

Abstract
We present a detailed study of the mechanism for oxidative heteroarylation, based on DFT calculations and experimental observations. We propose binuclear Au(II)-Au(II) complexes to be key intermediates in the mechanism for gold catalyzed oxidative heteroarylation. The reaction is thought to proceed via a gold redox cycle involving initial oxidation of Au(I) to binuclear Au(II)-Au(II) complexes by Selectfluor, followed by heteroauration and reductive elimination. While it is tempting to invoke a transmetalation/reductive elimination mechanism similar to that proposed for other transition metal complexes, experimental and DFT studies suggest that the key C-C bond forming reaction occurs via a bimolecular reductive elimination process (devoid of transmetalation). In addition, the stereochemistry of the elimination step was determined experimentally to proceed with complete retention. Ligand and halide effects played an important role in the development and optimization of the catalyst; our data provides an explanation for the ligand effects observed experimentally, useful for future catalyst development. Cyclic voltammetry data is presented that supports redox synergy of the Au···Au aurophilic interaction. The monometallic reductive elimination from mononuclear Au(III) complexes is also studied from which we can predict a ~15 kcal/mol advantage for bimetallic reductive elimination.
Keywords: Bimetallic reductive elimination, binuclear gold, gold catalysis, oxidative heteroarylation
Introduction
During the last few decades, group 10 transition-metal catalyzed cross couplings have become indispensable in organic synthesis.1 This has been enabled by the rich redox reactivity of Pd, Pt and Ni with organic molecules. In contrast, while homogeneous gold catalysis has emerged as one of the most active fields of organometallic chemistry,2 reports of transformations based on overall redox neutral gold(I) activation of π-bonds toward nucleophilic attack have almost exclusively dominated the recent literature.3 Developments by Hashmi,4 Zhang,5 Toste,6 and others7 have increased the scope of the already rich chemistry of Au(I) by developing oxidative transformations proposed to involve Au(I)/Au(III) cycles. In this regard, the gold-catalyzed oxidative heteroarylation of alkenes has attracted attention as a versatile transformation that may resemble traditional cross-coupling reactions. The original reactivity paradigm has been expanded to include various nucleophiles (oxy and aminoarylations), coupling partners (arylboronic acids and arylsilanes) and π-bonds (alkenes and alkynes). Moreover, both intra- and intermolecular variants have been described. Despite the significant progress recently achieved in the development of gold-catalyzed oxidative coupling reactions, there remain considerable uncertainties about the mechanism.
The few experimental reports on the oxidative heteroarylation of alkenes propose two distinct conflicting mechanisms. Zhang proposed as the first steps in the mechanism either oxidation of Au(I) to Au(III) by Selectfluor® followed by arylboronic acid transmetalation, or transmetalation followed by oxidation to form an aryl Au(III) complex. Both pathways produce a high oxidation state arylgold(III) intermediate that coordinates and activates the alkene substrate towards heteroauration. This proposed intermediate undergoes nucleophilic alkene addition, followed by traditional reductive elimination to regenerate the Au(I) catalyst. Based on Zhang’s proposed transmetalation/reductive elimination mechanism, a similar mechanism could be envisioned (Scheme 1b) for the gold catalyzed heteroarylation reaction. However, on the basis of experimental observations and preliminary computational evidence, we proposed a bimolecular reductive elimination step that does not involve transmetalation of the arylboronic acid coupling partner (Scheme 1a). To help resolve this and other discrepancies, we report herein a detailed computational and experimental investigation on the three main mechanistic stages of oxidative heteroarylation: oxidation, heteroauration, and reductive elimination.
Scheme 1.

Proposed pathways for the oxidative heteroarylation of alkenes. a) Proposed bimolecular reductive elimination pathway, and b) transmetalation/unimolecular reductive elimination pathway.
Results and Discussion
Oxidation
DFT analysis
During our ongoing mechanistic studies on reactions using LAuX as catalyst and Selectfluor as oxidant, our group identified several key mechanistic clues [Eq. (1) – (8)]. The failure to produce pyrrolidine B [Eq. (1)] from alkylgold(I) complex A with phenylboronic acid and Selectfluor® is consistent with oxidation preceding aminoauration. Furthermore, during catalyst optimization, we found binuclear catalyst dppm(AuBr)2 [dppm= bis(diphenylphosphanyl)methane] to exhibit superior performance compared to the mononuclear PPh3AuBr (81% vs. 47% yield). We speculated that a bimetallic catalytic process,8 might be operative especially considering the significant literature reports on binuclear Au(II) and Au(III) complexes9 and the recent reports by Ritter on the advantages of bimetallic reductive elimination in Pd catalyzed aromatic C–H oxidation. To investigate this, we performed calculations on the oxidative aminoarylation reaction using the binuclear catalyst.
