

Abstract
Cutaneous leishmaniasis (CL) is a neglected tropical disease that causes prominent skin scaring. No water soluble, non-toxic, short course and low cost treatment exists. We developed a new water soluble amphotericin B-polymethacrylic acid (AmB-PMA) using established and scalable chemistries. AmB-PMA was stable for 9 months during storage. In vitro, it was effective against Leishmania spp. promastigotes and amastigote infected macrophages. It was also less toxic and more effective than deoxycholate-AmB, and similar to liposomal AmB. Its in vivo activity was determined in both early and established CL lesion models of Leishmania major infection in genetically susceptible non-healing BALB/c mice. Intradermal AmB-PMA at a total dose of 18 mg of AmB/kg body weight led to rapid parasite killing and lesion healing. No toxicity was seen. No parasite relapse occurred after 80 days follow-up. Histological studies confirmed rapid parasite clearance from macrophages followed by accelerated fibroblast mediated tissue repair, regeneration and cure of the infection. Quantitative mRNA studies of the CL lesions showed that accelerated healing was associated with increased Tumor Necrosis Factor-α and Interferon-γ, and reduced Interleukin-10. These results suggest that a cost-effective AmB-PMA could be used to pharmacologically treat and immunotherapeutically accelerate the healing of CL lesions.
1. Introduction
Cutaneous leishmaniasis (CL) is a global vector borne neglected parasitic disease with increasing incidence. Most cutaneous disease is caused by Leishmania major, L. tropica, L. braziliensis and L. panamensis. It typically occurs as a single cutaneous lesion on exposed body parts (face and upper limb) and varies in size from nodules to large ulcers. It leads to disfiguring scars. This means that early and effective treatment is important for a good clinical outcome. To date, no effective vaccine exists, and the current drug formulary is limited by toxicity, increasing resistance, and costly long treatment regimens. The ideal low cost treatment should kill parasites rapidly and accelerate tissue repair by combining pharmacological with immuno-therapeutic approaches.
To date, there is no water soluble, short-term and highly effective treatment that is cost effective and also free of side-effects[1,2]. In this context, it is important to note that, in both animal models and man:- (i) water soluble deoxycholate-AmB is ineffective; (ii) water soluble high dose intravenous liposomal AmB leads only to the temporary resolution of CL lesions[3-7].
Leishmania sp. are obligate intracellular protozoa that infect, multiply and persist in macrophage endosomes. The organism has evolved to survive in the harsh environment of the phagolysosome, an organelle in which pathogens are usually destroyed. Amphotericin B is a polyene antibiotic produced by Streptomyces sp.. It is not water soluble and therefore has to be dissolved in neat DMSO. Parasite resistance to amphotericin B (AmB) has not occurred after 50 years of clinical use. AmB binds selectively to parasite membrane ergosterol and, to a lesser extent, human cholesterol. There is still a lack of detailed understanding of the relationship between its physico-chemical properties, pharmacokinetics and therapeutic activity[8-14]. The amphiphilic and ampholytic properties of AmB lead to the formation of aggregates in water which are toxic in man[8-12]. However, all existing water and lipid based formulations of AmB are ineffective at curing CL in controlled animal models[3-5]; (i) in the non-healing BALB/c mouse model of L. major, high dose intravenous LAmB (25 - 50 mg of AmB/kg for 6 - 12 days) led only to the temporary resolution of CL lesions; (ii) deoxycholate-AmB was ineffective; (iii) nanodisk associated AmB, when given intraperitoneally (5 mg of Amb/kg) over 21 days, led to the slow healing of CL lesions over 140 - 250 days.
There is also evidence to suggest that strengthening the host's immune responses with immuno-therapy could provide a therapeutically useful addition to the pharmacological activity of AmB[15]. This is because the clinical manifestations of leishmaniasis are a consequence of the balance between parasite defence mechanisms and host microbicidal and immunological responses. A T-helper type 1 (Th1) immune response that increases interferon (IFN)-γ production promotes parasite killing while a Th2 response enables parasite persistence[16-22].
We chose to work with polymethacrylic acid (PMA) because recent years have witnessed:- (i) the advent of PMA hydrogel capsules that are safe in animals: (ii) their development into a mature biomedical platform[23]. Studies have shown that under carefully defined conditions of stoichiometry, relative molecular mass, pH, anionic strength and temperature, mixtures of oppositely charged polyelectrolytes can form discrete, stable and water soluble macromolecules via hydrogen bonded self assembly[24-26]. This relies upon the self assembling ability of polycarboxylic acids; the hydrogen bonds formed act co-operatively to create strong oligomeric structures that have a polyanionic coat in water. In the case of PMA, there is the added advantage that its hydrogel swelling behaviour is pH dependent, and that it forms structures with properties that resemble biological tissues[27,28]. At alkaline pH (e.g., pH 8), its carboxylic acid groups are ionized and they repel each other; this means that the PMA hydrogel swells and small molecule drugs can be trapped within its matrix. However, at acid pH (e.g., pH 5), the carboxylic acid groups are not ionized and the PMA hydrogel collapses; the small molecule drug is released. This makes these pH responsive molecules with rapid and reversible switching between well defined size based conformations into useful candidates for miniaturized drug delivery for certain tissue micro-environments. In addition, anionic linear polymers with a MWt of 15 - 25 kDa preferentially accumulate in the endosomes of tissue macrophages in both animals and man, and have been shown to be non-toxic[29-33].
These physical properties of PMA and its widespread pharmaceutical use over many years[26,34] led us to investigate whether a new, low cost, heat stable and water soluble AmB based drug could be made for CL using established, scalable and low cost chemistries [35]. We now report our detailed results in both an early CL lesion and an established CL lesion non-healing BALB/c mouse model following infection with L. major organisms.
2. Materials and Methods
2.1. Ethics statement
All procedures involving animals were approved by and performed in accordance with the United Kingdom Government (Home Office) licence (PPL70/6712) and the requirements and regulations laid down by the ethical review committee of Imperial College London. Animals were kept in cages and provided with food and water ad libitum. The 3Rs of replacement, reduction and refinement were adhered to.
