

Abstract
RBP is a cellular protein that functions as a transcriptional repressor in mammalian cells. RBP has elicited great interest lately because of its established roles in regulating gene expression, in Drosophila and mouse development, and as a component of the Notch signal transduction pathway. This report focuses on the mechanism by which RBP represses transcription and thereby regulates expression of a relatively simple, but natural, promoter. The results show that, irrespective of the close proximity between RBP and other transcription factors bound to the promoter, RBP does not occlude binding by these other transcription factors. Instead, RBP interacts with two transcriptional coactivators: dTAFII110, a subunit of TFIID, and TFIIA to repress transcription. The domain of dTAFII110 targeted by RBP is the same domain that interacts with TFIIA, but is disparate from the domain that interacts with Sp1. Repression can be thwarted when stable transcription preinitiation complexes are formed before RBP addition, suggesting that RBP interaction with TFIIA and TFIID perturbs optimal interactions between these coactivators. Consistent with this, interaction between RBP and TFIIA precludes interaction with dTAFII110. This is the first report of a repressor specifically targeting these two coactivators to subvert activated transcription.
Keywords: RBP, transcriptional repression, TFIIA/TFIID targeting
The role of the cellular RBP protein in gene regulation has been established fairly recently. Earlier genetic studies demonstrated a pivotal role for the Drosophila homolog of RBP in development (Nash 1965, 1970; Furukawa et al. 1992; Schweisguth and Posakony 1992; Tun et al. 1994). Initial studies on mammalian RBP contributed biochemical and genetic characterizations, although RBP was thought to be a recombinase at that time (Hamaguchi et al. 1989, 1992; Matsunami et al. 1989; Kawaichi et al. 1992). Since then, RBP has been recognized to be a transcription factor that represses mammalian gene expression (Dou et al. 1994; Kannabiran et al. 1997; Plaisance et al. 1997; Oswald et al. 1998), but activates Drosophila gene expression (Brou et al. 1994). RBP has recently been implicated in the Notch signal transduction pathway in Drosophila, which may bridge the role of RBP in gene expression and development (Fortini and Artavanis-Tsakonas 1994; Bailey and Posakony 1995; Lecourtois and Schweisguth 1995; Hsieh et al. 1996; Eastman et al. 1997). That RBP has a pivotal regulatory role in gene expression is highlighted by its interaction with viral and cellular proteins that modulate its activity [Drosophila Hairless (Brou et al. 1994); Epstein–Barr encoded EBNA2 (Grossman et al. 1994; Henkel et al. 1994; Laux et al. 1994; Waltzer et al. 1994; Zimber-Strobl et al. 1994); EBNA3A,B,C (Robertson et al. 1996); and the mammalian proteins Notch (Fortini and Artavanis-Tsakonas 1994; Hsieh et al. 1996)] and KyoT2 (Taniguchi et al. 1998).
The identification of RBP as a transcriptional repressor in mammalian cells was established during studies of virus gene expression. The important role of RBP in regulating gene expression in the mature cell is highlighted by its sequestration during virus infection. Both adenovirus and Epstein-Barr virus evolved to sequester RBP for viral advantage. The adenoviral gene encoding the capsid protein polypeptide IX (pIX) is expressed only after viral DNA replication in infected cells. We found that the pIX promoter contained a repressive element that was specifically bound by cellular RBP. RBP was shown to repress pIX expression before viral DNA replication occurs; this repression was dependent on the presence of the RBP-specific DNA-binding site within the pIX promoter. Furthermore, purified RBP protein was shown to repress pIX transcription in a reconstituted transcription assay performed in vitro (Dou et al. 1994).
RBP was shown to be usurped during Epstein-Barr virus infection by the potent transcriptional activator EBNA2. EBNA2 lacks DNA-binding activity, but a complex formed between RBP and EBNA2 facilitates EBNA2 tethering to the DNA via the RBP-specific DNA-binding activity (Grossman et al. 1994; Henkel et al. 1994; Laux et al. 1994; Waltzer et al. 1994; Zimber-Strobl et al. 1994). Transcriptional activation by EBNA2 is expedited not only by RBP-mediated access to specific promoters, but also by EBNA2 masking of the RBP repression domain (Hsieh and Hayward 1995).
Transcriptional repression in eukaryotes has been appreciated relatively recently (for review, see Herschbach and Johnson 1993; Johnson 1995). Several repressors have been identified and their critical role in regulating gene expression has been established. Similar to the precedents set by studies of prokaryotic repressors, different eukaryotic repressors that target distinct components and stages of the transcription process have been documented recently. Some repressors target activated transcription by competing with activators for overlapping DNA-binding sites (e.g., Oct-1 and C/EBP; Wu et al. 1997). Other repressors interact with activators to mask activation domains (e.g., MDM2 and p53; Oliner et al. 1993). Activator–repressor interactions can also serve to tether the repressor to specific promoters. Then the repressor can effectively compete with other activators for basal transcription factors (e.g., MDM2 and TFIIE–TBP; Thut et al. 1997) or compete with a basal transcription factor for interaction with other activators (e.g., pRb and TBP; Weintraub et al. 1995). Some repressors complex with basal transcription factors to exclude interaction with other factors (Dr1 and TBP; Inostroza et al. 1992), whereas others can access promoters directly and target candidate basal transcription factors to silence gene expression (e.g., Kruppel and TFIIB–TFIIEβ; Sauer et al. 1995). RBP is a transcriptional repressor with specific DNA-binding activity, and RBP binding was known to be required for repression, yet the molecular basis of RBP-mediated repression was not known.
Our initial studies of the adenoviral pIX gene suggested that RBP-mediated repression does not involve competition between RBP and other transcription factors for promoter access (Dou et al. 1994). The pIX promoter is relatively simple, containing a single site for the cellular transcription factor Sp1 and a consensus TATA box (Babiss and Vales 1991). The RBP-specific DNA-binding site lies between these two positions immediately upstream of the TATA box. However, RBP binding did not dislodge either Sp1 or TBP from their respective sites. With this as a basis, we pursued the molecular mechanism by which RBP successfully silences pIX transcription.
Using a transcription assay reconstituted with recombinant factors and highly purified native factors, we established conditions for Sp1-activated pIX transcription and RBP-mediated repression. Repression was dependent on the presence of the RBP-binding site and was also dependent on the position of the RBP site within the pIX promoter. However, RBP-mediated repression could be thwarted when a transcription preinitiation complex intermediate composed of Sp1, TFIIA, and TFIID was formed on the pIX promoter. Consistent with our previous report showing that RBP does not occlude Sp1 or TBP binding, RBP does not occlude binding of Sp1, TFIIA, and TFIID. All four proteins bind the pIX promoter concomitantly. Instead of repressing by occlusion, our results show that RBP mediates repression by direct interaction with two coactivators: TFIIA and the TAFII110 subunit of TFIID. Moreover, the component of TFIID targeted by RBP is the same TAF that interacts with TFIIA as well as with the activation domains of Sp1, the only activator required for optimal pIX expression. However, our results also show that the domain of TAFII110 targeted by RBP is the same as that which interacts with TFIIA, but distinct from that which interacts with Sp1. Furthermore, interaction between RBP and TFIIA precludes interaction of TAFII110. Therefore, RBP interaction with TFIID and TFIIA alters optimal interaction between these two coactivators, not to dislodge them from the promoter, but instead to subvert activated transcription.
Results
Repression depends on the position of the RBP site in vivo
Figure 1a shows the simple adenoviral pIX promoter containing a single Sp1 site, a single RBP site, and a consensus TATA box. Our previous studies showed that this region is sufficient for pIX regulation, that RBP-mediated repression required the RBP-binding site, and that RBP binding did not dislodge Sp1 or TBP from the pIX promoter (Dou et al. 1994). We initiated our studies to identify the molecular basis of RBP repression by testing whether the position of the RBP site immediately upstream of the TATA box was fortuitous or important in repression.
Figure 1.


The position of the RBP-binding site determines RBP-mediated repression in vivo. (a) Schematic representation of the different pIX promoter constructs used in this study. The wild-type (WT) pIX promoter, the Sub9 promoter containing a linker substitution of the RBP-binding site, and the pIX promoter containing repositioned RBP-binding sites are shown (see text for description). (Arrows) Start sites of pIX transcription (+1); (boxes) position of the TATA box, Sp1, and RBP sites as indicated. In all cases, the distance between the Sp1 site and the TATA box was conserved so that Sp1-activated transcription would be expected to be the same in all cases. (b) Results from an RNase T2 protection assay with an antisense probe that spans the start site of pIX transcription and equivalent aliquots of RNA isolated from F9 cells transfected with the pIX constructs indicated at the top of the lanes. In all cases, cells were cotransfected with an expression vector for RBP and internal control for transfection efficiency (SV/dl17). The levels of pIX mRNA and internal control are scored within the same assay (see Materials and Methods). (Arrow) Protected fragment expected for pIX mRNA. The larger protected fragment derives from the internal control. The smaller protected fragment derives from transcription that initiates within vector sequences upstream of the pIX promoter and utilizes the acceptor splice site downstream of the pIX start site; this protected fragment serves as another internal control for transfection efficiency (see Materials and Methods).
The single RBP site was repositioned at several distinct sites within the promoter region of the pIX gene. In each case, the wild-type RBP site was substituted with linker sequence and the optimal distance between the Sp1 site and the TATA consensus was maintained (Fig. 1a). In two cases, the RBP site was repositioned upstream of the Sp1 site. RBP/20nt/Sp1 contains an RBP site 20 nucleotides upstream of the Sp1 site, whereas RBP/Sp1 contains an RBP site immediately upstream of the Sp1 site, similar to its normal position immediately upstream of the TATA box. In two other cases, the RBP site was maintained between Sp1 activator and TATA and placed either immediately downstream of Sp1 (Sp1/RBP) or centered between the two sites (RBP center). The repositioned RBP site restores RBP-specific DNA-binding activity in each case (data not shown). The Sub9 control contains only the linker substitution in lieu of the RBP site.
We first compared the levels of pIX expression from each of these constructs in transfected F9 cells (Fig. 1b). As shown previously with virus infections, the presence of the RBP site is repressive to pIX gene expression; wild-type pIX expression is barely detectable as opposed to the clearly detectable levels of pIX expression from the Sub9 construct that lacks the RBP-binding site (cf. lanes 1 and 2). The two constructs that maintained RBP binding between Sp1 and TATA retained susceptibility to repression (lanes 5, 6). On the other hand, the two constructs containing a single RBP site upstream of Sp1 gave rise to similar pIX expression as that observed with Sub9 (cf. lanes 2–4). This result suggested that repression was relieved when the RBP DNA target site was upstream of the activator.
Our previous results showed that RBP, Sp1, and TBP can co-bind the wild-type pIX promoter (Dou et al. 1994). However, the possibility existed that RBP binding may occlude the TFIID complex, thereby resulting in repression in the wild-type case and relief from repression in the RBP/Sp1 case in which RBP is distanced from the TATA box. We first tested RBP co-binding with TFIID and Sp1 at the wild-type pIX promoter using DNase I footprinting analyses (Fig. 2a). These results show that RBP and Sp1 co-bind the pIX promoter (cf. lanes 1–4), RBP and TFIID co-bind the pIX promoter (cf. lanes 1, 2, 5, and 6), and Sp1, TFIID, and RBP co-bind the pIX promoter (cf. lanes 1, 2, 7, and 8). Therefore, repression is not mediated by RBP occlusion of TFIID in the wild-type case.