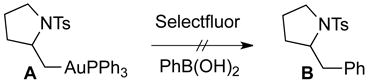 |
(1) |
| (2) |
| (3) |
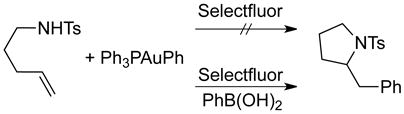 |
(4), (5) |
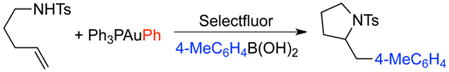 |
(6) |
| (7) |
| (8) |
We studied three potential oxidation pathways depending on the molecularity of the reaction (Figure 1, Scheme 2), all leading to a common intermediate presumed to lead to nucleophilic aminoauration. We envisioned initial 2-electron 1-center oxidation of LAu(I)X to LAu(III)XF(Solv)+ by Selectfluor followed by either coordination of the normally more abundant substrate (Scheme 2a) or interaction with LAu(I)X (Scheme 2b). We deem these two pathways as monometallic oxidations that lead to a binuclear complex. We also considered a possible bimetallic oxidation (2-electron 2-center) pathway characterized by a concerted oxidation of a weakly bound Au(I) dimer.10 Au(I) –Au(I) interactions or “aurophilic bonding”,11 especially in complexes with bidentate ligands10,12 (e.g. dppm) may favor the bimetallic oxidation pathway (Scheme 2c). We were able to find transition structures leading to the formation of a binuclear gold complex with a gold-gold bond for processes where substrate is bound to Au (Figure 1a, Scheme 3a), and without bound substrate (Figure 1b, Scheme 2b). Our calculations using the M06 functional and LACV3P**++(2f) basis set suggest that monometallic oxidation with binuclear substrate coordination (Scheme 2b) pathway is competitive with the bimetallic pathway (Scheme 2c). Given the challenge of computing accurate entropies,13 especially in cases with a change in the number of particles, we propose that for the case of a monodentate phosphine ligand, the monometallic binuclear pathway could be operative. On the other hand, the use of a bidentate phosphine, especially one that brings the gold atoms in close proximity (such as dppm), should favor the bimetallic oxidation pathway due to the minimal entropic cost. Notably, the binuclear complex is the lowest point in the enthalpy surface, and thus the pathway to its formation could be determined by the concentration of species.
Figure 1.

Transition structures 3, 5 and 9, including selected interatomic distances (Å) for the oxidative formation of binuclear Au species.
Scheme 2.

Mechanistic hypotheses for oxidation leading to a substrate coordinated binuclear complex.
Scheme 3.

Mechanistic hypothesis for aminoauration and bimetallic reductive elimination. Free energies at 298 K (ΔG298K) at M06/LACV3P**++(2f) level of theory.
Cyclic voltammetry
The superior performance of digold catalysts over monogold catalysts for so-called Au(I)/Au(III) catalysis6 is particularly interesting given recent proposals by Ritter and coworkers that cooperative “redox synergy” of Ag···Ag and Pd···Pd interactions qualitatively modulates redox potentials and thereby facilitates oxidative catalysis.8 We therefore sought to measure quantitatively how Au···Au interactions modulate redox potentials in catalytically relevant Au complexes. To accomplish this, we obtained cyclic voltammograms of complexes 15–20.

The monogold complexes 15 and 16 provided benchmark redox potentials in the absence of Au···Au interactions. An oxidative scan of 15 revealed two irreversible, 1-electron oxidations at approximately 1.48 V and 1.98 V,14 corresponding to Au(II)/Au(I) and Au(III)/Au(II) redox events, respectively (see Supporting Information). These oxidative peaks were absent for the fully oxidized 16, which instead exhibited an irreversible, 2-electron Au(III)/Au(I) reduction at −0.69 V with a corresponding re-oxidation at 0.72 V (Figure 3b).
Figure 3.

Cyclic voltammetry data: (a) oxidative scans for complexes 17 (solid) and 18 (dashed); (b) reductive scans for complexes 16 (solid) and 17 (dashed). Conditions: CH2Cl2 solvent, 0.1 M Bu4NPF6, 100 mV/s scan rate.
The known mixed-valent digold complex 17 has been shown to lack any significant Au···Au interaction in solution (based on UV-Vis) or in the solid state (based on X-ray crystallography).15 Indeed, an oxidative scan for 17 revealed two irreversible, 1-electron oxidation events with potentials similar to those for complex 15 (1.48 V and 1.96 V for Au(II)Au(III)/Au(I)Au(III) and Au(III)Au(III)/Au(II)Au(III), respectively), confirming that the distant presence of a coordinatively saturated Au(III) center has negligible effect on the oxidation potential of the Au(I) center in 17 (see Supporting Information for overlay). Interestingly, the irreversible, 2-electron reduction for 17 was observed at a less negative potential relative to that of 16 by approximately 160 mV (Figure 3b), suggesting that Au···Au interactions become important upon reduction of the Au(III) center in 17.
