toxic bile acids in our cohort as such samples were not collected in the initial study. However, absorption of UDCA is incomplete (30,31). Consequently, it would be expected that a proportion of the UDCA would be metabolized by bacterial flora to other bile acids. UDCA can be metabolized to both LCA and CDCA by intestinal flora (32). CDCA is typically metabolized to LCA by bacterial flora. Both CDCA and LCA have been shown to stimulate in vitro cell invasion in a dose-dependent mechanism (33,34). Furthermore, physiologic levels of CDCA and DCA can increase colon adenoma cell proliferation and reduce apoptosis (35). In the present study, our results revealed a trend toward higher levels of serum CDCA and LCA in patients with colorectal neoplasia although this was not statistically significant.
To our knowledge, this is the first study that looked at the long-term effects of high-dose UDCA on the development of colorectal neoplasia and the only one to suggest that UDCA may actually increase the risk of developing neoplasia. Th e primary limitation of this study is that we were not able to have all of the pathology specimens reviewed by a single pathologist and the diagnosis of dysplasia can be challenging. The majority of our cases were secondary to low-grade dysplasia and the management of low-grade dysplasia in UC is controversial and complex. Dysplasia is a marker for colorectal cancer risk (36). We believe that the diagnosis of low-grade dysplasia is clinically significant because individuals with low-grade dysplasia, particularly dysplasia-associated masses or lesions or flat lesions, may be screened more aggressively in clinical practice and it is generally accepted that there is an increased risk for developing colorectal cancer (17,37,38). Many individuals with low-grade dysplasia had recurrence or persistence of these findings on subsequent colonoscopies while on high-dose UDCA in this study. Our study was retrospective which does have some inherent limitations, particularly since the a priori hypothesis did not focus on the colorectal neoplasia development in this subset of UC patients within the initial trial. Consequently, some covariates, particularly with medications, family and social history (Table 1), were not universally collected in the initial study and available for our subsequent review. However, the proportion of missing information was similar between groups. In addition, 14 % of patients that may have been eligible for our review were excluded due to a lack of colonoscopy. Furthermore, some patients did not continue endoscopic surveillance within the study protocol after it was suggested that they stop the drug. However, endoscopic surveillance was balanced between the placebo and UDCA group as were the proportion in each group who did not have ongoing surveillance after the initial study. Finally, our serum bile acid analysis was unlikely to reach statistical significance due to the small sample size available for review.
Our results have several important clinical implications and raise new questions that should be addressed in subsequent studies. First, our study suggests that high-dose UDCA should not be used for the prevention of colorectal neoplasia in this population. Given the lack of long-term, prospective, randomized clinical studies, it is clear that more studies are needed before recommending the use of low-dose UDCA for the prevention of colorectal neoplasia in patients with concomitant PSC and UC. Furthermore, the role of fecal and serum bile acids in the pathogenesis of colorectal neoplasia in patients taking UDCA should be investigated further.
In conclusion, our results show that UDCA (28–30 mg/kg/day) is associated with an increased incidence of colorectal neoplasia and therefore should not be used for the prevention of colorectal neoplasia in this patient population.
StudyHighlights.
WHAT IS CURRENT KNOWLEDGE
-
✓
Individuals with primary sclerosing cholangitis (PSC) and ulcerative colitis are at a higher risk of developing colorectal neoplasia.
-
✓
Retrospective studies have shown mixed results for the use of ursodeoxycholic acid (UDCA) for the prevention of colorectal neoplasia in this population.
-
✓
High-dose UDCA (28–30 mg/kg/day) has been shown to increase the secondary bile acid, lithocholic acid.
-
✓
High-dose UDCA (28–30 mg/kg/day) has been shown to increase serious adverse events in patients with PSC.
WHAT IS NEW HERE
-
✓
High-dose ursodeoxycholic acid (UDCA) (28–30 mg/kg/day) is associated with the development of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis.
-
✓
The majority of patients developed colorectal neoplasia after ≥ 2 years of UDCA use.
-
✓
Serum levels of lithocholic acid were not significantly associated with colorectal neoplasia after receiving UDCA.