Figure 2.

RBP, Sp1, and TFIID co-bind the pIX promoter. (a) DNase I footprinting analysis on the coding strand of the wild-type pIX promoter as a function of rRBP, Sp1, and epitope-tagged TFIID proteins either alone or in combination as indicated on the top of each lane. (b) Similar analysis for rRBP and Sp1 on the coding strands of either wild-type or RBP/Sp1 pIX promoter constructs. Brackets enclose the protected region of the pIX promoter associated with binding by each protein. (G and G+A) Maxam–Gilbert sequencing reactions.
We similarly tested each of the constructs containing a repositioned RBP site. In the case of Sp1/RBP and RBP center, which exhibited repression in the presence of RBP in vivo (Fig. 1b), RBP binding at the repositioned site led to diminished Sp1 binding (data not shown). Therefore, the presence of RBP destabilized Sp1 binding in the Sp1/RBP and RBP center cases; this, no doubt, contributed to pIX repression from Sp1/RBP and RBP center in a manner disparate from the wild-type case in which all three factors co-bind. On the other hand, RBP and Sp1 did co-bind the RBP/Sp1 promoter similar to the wild-type case (Fig. 2b). Therefore, relief from RBP repression in the RBP/Sp1 case was not attributable to Sp1 occlusion of RBP binding, for example. This result strongly suggested that pIX repression was dependent on the position of the single RBP site within the pIX promoter.
Repression depends on the position of the RBP site in vitro
Because the comparative analysis shown in the previous section was performed in vivo, the possibility existed that chromatin structure may affect pIX gene expression from the constructs containing repositioned RBP sites, particularly RBP/20nt/Sp1 and RBP/Sp1, which were relieved from repression. To address this possibility and to establish conditions to examine RBP repression at the molecular level, each of these constructs was similarly compared for Sp1-activated pIX levels and susceptibility to RBP repression in transcription reactions performed in vitro. First, optimal levels of pIX transcription were reconstituted with purified recombinant transcription factors (TFIIB, TFIIE, and TFIIF), highly purified TFIIH and RNA polymerase II isolated from HeLa cells, purified epitope-tagged TFIID, and the purified recombinant coactivators TFIIA and PC4, in the absence and presence of Sp1 activator; each reaction included template with the wild-type pIX promoter and control template with the major late promoter, which does not contain Sp1 or RBP sites (see Materials and Methods). Figure 3a shows the levels of basal and Sp1-activated pIX transcription obtained under these conditions. Sp1 activation of pIX transcription is clearly detectable and dependent on the coactivators TFIIA and PC4.
Figure 3.

RBP represses pIX transcription in a highly purified in vitro transcription system. (a) Results of a transcription reaction performed in vitro with the wild-type pIX promoter. Transcription reactions contained two templates: the wild-type pIX promoter fused to a transcription unit containing a 112-nucleotide U-less cassette (pIX) and an internal control consisting of the adenovirus major late promoter fused to a transcription unit containing a 50-nucleotide U-less cassette (ML; see d and Materials and Methods). The ML promoter does not contain an Sp1- or RBP-binding site. (Arrows) pIX and ML transcription products. Basal and Sp1-activated pIX transcription are shown in lanes 1 and 2, respectively. Sp1 activation depends on the presence of cofactors TFIIA (lane 3) and PC4 (lane 4). (b) Purification scheme for recombinant RBP protein. After induction, GST–RBP fusion protein was purified with GST beads, treated with bovine thrombin, and purified further with Mono S (FPLC) and Benzamidine Sepharose. The protein was concentrated on Mono S (SMART system, Pharmacia). (c) Silver-stain analysis of purified protein fractions after separation by SDS-PAGE. Fractions containing recombinant RBP as a function of purification as well as native RBP protein purified from HeLa cells are indicated at the top of the lanes (see Materials and Methods). (MWM) Protein molecular weight markers. (d) Schematic representation of the templates used in the in vitro transcription reactions and described in a. (Right) Results of transcription reactions performed with increasing amounts of recombinant RBP (rRBP) and pIX template containing the wild-type (lanes 1–6) or Sub9 mutant promoter (lanes 7–10) as indicated at the bottom of the lanes. The amounts of rRBP used are indicated at the top of the lanes. Repression is not detected in the Sub9 case in which the RBP-binding site is mutated. Both rRBP and native RBP have similar DNA-binding and repression activities.
Figure 3b shows the purification scheme followed for the isolation of recombinant RBP. Figure 3c shows the preparations of recombinant (lane 4) and native RBP (lane 6) utilized in the transcription assays. The addition of increasing amounts of purified human (data not shown) or recombinant RBP to the transcription reactions resulted in pIX repression (Fig. 3d). On the other hand, although the levels of Sp1 activation of pIX transcription from the Sub9 construct, mutant in RBP binding, were similar to those of the wild-type, the addition of increasing amounts of RBP was ineffectual in this case. This result established optimal conditions of activated pIX transcription, optimal conditions of RBP-mediated repression, and verified the requirement of the RBP site in mediating pIX repression in vitro.
Next, we compared the levels of Sp1-activated pIX transcription and susceptibility to repression from the RBP/Sp1 and RBP center constructs relative to wild-type and Sub9 cases (Fig. 4). All of these constructs gave rise to similar levels of Sp1-activated transcription. Similar to the results obtained in vivo, RBP repressed pIX transcription from the wild-type (lanes 1–4) and RBP center (lanes 13–16) constructs, but not from the Sub9 (lanes 5–8) or RBP/Sp1 (lanes 9–12) constructs. In addition, the orientation of the RBP site immediately upstream of the TATA box (RBP reverse) was inconsequential to repression in vitro (lanes 17–20) and in vivo (data not shown). Although repression in the RBP center case is likely attributable to RBP-mediated occlusion of Sp1 binding, as opposed to the wild-type case, relief from repression in the RBP/Sp1 case cannot be attributed to Sp1 blocking of RBP binding (see previous section). Therefore, repression is achieved dependent on both the presence and position of the RBP site, as Sub9 that lacks the RBP site is unaffected by RBP addition in vivo and in vitro and RBP/Sp1 is also relieved from repression in vivo and in vitro even though RBP and Sp1 co-bind. Most importantly, the pattern of repression observed in vitro as a function of the position of the RBP-specific DNA-binding site mimics that observed in vivo.
Figure 4.

The position of the RBP-binding site determines RBP-mediated repression in vitro. (Top) pIX promoter and start site for pIX transcription (+1). (Bottom) pIX promoters fused to the transcriptional unit containing a 112-nucleotide U-less cassette (see Materials and Methods). The levels of transcription from each construct were compared under the following conditions: in the absence of Sp1 (basal), in the presence of Sp1, and in the presence of Sp1 and increasing amounts of RBP repressor as indicated at the top of the lanes. Although the levels of Sp1-activated transcription are similar in all cases, susceptibility to RBP-mediated repression depends on the position of the RBP site. The levels of Sp1-activated transcription from the wild-type (lanes 1–4), RBP center (lanes 13–16), and RBP reverse (lanes 17–20) constructs were decreased in the presence of increasing amounts of RBP. The addition of RBP had no detectable affect on Sp1-activated transcription from Sub9 (lanes 5–8) and RBP/Sp1 (lanes 9–12) constructs. The pattern of pIX repression dependent on the position of the RBP-binding site is consistent with the results obtained from these constructs in vivo (Fig. 1b).
Repression as a function of transcription preinitiation subcomplex formation
The establishment of this reconstituted transcription assay validated the interpretation of the results we obtained in vivo. In addition, this assay allowed us to explore two aspects of RBP repression at the molecular level: First, the stage of transcription preinitiation complex formation in which RBP repression is functional; and second, the components within the complex that may be susceptible to repression. To this end, we first examined whether RBP-mediated repression in vitro was dependent on the order of RBP addition during transcription preinitiation complex formation. When RBP is added concomitantly with all of the transcription factors required for preinitiation complex formation, pIX repression is apparent (Fig. 5a, lanes 1–3). However, if RBP is added subsequent to preinitiation complex formation, repression was no longer observed (lane 4). This result suggested that stable preinitiation complexes formed in the absence of RBP can thwart repression.
Figure 5.

RBP mediates repression at the early stages of preinitiation complex formation. (a) Conditions used to obtain single round transcription with linearized templates in these assays (see Materials and Methods). In all cases, Sp1 and PC4 were added together with the general transcription factors (GTFs). The levels of pIX transcription obtained during preinitiation complex formation are shown as a function of time of addition of RBP repressor (R), or time of addition of TFIIA (IIA) or TFIIH (IIH) in the absence of or presence of RBP repressor as indicated at the top of the lanes. (Vertical arrows) Preincubation times of 30 min before the addition of NTPs. pIX and ML transcripts are indicated on the left. Preinitiation complexes formed in the absence of RBP are protected from repression. Coaddition of RBP during complex formation results in repression. Repression is apparent on later addition of RBP as a function of the presence of TFIIA, but not TFIIH. (b) Conditions of single round transcription as in a but in this case, subcomplexes were preformed for 30 min with specific candidate transcription factors as indicated: TFIID (D), TFIIA (A), and/or TFIIB (B). In all cases, Sp1 and PC4 were added together with the candidate transcription factors. The levels of pIX transcription obtained during preinitiation complex formation are shown as a function of the presence of the specific candidate transcription factors and time of addition of RBP repressor. Arrows at the top of the lanes indicate preincubation times of 30 min before addition of NTPs. pIX and ML transcripts are indicated on the left. Subcomplexes containing TFIID are partially resistant to the later addition of RBP, but are made completely resistant when preformed in the presence of TFIIA, but not TFIIB.
We next sought to identify the minimal transcription components that can resist the repressive activity of RBP when a subcomplex containing these components is preformed prior to RBP addition. Binding of TFIID to the TATA motif nucleates transcription preinitiation complex formation (Buratowski et al. 1989; Orphanides et al. 1996; Roeder 1996). Therefore, we tested subcomplexes formed in the presence of Sp1 with either TFIID alone or with additional candidate transcription factors for resistance to repression upon subsequent addition of RBP. In all cases, Sp1 and the candidate transcription factors were preincubated with pIX and control templates, and, then, the remaining factors required for optimal pIX transcription were added. Each of the candidates was tested in sets of three with respect to RBP addition: In the complete absence of RBP to gauge optimal pIX transcription under these conditions; in the presence of RBP added during preincubation; or in the presence of RBP added subsequently along with the remaining factors. Figure 5b shows the results of this analysis.
The levels of Sp1-activated pIX transcription in the absence of RBP were similar in all of the preincubation trials (Fig. 5b, cf. lane 1 with lanes 2, 5, 8, and 11). Similar to the results obtained with the complete complex, all of the subsets of candidate factors examined were susceptible to repression when RBP was added during complex formation (cf. lanes 3, 6, 9, and 12). Subcomplex containing Sp1 and TFIID alone was partially resistant to later RBP addition (cf. lanes 3–4, see below). On the other hand, preincubation with Sp1, TFIID, and TFIIA was completely resistant to later addition of RBP, similar to the results shown above when the complex was preformed with all of the factors (lanes 5–7). This result suggested that stable complex formed in the presence of TFIIA and Sp1–TFIID was able to impede the repressive activity of RBP. This finding was specific to TFIIA as preformed complex containing Sp1, TFIID, and TFIIB was as equally susceptible to later RBP addition as was Sp1–TFIID alone (lanes 8–10). On the other hand, when TFIIA was present, the Sp1–TFIID–TFIIB subcomplex was now made resistant to RBP repression (lanes 11–13). This result strongly suggested that the presence of TFIIA rendered a subcomplex completely resistant to later RBP addition.