Though complexes similar to the digold(I) complex 18 exhibit aurophilic Au(I)···Au(I) interactions in solution (based on EXAFS),16 18 itself has been characterized both with and without a short Au···Au contact in the solid state depending on the crystallization method.17 Thus, the Au(I)···Au(I) interaction may be of the same order of magnitude as crystal packing forces. In this context, we sought to determine whether this weak aurophilic bonding measurably affects redox potentials. The cyclic voltammogram of 18 featured two irreversible, 1-electron oxidations that were shifted cathodically from complexes 15 and 17 by approximately 140 mV (Figure 3a). A corresponding re-reduction peak (that was absent during reductive scans) was observed at −0.45 V, approximately where reduction of 17 occurred (Figure 3a). The 140 mV cathodic shift in oxidation potential of 18 relative to 15 and 17, as well as the 160 mV anodic shift in reduction potential of 17 relative to 16, are to our knowledge the first quantitative measurements evaluating the extent that aurophilic interactions modulate redox potentials. The observed trend in redox potentials is fully consistent with early observations by Fackler and others that aurophilic Au(I)/Au(I) complexes are able to activate organohalide substrates that are inert towards mononuclear Au(I) complexes, through a bimetallic oxidative addition driven by Au(II)/Au(II) bond formation.18 This concept has catalytic relevance given that the first step in the proposed heteroarylation catalytic cycle represents a 2-electron oxidation of the catalyst, and that the turnover step represents a 2-electron reduction of the catalyst.
We next determined that this concept holds for carbene-ligated gold complexes as well as for phosphine-ligated gold complexes. The digold(I) complex DIMes(AuCl)2 (20, DIMes = 1,1′-di(mesityl)-3,3′-methylenediimidazol-2,2′-diylidene) was synthesized by acidolysis of DIMes(AuMe)2 with HCl. The crystal structures of both DIMes(AuMe)2 and 20 are provided in the Supporting Information; neither complex provided evidence of short Au···Au contacts in the solid state. Nonetheless, both 20 and its monogold analogue 19 exhibited irreversible, 2-electron oxidation events by cyclic voltammetry, with the 2-electron oxidation of 19 occurring at a higher potential (1.96 V) than that of 20 (1.64 V).19 Thus, though the aurophilic interaction proved unobservable by X-ray crystallography in the case of 20, our results suggest the existance of a weak and fluxional interaction in solution with a measurable impact on redox potential. This causes digold(I) complex 20 to be oxidized at a milder potential than monogold complex 19. The compiled electrochemistry results presented herein are summarized in Table 1. Lastly, we note that DFT calculations using the M06 functional and LACV3P**++(2f) basis set correctly reproduce the observed trend in redox potentials and predict a difference of 0.26 V in the oxidation potentials of complexes 19 and 20 (see Supporting Information), which can be viewed as a validation of this DFT method for analyzing bimetallic gold complexes.
Table 1.
Summary of electrochemistry data.
| Complex | Eox for Au(I) (V)a | Ered for Au(III) (V)a |
|---|---|---|
| 15 | 1.48b | / |
| 16 | / | −0.69 |
| 17 | 1.48b | −0.53 |
| 18 | 1.34b | / |
| 19 | 1.96c | / |
| 20 | 1.64c | / |
Referenced to FeCp2+/FeCp2.
Potential of first 1-electron oxidation.
Potential of 2-electron oxidation.
Aminoauration
Binuclear intermediates also appear to be key to aminoauration. For example, our calculations showed that using [Ph3PAu]+, aminoauration does not lead to a stable cyclized intermediate. Relaxed coordinate scans of the cyclization reaction using [Ph3PAu]+, exhibited an uphill process with no minimum for the cyclized intermediate (see Supporting Information). This may seem counterintuitive since aminoauration has been described before;20 however, the substrates that cyclize successfully involve stronger nucleophiles than amides. Because ionization by solvation (heterolysis) of the gold halide LAuCl is unfavorable (predicted to be endothermic by ~30 kcal/mol in acetonitrile),21 we envision that a mononuclear mechanism would involve oxidation of LAuX, followed by exothermic (by 12.9 kcal/mol) coordination of the resulting gold(III) complex with the alkene. Subsequent intramolecular base-assisted (amine originating from the reduction byproduct of Selectfluor) addition of the amide nucleophile then would lead to a cyclized phosphinegold(III) intermediate.
On the other hand, bimetallic aminoauration was indeed found to be a downhill process according to DFT (Scheme 3). A transition state for the aminoauration pathway was located using the binuclear catalysts with PPh3 and dppm ligands. We find the aminoauration barrier using both PPh3 and dppm ligands to be ~8 kcal/mol (8.8 kcal/mol for PPh3 and 7.7 kcal/mol for dppm).