2.2. Chemical synthesis & analytical characterisation
A fully hydrolysed polymethacrylic acid sodium salt (PMA; Mp 18.5 kDa, Mw 18.6 kDa, pKa 7.3, Sigma {i.e., the gas phase chromatography standard used for polymer characterisation}) was treated with activated charcoal to remove lipopolysaccharide. Endotoxin free AmB (amine pKa 10.0) and glassware, and water for injection were used to make the AmB-PMA. AmB is insoluble in water. It is soluble in neat DMSO. PMA was dissolved in water at 5 mg/ml. The water soluble PMA was then added to the AmB dissolved in DMSO (5 mg/ml; i.e., soluble and monomeric AmB). Sodium hydroxide (1 N) followed by water were added slowly and the mixture stirred at pH 9.2 for 1 h at 20°C. Under these conditions, the ampholytic character of AmB enabled its hydrogen bond mediated self assembly with ionized carboxylic acids in the swollen PMA hydrogel's matrix. In contrast, the simple mixing of AmB with PMA led only to the formation of an insoluble mass that could not be drawn up and administered by injection. The solution was dialysed against water to remove DMSO and free AmB, and then freeze dried. The AmB-PMA's endotoxin content was <0.06 EU units/ml; i.e., the EU standard for water for injection. It was stored in the dark under argon at 4°C. The percentage loading of AmB was determined in 50% methanol by UV spectroscopy at 409 nM, and the relative distribution of the different forms of AmB determined in water by UV spectroscopy from 300 to 450 nm[11,36].
2.3. In vitro biological evaluation
The haemolytic activity of PMA and AmB-PMA was determined by the degree of erythrocyte cell membrane lysis of a 2% v/v solution of washed human erythrocytes at 1 h[9]. The compounds were dissolved in serum free RPMI media. A 1% (v/v) solution of the detergent Triton X-100 was used as the 100% cell lysis positive control. After incubating the compound with the cells in triplicate for 1 h, the tissue culture plate was centrifuged and the supernatant's optical density determined at 409 nm.
The cellular toxicity of PMA and AmB-PMA was determined by culturing each molecule with the macrophage cell line U937, and with mouse bone marrow derived macrophages, and with human monocyte derived macrophages. Cells were suspended at 1 × 106/ml and serial dilutions of the compound (0 - 500 μg/ml of PMA and 0 - 500 μg/ml of AmB for AmB-PMA) added in triplicate. A 2% (v/v) solution of Triton X-100 was used as the 100% cell lysis positive control. The plates were incubated at 37°C, 5% CO2 for 3 days after which MTT was added and cell viability determined.
The immuno-modulatory properties of PMA and AmB-PMA were determined by culturing each molecule with the macrophage cell line U937, and with human monocyte derived macrophages. Cells in RPMI/10% FCS were plated at 1 × 106/ml and left to adhere overnight at 37°C, 5%, CO2. Compounds were then added in triplicate and in serial dilution (0 - 500 μg/ml of PMA and 0 - 500 μg/ml of AmB for AmB-PMA) for 1 h and the cells cultured for 3 h. The plate was then centrifuged at 1,800 rpm for 5 min and the supernatant removed. For each assay, the negative control was cells cultured without compound and the positive control was 25 ng/ml of highly purified LPS because it induced a maximal chemokine and cytokine response[37].
The cells were lysed with Trisol (Tri Reagent, Sigma Aldrich) and the cell pellet harvested. RNA was isolated using Max Tract tubes (Qiagen) and the phenol/chloroform method for precipitating RNA. It was then dissolved in RNase-free water ready for reverse transcription. Quantitative real time RT-PCR for MIP-1β (CCL4), TNF-α, IL-2, IL-4, IL-6, IL-10, IL-12p40, IFN-γ and iNOS mRNA was performed as previously described[37] (and see also the additional details provided below). The 3 h time point for harvesting mRNA was chosen because it was optimal for detecting the cytokines listed.
The in vitro anti-leishmanial activity of PMA and AmB-PMA was determined against L. major and L. donovani stationary phase promastigotes using an MTT assay[4]. One hundred thousand promastigotes in supplemented SD media were seeded per well and the compound added in triplicate. The plates were incubated in a humidified chamber at 26°C, 5 % CO2 for 24 h. A 2% (v/v) solution of Triton X-100 was used as the 100% cell lysis positive control. MTT was added, the plate incubated for 24 h, and then centrifuged at 1,600 rpm for 5 min. The supernatant was removed, DMSO added, and promastigote viability determined at an optical density of 570 nm.
The in vitro anti-leishmanial activity of PMA and AmB-PMA was determined using mouse bone marrow derived macrophages[4]. Cells were plated at 5 × 105/ml onto chamber slides (Lab-Tek) and left to adhere for 24 h. Healthy dividing L. major promastigotes were added at a multiplicity of infection (MOI) of 5:1 to both resting macrophages, and to activated macrophages which had been treated with both recombinant mouse IL-4 (20 Units/ml, PeproTech, UK) and recombinant mouse IL-10 (10 U/ml, PeproTech, UK). The compound was then added and the cells cultured at 37°C, 10% CO2 for 3 days. The optimum end point for the experiment was determined by collecting cells at various time points and staining them with Giemsa May Grunwald. The average intracellular parasite load was determined by oil immersion microscopy of 200 stained macrophages and performed in duplicate. The infection index was expressed as the average number of intracellular parasites per 100 infected macrophages.
The in vitro anti-leishmanial activity of PMA and AmB-PMA was also determined against both L. donovani amastigote infected mouse bone marrow derived macrophages and L. donovani amastigote infected human monocyte derived macrophages[4]. Cells were plated at 7.5 × 105/ml onto chamber slides (Lab-Tek) and left to adhere for 24 h. Healthy dividing amastigote cultures were prepared, the amastigotes pelleted, and then washed with PBS before resuspending them in serum free media; Leishmania spp. are sensitive to complement mediated lysis. Amastigotes were then added to the macrophages at a MOI of 5:1 and the cells cultured at 37°C, 10 % CO2 for 3 days.