Coaddition of RBP and TFIIA is required for functional repression
Because subcomplex containing Sp1 and TFIID could be rendered as resistant to RBP as the complete complex as long as TFIIA was present, we next tested whether RBP was specifically targeting TFIIA during preinitiation complex formation. The TFIIA coactivator can be added at any time during transcription preinitiation complex formation without an effect on activated transcription (Fig. 5a, cf. lanes 2 and 5). In this case, we tested whether or not subcomplexes devoid of TFIIA are resistant to the subsequent addition of RBP when TFIIA is also added later with RBP. As a control, we similarly tested TFIIH, which is required for transcription (lane 9) and is the last factor that assembles into the preinitiation complex (cf. lanes 1 and 7). As described above, complex containing all the factors is susceptible to concomitant, but not subsequent, RBP addition (lanes 1–4). However, when a similar complex is formed in the absence of TFIIA, the later addition of RBP with TFIIA now renders the complex susceptible to repression (lane 6). This is not the case with TFIIH, however. Complex formed with all the factors including TFIIA, but in the absence of TFIIH, is resistant to RBP when RBP is added later with TFIIH (lanes 7–9). Therefore, not only does the presence of TFIIA render a subcomplex resistant to RBP as shown in the previous section, but the omission of TFIIA during preinitiation complex formation renders the complex susceptible to repression when complemented later for TFIIA in the presence of RBP. This result strongly suggests that RBP is functionally targeting TFIIA in mediating repression of activated pIX transcription.
RBP, Sp1, TFIID, and TFIIA co-bind the pIX promoter
Thus far, our results show that the complex must contain TFIIA to fully resist repression and that TFIID alone renders the complex partially resistant to RBP activity. The functional relevance of this finding to repression is dependent on the ability of RBP to co-bind with Sp1–TFIID and with Sp1–TFIID–TFIIA. The footprinting results shown in Figure 2a demonstrated that RBP binding immediately upstream of the TATA box in the pIX promoter does not occlude TFIID or Sp1 binding and, therefore, repression does not entail overlapping DNA-binding sites for these three factors.
Previous studies showed that TFIIA can extend the DNA coverage of TFIID by several nucleotides upstream of the TATA box at some promoters (Van Dyke et al. 1988; Buratowski et al. 1989; Cortes et al. 1992). If such an extension were relevant at the pIX promoter, TFIIA binding may overlap with that of RBP, which binds immediately upstream of the TATA box. Repression may then entail RBP-mediated occlusion of TFIIA binding. This possibility would be consistent with RBP targeting of TFIIA in the subcomplex experiments shown above. For example, stable complexes formed in the presence of TFIIA may occlude RBP binding upon its later addition, while complexes formed in the presence of RBP may result in RBP occlusion of TFIIA binding and hence, repression. Given this possibility, we tested for co-binding of RBP, Sp1, TFIID, and TFIIA. We did not observe any detectable difference in footprinting analyses using TFIID in the absence or presence of TFIIA on the pIX promoter. Therefore, we could not use this assay to test for co-binding of TFIIA with the other factors including RBP. Instead, we employed two additional assays.
First, we tested directly for the presence of RBP with TFIIA at the pIX promoter under conditions that produce a transcriptionally competent complex that is resistant to RBP repression. Figure 6a shows a schematic representation of the experiment performed. The pIX template was immobilized on beads under complete transcription conditions in the presence of RBP added either concomitant with or subsequent to the other factors. Then, aliquots of the beads were analyzed independently for the levels of pIX transcription attained and for the presence of candidate transcription factors (TFIIA, RBP, and TFIIB as control, see Materials and Methods).
Figure 6.

RBP-mediated repression does not affect TFIIA binding and preinitiation complex formation does not inhibit RBP binding. (a) Schematic representation of the experimental design employing immobilized pIX template. Transcription preinitiation complexes were bound to the pIX promoter immobilized on magnetic beads. RBP was added at time 0 or after 40 min of preincubation time. Reactions were then incubated for an additional 40 min, immobilized templates were washed, and, then, aliquots were tested for levels of pIX transcription (b), or for proteins bound to the pIX template by use of Western blot analyses (c); (for details, see Materials and Methods). (b) (Lanes 3–6) Results of in vitro transcription assays using conditions for single round transcription with aliquots of immobilized templates prepared as described in a. The order of addition of specific factors is indicated on top of each lane. (Vertical arrows) Preincubation times of 30 min before the addition of NTPs. Preinitiation complexes formed on immobilized templates in the absence of TFIID do not give rise to detectable levels of transcription (lanes 5, 6). Susceptibility to repression is apparent on coaddition of RBP, but resistance to repression is apparent upon later addition of RBP (lanes 3, 4). These results are similar to those shown in Fig. 4a and repeated here in lanes 1 and 2 with nonimmobilized pIX template. (c) Western blot analyses of proteins bound to the immobilized pIX templates prepared as described in a. (Lanes 1–3) Controls for the presence of TFIIA, TFIIB, and RBP, respectively. The protein specificity of each antibody used is shown at left. Similar levels of either TFIIA or RBP are detected on immobilized pIX templates irrespective of the time of addition of RBP during complex formation (lanes 4, 5). The level of RBP protein detected is not affected by the absence of TFIID during complex formation (lane 6); however, TFIIA is not detectable in the absence of TFIID (lanes 6,7). Detection of RBP is dependent on its addition (lane 7).
We observed that a preformed complex on an immobilized pIX template exhibited similar resistance to subsequent, but not concomitant, RBP addition, as in the case of the nonimmobilized, control template (Fig. 6b, cf. lanes 1–4). In addition, transcription under these conditions exhibited the expected requirement for TFIID (cf. lanes 3–6). Although the complete complex was susceptible to repression dependent on the order of RBP addition, similar levels of RBP, TFIIA, and TFIIB were found to be associated with the immobilized pIX template, irrespective of the order of RBP addition (Fig. 6c, lanes 4, 5). The detection of these factors was specific, as shown in the case of complexes formed in the absence of TFIID. Because TFIID tethers the preinitiation complex to the promoter, the omission of TFIID resulted in undetectable levels of TFIIB and TFIIA, as expected (lanes 6, 7). On the other hand, because RBP binds the pIX promoter independently, the omission of TFIID had no affect on the levels of RBP found complexed to the pIX promoter (cf. lanes 4–6). Because the levels of RBP detected in the case of the complete complex were unaffected by the order of RBP addition, the inability of RBP to repress at later times appears not to be attributable to its occlusion by stable preinitiation complex formation. Moreover, because the levels of TFIIA detected in the presence of TFIID were similar when RBP was added together with TFIIA or at a later time, these results also show that TFIIA is apparently not occluded during complex formation in the presence of RBP. Therefore, complex susceptibility or resistance to repression appears not to correlate with TFIIA or RBP occlusion, respectively.
Second, we assayed directly for the presence of RBP, Sp1, TFIID, and TFIIA at the pIX promoter using agarose gel retardation assays and radiolabeled probe containing the pIX promoter region (Fig. 7; see Materials and Methods). To optimize visualization of complexes containing the multiple factors, conditions of limiting probe were employed. The controls for this experiment are contained in panel b. First, complex containing Sp1 and RBP migrated slower than complex containing Sp1 alone (cf. lanes 2 and 3) and more diffusely than complex containing RBP alone (cf. lanes 1 and 3). This complex contained both proteins as evidenced first by its complete supershifting with antibody specific to RBP (cf. lanes 3 and 6). Second, the mobility of the complex reverted to that of RBP alone in the presence of excess Sp1 oligonucleotide (cf. lanes 1, 3, and 4), but not in the presence of excess TATA oligonucleotide (cf. lanes 3 and 5). This result confirms that RBP and Sp1 can co-bind the pIX promoter under these conditions.
Figure 7.
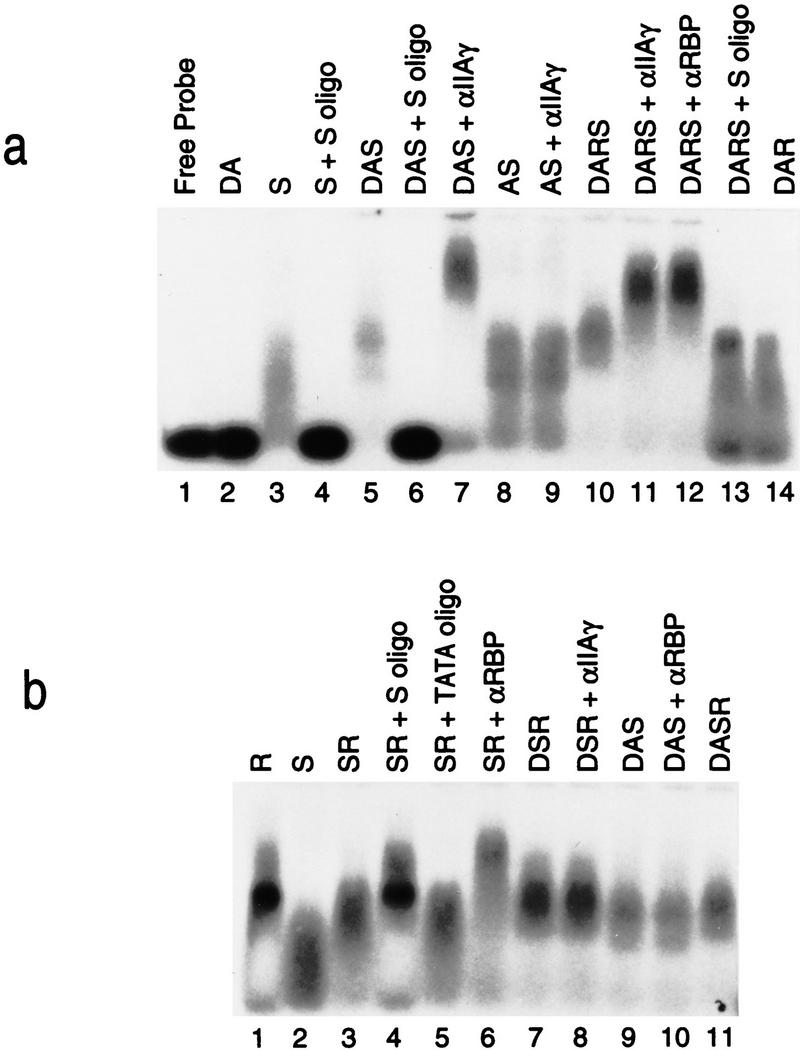
RBP binding does not occlude TFIIA. (a,b) Results of Mg2+–agarose gel-shift assays performed with radiolabeled probe containing the wild-type pIX promoter (nucleotides −60 to +140 relative to the start site for pIX transcription) and the proteins indicated at the top of each lane; the addition of specific antibodies is also indicated. See Materials and Methods for details. (S) Sp1; (R) RBP; (D) TFIID; and (A) TFIIA. Specific oligonucleotide competitors are Sp1 consensus site (S oligo); TATA consensus (TATA oligo). Antibodies used are antibody specific to RBP (α-RBP); antibody specific to the γ-subunit of TFIIA (α-IIAγ).