ACKNOWLEDGMENTS
We thank the Center for Translational Science Activities and Ross Dierkhising for providing advice and assistance regarding the statistical analyses in this manuscript. We also thank Jill Keach and Jeffery Schmoll for their work in the original high-dose ursodeoxycholic acid study and logistical support for the present study.
Financial support: This study was supported by M01RR000065 awarded to Virginia Commonwealth University from the National Center for Research Resources.
Footnotes
CONFLICT OF INTEREST Guarantor of the article: John E. Eaton, MD.
Potential competing interests: Keith D. Lindor has previously received funding from Axcan Pharma. Marina G. Silveira was the 2009–2010 AASLD advanced–transplant fellowship awardee. John E. Eaton was the recipient of the PSC Partners Seeking a Cure Award for work presented from this manuscript at the 2010 AASLD meeting as an oral presentation. Velimir A.C. Luketic has previously received funding from Globe-Immune, Intercept, Vertex, Pharmasset, GlaxoSmithKline, Biolex, Merck (formerly Schering Plough Research Institute), and Johnson&Johnson.
Specific author contributions: Study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, and critical revision of the manuscript: John E. Eaton; study concept and design, acquisition of data, analysis and interpretation of data, and critical revision of the manuscript: Marina G. Silveira and Emmanouil Sinakos; study concept and design, reviewing the data, analysis and interpretation of data, and critical revision of the manuscript: Darrell S. Pardi; critical revision of manuscript: Kris V. Kowdley, Velimir A.C. Luketic, M. Edwyn Harrison, Timothy McCashland, Alex S. Befeler, Denise Harnois, Jan Petz, and Roberta Jorgensen; study concept and design, interpretation of data, critical revision of the manuscript, and study supervision: Keith D. Lindor. All authors take responsibility for the integrity of the data and the accuracy of the analysis.
REFERENCES
- 1.Lewis JD, Deren JJ, Lichtenstein GR. Cancer risk in patients with inflammatory bowel disease. Gastroenterol Clin N Am. 1999;28:459–77. doi: 10.1016/s0889-8553(05)70065-0. [DOI] [PubMed] [Google Scholar]
- 2.Silveira MG, Lindor KD. Primary sclerosing cholangitis. Can J Gastroenterol. 2008;22:689–98. doi: 10.1155/2008/824168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchesa P, Lashner BA, Lavery IC, et al. The risk of cancer and dysplasia among ulcerative colitis patients with primary sclerosing cholangitis. Am J Gastroenterol. 1997;92:1285–8. [PubMed] [Google Scholar]
- 4.Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for treatment of primary sclerosing cholangitis. Hepatology. 2009;50:808–14. doi: 10.1002/hep.23082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis Ursodoexycholic Acid Study Group. N Engl J Med. 1997;336:691–5. doi: 10.1056/NEJM199703063361003. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell SA, Bansi DS, Hunt N, et al. A preliminary trial of high dose ursodoexycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001;121:900–7. doi: 10.1053/gast.2001.27965. [DOI] [PubMed] [Google Scholar]
- 7.Harnois DM, Angulo P, Jorgensen RA, et al. High-dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. Am J Gastroenterol. 2001;96:1558–62. doi: 10.1111/j.1572-0241.2001.03777.x. [DOI] [PubMed] [Google Scholar]
- 8.Khare S, Mustafi R, Cerda S, et al. Ursodeoxycholic acid suppresses Cox-2 expression in colon cancer: roles of ras, p38, and ccaat/enhancer-binding protein. Nutr Cancer. 2008;60:389–400. doi: 10.1080/01635580701883003. [DOI] [PubMed] [Google Scholar]