Reductive elimination
DFT study
Probably, the largest discrepancy between the proposed mechanisms is transmetalation. Zhang initially proposed a mechanism involving arylboronic acid transmetalation (before or after oxidation) based on the reactivity that boronates display in analogous to d10 metal-catalyzed cross coupling reactions.22 We were unable to observe transmetallation of arylboronic acids, arylsilanols, and aryltrimethylsilanes with LAuX under relevant non-catalytic reaction conditions [Eq. (2) and (3)].6 However, these tests do not rule out a reversible process with a Keq that greatly favors reactants. Thus, transmetalation was tested under catalytic conditions by subjecting substrate and active catalyst PPh3AuPh to Selectfluor in acetonitrile [Eq. (4)]. The lack of reactivity in the absence of an exogenous arylboronic acid, together with the recovered reactivity upon addition of PhB(OH)2 [Eq. (5)] argues against a pathway involving direct transmetalation. Notably, it was observed that the transferred aryl group originates from the arylboronic acid and not from the phenylgold species [Eq. (6)], conclusively establishing the absence of transmetalation under the present reaction conditions. We investigated these observations computationally; relaxed coordinate scans for transmetalation to Au(III) fluoride species were not able to locate a viable pathway and exhibited high energies (>80 kcal) without formation of a Au-Ph bond. These data suggest that transmetalation to Au(I) and Au(III) species within the catalytic cycle of oxidative heteroarylation is not operative.
Given the absence of direct transmetalation, we posited that C-aryl bond formation resulting from traditional reductive elimination should also be inoperative.23 We therefore focused on investigating likely C–C bond formation steps. Considering the exothermicity provided by the formation of a B–F (or Si–F) bond, we examined the minimum energy pathway by decreasing the distance between arylboronic acid and Au(III) fluoride species. Relaxed coordinate scans exhibited concerted reductive elimination with formation of both C–C and B–F bonds, effectively achieving regeneration of the catalyst and product demetalation in one step. We found a concerted but asynchronous five-membered cyclic transition structure (Figure 4a), in which the B–F bond is formed prior to the C–C bond. The polarized Au–F bond and the B–F interaction are key for reductive elimination.
Figure 4.

Transition structures for the bimolecular reductive elimination from a) mononuclear PPh3Au, b) binuclear [PPh3Au]2, and c) binuclear (dppm)Au2 complexes.
Stereochemical analysis
To further characterize the nature of C–C reductive elimination, we sought to determine the stereochemical course of this reaction. Stereochemical analyses of the overall catalytic aminoarylation reaction have been performed both by Zhang5b and Toste6b, but different conclusions regarding the stereochemical course of C–C bond formation were reached depending on whether syn- or anti-aminoauration was assumed. To gain further insight, herein we focus now on exploiting our previously reported ability to isolate6c alkylgold(III) fluoride complexes [(IPr)Au(R)F2] (IPr = 1,3-bis(2,6-di-iso-propylphenyl)imidazol-2-ylidene) and thus study C–C reductive elimination as a stoichiometric transformation. In particular, we chose to examine the coupling of PhB(OH)2 with [(IPr)Au(R)F2] bearing the neo-hexyl-d2 substituent (i.e., R = syn- or anti-CHDCHDtBu), whose relative stereochemistry can be easily monitored by measuring the vicinal 3JHH coupling in routine 1H NMR experiments (Scheme 4).24
Scheme 4.

Stereochemistry of C–C reductive elimination.
Reaction of syn-tBuCHDCHDB(OH)2 (3JHH = 4.2 Hz) with (IPr)AuOH 25 resulted in clean transmetalation with inversion of stereochemistry, producing anti-tBuCHDCHDAu(IPr) (3JHH = 11.2 Hz) in good yield. Because alkyl transfer from boron to gold is unprecedented,26 connectivity of the unlabeled analogue tBuCH2CH2Au(IPr) was established by X-ray crystallography (see Supporting Information). To our knowledge, the stereochemistry of B-to-Au transmetalation has not been studied previously. The stereochemistry of B-to-Pd transmetalation has been studied previously by analysis of catalytic reaction products, with inversion of stereochemistry27 during transmetalation being postulated less commonly than retention of stereochemistry28.
In order to test the validity of our experimental design, we chose to examine the stereochemical course of C–I reductive elimination from Au(III). Scott et al. have shown29 that reaction of (IPr)AuMe with I2 results in oxidative addition to form trans-(IPr)AuMeI2 as a transient intermediate, followed by reductive elimination of MeI. A detailed kinetics analysis in that report indicated inner-sphere C–I reductive elimination rather than SN2-like attack of outer-sphere I− on a cationic methylgold(III) intermediate. Thus, C–I reductive elimination is expected to proceed with retention of stereochemistry. Indeed, we found that reaction of anti-tBuCHDCHDAu(IPr) with I2 resulted in quantitative conversion to anti-tBuCHDCHDI (3JHH = 12.8 Hz).