Next, we tested for co-binding of Sp1, TFIIA, and TFIID. Among these factors, only TFIIA does not contain specific DNA-binding activity; the association of TFIIA with the promoter is completely dependent on the presence of TFIID (as shown above in Fig. 6c). Although a complex containing only TFIID and TFIIA was not stably formed under these assay conditions, a complex formed in the presence of these two factors and Sp1 gave rise to a supershifted complex relative to Sp1 alone (Fig. 7a, cf. lanes 1–5). The dependence of this complex on Sp1 was evident by its inhibition with excess Sp1 oligonucleotide (cf. lanes 5 and 6). The presence of TFIIA was evidenced by reactivity of this complex with antibody specific to TFIIA (cf. lanes 5 and 7). The presence of TFIID was required to obtain this complex as evidenced by the lack of reactivity to antibody specific to TFIIA when the complex was formed with only Sp1 and TFIIA (lanes 8, 9). The observed supershift of this complex with anti-TFIIA antibody was specific as a complex formed in the presence of Sp1, TFIID, and RBP, without TFIIA, was unaffected by the addition of anti-TFIIA antibody (Fig. 7b, lanes 7, 8).
Having established these conditions, we next analyzed complex formation in the presence of all four proteins: Sp1, TFIID, TFIIA, and RBP (Fig. 7a, lane 10). The presence of RBP during this complex formation resulted in a species that migrated only slightly more slowly than a complex formed in its absence (cf. lanes 5 and 10). On the other hand, the presence of Sp1 during this complex formation substantially decreased its mobility relative to the complex formed in its absence (cf. lanes 10 and 14). That this complex did indeed contain TFIIA, RBP, and Sp1 was confirmed by its reactivity with antibody specific to TFIIA (lanes 10, 11) or to antibody specific to RBP (lanes 10, 12) and to its inhibition by excess Sp1 oligonucleotide such that the species remaining migrated similar to a complex containing only RBP, TFIID, and TFIIA (cf. lanes 10, 13, and 14). This result shows that all four proteins can coexist on the pIX promoter. Together, the results shown in Figures 6 and 7 establish that the presence of RBP does not occlude TFIIA binding, thereby eliminating this possible mechanism of repression.
RBP interacts specifically with TFIIA and with TFIID
On the basis of our results thus far, TFIIA, TFIID, and/or Sp1 were likely targets for RBP in repression. Because RBP binding does not disrupt nucleation of these factors at the pIX promoter, we next tested whether RBP mediates repression by direct interaction with any of these candidates. We found that RBP inhibited both Sp1-activated pIX transcription (see above) as well as GAL4–Sp1-activated transcription from a pIX template containing two GAL4 sites in lieu of the Sp1 site (data not shown). GST–RBP did not exhibit detectable interaction with GAL4–Sp1 activator in GST pull-down experiments (data not shown). In addition, the coactivator PC4 that was added with Sp1 during complex formation and in functional transcription assays did not show detectable interaction with GST–RBP (data not shown). On the other hand, GST–RBP did interact with both human TFIID and recombinant TFIIA (Fig. 8b,c). This interaction was specific, as GST alone was ineffectual. Moreover, GST-RBP did not exhibit detectable interaction with TFIIB (Fig. 8d; see below). As shown above, the presence of TFIID during subcomplex formation exhibited partial resistance to subsequent RBP addition, and this partial resistance was increased in the presence of TFIIA, but not TFIIB. Interestingly, these experiments demonstrate that RBP interacts specifically with the same factors required for subcomplex resistance to repression.
Figure 8.

RBP interacts specifically with TFIIA and TFIID. The results of GST pull-down assays analyzed by Western blot with an antibody specific to the protein indicated at the top of a–d. GST pull-down assays were performed with GST or GST–RBP as indicated at the top of a–d and the protein indicated at the top as follows: (a) hTBP, human recombinant TBP; (b) eTFIID, epitope-tagged TFIID; (c) rTFIIA, recombinant TFIIA; and (d) rTFIIB, recombinant TFIIB. (Lanes 1) An aliquot of the protein specified as a positive control for the Western analysis. (M) Protein molecular weight markers right.
Because TFIIA and TFIID contain several subunits, we tested which specific components of these factors interact with RBP. TFIIA consists of the α, β, and γ subunits; α, β, and γ are required for activation, whereas β and γ are sufficient for antirepression (Sun et al. 1994; Ma et al. 1996; for review, see Orphanides et al. 1996). The recombinant form of the α,β subunits of TFIIA (TFIIA α,β) interacted with GST–RBP and not with GST alone (data not shown, see below). Our results with the γ subunit alone were not conclusive as TFIIA γ exhibited similar low levels of interaction with GST–RBP as with GST alone (data not shown).
The complex TFIID consists of TBP and the TAFIIs (for review, see Burley and Roeder 1996). GST–RBP interaction with TFIID is not mediated by interaction with the TBP component, however (Fig. 8a). Next, we examined whether GST–RBP targets one or more of the TAFIIs.
The candidate TAFIIs examined in this section were derived from Drosophila, whereas the functional assays performed in the previous sections utilized human TFIID. Nonetheless, substitution of dTFIID in the transcription reaction performed in vitro gave rise to similar levels of Sp1-activated pIX transcription, as expected, and similar levels of RBP-mediated repression (data not shown). Therefore, functional repression is not dependent on the source of TFIID. Furthermore, the highly purified preparation of TFIID that was used in the functional transcription assays presented above, contained detectable levels of all the human homologs of the dTAFIIs identified (data not shown).
Of the several candidate dTAFIIs examined, GST–RBP interacted only with dTAFII110 (Fig. 9a, lanes 7–9). This interaction was specific as GST alone was ineffectual. Next, we verified this interaction using Far Western analyses (Fig. 9b,c). RBP showed specific interaction with dTAFII110; no interaction was detectable between RBP and hTBP, hTFIIB, dTAFII150, the BSA control, or the molecular weight markers. These results demonstrate that the interaction between RBP and TFIID shown above specifically involves the dTAFII110 component of TFIID.
Figure 9.

RBP interacts specifically with dTAFII110. (a) GST pull-down assays with 35S-radiolabeled TAF candidates prepared by translation in vitro and GST or GST–RBP as indicated at the top of the lanes. The first lane of each set of three shows the radiolabeled TAFII examined (10% of input) and the second and third lanes show the results from GST pull-down assays with this TAFII and either GST or GST–RBP, respectively. (b) Far Western analysis with ∼0.5 μg of protein indicated at the top and biotinylated rRBP. (c) SDS-PAGE analysis of ∼0.5 μg of protein indicated at the top and visualized by Coomassie blue staining. (MWM) Protein molecular weight markers, the sizes of which are indicated right.
The results in this section showed that RBP interacts with TFIIA and TFIID, that the interaction between RBP and TFIIA involves the α,β subunits (see below), and that the interaction between RBP and TFIID directly involves dTAFII110. Previous results showed that dTAFII110 interacts with the activation domains of Sp1 (Hoey et al. 1993; Gill et al. 1994), the only activator required for optimal pIX expression. Interestingly, previous results showed that the interaction between TFIIA α,β and TFIID also directly involves dTAFII110, as well as TBP (Yokomori et al. 1993). Therefore, RBP interacts with the TAF component of TFIID that mediates its interaction with Sp1 and with TFIIA. Sp1 has been reported to interact specifically with the amino terminus of dTAFII110 (Hoey et al. 1993). However, the region of dTAFII110 interaction with TFIIA α,β has not been reported. Therefore, to identify factor interactions involving dTAFII110 that may be impeded in the presence of RBP, we examined discrete regions of dTAFII110 for interaction with RBP and for interaction with TFIIA α,β.
Distinct from Sp1, RBP and TFIIA α,β interact specifically with the carboxyl terminus of dTAFII110
Figure 10a shows the results of GST pull-down assays with either GST–RBP or a GST-fusion protein containing the α,β subunits of TFIIA (GST–IIA α,β) and candidate truncation mutants of dTAFII110. Both GST–RBP and GST–IIA α,β exhibited interaction with full-length dTAFII110, as expected (amino acids 1–921, lanes 1–3 and lanes 13–15, respectively). Both GST–RBP and GST–IIA α,β interacted with regions of dTAFII110 spanning amino acids 1–684 (lanes 4–6 and lanes 16–18, respectively) and the carboxyl terminus of dTAFII110 spanning amino acids 666–921 (lanes 10–12 and lanes 22–24, respectively). The interactions were specific as GST alone was ineffectual. On the other hand, neither GST–RBP nor GST–IIA α,β exhibited detectable interaction with the amino terminus of dTAFII110 (1–307), which has been reported to interact with Sp1 (lanes 7–9 and lanes 19–21, respectively). This result showed that RBP and TFIIA α,β interact with the same region of dTAFII110, that is, the carboxyl terminus, which is distinct from that reported for interaction with Sp1, that is, the amino terminus.
Figure 10.



Both RBP and TFIIA α,β interact specifically with the carboxy-terminal domain of dTAFII110, which is inhibited from interaction when RBP complexes with TFIIA α,β. (a) GST pull-down assays with different 35S-labeled dTAFII110 truncated proteins translated in vitro, as indicated at the top of the lanes. Analyses using either GST or GST–RBP (lanes 1–12) and either GST or GST–IIAαβ (lanes 13–24) are shown. The first lane of each set of three shows 10% of the input radiolabeled truncated dTAFII110 examined, the second lane shows the pull-down result with GST, and the third lane shows the pull-down result with either GST–RBP or GST–IIAαβ, as indicated. (b) GST pull-down experiments with GST–RBP in the absence or presence of increasing amounts of either TFIIA α,β or TFIIB, as indicated, and subsequent addition of 3 μl of 35S-labeled dTAFII110 truncation protein containing amino acids 666–921, as indicated. A Western blot analysis of TFIIA α,β interaction with GST–RBP as a function of the addition of increasing amounts of TFIIA α,β is also shown. (+) 10% of the input protein. (c) Similar analysis as performed in b, but with the highest level of TFIIA α, β or TFIIB shown in b and increasing amounts of 35S-labeled dTAFII110 truncated protein containing amino acids 666–921, as indicated. A Western blot analysis of TFIIA α,β interaction with GST–RBP under these conditions is also shown.
RBP interaction with TFIIA inhibits dTAFII110 interaction
The results in the previous section showed that both RBP and TFIIA α,β interact specifically with the carboxyl terminus of dTAFII110 (C-TAFII110, 666–921). To determine the functional relevance of these interactions with respect to repression, we tested whether complexes containing GST–RBP and TFIIA α,β retain the ability of either protein to interact with C-TAFII110 or if subsequent C-TAFII110 interaction is now impeded (Fig. 10b). The levels of C-TAFII110 binding to GST–RBP were examined as a function of preincubation of GST–RBP with increasing amounts of either TFIIA α,β or TFIIB. The levels of C-TAFII110 binding to GST–RBP were found to markedly decrease as a function of increasing TFIIA α,β preincubation (cf. lanes 2–6). This was not the case with TFIIB, however, which does not interact with RBP (Fig. 8). The levels of C-TAFII110 binding to GST–RBP were unaffected by increasing levels of TFIIB, comparable to those used for TFIIA α,β (lanes 8–12).