Infection was confirmed at day 1 by fixing some cells, staining with Giemsa May Grunwald, and microscopically counting the number of intracellular amastigotes. The media was then aspirated and the macrophages washed with PBS. The compound was dissolved in culture media, added to the cells, and incubated for 3 days. The macrophages were washed to remove any extracellular amastigotes and the slides dried, fixed with methanol, and stained with Giemsa May Grunwald. The average intracellular parasite load was determined by oil-immersion microscopy of 200 Giemsa stained macrophages and performed in duplicate. The infection index was expressed as the average number of intracellular parasites per 100 infected macrophages.
2.4. In vivo biological evaluation
Animal studies were performed under UK Home Office and Imperial College licences for animal experimentation. The in vivo efficacy of the PMA and AmB-PMA was determined in the genetically susceptible BALB/c mouse model of CL[38]. In this non-healing mouse model, an inability to activate macrophages and a poor cell mediated immune response results in chronic ulceration at the site of cutaneous parasite inoculation[39,40]. Disease was induced by subcutaneous injection into the hind footpad of 2 × 106 stationary phase L. major LV 39 (MRHO/SU/59/P-strain) promastigotes that had been cultured directly from lesion amastigotes to ensure their virulence. The parasite load in footpad lesions from killed mice was determined by spraying the footpad with 70 % ethanol followed by its sterile removal. The skin was dissected away and the footpad cut into smaller pieces before homogenising it through a metal sieve. Single cell suspensions were prepared using a pestle in 2 ml of PBS. A 10% dilution in PBS was then made before adding 10 μl to a calibrated Neubauer haemocytometer and counting the number of L. major parasites present[41].
AmB-PMA was administered with a very fine 29G needle. In the early lesion model, 3 intradermal footpad injections were given on days 7, 14 and 21 post infection. In each injection of 50 μl, the concentration of AmB was 2.7 mg/ml making a total of 400 μg of AmB/mouse (i.e., 18 mg of AmB/kg body weight). In the established lesion model, AmB-PMA was started at day 21 following confirmation of an established infection in a subgroup of 4 footpads; i.e., >2 × 106 amastigotes/footpad[41,42]. Three intradermal footpad injections were given over the course of one week on days 21, 25 and 28 post-infection. In each injection of 50 μl, the concentration of AmB was 2.7 mg/ml making a total dose of 400 μg of AmB/mouse (i.e., 18 mg of AmB/kg body weight). Body weight and lesion size were monitored regularly until day 80 by determining the increase in footpad thickness of the infected footpad as compared to the control contralateral footpad using a dial gauge calliper (Kröplin Schnelltaster, Schlüchtern, Germany). In both the early lesion model and in the established lesion model, at least three independent experiments were performed. Control mice were culled when their footpad lesion reached 4 mm in diameter and were ulcerated, and in accordance with the terms of our animal licence. Healing and cure were defined as the disappearance of induration and complete re-epithelialization of the footpad with no lesion relapse at day 80 post infection.
2.5. Delayed type hypersensitivity (DTH) response to reinfection
The delayed type hypersensitivity (DTH) response to reinfection after a therapeutic response to AmB-PMA treatment was also evaluated. Sixty days after the primary infection, 12 BALB/c mice with healed lesions were challenged with 2 × 106 stationary phase L. major (LV 39) promastigotes in the uninfected contralateral footpad. Mice with healed lesions by day 35 were compared with mice with an unhealed primary infection at day 50. Both footpads were monitored. The change in size of each footpad was recorded as the increase in footpad thickness after its inoculation relative to the size of the same footpad just before its inoculation. Daily measurements were made using a dial gauge calliper (Kröplin Schnelltaster, Schlüchtern, Germany) for 7 days.
2.6. Animal studies with free AmB, deoxycholate-AmB or liposomal-AmB
We were restricted in the additional animal model based studies that could be performed using free AmB, Fungizone (Squibb; i.e., deoxycholate-AmB) or AmBisome (Gilead; i.e., liposomal-AmB) for the following toxicity related reasons. Firstly, free AmB is not absorbed through the skin, is insoluble in water, and will only dissolve in an organic solvent like neat DMSO. Although DMSO can be used on the skin at a 10% concentration, we found that free AmB was insoluble in 10% AmB. Secondly, Fungizone and AmBisome have been shown to be biologically ineffective in published animal model based studies of CL caused by L. major, irrespective of the route of drug administration[3-5]. Thirdly, Fungizone is made up of 33% AmB and 66% deoxycholic acid, which is a detergent. When injected at a concentration of only 2.5% into subcutaneous tissues, deoxycholic acid causes intense pain, swelling, acute inflammatory lympho-mononuclear infiltration, adipocyte apoptosis with fat necrosis, intense phagocytosis of fat cells by subcutaneous tissue macrophages, and necrosis of small blood vessels[43,44]. Fourthly, the product monograph for Ambisome states that it can cause significant tissue inflammation when injected subcutaneously in both rats and man (i.e., accidental drug extravasation from an intravenous infusion). Fifthly, the extensive studies of liposomes by Oussoren et al. have shown that both the route of administration and the composition of liposomes are important determinants of their toxicity[45]. For example, liposomes given by intramuscular injection have a faster and more complete clearance of the drug from the injection site (and therefore less local toxicity) than those given by subcutaneous injection. Kadir et al. found that intramuscular injections of liposomes led to macrophage and fat cell infiltration which, in time, came to be replaced by loose connective tissue[46]. This has meant that, in recent years, liposome based drug delivery to the skin and subcutaneous tissue has not been proactively developed by the pharmaceutical industry as a useful new drug delivery method for liposomes. For all of these toxicity related reasons, we were unable to perform comparator animal studies with either free AmB or with the licensed products Fungizone and AmBisome.