- 9.Batta AK, Salen G, Holubec H, et al. Enrichment of the more hydrophilic bile acid ursodeoxycholic acid in the fecal water-soluble fraction after feeding to rats with colon cancer polyps. Cancer Res. 1998;58:1684–7. [PubMed] [Google Scholar]
- 10.Wall RK, Frawley BP, Hartmann S, et al. Mechanism of action of chemo-protective ursodeoxycholate in the azoxymethane model of colon carcinogenesis: potential roles of protein kinase C-α, -βII, and –ζ. Cancer Res. 1995;55:5257–64. [PubMed] [Google Scholar]
- 11.Rodrigues CMP, Kren BT, Steer CJ, et al. The site-specific delivery of ursodeoxycholic acid to the rat colon by sulfate conjugation. Gastroenterology. 1995;9:1835–44. doi: 10.1016/0016-5085(95)90750-5. [DOI] [PubMed] [Google Scholar]
- 12.Im E, Martinez JD. Ursodeoxycholic acid (UDCA) can inhibit deoxcholic acid (DCA) induced apoptosis via modulation of EGFR/RAF-1/ERK signaling in human colon cancer cells. J Nutr. 2004;134:483–6. doi: 10.1093/jn/134.2.483. [DOI] [PubMed] [Google Scholar]
- 13.Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med. 2001;134:89–95. doi: 10.7326/0003-4819-134-2-200101160-00008. [DOI] [PubMed] [Google Scholar]
- 14.Pardi DS, Loftus EV, Kremers WK, et al. Ursodeoxycholic acid as a chemo-preventative agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889–93. doi: 10.1053/gast.2003.50156. [DOI] [PubMed] [Google Scholar]
- 15.Wolf JM, Rybicki LA, Lashner BA. The impact of ursodeoxycholic acid on cancer, dysplasia and mortality in ulcerative colitis patients with primary sclerosing cholangitis. Aliment Pharmacol Ther. 2005;22:783–8. doi: 10.1111/j.1365-2036.2005.02650.x. [DOI] [PubMed] [Google Scholar]
- 16.Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51:660–78. doi: 10.1002/hep.23294. [DOI] [PubMed] [Google Scholar]
- 17.Kornbluth A, Sachar DB. The Practice Parameters Committee of the American Collegeof Gastroenterology. Ulcerative Colitis Practice Guidelines in Adults: American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2010;105:501–23. doi: 10.1038/ajg.2009.727. [DOI] [PubMed] [Google Scholar]
- 18.Riddell RH, Goldman H, Ransohoff DF, et al. Dysplasia in inflammatory bowel disease: standard classification with provisional clinical applications. Hum Pathol. 1983;14:931–68. doi: 10.1016/s0046-8177(83)80175-0. [DOI] [PubMed] [Google Scholar]
- 19.Rubin PH, Friedman S, Harpaz N, et al. Colonoscopic polypectomy in chronic colitis: conservative management after endoscopic resection of dysplastic polyps. Gastroenterology. 1999;117:1295–300. doi: 10.1016/s0016-5085(99)70279-9. [DOI] [PubMed] [Google Scholar]
- 20.Engelsgjerd M, Farraye FA, Odze RD. Polypectomy may be adequate treatment for adenoma-like dysplastic lesions in chronic ulcerative colitis. Gastroenterology. 1999;117:1288–94. doi: 10.1016/s0016-5085(99)70278-7. [DOI] [PubMed] [Google Scholar]
- 21.Sinakos E, Marschall HU, Kowdley KV, et al. Bile acid changes after high-dose ursodeoxycholic acid treatment in primary sclerosing cholangitis: relation to disease progression. Hepatology. 2010;52:197–203. doi: 10.1002/hep.23631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yui S, Saeki T, Kanamoto R, et al. Characteristics of apoptosis in HCT116 colon cancer cells induced by deoxycholic acid. J Biochem. 2005;138:151–7. doi: 10.1093/jb/mvi106. [DOI] [PubMed] [Google Scholar]
- 23.Kay RM. Effects of diet on fecal excretion and bacterial modification of acidic and neutral steroids, and implications for colon carcinogenesis. Cancer Res. 1981;41:3774–7. [PubMed] [Google Scholar]