A similar result was obtained with GST–RBP in the presence of a high level of either TFIIA α,β or TFIIB and subsequent addition of increasing amounts of C-TAFII110 (Fig. 10c). The levels of C-TAFII110 interaction with GST–RBP were consistently reduced in the case of preincubation with TFIIA α,β, relative to the control case using TFIIB (cf. lanes 2–6 with lanes 8–12). These results showed that interaction between GST–RBP and TFIIA α,β specifically inhibits subsequent C-TAFII110 interaction with either GST–RBP or TFIIA α,β.
Activator-induced resistance to repression
Our results thus far have shown that RBP targets TFIIA and TFIID by direct interaction, but that RBP, TFIIA, and TFIID can co-bind the pIX promoter. The interaction between TFIID and TFIIA presumably mediates activation by altering TFIID conformation. This conformational change may be induced/stabilized in the presence of an activator (Lee et al. 1992; Lieberman and Berk 1994; Chi et al. 1995; Chi and Carey 1996; Oelgeschläger et al. 1996), but prevented/destabilized in the presence of RBP repressor, which interacts with both TFIID and TFIIA. Consistent with this, we showed in the previous section that RBP interaction with TFIIA α,β disrupts interaction with the relevant domain of dTAFII110. We also showed in previous sections that transcription preinitiation complexes were resistant to repression upon later addition of RBP, as long as TFIIA was present during complex formation. Even though later addition of TFIIA did not affect the levels of Sp1 activation, later addition of TFIIA with RBP now rendered the complex susceptible to repression. This suggests that the presence of TFIIA during complex formation with the other factors alters the nature of the complex with respect to RBP-mediated repression. Therefore, we tested the functional relevance of RBP-mediated disruption of dTAFII110/TFIIA α,β interaction during preinitiation complex formation, this time as a function of Sp1 activator addition.
We first examined the levels of activated pIX transcription obtained as a function of time of Sp1 addition during preinitiation complex formation, in the absence of RBP (Fig. 11). The addition of Sp1 after the transcription complex was formed gave rise to activated pIX transcription; however, the levels were markedly reduced relative to those obtained when Sp1 was present during preinitiation complex formation (cf. lanes 1, 2, and 5). Experiments involving preinitiation complex formation in the studies presented here were performed under conditions of excess amounts of the tested transcription factors. In addition, the studies in the previous sections showed that Sp1, TFIID, and TFIIA co-bind the pIX promoter. Therefore, the reduced levels of activated pIX transcription observed upon later Sp1 addition is not consistent with Sp1 occlusion. Instead, complexes that are preformed in the presence of TFIID and TFIIA, in the absence of activator, appear to be refractory to optimal activation when Sp1 is added later.
Figure 11.

Sp1 and RBP target the same step in complex formation. The levels of pIX transcription obtained during preinitiation complex formation are shown as a function of time of addition of Sp1 and RBP as indicated at the top of the lanes. Conditions of single round transcription were used. (Vertical arrows) Preincubation times of 30 min each before addition of NTPs. (S) Sp1; (R) RBP. Later addition of Sp1 gives rise to reduced levels of activated pIX transcription. Sp1 is required during complex formation to inhibit repression by RBP.
As shown in a previous section and again here, preinitiation complexes formed in the presence of Sp1 were susceptible to repression upon concomitant, but not subsequent, RBP addition (lanes 1–4). However, a different result was obtained during conditions of suboptimal Sp1 activation resulting from later Sp1 addition. In this case, the complexes that gave rise to suboptimal activation were now susceptible to repression when RBP was added subsequent to Sp1 (cf. lanes 5 and 6). This result strongly suggests that the complexes containing TFIID and TFIIA that were preformed in the absence of Sp1 are not only refractory to activation but are now susceptible to repression. Furthermore, the later addition of Sp1 cannot restore complex resistance to subsequent RBP-mediated repression. Therefore, resistance to repression is dependent on the presence of Sp1 along with the two targets of RBP: TFIIA and TFIID. Our results are consistent with RBP interaction with TFIIA and TFIID to destabilize conformational changes that are normally stabilized in the presence of Sp1 and thereby facilitate activated transcription.
Discussion
The studies presented here show that RBP is distinct in mediating repression with respect to the previously reported repressors. Although RBP binds very closely to transcription factors that are crucial for activated transcription, occlusion of adjacent factor binding has been eliminated as a possible repressive mechanism at the pIX promoter (Dou et al. 1994; this study). RBP does not interact with components of the basal transcriptional machinery such as TBP or TFIIB, nor does it functionally interact with the Sp1 activator. Instead, RBP evolved to silence activated transcription by interacting directly with two transcription coactivators. Our findings reported here are the first example of a transcription repressor directly targeting, by interaction, TFIIA and a TAFII component of TFIID. As shown here, repression can be relieved when stable preinitiation complexes are formed before addition of RBP repressor. The resistant subcomplexes are Sp1–TFIID–TFIIA dependent.
TFIID has been shown to be required for activated transcription in vitro by use of highly purified transcription factors. RBP interacts specifically with the TAFII110 component of TFIID. This same TAFII has been shown to interact with the activation domains of Sp1 (Hoey et al. 1993; Gill et al. 1994). The possibility existed that RBP may compete with Sp1 for TAFII110. However, RBP also represses GAL4–VP16-mediated activation in vivo and in vitro (Dou et al. 1994; data not shown). VP16 interaction with TFIID involves dTAFII40, not dTAFII110 (Goodrich et al. 1993). Therefore, the interaction between TFIID and TFIIA was the more likely target for RBP in repression. In fact, the studies presented here show that both RBP and the α,β subunits of TFIIA interact specifically with the carboxyl terminus of dTAFII110 as opposed to Sp1 which interacts with the amino terminus (Hoey et al. 1993). Furthermore, interaction between RBP and TFIIA α,β disrupts interaction with the relevant domain of dTAFII110. Although our results do not eliminate the possibility that RBP interaction with dTAFII110 and with TFIIA may also result in disruption of Sp1 and TAFII110 interaction, the direct target of RBP appears to be interaction between TFIIA and dTAFII110.
TFIIA has been directly implicated in mediating transcription activation in two ways (for review, see Burley and Roeder 1996; Orphanides et al. 1996). First, interaction between TFIIA and TBP precludes TBP targeting by other repressors (Meisterernst and Roeder 1991; Inostroza et al. 1992; Auble and Hahn 1993; Merino et al. 1993). Second, interaction between TFIIA and TFIID stabilizes TFIID promoter interaction and is believed to induce conformational changes in the presence of an activator that are conducive to transcription activation (Lieberman and Berk 1994; Chi et al. 1995; Kobayashi et al. 1995; Chi and Carey 1996; Oelgeschläger et al. 1996; Lieberman et al. 1997). The documented roles of TFIIA in activated transcription suggested several possible means by which RBP may be repressive. RBP interaction with TFIIA may impede activation by exposing TBP to other repressors. Because the reconstituted transcription reaction used here contains recombinant factors or highly purified native factors, we believe the presence of other putative repressors is unlikely. On the other hand, RBP interaction with TFIIA may completely disrupt TFIIA interaction with TFIID. However, because TFIIA is tethered to the promoter by interaction with TFIID, and our studies show that RBP binding to the pIX promoter does not dislodge TFIIA, we believe this possibility is also unlikely. Instead, our results strongly support that RBP interaction with TFIIA specifically disrupts interaction with the TAFII110 component of TFIID. This would not necessitate TFIIA occlusion from the promoter as TFIIA also interacts with TFIID via the TBP component that is not targeted by RBP.
TFIIA interaction with TFIID has been shown to alter TFIID conformation, which may mediate activated transcription (Chi et al. 1995; Oelgeschläger et al. 1996). This phenomenon appears to be the most likely target for RBP in repression. RBP interaction with TFIIA and TFIID may impede the induction of conformational changes in TFIID, which, in turn, may subvert activated transcription. Our results showed that transcription preinitiation subcomplexes are completely resistant to repression when RBP is added subsequent to Sp1–TFIIA–TFIID. When subcomplexes are formed in the presence of TFIID and Sp1 activator, repression is achieved only when RBP is added along with TFIIA. However, complexes formed in the presence of TFIIA and TFIID, but in the absence of activator, are not sufficient to thwart repression. Our results also show that formation of TFIIA–TFIID complexes in the presence of Sp1 is required not only for optimal levels of activation, but also to render TFIIA–TFIID resistant to RBP.
In the absence of repressor, later addition of Sp1 resulted in markedly reduced levels of activated transcription. That optimal activation requires the coaddition of Sp1 during subcomplex formation is consistent with activator-stabilized/induced TFIIA–TFIID interactions that facilitate increased transcription. The nature of the TFIIA–TFIID complexes formed before Sp1 addition correlates with reduced accessibility to Sp1 and, therefore, with increased accessibility to RBP, even though RBP was added after the activator. Therefore, activator-induced complexes are required for protection from RBP repression. Our findings showed that RBP and the α,β subunits of TFIIA both interact with the carboxyl terminus of TAFII110 and that RBP interaction with TFIIA α,β disrupts interaction with the relevant domain of TAFII110. Taken together, these results strongly suggest that the TFIIA–TFIID interaction in the presence of Sp1 is stabilized from interaction with RBP, but in the absence of Sp1, RBP disrupts TFIIA–TFIID interactions that are required for activated transcription. A model depicting this mechanism of repression is shown in Figure 12.
Figure 12.
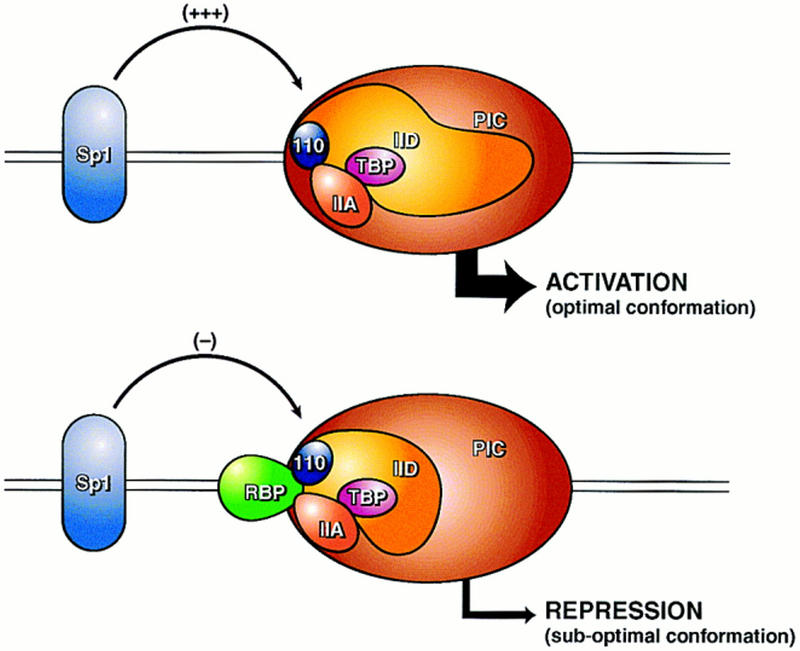
Diagram of the molecular basis for RBP repression showing competition between RBP and Sp1 for the TFIIA–TFIID subcomplexes. RBP interacts with TFIIA and with the dTAFII110 component of TFIID. This interaction disrupts TFIID–TFIIA interactions that involve dTAFII110 but does not dislodge TFIIA, which may still interact with the TBP component of TFIID. Disruption of TFIID–TFIIA interactions by RBP is presumed to inhibit Sp1-induced conformational changes in TFIID mediated by TFIIA that are required for increased transcription. On the other hand, Sp1-induced conformational changes in TFIID–TFIIA during complex formation provide protection from subsequent RBP interaction.