2.7. Quantitative real time mRNA RT-PCR
Trisol (Tri Reagent, Sigma Aldrich) was used to lyse the cells and the cell pellet harvested for RNA extraction. RNA was isolated using Max Tract tubes (Qiagen) and the phenol/chloroform method for precipitating RNA. It was then dissolved in RNase-free water and 400 ng reverse transcribed with a Qiagen RT kit. Quantitative RT-PCR was performed at least twice and in triplicate on each sample using Sybr Green Jumpstart PCR mix (Sigma) and a Corbett Rotorgene 3000. Quantification was performed using a multi-gene plasmid that contained cloned gene sequences for chemokines and cytokines and the reference gene HPRT[37,47]. Standards were diluted in the range 20 - 2 × 107 copies. The PCR assays had good linearity (mean r2 = 0.997) for all standard curves and a mean coefficient of variation of 8%. Results were determined as the absolute mRNA copies/105 HPRT copies. Using this assay, 3-fold differences in mRNA copy number could be reliably and reproducibly determined.
The primer sequences used were:-
HPRT forward (f)GTAATGATCCAGTCAACGGGGGAC
HPRT reverse (r)GCAAGCTTGCAACCTTAACCA;
MIP-1β (CCL4) (f)CCTCTCTCTCCTCTTGCTCGT, (r)GCAAAGACTGCTGGTCTCATAG;
TNF-α (f)CACCACGCCTCTGTGTCTACTG, (r)TGAGAAGATGATCTGACTGTGA;
IL-2 (f)CTTCAGTGTCTAGAAGAAGAACT, (r)AATGTTGTTTCAGATCCCTTTAGTT;
IL-4 (f)TCAACCCCCAGCTAGTTGTC, (r)TGTTCTTCGTTGCTGTGAGG;
IL-6 (f)ACCGCTATGAAGTTCCTCTCTG, (r)AATTAAGCCTGCGAGACTTGTGAA;
IL-10 (f)GGTTGCCAAGCCTTATCGGA, (r)ACCTGCTCCACTGCCTTGCT;
IL12 p40 (f)CCTGAAGTGTGAAGCACCAA, (r)AGTCCCTTTGGTCCAGTGTG;
IFN-γ (f)TCAAGTTCAAGTGGCATAGATGTGG, (r)TGGCTCTGCAGGTTTTCATG;
iNOS (f)GTTCTCAGCCCAACAATACAAGA, (r)GTGGACG GGTCGATGTCAC.
2.8. Histological studies
Tissues were fixed with 10% formalin saline for 24 h and embedded in paraffin wax. One micron thick sections were cut from the paraffin blocks and stained with haematoxylin and eosin (H & E) and Periodic acid Schiff (PAS) stains.
2.9. Data analysis
Data analysis was performed using Graphpad Prism 4 software. Results are given as the mean ± sem. p values were determined using a Mann-Whitney test.
3. Results
3.1. Analytical characterisation of AmB-PMA
Under the alkaline synthesis conditions used, the ampholytic (i.e., able to react as either an acid or base) character of AmB enabled its amine (pKa 10.0) to complex with the ionized carboxylic acids in the swollen PMA hydrogel (pKa 7.3) via co-operative hydrogen bonds. Initial structure-function studies established that an AmB loading of 28 ± 2 % was optimum for ensuring the long-term stability (i.e., 9 m) of AmB-PMA at physiological pH (i.e., 7.3); free AmB has a shelf-life of 12 months[47,48]. The median diameter of AmB-PMA was 61 nm by dynamic light scattering as compared to 53 nm for deoxycholate-AmB (Fungizone, Squibb) and 78 nm for liposomal-AmB (LAmB, AmBisome, Gilead). The circular dichroism (CD) spectra from 250 to 450 nm of AmBPMA was similar to deoxycholate-AmB[47,48]. The UV spectroscopic distribution of AmB in AmB-PMA was also similar to that in deoxycholate-AmB and liposomal-AmB (Figure 1a).
Figure 1a. UV spectra.

UV spectra (300 to 450 nm) of AmB-PMA in water (solid line) as compared to deoxycholate-AmB [Fungizone, Squibb] (dotted line) and liposomal AmB [AmBisome, Gilead] (dashed line) at a concentration of 10 μg of AmB/ml (n = 20).
The AmB-PMA dissolved easily at 4 mg/ml in water for injection at pH 7.3 to give a clear, yellow and water soluble solution. It remained in solution after centrifugation at 30,000 rpm for 30 min. However, when the solution's pH was decreased from 6.6 to 6.3, the AmB was released from PMA as confirmed by UV spectroscopy, and the solution became turbid because of the presence of free and water insoluble AmB. The free AmB could now be pelleted by centrifugation at 1,800 rpm for 10 min (Figure 1b). The same result was obtained when this experiment was repeated by dissolving the AmB-PMA in RPMI media containing 10% serum.
Figure 1b. pH related solubility.

pH related solubility of the AmB-PMA in water. AmB-PMA was dissolved in water for injection (pH 7.3) at 1 mg of AmB/ml. On centrifugation, the AmB-PMA remained in solution. The solution pH was then adjusted to 5. The clear solution went turbid and, when centrifuged at 1,800 rpm for 10 min, gave an orange-yellow precipitate. The orange-yellow pellet dissolved in DMSO thereby confirming that it was free AmB. The optical density of the supernatants was measured at 409 nm and the result expressed as the percentage water solubility of the AmB in AmB-PMA when the solubility at pH 7.3 was 100% (n = 6).
3.2. In vitro activity and toxicity of PMA and AmB-PMA
PMA was not toxic to erythrocytes or macrophages at 2 mg/ml after 24 h in an MTT assay. It was also not toxic to either cell type at 500 μg/ml after 3 days. PMA had no intrinsic anti-leishmania activity. Endotoxin free (i.e., <0.06 EU/ml) PMA did not stimulate Macrophage Inflammatory Protein (MIP) -1β (CCL4), TNF-α, IL-2, IL-4, IL-6, IL-10, IL-12p40, IFN-γ or iNOS synthesis in macrophages up to 2 mg/ml.