- 24.Nagengast FM, Grubben MJ, van Munster IP. Role of bile acids in colorectal carcinogenesis. Eur J Cancer. 1995;31:1067–70. doi: 10.1016/0959-8049(95)00216-6. [DOI] [PubMed] [Google Scholar]
- 25.Bernstein H, Bernstein C, Payne CM, et al. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol. 2009;15:3329–40. doi: 10.3748/wjg.15.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Booth LA, Gilmore IT, Bilton RF. Secondarybile acid induced DNA damage in HT29 cells: are free radicals involved? Free Radic Res. 1997;26:135–44. doi: 10.3109/10715769709097792. [DOI] [PubMed] [Google Scholar]
- 27.Kulkarni MS, Cox BA, Yielding KL. Requirements for induction of DNA strand breaks by lithocholic acid. Cancer Res. 1982;42:2792–5. [PubMed] [Google Scholar]
- 28.Payne CM, Weber C, Crowley-Skillicorn C, et al. Deoxycholate induces mitochondrial oxidative stress and activates NF-kappaB through multiple mechanisms in HCT-116 colon epithelial cells. Carcinogenesis. 2007;28:215–22. doi: 10.1093/carcin/bgl139. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein H, Bernstein C, Payne CM, et al. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. 2005;589:47–65. doi: 10.1016/j.mrrev.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 30.Walker S, Rudolph G, Raedsch R, et al. Intestinal absorption of ursodeoxycholic acid in patients with extrahepatic biliary obstruction and bile drainage. Gastroenterology. 1992;102:810–5. doi: 10.1016/0016-5085(92)90162-r. [DOI] [PubMed] [Google Scholar]
- 31.Fedorowski T, Salen G, Colallilo A, et al. Metabolism of ursodeoxycholic acid in man. Gastroenterology. 1977;73:1131–7. [PubMed] [Google Scholar]
- 32.Higashi S, Setoguchi T, Katsuki T. Conversion of 7-ketolithocholic acid to ursodeoxycholic acid by human intestinal anaerobic microorganisms: interchangeability of chenodeoxycholic acid and ursodeoxycholic acid. Gastroenterol Jpn. 1979;14:417–24. doi: 10.1007/BF02773728. [DOI] [PubMed] [Google Scholar]
- 33.Debruyne PR, Bruynell EA, Karaguni I-M, et al. Bile acids stimulate invasion and haptotaxis in human colorectal cancer cells through activation of multiple oncogenic signaling pathways. Oncogene. 2002;21:6740–50. doi: 10.1038/sj.onc.1205729. [DOI] [PubMed] [Google Scholar]
- 34.Baek MK, Park JS, Park JH, et al. Lithocholic acid upregulates uPAR and cell invasiveness via MAPK and AP-1 signaling in colon cancer cells. Cancer Lett. 2010;290:123–8. doi: 10.1016/j.canlet.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 35.McMillan L, Butcher S, Wallis Y, et al. Bile acids reduce the apoptosis effects of sodium butyrate on human colon adenoma (AA/C1) cells: implications for colon carcinogenesis. Biochem Biophys Res Commun. 2000;273:45–9. doi: 10.1006/bbrc.2000.2899. [DOI] [PubMed] [Google Scholar]
- 36.Farraye FA, Odze RD, Eaden J, AGA Institute Medical Position Panel on Diagnosis and Management of Colorectal Neoplasia in Inflammatory Bowel Disease et al. AGA medical position statement on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. 2010;138:738–45. doi: 10.1053/j.gastro.2009.12.037. [DOI] [PubMed] [Google Scholar]
- 37.Ullman TA, Loftus EV, Jr, Kakar S, et al. The fate of low grade dysplasia in ulcerative colitis. Am J Gastroenterol. 2002;97:922–7. doi: 10.1111/j.1572-0241.2002.05610.x. [DOI] [PubMed] [Google Scholar]
- 38.Thomas T, Abrams KA, Robinson RJ, et al. Meta-analysis: cancer risk of low-grade dysplasia in chronic ulcerative colitis. Aliment Pharmacol Ther. 2007;25:657–68. doi: 10.1111/j.1365-2036.2007.03241.x. [DOI] [PubMed] [Google Scholar]