Our results described here also showed that the position of the single RBP site in the pIX promoter is determinant to repression, but that this effect is not attributable to overlap with other transcription factor binding sites. If the RBP site is situated upstream of the Sp1 consensus site, repression is inoperative in vivo and in vitro. In the case of simple promoters like pIX and E1B, the distance of the single Sp1 site from the TATA box is crucial to activation (Wu and Berk 1988). Stable complexes between Sp1 and TFIID–TFIIA appear to be restricted by the separation between activator and coactivators in these simple promoters. RBP may only disrupt Sp1 interaction with TFIID–TFIIA efficiently when situated close to the coactivators or alternatively, stable complexes between RBP and TFIID–TFIIA may also be restricted by distance. This restriction on repression in the natural case may ultimately reveal an important facet to activated transcription, which is targeted by RBP.
Although the position of RBP immediately upstream of Sp1 relieved repression, this was not attributable to Sp1-mediated occlusion of RBP binding. Also, repositioning of the RBP site 20 nucleotides upstream of Sp1 relieved repression. Although we have not examined an RBP site positioned further upstream of Sp1 for repression in this study, the possibility exists that repression will be restored if the RBP site is positioned at greater distances. In this case, RBP may effectively compete with Sp1 for TFIIA–TFIID as a function of DNA folding, which may bring RBP in closer proximity to its targets. Previous studies have shown that GAL4–RBP represses transcription irrespective of the position of multiple GAL4-binding sites within an artificial promoter construct (Hsieh and Hayward 1995). Because our results show that RBP interacts with TFIID and TFIIA, the presence of multiple RBP repressor molecules, as in the GAL4 case, may effectively increase the chances of RBP interaction with these targeted factors with resultant repression irrespective of position. However, our results described here show that there are restrictions to the position of a single RBP site, as exists in the natural pIX promoter, with respect to pIX repression in vivo and in vitro.
The disparate role of RBP in repression versus activation in mammalian and Drosophila cells, respectively, has yet to be clarified. However, RBP/Su(H) is targeted by a number of viral and Drosophila proteins that modulate its activity (see introductory section). Therefore, the possibility exists that an as-yet-unidentified endogenous factor present in Drosophila cells may also modulate RBP activity and result in transcription activation. This possibility is more likely than RBP conversion to a repressor by a possible endogenous factor present in mammalian cells, as purified RBP protein functions as a repressor in transcription assays performed in vitro with partially purified (Dou et al. 1994) and highly purified (this study) factors.
Transcriptional repressors have been studied extensively in prokaryotes, thereby providing a strong precedent for the more recently appreciated role of repressors in eukaryotes. These previous studies revealed that almost all stages of the transcription process can be targeted by different repressors; indeed, eukaryotic repressors have been documented to target specific activators and specific components of the basal transcription machinery (for review, see Herschbach and Johnson 1993; Johnson 1995). In this report, we show yet another stage in the transcription process to be targeted by RBP—that involving TFIIA and TFIID-mediated activation. Future studies of the molecular basis of repression may serve to provide further insight into the activation process itself.
Materials and methods
Transient expression assays and RNA analyses
Undifferentiated F9 cells were grown in DMEM containing 10% defined calf serum (Hyclone) on gelatin-treated 100-mm tissue culture plates. Cells were transfected by use of the calcium-phosphate precipitation technique as described previously (Babiss and Vales 1991). Each transfection was performed with 10 μg of construct containing the pIX gene, 10 μg of CMV/RBP expression vector, 10 μg of carrier pGEM-1, and 10 μg of SV/dl17 internal control for transfection efficiency (Kannabiran et al. 1997). The isolation of RNA and analysis by RNase T2 protection with an antisense probe that spans the start site for pIX transcription were as described previously (Babiss and Vales 1991). In this case, template for SP6 probe isolation was digested with AflII so that the probe spans nucleotides 3535–3785 of adenovirus type 5. The start site for pIX transcription is nucleotide 3580 so that pIX mRNA is expected to protect a fragment of 205 nucleotides. The SV/dl17 internal control contains the SV40 promoter fused to an internal portion of the pIX gene contained within nucleotides 3560–5640 SV/dl17-derived mRNA is expected to protect a fragment of 225 nucleotides. Transcripts that initiate within vector sequences upstream of the pIX promoter utilize the adenovirus E1B acceptor splice site at nucleotide 3595 and protect a fragment of 190 nucleotides.
Derivation of plasmid constructs
The pIX constructs used in transient expression assays were derived as follows. Oligonucleotides containing the pIX promoter with a repositioned RBP site as delineated below were ligated into HindIII- and BamHI-digested plasmid DNA containing −20 nucleotides upstream of the pIX cap sites as described previously for the wild-type and Sub9 constructs (Babiss and Vales 1991). The DNA sequences of the oligonucleotides were as follows: RBP/Sp1, 5′-AGCTTGGTGGGAAAGAA-GTGGGCGTGGCTTAAGGGCTCGAGCTCATATATAA-3′; RBP center, 5′-AGCTTGGGCGTGGCTTATGGGAAAGAAGATCTATATAA-3′; Sp1/RBP, 5′-AGCTTGGGCGTGGCT-GGGAAAGAAGAGCTCATATATAA-3′; RBP reverse, 5′- AG-CTTGGGCGTGGCTTAAGGGTTCTTTCCCATATATAA-3′; the core RBP site is shown in bold, the core Sp1 site is underlined, and the linker substitution of the normal RBP site is shown in italics. The opposite strands contained a BamHI linker for ligation. The Sub9 construct used here contained an XhoI linker substitution of the RBP site as delineated in RBP/Sp1 above.
The analogous pIX promoter constructs containing the 112 nucleotide U-less cassette that were analyzed in transcription assays performed in vitro were derived as follows. DNA fragments containing each of the pIX promoters were generated by PCR with the appropriate pIX plasmids described above as templates and the primers delineated below. Then, the PCR-derived pIX promoter fragments were inserted into plasmid pMLP-U112 after digestion of the fragments and plasmid DNAs with EcoRI and ApaI. The specific 5′ primers used to generate the PCR products were as follows; for pIX/wt, Sub9, and RBP reverse, 5′-TAGATGGAATTCTGGGCGTGGCTTAAGGT-3′; for RBP/SP1, 5′-TAGATGGAATTCGGTGGGAAGAAGTGGGCG-3′; and for RBP center, 5′-TAGATGGAATTCTGGGCGTGGCTTATGGGA-3′; the EcoRI site is shown in bold print. The 3′ primer used in all cases was as follows: 5′-GTGCTAGGGCCCGTTGGTGTGCAAAACTACATAAGACCC-3′; the ApaI site is shown in bold. The pMLP-U50 template used as internal control contains the adenovirus major late core promoter sequences fused to a 50-nucleotide U-less cassette.
Purification of native and recombinant RBP
Purification of native RBP from HeLa cell nuclear extract was performed as described previously (Dou et al. 1994). Induction of GST–RBP fusion protein and its purification with GST beads was performed as described previously (Yeung et al. 1994). Once GST–RBP was bound to the beads, 40 NIH units of thrombin (Calbiochem) was added and the sample incubated 4 hr at room temperature to release RBP from the GST moiety following procedures described for the GST-Gene Fusion System (Pharmacia). RBP protein (1.6 mg) was dialyzed at 4°C against buffer C (20 mm Tris-HCl at pH 7.9, 0.2 mm EDTA, 10 mm β-mercaptoethanol, 10% glycerol) with the addition of 80 mm KCl and then loaded onto a Mono S HR 5/5 column (FPLC, Pharmacia) equilibrated in buffer C. Protein was eluted with a linear gradient between 80 and 1000 mm KCl in the same buffer. RBP elutes with 0.3 m KCl. Fractions were pooled (1.3 mg), dialyzed at 4°C against buffer E (50 mm Tris-HCl at pH 8.0, 500 mm NaCl), and loaded onto a Benzamidine–Sepharose column equilibrated in the same buffer. The flowthrough was collected and reloaded five times onto the column before dialysis at 4°C against buffer C containing 80 mm KCl and 0.2 mm PMSF. Finally, RBP (0.7 mg, 9.5 ml) was concentrated on a Mono S column (SMART System, Pharmacia) equilibrated in the same buffer. RBP was step eluted with buffer C containing 1000 mm KCl, dialyzed in buffer C containing 100 mm KCl, and stored frozen at −80°C.
In vitro transcription assays
Transcription assays (20-μl reactions) were performed as indicated previously (Ma et al. 1996) with two DNA templates (100 ng each) as indicated in the figures. Briefly, transcription factors used in these assays were: epitope-tagged holo-TFIID (eTFIID, 5 ng of TBP determined by quantitative Western blots, Zhou et al. 1992), rTFIIA (α,β and γ subunits, 75 ng), rTFIIB (20 ng), RNA polymerase II (alkyl–Superose fraction, 40 ng), rTFIIF (20 ng), rTFIIE (15 ng), and TFIIH (phenyl–Superose fraction, 65 ng). Activation reactions additionally contained rPC4 (30 ng) and rSp1 (0.5 fpu; Promega). For RBP-mediated repression, the amounts of RBP added to the reactions are indicated in each figure. Factors and templates were preincubated for 30 min at 30°C and nucleotides were added and incubated for another 45 min at 30°C. The RNA products were separated by electrophoresis on 8% denaturing polyacrylamide gels.
Single round transcription conditions were established by pulse and chase procedures with nucleotide starvation. Briefly, after preincubation of the factors with the template as described above, transcription reactions were pulsed by addition of cold ATP and GTP (0.1 mm final) plus [α-32P]CTP (0.625 μm). After 2 min of incubation, the reactions were chased for 30 min by the addition of cold CTP (0.1 mm) and the products separated as above.
Immobilized templates
Formation of transcription competent complexes on immobilized templates was performed as described previously (Zawel et al. 1995) with the following modifications: The immobilized template used was a HindIII–NdeI fragment containing the pIX promoter (∼400 bp) derived from the pIX U112 plasmid. The template was biotinylated by Klenow enzyme with biotin–dATP at the NdeI site at 220 bp upstream of the transcription initiation site. The amount of DNA and factors used for each reaction was equivalent to seven standard transcription reactions described above. Biotinylated DNA (0.7 μg; 0.382 pmole) was bound to 100 μg of streptavidin-coated magnetic beads according to the manufacturer (Dynabeads M280, Dynal, Inc.). The DNA-bound beads were washed twice with transcription buffer (Zawel et al. 1995) containing 5 μg/μl of BSA, then incubated for 20 min in the same buffer and finally washed three times with buffer without BSA. Transcription factors were added to the template in the presence of 0.4 μg of pBR322 as competitor DNA. Bound complexes were washed three times with transcription buffer containing 0.05% sarkosyl, and a magnetic stand (Promega) was used to concentrate the beads to the wall of the tube. The same washing procedure was repeated omitting Sarkosyl. Finally, the amount of beads equivalent to two transcription reactions was assayed for transcription by use of single round transcription as described above, and the remaining beads were used to analyze the factors bound to the template by Western blotting.