Erythrocyte lysis by endotoxin free AmB-PMA (at 30 μg of AmB/ml) was 5 ± 1 % compared to 100 ± 5 % for deoxycholate-AmB, and 4 ± 1 % for LAmB after 1 h (Figure 2). No toxicity was seen in macrophages with AmB-PMA at 400 μg of AmB/ml after 3 days. In stability studies, lyophilised AmB-PMA powder was chemically stable (as determined by its loading of AmB, UV spectroscopic distribution, pH responsiveness and CD spectra) (see Supplementary Figure 1) and bioactive (as determined by erythrocyte lysis, macrophage toxicity and anti-leishmania activity) after 9 months storage at 4°C[47,48].
Figure 2. Erythrocyte lysis.

Erythrocyte lysis for AmB-PMA (▲) compared to deoxycholate-AmB (○), and liposomal AmB (□). (n = 20). PMA did not cause erythrocyte lysis.
The 50% lethal dose (LD50) of endotoxin free AmB-PMA for L. major promastigotes was 0.3 ± 0.1 μg of AmB/ml and for L. donovani promastigotes was 0.12 ± 0.1 μg of AmB/ml. For L. donovani amastigotes in macrophages, the LD50 of AmB-PMA was 0.07 ± 0.1 μg of AmB/ml compared to 0.22 ± 0.1 μg of AmB/ml for deoxycholate-AmB, and 0.11 ± 0.1 μg of AmB/ml for liposomal AmB (Figure 3).
Figure 3. In vitro anti-leishmanial activity.

Anti-leishmanial activity of AmB-PMA (▲; ED50 = 0.07 ± 0.1 μg of AmB/ml) as compared to deoxycholate AmB (○; ED50 = 0.22 ± 0.1 μg AmB/ml) and liposomal AmB (□; ED50 = 0.11 ± 0.1 μg AmB/ml) in L. donovani amastigote infected macrophages (n = 9). PMA had no anti-leishmanial activity.
In the human monocyte derived macrophage based model of chemokine and cytokine expression that we have previously described in detail, the maximal pro-inflammatory response occurred with 25 ng/ml of ultrapure Salmonella minnesota lipopolysaccharide (LPS)[37]. In the macrophage cell line U937, neither PMA (12.5 - 200 μg/ml) nor AmBPMA (12.5 - 200 μg of AmB/ml in the AmB-PMA) had any effect on MIP-1β, TNF-α, IL-10 or IFN-γ. The maximal TNF-α response that could be induced in human monocyte derived macrophages with endotoxin free AmB-PMA (50 μg of AmB/ml) was 30 ± 7% of the maximal response seen with LPS (Figure 4). This contrasted with 107 ± 22% for deoxycholate-AmB. Liposomal AmB did not stimulate a TNF-α response. In the case of IFN-γ, the maximal response with endotoxin free AmB-PMA was 88 ± 13% of the maximal response seen with LPS, and 98 ± 12% for deoxycholate-AmB (Figure 4). Liposomal AmB did not stimulate an IFN-γ response. Finally, endotoxin free AmB-PMA did not stimulate IL-10, MIP -1β, IL-2, IL-4, IL-6, IL-12p40 or iNOS. These in vitro results showed that AmB-PMA was stable, non-toxic, efficiently killed Leishmania amastigotes in macrophages, and had immuno-modulatory properties.
Figure 4. In vitro immuno-modulatory activity.

TNF-α and IFN-γ mRNA as determined by quantitative real-time RT-PCR in macrophages treated with AmBPMA (50 μg of AmB/ml) and compared to deoxycholate-AmB and liposomal AmB. Control cells were cultured in media only. Ultrapure Salmonella minnesota lipopolysaccharide (LPS) was used at 25 ng/ml. Endotoxin free PMA was used at 2 mg/ml and had no immuno-modulatory activity (n = 6).
3.3. AmB-PMA in BALB/c mice with CL lesions
The in vivo activity of AmB-PMA was determined in both early and established non-healing CL footpad lesions in genetically susceptible BALB/c mice infected with L. major promastigotes. In this non-healing model of CL, an inability to activate macrophages and a poor cell mediated immune response results in parasite persistence and a sustained neurophil response that leads to chronic tissue ulceration. This is because the parasites modify their local cytokine environment such that the chemoattractants required for effector immune cell recruitment do not accumulate[49,50,51]. This immunological profile is similar in the subcutaneous (footpad) and intradermal (ear) models of L. major infection in BALB/c mice[52].
In control experiments, endotoxin free PMA reconstituted in water for injection was not toxic to mice, and had no effect on either parasite count or cytokine levels[47,48]. In preliminary dose-response studies with AmB-PMA in the early lesion mouse model, several treatment schedules and doses were evaluated[47,48]. These studies showed that:- (i) a total dose of 150 μg of AmB/mouse led to a 95% reduction in parasite load but only a 75% reduction in lesion size; (ii) a total dose of 250 μg of AmB/mouse led to a 95% reduction in parasite load and a 95% reduction in lesion size. These observations led us to perform detailed studies with a total dose of 400 μg of AmB/mouse (i.e., 18 mg of AmB/kg body weight) administered as three intradermal footpad injections of 50 μl each.
3.4. AmB-PMA in BALB/c mice – early CL lesion model
In the early lesion model, 24 mice were treated over a 3 week period on days 7, 14 and 21 post-infection. Six control mice received water injections of 50 μl each. Eighteen treated and 6 control mice were culled at day 35 (Figure 5a & Supplementary Figure 2a). The remaining 6 treated mice continued to be monitored twice weekly until day 80 and were then culled.
Figure 5a. In vivo anti-leishmanial activity – acute model.

In vivo activity of AmB-PMA in the acute non healing cutaneous lesion model in BALB/c mice infected with L. major promastigotes. Three intradermal injections were given over a 3 week period on days 7, 14 and 21 post infection making a total dose of 400 μg of AmB/mouse (i.e., 18 mg of AmB/kg body weight). The footpad lesion size for AmB-PMA treated (□; open square; n = 24 mice/time point) and control (▲; closed triangle; n = 6 mice/time point) animals is shown. PMA had no anti-leishmanial activity. Grey shading on X-axis:- time period during which 3 intradermal injections of AmB-PMA were given.