We have previously shown that oxidation of (IPr)AuMe with XeF2 produces cis-(IPr)AuMeF2, which reacts with PhB(OH)2 to produce toluene.6c In order to test the stereochemical course of this C–C reductive elimination process, we oxidized anti-tBuCHDCHDAu(IPr) with XeF2 to generate cis-[antitBuCHDCHDAuF2(IPr)]. 19F NMR properties of this oxidized intermediate were consistent with those previously reported for cis-(IPr)AuMeF2. Subsequent reaction with PhB(OH)2 proceeded with retention of stereochemistry, producing anti-tBuCHDCHDPh (3JHH = 12.4 Hz) as the sole C–C coupling product (Scheme 4). Analogous results were obtained by performing the C–C coupling with syntBuCHDCHDAu(IPr) (3JHH = 5.4 Hz) to yield syn-tBuCHDCHDPh (3JHH = 5.0 Hz).
Electronic structure and halide effect
Given the importance of the concerted formation of the B–F (or Si–F) bond in the cross coupling step, we expect that the more polarized the Au–F will lower the barrier for the reductive carbon-carbon bond formation. We observe this structurally with calculated bond distances and electronically with NBO analyses (Table 2).30 The Au–F bond distance for the intermediate that precedes reductive elimination increases from 2.02 Å for the PPh3 mononuclear complex, to 2.07 Å for the PPh3 binuclear complex. Electronically, the natural charge on F atom is −0.70 e for the mononuclear complex and −0.81 e for the binuclear complex. This effect has been observed before as a structural trans effect and trans influence that is amplified by the Au–Au bonding system.31 In addition, it is expected that a bromide ligand further polarizes the trans Au–F bond relative to a chloride. Indeed, we find that the Au–F bond is elongated by ~0.01 Å with respect to the Cl complex (Table 2). Moreover, the charge on fluorine is 0.01e more negative in natural charge in the F–Au–Au–Br complex compared to the F–Au–Au–Cl tetrad. The natural atomic orbital bond orders follow this trend; the Au–F natural bond order decreases from 0.233 for F–Au–Cl to 0.218 for F–Au–Br to 0.169 for F–Au–Au–Cl to 0.160 for F–Au–Au–Br. Taken together, these observations suggest that bromide ligand increases the polarization of the Au–F bond, which is key for the reductive elimination to occur; this hypothesis is in agreement with the higher activity of the dppm/bromide catalyst. The molecular orbitals for the intermediate that undergoes reductive elimination support this view (Figure 5). The HOMO is bonding for C–Au–Au and antibonding for F–Au and Au–Cl. Using NBO analyses, we also characterized the Au–Au bond as a polarized interaction that involves the 5dz2 orbital with partial 6s (~7%) character on the donor Aub atom to an empty orbital on Aua that is mostly 6s character (~87%) with partial 5dz2 character (~11%). This molecular orbital (HOMO-54) is shown in Figure 5.
Table 2.
Selected bond distances, natural bond orders and natural atomic charges of mononuclear [(PPh3)(AuX)F(Cyclized Substrate)]and binuclear Au intermediates [(PPh3)2(AuX)2F(Cyclized Substrate)].
| Bond Distances and Natural Bond Orders | ||||
|---|---|---|---|---|
| F–Au–Cl | F–Au–Br | F–Aua–Aub–Cl | F–Aua–Aub–Br | |
| Au–F | 1.98 Å (0.233) | 1.99 Å (0.218) | 2.06 Å (0.175) | 2.07 Å (0.160) |
|
| ||||
| Au–X | 2.30 Å (0.418) | 2.48 Å (0.394) | 2.49 Å (0.260) | 2.67 Å (0.228) |
|
| ||||
| Au–Au | – | – | 2.64 Å (0.232) | 2.66 Å (0.236) |
|
| ||||
| Natural Atomic Charges | ||||
|
| ||||
| Aua | +0.905 | +0.850 | +0.819 | +0.806 |
| Aub | / | / | +0.615 | +0.510 |
|
| ||||
| X | −0.446 | −0.354 | −0.545 | −0.561 |
|
| ||||
| F | −0.667 | −0.675 | −0.732 | −0.729 |
Figure 5.

Selected molecular orbitals for key intermediate 12-dppm [(dppm)(AuCl)2F(Cyclized Substrate)].