Gel retardation and footprinting assays
Reaction conditions for gel mobility shift assay were as described previously (Maldonado et al. 1990). The probe was prepared as follows from a plasmid containing the wild-type pIX gene that was described previously (Dou et al. 1994). pIX plasmid DNA digested with NcoI was radiolabeled with Klenow in the presence of all four [α-32P]dNTPs followed by subsequent digestion with HindIII. The resultant radiolabeled 160-bp fragment contained the pIX promoter from nucleotide −60 to nucleotide +100 relative to the start site for pIX transcription. Binding assays containing eTFIID were performed in 20 μl reactions with Mg2+-agarose gels as described previously (Lieberman and Berk 1994). Incubations were performed at 30°C for 1 hr as described above using poly(A-U) (0.5 μg) as a nonspecific competitor and 5 mm ZnCl2. The amounts of protein in the reactions were as follows: eTFIID (5 μl, containing 25 ng of TBP), rTFIIA (75 ng), rSp1 (2 fpu; Promega), and rRBP (71 ng). Anti-TFIIAγ antibodies (1.8 μg) or protein-A-purified anti-GST–RBP antibodies (1.7 μg) were added when indicated and the sample incubated for an additional 30 min at 30°C. The competitor oligonucleotides Sp1 and TATA (20 ng) have been described previously (Dou et al. 1994). Then, the complexes were resolved in a 0.7% low EEO–agarose gel (Promega) containing 3% glycerol in 1× G buffer (5 mm Mg-acetate, 3% glycerol, 0.5× TBE) after electrophoresis at 55 V in the same buffer for 6 hr at 4°C.
Footprinting assays were performed in 40-μl reactions containing 2.5 fmoles of radiolabeled probe. The probe was an NcoI–NheI fragment (∼ 390 nucleotides) obtained from plasmid DNA containing either the wild-type or the RBP/Sp1 promoter constructs as described previously (Dou et al. 1994). The following amounts of protein were used: eTFIID (15 μl, containing 5 ng of TBP per microliter), rRBP (43 ng), and rSp1 (2 fpu; Promega). Binding reactions were performed as described above and were incubated for 1.5 hr at 30°C. DNase I (BRL, 10 U/μl) digestions were performed with 0.01 units for 30 sec. The reaction was stopped, phenol–chloroform extracted, ethanol precipitated with carrier RNA, dried, resuspended in formamide loading buffer, heated for 2 min at 90°C, and resolved on a 6% denaturing polyacrylamide gel.
Protein–protein interactions performed in vitro
Approximately 20 μl of glutathione–agarose beads containing 2 μg of either GST or GST–RBP fusion protein were mixed with 20 μl of eTFIID (containing 5 ng of TBP/μL, determined by quantitative Western blot analysis), 0.35 μg of rTFIIA, 0.5 μg of rTFIIB, or 10 μl of hTBP (induced Escherichia coli extract; the equivalent of 2 μg of hTBP was used). Incubation was performed for 1 hr at 4°C in a total volume of 0.3 ml with buffer C (20 mm Tris-HCl pH 7.9, 0.2 mm EDTA, 0.1 mm PMSF, 10% glycerol) containing 100 mm KCl and 0.2% NP-40. The beads were then washed four times with buffer C containing 500 mm KCl and 0.4% NP-40. The bound proteins were then eluted with SDS-PAGE loading buffer, resolved by SDS-PAGE, and analyzed by Western blotting. eTFIID interaction was scored by use of 12CA5 mAb, TFIIA with 7C12/E2 mAb, and hTBP and TFIIB with affinity-purified antibodies.
For assays with proteins translated in vitro (candidate dTAFIIs, Fig. 9), equal amounts of protein were used as determined by fluorography. Positive lanes show 10% of the proteins translated in vitro that were used in the interaction studies.
Two micrograms of GST–RBP or GST–TFIIA α,β was used to analyze the interaction with different truncated forms of 35S-dTAFII110 (Fig. 10a). Beads were first preincubated for 1 hr at 4 °C with 50 μg of BSA in 0.1 ml of buffer C containing 0.1% NP-40, and then 3 μl of the different truncated mutant proteins was added and the sample incubated for another hour at 4°C. The beads were then washed four times with 0.5 ml of buffer C containing 500 mm KCl and 0.2% NP-40, and once with water. The bound proteins were eluted with SDS-PAGE loading buffer, resolved by SDS-PAGE, the gels treated with 3ENHANCE (NEN) following the manufacterer indications, dried, and exposed overnight.
For competiton experiments (Fig. 10b,c), 2 μg of GST–RBP protein was incubated overnight at 4°C with the indicated amounts of rTFIIA α,β or rTFIIB in 0.1 ml of buffer C containing 0.1% NP-40 and 50 μg of BSA. Beads were washed three times, resuspended in 0.1 ml of the same buffer, 3 μl (Fig. 10b) or the amounts indicated (Fig. 10c) of 35S-labeled dTAFII110 added, and the reactions incubated for another hour at 4°C. The beads were then treated as specified above.
Far Western analyses of protein–protein interactions were performed as described previously (Inostroza et al. 1992). Briefly, ∼0.5 μg of each candidate protein was electrophoresed in two SDS-PAGE mini gels. One was stained with Coomassie blue and the other blotted to PVDF membrane. Proteins in the membrane were denatured and renatured as described and then incubated overnight with biotinylated-rRBP (1 μg/ ml). The blots were then washed, incubated with streptavidin–alkaline phosphatase for 1 hr, washed, and developed with NBT/BCIP as described previously.
Acknowledgments
We gratefully acknowledge the members of the laboratories of D.R. and L.D.V. for their ideas and helpful discussions during the progress of this work. We express special gratitude to Dr. Jin-Long Chen and Dr. Ludger Klein-Hitpass for their generous donations of the dTAFII110 truncation mutants, which greatly expedited our research studies. This work was supported by grants from the National Science Foundation (MCB-9407333) and from the National Institutes of Health (GM54890) to L.D.V. and by a grant from the National Institutes of Health (GM-48518) and by funding from the Howard Hughes Medical Institute to D.R.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL valesly@umdnj.edu; FAX (732) 235-4783.
References
- Auble DT, Hahn S. An ATP-dependent inhibitor of TBP binding to DNA. Genes & Dev. 1993;7:844–856. doi: 10.1101/gad.7.5.844. [DOI] [PubMed] [Google Scholar]
- Babiss LE, Vales LD. Promoter of the adenovirus polypeptide IX gene: Similarity to E1B and inactivation by substitution of the simian virus 40 TATA element. J Virol. 1991;65:598–605. doi: 10.1128/jvi.65.2.598-605.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AM, Posakony JW. Suppressor of Hairless directly activates transcription of Enhancer of split Complex genes in response to Notch receptor activity. Genes & Dev. 1995;9:2609–2622. doi: 10.1101/gad.9.21.2609. [DOI] [PubMed] [Google Scholar]
- Brou C, Logeat F, Lecourtois M, Vandekerckhove J, Kourilsky P, Schweisguth F, Israel A. Inhibition of the DNA-binding activity of Drosophila Suppressor of Hairless and its human homolog, KBF2/RBP-Jκ, by direct protein–protein interaction with Drosophila Hairless. Genes & Dev. 1994;8:2491–2503. doi: 10.1101/gad.8.20.2491. [DOI] [PubMed] [Google Scholar]
- Buratowski S, Hahn S, Guarentee L, Sharp PA. Five intermediate complexes in transcription initiation by RNA polymerase II. Cell. 1989;56:549–561. doi: 10.1016/0092-8674(89)90578-3. [DOI] [PubMed] [Google Scholar]
- Burley SK, Roeder RG. Biochemistry and structural biology of transcription factor IID (TFIID) Annu Rev Biochem. 1996;65:769–799. doi: 10.1146/annurev.bi.65.070196.004005. [DOI] [PubMed] [Google Scholar]
- Chi T, Carey M. Assembly of the isomerized TFIIA–TFIID–TATA ternary complex is necessary and sufficient for gene activation. Genes & Dev. 1996;10:2540–2550. doi: 10.1101/gad.10.20.2540. [DOI] [PubMed] [Google Scholar]
- Chi T, Lieberman P, Ellwood K, Carey M. A general mechanism for transcriptional synergy by eukaryotic activators. Nature. 1995;377:254–257. doi: 10.1038/377254a0. [DOI] [PubMed] [Google Scholar]
- Cortes P, Flores O, Reinberg D. Factors involved in specific transcription by mammalian RNA polymerase II: Purification and analysis of transcription factor IIA and identification of transcription factor IIJ. Mol Cell Biol. 1992;12:413–421. doi: 10.1128/mcb.12.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou S, Zeng X, Cortes P, Erdjument-Bromage H, Tempst P, Honjo T, Vales LD. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol Cell Biol. 1994;14:3310–3319. doi: 10.1128/mcb.14.5.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman DS, Slee R, Skoufos E, Bangalore L, Bray S, Delidakis C. Synergy between Suppressor of Hairless and Notch in regulation of Enhancer of split mγ and mδ expression. Mol Cell Biol. 1997;17:5620–5628. doi: 10.1128/mcb.17.9.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortini ME, Artavanis-Tsakonas S. The Suppressor of Hairless protein participates in Notch receptor signaling. Cell. 1994;79:273–282. doi: 10.1016/0092-8674(94)90196-1. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Maruyama S, Kawaichi M, Honjo T. The Drosophila homolog of the immunoglobulin recombination signal-binding protein regulates peripheral nervous system development. Cell. 1992;69:1191–1197. doi: 10.1016/0092-8674(92)90640-x. [DOI] [PubMed] [Google Scholar]
- Gill G, Pascal E, Tseng ZH, Tjian R. A glutamine-rich hydrophobic patch in transcription factor Sp1 contacts the dTAFII110 component of the Drosophila TFIID complex and mediates transcriptional activation. Proc Natl Acad Sci. 1994;91:192–196. doi: 10.1073/pnas.91.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JA, Hoey T, Thut CJ, Admon A, Tjian R. Drosophila TAFII40 interacts with both a VP16 activation domain and the basal transcription factor TFIIB. Cell. 1993;75:519–530. doi: 10.1016/0092-8674(93)90386-5. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the Jκ recombination signal sequence binding protein. Proc Natl Acad Sci. 1994;91:7568–7572. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi Y, Matsunami N, Yamamoto Y, Honjo T. Purification and characterization of a protein that binds to the recombination signal sequence of the immunoglobulin Jκ segment. Nucleic Acids Res. 1989;17:9015–9026. doi: 10.1093/nar/17.22.9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi Y, Yamamoto Y, Iwanari H, Maruyama S, Furukawa T, Matsunami N, Honjo T. Biochemical and immunological characterization of the DNA binding protein (RBP-Jκ) to mouse Jκ recombination signal sequence. J Biochem. 1992;112:314–320. doi: 10.1093/oxfordjournals.jbchem.a123898. [DOI] [PubMed] [Google Scholar]