3.5. AmB-PMA in BALB/c mice – established CL lesion model
In the established and shortened one week treatment lesion model, the presence of an established infection was first confirmed by harvesting 4 control mice for parasite load determination and histological studies. Sixteen mice were then treated with AmB-PMA over a one week period on days 21, 25 and 28 post-infection. Six control mice received water injections of 50 μl each; all were culled by day 52 because their footpad lesions reached the maximum size allowed by our animal license (Figure 5b). The 16 treated mice were monitored twice weekly until day 80 and then culled (Supplementary Figure 2b).
Figure 5b. In vivo anti-leishmanial activity – established model.

In vivo activity of AmB-PMA in the established non healing cutaneous lesion model in BALB/c mice infected with L. major promastigotes. The presence of an established lesion was first confirmed by harvesting 4 control mice for parasite load quantification and histological studies. A shortened treatment course was given; 3 intradermal injections over a one week period on days 21, 25 and 28 post infection making a total dose of 400 μg of AmB/mouse (i.e., 18 mg of AmB/kg body weight). The footpad lesion size for AmB-PMA treated (□; open square; n = 16 mice/time point) and control (▲; closed triangle; n = 6 mice/time point) animals is shown. PMA had no anti-leishmanial activity. Grey shading on the X-axis:- time period during which 3 intradermal injections of AmB-PMA were given.
In these CL lesion models, AmB-PMA led to efficient parasite killing and resolution of non-healing cutaneous lesions in L. major infected BALB/c mice. The parasite number fell by >3 log10 as determined by both ex-vivo counting (Figure 6) and detailed histological studies performed at regular time points (Figure 8). In addition, there was no relapse of CL lesions during long-term follow up from day 35 to day 80 in the established lesion model (Figure 5b).
Figure 6. L. major parasite load in footpad lesions.

L. major parasite load by ex-vivo counting for AmB-PMA treated (□; open square) and control (▲; closed triangle) BALB/c mice in the acute lesion model (n = 6 mice/time point). The ○ symbol at day 0 is the infectious inoculum of L. major promastigotes used.
Figure 8. Footpad histology.






Histological sections (H&E stain, × 1,000 magnification) showing the L. major footpad lesion in an infected and untreated BALB/c mouse footpad biopsy as compared to an infected and AmB-PMA treated (400 μg of AmB/mouse) BALB/c mouse footpad biopsy. (a) Infected and untreated at day 14. L. major amastigotes are seen in macrophages. (b) Infected and untreated at day 28. Numerous amastigotes are seen in the phagolysosomes of macrophages. (c) Infected and untreated at day 50. Numerous amastigotes persist in macrophage phagolysosomes. (d) Infected and treated at day 28. Neutrophils are present. There are no amastigotes. (e) Infected and treated at day 50. There are no neutrophils or amastigotes present. The lesion shows an infiltrate of histiocytes, lymphoid cells and plasma cells. (f) Infected and treated at day 80. The presence of myofibroblasts indicates that lesion healing is in its final stages. There are no amastigotes.
3.6. AmB-PMA in BALB/c mice – toxicity studies
There was no clinical (i.e., weight and general well being as assessed by feeding and drinking behaviour, and the state of the animal's fur), haematological (i.e., haemoglobin, white cell count, platelet count) clotting (i.e., prothrombin time, kaolin partial thromboplastin time, thrombin time), renal biochemistry (i.e., plasma sodium, potassium, bicarbonate, urea, creatinine) or hepatic biochemistry (i.e., plasma aspartate transaminase, alkaline phosphatise, albumin, calcium, phosphate) evidence of AmB-PMA toxicity. These in vivo studies showed that AmB-PMA led to the rapid control of parasite load in genetically susceptible BALB/c mice with non-healing lesions, and to the accelerated healing and cure of both early and established CL lesions.
3.7. Delayed type hypersensitivity (DTH) response to reinfection
Animals cured of CL with AmB-PMA in one footpad were then challenged with infectious parasites in the contralateral footpad. Sixty days after the primary infection, 12 BALB/c mice with healed primary lesions were challenged with 2 × 106 stationary phase L. major (LV 39) promastigotes on three separate occasions. Mice with healed lesions by day 35 were compared with mice with an unhealed primary infection at day 50. A delayed type hypersensitivity (DTH) response was only seen in those mice whose primary footpad lesion had healed by day 35 (Figure 7). This result is interesting because it suggests that Leishmania specific T-cells were being recalled to the challenge site. The observation is notable because inoculation with live parasites is the only intervention that has previously been shown to provide effective immunity; it enabled the recall of an early and sustained IFN-γ response[53,54].
Figure 7. Rechallenge experiment.

Challenge with parasites of BALB/c mice with a healed primary lesion by day 35 compared to mice with an unhealed primary infection at day 50. These mice were challenged with 2 × 106 stationary phase L. major promastigotes in the contralateral footpad on day 60. A classical delayed type hypersensitivity response was only seen in the footpads of the mice with AmB-PMA healed lesions (■; closed square, mice with an AmBPMA healed lesion by day 35; ○; mice with an unhealed primary infection at day 50).(n = 12 mice/time point).
3.8. Histopathological studies
Multiple, well established and histologically circumscribed areas of infection with central foci of necrosis were present in CL lesions by day 7 after parasite inoculation. Neutrophils were present in established lesions (Figure 8a & b). Macrophages containing L. major amastigotes were present in untreated established lesions, with plasma cells at the lesion's periphery (Figure 8c). They did not organise into granulomas. In AmB-PMA treated mice, amastigotes disappeared rapidly from the endosomes of macrophages (Figure 8d & e), and the acute inflammatory infiltrate started to resolve after the first dose of AmB-PMA. By day 80, organising fibroblasts were the predominant cell type present with only occasional macrophages seen. No amastigotes were present (Figure 8f)[55]. The macroscopic appearance of the untreated footpad lesion on day 35 and that of the treated and healed footpad lesion on day 80 is shown in Supplementary Figure 2.