Conclusions
In summary, based on combining both experimental observations and computational investigations, we propose a mechanism for gold-catalyzed heteroarylation reactions that is composed of three main steps: 1) oxidation of Au(I) by Selectfluor, 2) activation of the alkene followed by nucleophilic addition, and 3) bimolecular reductive elimination. Our calculations show that that access to binuclear Au(II)–Au(II) intermediates lowers the barriers for all three of these steps, supported by our cyclic voltammetry data. In accord with our experimental findings, our computational studies indicate that the Au–F bond plays a key role in the bimolecular reductive elimination. Thus, we conclude that the spectator halide (Br vs Cl) in the gold catalysts impacts reactivity by changing the nature of the Au–F bond.
An overall proposed catalytic cycled based on all experimental and computational data presented herein is shown in Scheme 5 for the catalyst dppm(AuBr)2. Facile oxidation of the aurophilic Au(I)Au(I) catalyst with Selectfluor is partially driven by Au(II)-Au(II) σ-bond formation, yielding cationic [dppm(AuBr2)(AuF)]+. Heteroauration then occurs in an anti sense to give a neutral alkylgold species. Concerted bimolecular reductive elimination with an arylboronic acid coupling partner then occurs with retention of stereochemistry to give a catalytic process with overall inversion of stereochemistry as has been noted before.5b–c,6b
Scheme 5.

Revised mechanism for heteroarylation of alkenes catalyzed by dppm(AuBr)2.
Supplementary Material
Acknowledgments
This material is based upon work supported as part of the Center for Catalytic Hydrocarbon Functionalization, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001298. Computational facilities were funded by grants from ARO-DURIP and ONR-DURIP. N.P.M. was supported by an NIH Kirchstein-NRSA postdoctoral fellowship. Prof. Chris Chang provided access to a potentiostat for cyclic voltammetry measurements.
Footnotes
Supporting Information Available: Synthetic details, computational details and XYZ coordinates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem-Int Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]
- 2.For general reviews of gold catalysis see: Shapiro ND, Toste FD. Synlett. 2010:675. doi: 10.1055/s-0029-1219369.Fürstner A. Chem Soc Rev. 2009;38:3208. doi: 10.1039/b816696j.Jiménez-Núñez E, Echavarren AM. Chem Rev. 2008;108:3326. doi: 10.1021/cr0684319.Shen HC. Tetrahedron. 2008;64:7847.Gorin DJ, Sherry BD, Toste FD. Chem Rev. 2008;108:3351. doi: 10.1021/cr068430g.Li Z, Brouwer C, He C. Chem Rev. 2008;108:3239. doi: 10.1021/cr068434l.Arcadi A. Chem Rev. 2008;108:3266. doi: 10.1021/cr068435d.Hashmi ASK. Chem Rev. 2007;107:3180. doi: 10.1021/cr000436x.
- 3.For early examples of this reactivity using cationic phosphinegold(I) complexes, see: Teles JH, Brode S, Chabanas M. Angew Chem, Int Ed. 1998;37:1415. doi: 10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.0.CO;2-N.Mizushima E, Sato E, Hayashi K, Tanaka TM. Angew Chem, Int Ed. 2002;41:4563. doi: 10.1002/1521-3773(20021202)41:23<4563::AID-ANIE4563>3.0.CO;2-U.Reetz MT, Sommer K. Eur J Org Chem. 2003:3485.Mizushima E, Hayashi T, Tanaka M. Org Lett. 2003;5:3349. doi: 10.1021/ol0353159.Nieto-Oberhuber C, Munuz MP, Bunuel E, Nevado C, Cardenas DJ, Echavarren AM. Angew Chem, Int Ed. 2004;43:2403. doi: 10.1002/anie.200353207.Kennedy-Smith JJ, Staben ST, Toste FD. J Am Chem Soc. 2004;126:4526. doi: 10.1021/ja049487s.Mamane V, Gress T, Krause H, Fürstner A. J Am Chem Soc. 2004;126:8654. doi: 10.1021/ja048094q.Luzung MR, Markham JP, Toste FD. J Am Chem Soc. 2004;126:10858. doi: 10.1021/ja046248w.