- Henkel T, Ling PD, Hayward SD, Peterson MG. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein Jκ. Science. 1994;265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- Herschbach BM, Johnson AD. Transcriptional repression in eukaryotes. Annu Rev Cell Biol. 1993;9:479–509. doi: 10.1146/annurev.cb.09.110193.002403. [DOI] [PubMed] [Google Scholar]
- Hoey T, Weinzierl ROJ, Gill G, Chen J-L, Dynlacht BD, Tjian R. Molecular cloning and functional analysis of Drosophila TAF110 reveal properties expected of coactivators. Cell. 1993;72:247–260. doi: 10.1016/0092-8674(93)90664-c. [DOI] [PubMed] [Google Scholar]
- Hsieh JJ-D, Hayward SD. Masking of the CBF1/RBPJκ transcriptional repression domain by Epstein-Barr virus EBNA2. Science. 1995;268:560–563. doi: 10.1126/science.7725102. [DOI] [PubMed] [Google Scholar]
- Hsieh JJ-D, Henkel T, Salmon P, Robey E, Peterson MG, Hayward SD. Truncated mammalian Notch1 activates CBF1/RBPJκ-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol Cell Biol. 1996;16:952–959. doi: 10.1128/mcb.16.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inostroza JA, Mermelstein FH, Ha I, Lane WS, Reinberg D. Dr1, a TATA-binding protein-associated phosphoprotein and inhibitor of class II gene transcription. Cell. 1992;70:477–489. doi: 10.1016/0092-8674(92)90172-9. [DOI] [PubMed] [Google Scholar]
- Johnson AD. The price of repression. Cell. 1995;81:655–658. doi: 10.1016/0092-8674(95)90524-3. [DOI] [PubMed] [Google Scholar]
- Kannabiran C, Zeng X, Vales LD. The mammalian transcriptional repressor RBP (CBF1) regulates interleukin-6 gene expression. Mol Cell Biol. 1997;17:1–9. doi: 10.1128/mcb.17.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaichi M, Oka C, Shibayama S, Koromilas AE, Matsunami N, Hamaguchi Y, Honjo T. Genomic organization of mouse Jκ recombination signal binding protein (RBP-Jκ) gene. J Biol Chem. 1992;267:4016–4022. [PubMed] [Google Scholar]
- Kobayashi N, Boyer TG, Berk AJ. A class of activation domains interacts directly with TFIIA and stimulates TFIIA-TFIID-promoter complex assembly. Mol Cell Biol. 1995;15:6465–6473. doi: 10.1128/mcb.15.11.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laux G, Adam B, Strobl LJ, Moreau-Gachelin F. The Spi-1/PU.1 and Sp1-B ets family transcription factors and the recombination signal sequence binding protein RBP-Jκ interact with an Epstein-Barr virus nuclear antigen 2 responsive cis-element. EMBO J. 1994;13:5624–5632. doi: 10.1002/j.1460-2075.1994.tb06900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecourtois M, Schweisguth F. The neurogenic Suppressor of Hairless DNA-binding protein mediates the transcriptional activation of the Enhancer of Split Complex genes triggered by Notch signaling. Genes & Dev. 1995;9:2598–2608. doi: 10.1101/gad.9.21.2598. [DOI] [PubMed] [Google Scholar]
- Lee DK, DeJong J, Hashimoto S, Horikoshi M, Roeder RG. TFIIA induces conformational changes in TFIID via interactions with the basic repeat. Mol Cell Biol. 1992;12:5189–5196. doi: 10.1128/mcb.12.11.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman PM, Berk A. A mechanism for TAFs in transcriptional activation: Activation domain enhancement of TFIID–TFIIA–promoter DNA complex formation. Genes & Dev. 1994;8:995–1006. doi: 10.1101/gad.8.9.995. [DOI] [PubMed] [Google Scholar]
- Lieberman PM, Ozer J, Gursel DB. Requirement for transcription factor IIA (TFIIA)-TFIID recruitment by an activator depends on promoter structure and template competition. Mol Cell Biol. 1997;17:6624–6632. doi: 10.1128/mcb.17.11.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Olave I, Merino A, Reinberg D. Separation of the transcriptional coactivator and antirepression function of transcription factor IIA. Proc Natl Acad Sci. 1996;93:6583–6588. doi: 10.1073/pnas.93.13.6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado E, Ha I, Cortes P, Weis L, Reinberg D. Factors involved in specific transcription by mammalian RNA polymerase II: Role of transcription factors IIA, IID, and IIB during formation of a transcription-competent complex. Mol Cell Biol. 1990;10:6335–6347. doi: 10.1128/mcb.10.12.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunami N, Hamaguchi Y, Yamamoto Y, Kuze K, Kangawa K, Matsuo H, Kawaichi M, Honjo T. A protein binding to the Jκ recombination sequence of immunoglobulin genes contains a sequence related to the integrase motif. Nature. 1989;342:934–937. doi: 10.1038/342934a0. [DOI] [PubMed] [Google Scholar]
- Meisterernst M, Roeder RG. Family of proteins that interact with TFIID and regulate promoter activity. Cell. 1991;67:557–567. doi: 10.1016/0092-8674(91)90530-c. [DOI] [PubMed] [Google Scholar]
- Merino A, Madden KR, Lane WS, Champoux JJ, Reinberg D. DNA topoisomerase I is involved in both repression and activation of transcription. Nature. 1994;365:227–232. doi: 10.1038/365227a0. [DOI] [PubMed] [Google Scholar]
- Nash D. The expression of Hairless in Drosophila and the role of two closely linked modifiers of opposite effect. Genet Res. 1965;6:175–189. doi: 10.1017/s0016672300004079. [DOI] [PubMed] [Google Scholar]
- ————— The mutational basis for the “allelic” modifier mutants, Enhancer and Suppressor of Hairless, of Drosophila melanogaster. Genetics. 1970;64:471–479. doi: 10.1093/genetics/64.3-4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelgeschläger T, Chiang C-M, Roeder RG. Topology and reorganization of a human TFIID-promoter complex. Nature. 1996;382:735–738. doi: 10.1038/382735a0. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingram S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumor suppressor p53. Nature. 1993;364:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- Orphanides G, Lagrange T, Reinberg D. The general transcription factors of RNA polymerase II. Genes & Dev. 1996;10:2657–2683. doi: 10.1101/gad.10.21.2657. [DOI] [PubMed] [Google Scholar]
- Oswald F, Liptay S, Adler G, Schmid RM. NF-κB2 is a putative target gene of activated Notch-1 via RBP-Jκ. Mol Cell Biol. 1998;18:2077–2088. doi: 10.1128/mcb.18.4.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisance S, Berghe WV, Boone E, Fiers W, Haegeman G. Recombination signal sequence binding protein Jκ is constituitively bound to the NF-κB site of the interleukin-6 promoter and acts as a negative regulatory factor. Mol Cell Biol. 1997;17:3733–3743. doi: 10.1128/mcb.17.7.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson ES, Lin J, Kieff E. The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJκ. J Virol. 1996;70:3068–3074. doi: 10.1128/jvi.70.5.3068-3074.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder RG. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci. 1996;21:327–334. [PubMed] [Google Scholar]
- Sauer F, Fondell JD, Ohkuma Y, Roeder RG, Jackle H. Control of transcription by Kruppel through interactions with TFIIB and TFIIEβ. Nature. 1995;375:162–164. doi: 10.1038/375162a0. [DOI] [PubMed] [Google Scholar]
- Schweisguth F, Posakony JW. Suppressor of Hairless, the Drosophila homolog of the mouse recombination signal-binding protein gene, controls sensory organ cell fates. Cell. 1992;69:1199–1212. doi: 10.1016/0092-8674(92)90641-o. [DOI] [PubMed] [Google Scholar]
- Sun X, Ma D, Sheldon M, Kam Y, Reinberg D. Reconstitution of human TFIIA activity from recombinant polypeptides: A role in TFIID-mediated transcription. Genes & Dev. 1994;8:2336–2348. doi: 10.1101/gad.8.19.2336. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Furukawa T, Tun T, Han H, Honjo T. LIM protein KyoT2 negatively regulates transcription by association with the RBP-J DNA-binding protein. Mol Cell Biol. 1998;18:644–654. doi: 10.1128/mcb.18.1.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thut CJ, Goodrich JA, Tjian R. Repression of p53-mediated transcription by MDM2: A dual mechanism. Genes & Dev. 1997;11:1974–1986. doi: 10.1101/gad.11.15.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T, Kawaichi M. Recognition sequence of a highly conserved DNA binding protein RBP-Jκ. Nucleic Acids Res. 1994;22:965–971. doi: 10.1093/nar/22.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke MW, Roeder RG, Sawadogo M. Physical analysis of transcription preinitiation complex formation on class II gene promoter. Science. 1988;241:1335–1338. doi: 10.1126/science.3413495. [DOI] [PubMed] [Google Scholar]
- Waltzer L, Logeat F, Brou C, Israel A, Sergeant A, Manet E. The human Jκ recombination signal sequence binding protein (RBP-Jκ) targets the Epstein-Barr virus EBNA2 protein to its DNA responsive elements. EMBO J. 1994;13:5633–5638. doi: 10.1002/j.1460-2075.1994.tb06901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub SJ, Chow KNB, Luo RX, Zhang SH, He S, Dean DC. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature. 1995;375:812–815. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- Wu GD, Lai EJ, Huang N, Wen X. Oct-1 and CCAAT/Enhancer binding protein (C/EBP) bind to overlapping elements within the interleukin-8 promoter. J Biol Chem. 1997;272:2396–2403. [PubMed] [Google Scholar]
- Wu L, Berk A. Constraints on spacing between transcription factor binding sites in a simple adenovirus promoter. Genes & Dev. 1988;2:403–411. doi: 10.1101/gad.2.4.403. [DOI] [PubMed] [Google Scholar]
- Yeung KC, Inostroza JA, Mermelstein FH, Kannabiran C, Reinberg D. Structure-function analysis of the TBP-binding protein Dr1 reveals a mechanism for repression of class II gene transcription. Genes & Dev. 1994;8:2097–2109. doi: 10.1101/gad.8.17.2097. [DOI] [PubMed] [Google Scholar]
- Yokomori K, Admon A, Goodrich JA, Chen J-L, Tjian R. Drosophila TFIIA-L is processed into two subunits that are associated with the TBP/TAF complex. Genes & Dev. 1993;7:2235–2245. doi: 10.1101/gad.7.11.2235. [DOI] [PubMed] [Google Scholar]
- Zawel L, Kumar PK, Reinberg D. Recycling of the general transcription factors during RNA polymerase II transcription. Genes & Dev. 1995;9:1479–1490. doi: 10.1101/gad.9.12.1479. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Lieberman PM, Boyer TG, Berk AJ. Holo-TFIID supports transcriptional stimulation by diverse activators and from a TATA-less promoter. Genes & Dev. 1992;6:1964–1974. doi: 10.1101/gad.6.10.1964. [DOI] [PubMed] [Google Scholar]
- Zimber-Strobl U, Strobl LJ, Meitinger C, Hinrichs R, Sakai T, Furukawa T, Honjo T, Bornkamm GW. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-Jκ, the homologue of Drosophila Suppressor of Hairless. EMBO J. 1994;13:4973–4982. doi: 10.1002/j.1460-2075.1994.tb06824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]