3.9. Quantitative mRNA real time RT-PCR studies
The highest cytokine mRNA copy number and the largest changes with AmB-PMA treatment were seen at the site of parasite growth and lesion development in the inoculated footpad. TNF-α and IFN-γ were both elevated in AmB-PMA treated footpad lesions at the first point sampled (day 14). TNF-α then fell to low levels but IFN-γ remained elevated until the cutaneous lesion had healed (p<0.001) (Figures 9a & b). This cytokine increase was associated with a sustained increase in footpad iNOS during the lesion's healing phase (p<0.001) (Figure 10). AmB-PMA also led to a fall in footpad IL-10 to control lymph node levels (p<0.001) (Figure 9c). There was no significant change in MIP-1β (CCL4) (Figure 9d). AmB-PMA also had no effect on footpad IL-2, IL-4, IL-6 or IL-12p40.
Figures 9 a - d. Cytokine and chemokine responses - 1.


Quantitative real-time mRNA RT-PCR for TNF-α, IFN-γ, IL-10 and MIP-1β in BALB/c mice. AmB-PMA treated footpad (□; open square, solid line); control footpad (▲; closed triangle, dashed line); AmB-PMA treated (and normal size) draining lymph node (○; open circle, solid line); untreated (and enlarged) draining lymph node (●; closed circle, dashed line); Cytokine/chemokine in normal BALB/c mouse spleen (+; S) and lymph node (x; L). n = 6 mice/time point.
Figure 10. Cytokine and chemokine responses - 2.

Quantitative real time mRNA RT-PCR for iNOS in BALB/c mice. AmB-PMA treated footpad (□; open square, solid line); control footpad (▲; closed triangle, dashed line); AmB-PMA treated (and normal size) draining lymph node (○; open circle, solid line); control (and enlarged) draining lymph node (●; closed circle); iNOS in normal BALB/c mouse lymph node (x; L). n = 6 mice/time point.
In the lesion's draining lymph nodes, chemokine and cytokine levels were much lower than those found at the site of footpad infection. There was a modest increase in lymph node TNF-α as footpad TNF-α fell (Figure 9a). In contrast, draining lymph node IFN-γ, IL-10 and MIP-1β (CCL4) did not show a substantial change (Figures 9b - d). iNOS showed a modest increase during the latter stages of lesion healing. There were no differences in spleen TNF-α, IFN-γ, IL-10 and IL-12p40 between AmB-PMA treated and control animals.
4. Discussion
Our results show that established, scalable and low cost synthetic chemistries can be used to make a water soluble AmB-PMA drug that is both safe and stable. In an established and non-healing mouse model of CL lesions, a one week course of 3 intradermal injections led to the rapid pharmacological killing of parasites followed by accelerated healing and cure of the lesion. The accelerated healing was associated with increased TNF-α, IFN-γ and iNOS in the lesions, and reduced IL-10. In mice with an AmB-PMA healed primary infection that were rechallenged with infectious parasites, a DTH response was seen. Taken together, these results suggest, for the first time, that it may be possible to combine the anti-parasite pharmacology of AmB with host immune responses to enhance the cost-effective healing and cure of CL lesions.
Although several studies over many years have suggested that some macromolecules can have immuno-modulatory properties[55-59], we found that this was not the case for 18.5 kDa PMA. Macrophages cultured with PMA over a wide concentration range (0 – 2 mg/ml ) did not alter their homeostatic chemokine or cytokine production (Figure 4), and no immuno-modulatory activity was seen when PMA was administered to mice by a variety of routes[47,48]. This was an important new finding because other macromolecules with immuno-modulatory properties have proved difficult to progress as pharmaceutical drugs; this reflects their lack of homogeneity, irrespective of whether they are derived from natural or synthetic chemistry sources.
Cohen et al. have recently suggested that an ideal drug delivery system should have a diameter of 25 - 100 nm[60]; in our case the AmB-PMA has a diameter of 61 nm. Another of our aims was to safely deliver AmB to parasitized macrophage endosomes in established CL lesions. This was made possible by the different pKa values of PMA and AmB. At physiological pH (i.e., 7.3), the degree of ionization of the carboxylic acids of PMA is small (pKa 7.3), and of the amine of AmB is large (pKa 10.0). This means that during the alkaline synthesis of AmB-PMA (i.e., when the solution pH >PMA's pKa), the ionized carboxylic acids of PMA led to the formation of a hydrogel based matrix within which a defined concentration of AmB could be trapped and retained via hydrogen bonded self assembly. This enabled the non-toxic transport of AmB through tissues and into the cytoplasm. However, in the acidic micro-environment of the endosomallysosomal compartment (pH 5 - 5.5) in which the parasite survives and persists, the carboxylic acids were no longer ionized, the hydrogen bonds disappeared, and the hydrogel's matrix collapsed. As a result, AmB was released. This transition of PMA from its swollen to its collapsed state occurred over the narrow pH range of 6.5 - 6.7. Endosomal AmB disrupted the amastigote's membrane integrity, exposed parasite cytoplasm to a lethal influx of protons, and deprived it of essential substrates (Figure 11).
Figure 11. Proposed mechanism of action.