- 4.Hashmi SK, Ramamurthi TD, Rominger F. J Organomet Chem. 2009;694:592. [Google Scholar]
- 5.(a) Zhang GZ, Peng Y, Cui L, Zhang LM. Angew Chem-Int Ed. 2009;48:3112–3115. doi: 10.1002/anie.200900585. [DOI] [PubMed] [Google Scholar]; (b) Zhang GZ, Cui L, Wang YZ, Zhang LM. J Am Chem Soc. 2010;132:1474–1475. doi: 10.1021/ja909555d. [DOI] [PubMed] [Google Scholar]; (c) Zhang G, Luo Y, Wang Y, Zhang L. Angew Chem Int Ed. 2011;50:4450. doi: 10.1002/anie.201100293. [DOI] [PubMed] [Google Scholar]
- 6.(a) Melhado AD, Brenzovich WE, Lackner AD, Toste FD. J Am Chem Soc. 2010;132:8885–8886. doi: 10.1021/ja1034123. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brenzovich WE, Benitez D, Lackner AD, Shunatona HP, Tkatchouk E, Goddard WA, Toste FD. Angew Chem-Int Ed. 2010;49:5519–5522. doi: 10.1002/anie.201002739. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mankad NP, Toste FD. J Am Chem Soc. 2010;132:12859–12861. doi: 10.1021/ja106257n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Brenzovich WE, Brazeau JF, Toste FD. Org Lett. 2010;12:4728. doi: 10.1021/ol102194c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ball LT, Green M, Lloyd-Jones GC, Russel CA. Org Lett. 2010;12:4724. doi: 10.1021/ol1019162.Wang W, Jasinski J, Hammond GB, Xu B. Angew Chem Int Ed. 2010;49:7247. doi: 10.1002/anie.201003593.Hopkinson MN, Tessier A, Salisbury A, Giuffedi GT, Combettes LE, Gee AD, Gouverneur V. Chem Eur J. 2010;16:7443. doi: 10.1002/chem.201000322.de Haro T, Nevado C. Angew Chem-Int Ed. 2011;50:906–910. doi: 10.1002/anie.201005763.For a recent reviews see: Engle KM, Mei TS, Wang X, Yu JQ. Angew Chem-Int Ed. 2011;50:1478. doi: 10.1002/anie.201005142.Hopkins MN, Gee AD, Gouverneur V. Chem Eur J. doi: 10.1002/chem.201100736.
- 8.(a) Chuang GJ, Wang W, Lee E, Ritter T. J Am Chem Soc. 2011;133:1760. doi: 10.1021/ja108396k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Powers DC, Xiao DY, Geibel MAL, Ritter T. J Am Chem Soc. 2010;132:14530–14536. doi: 10.1021/ja1054274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Powers DC, Benitez D, Tkatchouk E, Goddard WA, Ritter T. J Am Chem Soc. 2010;132:14092–14103. doi: 10.1021/ja1036644. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Powers DC, Geibel MAL, Klein JEMN, Ritter T. J Am Chem Soc. 2009;131:17050–17051. doi: 10.1021/ja906935c. [DOI] [PubMed] [Google Scholar]; (e) Tang P, Furuya T, Ritter T. J Am Chem Soc. 2010;132:12150–12154. doi: 10.1021/ja105834t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Powers DC, Ritter T. Nat Chem. 2009;1:302–309. doi: 10.1038/nchem.246. [DOI] [PubMed] [Google Scholar]
- 9.(a) Yam VWW, Li CK, Chan CL, Cheung KK. Inorg Chem. 2001;40:7054–7058. doi: 10.1021/ic010744t. [DOI] [PubMed] [Google Scholar]; (b) Bhargava SK, Mohr F, Bennett MA, Welling LL, Willis AC. Organomet. 2000;19:5628–5635. [Google Scholar]; (c) Bennett MA, Bhargava SK, Mirzadeh N, Priver SH, Wagler J, Willis AC. Dalton Trans. 2009:7537–7551. doi: 10.1039/b906769h. [DOI] [PubMed] [Google Scholar]; (d) Jiang Y, Alvarez S, Hoffmann R. Inorg Chem. 1998;37:3018. [Google Scholar]; (e) Pyykko P, Mendizabal F. Inorg Chem. 1998;37:3018. [Google Scholar]; (f) Melgarejo DY, Chiarella GM, Fackler JP, Jr, Perez LM, Rodrigue-Witchel A, Reber C. Inorg Chem. 2011;50:4238. doi: 10.1021/ic200394q. [DOI] [PubMed] [Google Scholar]
- 10.(a) Fernandez EJ, Hardacre C, Laguna A, Lagunas MC, Lopez-de-Luzuriaga JM, Monge M, Montiel M, Olmos ME, Puelles RC, Sanchez-Forcada E. Chem Eur J. 2009;15:6222–6233. doi: 10.1002/chem.200900167. [DOI] [PubMed] [Google Scholar]; (b) Wu J, Kroll P, Dias HVR. Inorg Chem. 2009;48:423–425. doi: 10.1021/ic8020854. [DOI] [PubMed] [Google Scholar]
- 11.Schmidbaur H, Schier A. Chem Soc Rev. 2008;37:1931–1951. doi: 10.1039/b708845k. [DOI] [PubMed] [Google Scholar]; (b) Partyka DV, Updegraff JB, Zeller M, Hunter AD, Gray TG. Dalton Trans. 2010:5388–5397. doi: 10.1039/b920717a. [DOI] [PubMed] [Google Scholar]
- 12.Pan QJ, Zhou X, Guo YR, Fu HG, Zhang HX. Inorg Chem. 2009;48:2844–2854. doi: 10.1021/ic801687w. [DOI] [PubMed] [Google Scholar]
- 13.To obtain the enthalpy and free energy at 298K, we combine the QM electronic energy (including solvation) with QM vibrational frequencies (unscaled) inserted into the appropriate quantum statistical relations. For the small organic species we use the full ideal gas vibrational, rotational, and translational components. For the metal complexes, we use the vibrational components to the entropy plus 6R (Stot= Svib + 6R) to account for the change of rotational and translational degrees of freedom due to formation of a product molecule. We have found this methodology to be adequate for solution phase free energy calculations, see: Benitez D, Tkatchouk E, Goddard WA. Organomet. 2009;28:2643–2645.
- 14.All potentials are referenced to the FeCp2+/FeCp2 couple.
- 15.Teets TS, Nocera DG. J Am Chem Soc. 2009;131:7411–7420. doi: 10.1021/ja9009937. [DOI] [PubMed] [Google Scholar]
- 16.de la Riva H, Pintado-Alba A, Nieuwenhuyzen M, Hardacre C, Lagunas MC. Chem Commun. 2005:4970. doi: 10.1039/b508863a. [DOI] [PubMed] [Google Scholar]
- 17.(a) Schmidaur H, Wohllenben A, Wagner F, Orama O, Huttner G. Chem Ber. 1977;110:1748. [Google Scholar]; (b) Healy PC. Acta Cryst E. 2003;59:m1112. [Google Scholar]
- 18.Fackler JP., Jr Inorg Chem. 2002;41:6959. doi: 10.1021/ic025734m. [DOI] [PubMed] [Google Scholar]
- 19.In the case of these carbene complexes, we hypothesis that the observed 2-electron oxidation is a result of two unresolved 1-electron redox events.
- 20.(a) LaLonde RL, Brenzovich WE, Benitez D, Tkatchouk E, Kelley K, Goddard WA, Toste FD. Chem Sci. 2010;1:226–233. doi: 10.1039/C0SC00255K. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang ZJ, Benitez D, Tkatchouk E, Goddard WA, III, Toste FD. J Am Chem Soc. 2010;132:13064–13071. doi: 10.1021/ja105530q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Less coordinating anions exhibited lower yields, see ref 6b.
- 22.Ph3PAuBr transmetalates with PhB(OH)2 to 59% yield in the presence of Cs2CO3 at 50°C after 24h, see: Partyka DV, Zeller M, Hunter AD, Gray TG. Angew Chem-Int Ed. 2006;45:8188–8191. doi: 10.1002/anie.200603350.
- 23.Our studies do not exclude the possibility of reductive elimination from binuclear Au(II) or mononuclear (III) centers as proposed by Gouverneur and co-workers, see: Hopkinson MN, Ross JE, Giuffredi GT, Gee AD, Gouverneur V. Org Lett. 2010;12:4904–4907. doi: 10.1021/ol102061k.
- 24.Whitesides GM, Boschetto DJ. J Am Chem Soc. 1969;91:4313. [Google Scholar]
- 25.Gaillard S, Slawin AMZ, Nolan SP. Chem Commun. 2010;46:2742. doi: 10.1039/c0cc00018c. [DOI] [PubMed] [Google Scholar]
- 26.Hashmi, et al. have reported the preparation of Ph3PAuEt from Ph3PAuCl and EtB(OH)2 in the presence of Cs2CO3, but the reported characterization data does not match that of independently synthesized Ph3PAuEt. See ref. 4.
- 27.Selected references: Ohmura T, Awano T, Suginome M. J Am Chem Soc. 2010;132:13191. doi: 10.1021/ja106632j.Sandrock DL, Jean-Gérard L, Chen C-y, Dreher SD, Molander GA. J Am Chem Soc. 2010;132:17108. doi: 10.1021/ja108949w.
- 28.Selected references: Ridgway BH, Woerpel KA. J Org Chem. 1998;63:458. doi: 10.1021/jo970803d.Matos K, Soderquist JA. J Org Chem. 1998;63:461. doi: 10.1021/jo971681s.Wendt OF. Curr Org Chem. 2007;11:1417.
- 29.Scott VJ, Labinger JA, Bercaw JE. Organometallics. 2010;29:4090. [Google Scholar]
- 30.Reed AE, Weinhold F. J Chem Phys. 1985;83:1736–1740. [Google Scholar]; (b) Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735–746. [Google Scholar]
- 31.(a) Basil JD, Murray HH, Fackler JP, Tocher J, Mazany AM, Trzcinskabancroft B, Knachel H, Dudis D, Delord TJ, Marler DO. J Am Chem Soc. 1985;107:6908–6915. [Google Scholar]; (b) Flemming JP, Pilon MC, Borbulevitch OY, Antipin MY, Grushin VV. Inorg Chim Acta. 1998;280:87–98. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.