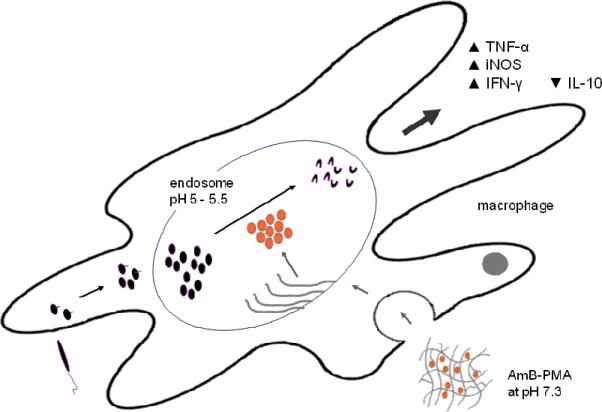
The macrophage is the host cell for Leishmania replication and also the effector for parasite killing. The parasite survives and persists in the late endosomes whose pH is 5 - 5.5. Infection of macrophages does not trigger inflammation and parasite carrying macrophages are not activated. AmB-PMA accumulates in endosomes with a pH of 5 - 5.5. The change from physiological pH (7.3) to that of the endosomal lysosomal compartment pH (5 - 5.5) results in the collapse of the PMA hydrogel matrix. This leads to the release of free AmB. This endosomal AmB rapidly kills parasites, a process that is accelerated by the production of TNF-α, IFN-γ and iNOS within the CL lesion itself.
Macrophages are both the host cell for Leishmania replication and the effector cell for parasite killing. The parasite survives and persists in these late endosomes whose pH is 5 - 5.5. Infection of macrophages does not trigger a pro-inflammatory response, and parasite carrying macrophages inhibit both apoptosis and necrosis[61-64]. The non-toxic delivery of AmB to these macrophage endosomes enables parasite killing by apoptosis[65]. This can be enhanced by co-administration of recombinant IFN-γ or an anti-IL-10 receptor antibody[15,66]. It means that leishmania antigen needs to be:- (i) supplemented with TNF-α: (ii) iNOS has to be induced [67]; and (iii) IL-10 has to be neutralized in order to activate tissue macrophages to produce INF-γ[16]. Our results show that AmB-PMA treatment of CL lesions led to the rapid killing of parasites in macrophages, and to increased TNF-α, IFN-γ and iNOS in the lesion itself (Figures 9, 10 & 11).
Lecoeur et al. has recently shown that two key endpoints should be expected of any new treatment for CL[68]. The first is accelerated clearance of the maximal number of parasites. The second is rapid and stable tissue repair and regeneration. Parasites needed to be exposed to drug for 5 or more days to ensure the long term control of parasite load. This was because duration of parasite drug exposure was a stronger determinant of a good long term outcome than total drug dose. Discontinuous drug applications were also better than several daily applications over a few days. The further clinical development of AmB-PMA as a potential pharmaceutical drug would benefit from the recent description of a transdermal delivery patch that consists of 600 μm long dissolving microneedles. This minimally invasive alternative to hypodermic injection would enable the simple and painless self-administration of AmB-PMA. Furthermore, there is no medical waste because the patch's backing is made of a water soluble polymer that dissolves in a bowl of water that can simply be flushed away[69-71].
5. Conclusion
We report the chemical synthesis of a new water soluble non-toxic AmB-PMA that is chemically simple to make and is also stable during long term storage. Administration by fine needle intradermal injection into CL lesions led to the rapid healing of both early and established L. major infections in the non-healing BALB/c mouse model of CL. A one week course of 3 injections was sufficient to achieve rapid healing and cure. The pharmacologically mediated activity of AmB was enhanced by host immune responses that accelerated healing and led to cure of the infection. This cost-effective AmB-PMA molecule has the potential to be developed into a suitable (and potentially self-administered) transdermally delivered treatment for CL lesions for use in low income countries with limited healthcare resources.
Supplementary Material
6. Acknowledgements
S.S. is grateful to NIH (US) for project grant 1U01A1075726, the UK Williams Trust Fund for project grant AP-117, DNDi Geneva, and the NIHR Biomedical Research Fund for funding support. M.R. is the recipient of the UK Wellcome Trust Fellowship 078223MA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. Competing financial interests:
The authors declare that they have no competing financial interests.
References
- 1.Mahajan VK, Sharma NL. Therapeutic options for cutaneous leishmaniasis. J Dermatol Treat. 2007;18:97–104. doi: 10.1080/09546630601159474. [DOI] [PubMed] [Google Scholar]
- 2.Reithinger R, Dujardin J-C, Louzir H, Pirmez C, Alexander B, Brooker S, et al. Cutaneous leishmaniasis. Lancet Inf Dis. 2007;7:581–596. doi: 10.1016/S1473-3099(07)70209-8. [DOI] [PubMed] [Google Scholar]
- 3.Frankenburg S, Glick D, Klaus S, Barenholz Y. Efficacious topical treatment for murine cutaneous leishmaniasis with ethanolic formulations of AmB. Antimicrob Agents Chemother. 1998;42:3092–3096. doi: 10.1128/aac.42.12.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yardley V, Croft SL. A comparison of the activities of three AmB lipid formulations against experimental visceral and cutaneous leishmaniasis. Int J Antimicrob Agents. 2000;13:243–248. doi: 10.1016/s0924-8579(99)00133-8. [DOI] [PubMed] [Google Scholar]
- 5.Nelson KG, Bishop JV, Ryan RO, Titus R. Nanodisk associated AmB clears L. major cutaneous infection in susceptible BALB/c mice. Antimicrob Agents Chemother. 2006;50:1238–1244. doi: 10.1128/AAC.50.4.1238-1244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wortmann G, Zapor M, Ressner R, Fraser S, Hartzell J, Pierson J, et al. Lipsosomal AmB for treatment of cutaneous leishmania. Am J Trop Med Hyg. 2010;83:1028–1033. doi: 10.4269/ajtmh.2010.10-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solomon M, Pavlotsky F, Leshem E, Ephros M, Trau H, Schwartz E. Liposomal AmB treatment of cutaneous leishmaniasis due to L. tropica. J Eur Acad Dermatol Venereol. 2010 Dec 5; doi: 10.1111/j.1468-3083.2010.03908.x. doi: 10.1111/j.1468-3083.2010.03908.x. [DOI] [PubMed] [Google Scholar]
- 8.Bolard J, Legrand P, Heitz F, Cybulska B. One sided action of AmB on cholesterol containing membranes is determined by its association in media. Biochemistry. 1991;30:5707–5715. doi: 10.1021/bi00237a011. [DOI] [PubMed] [Google Scholar]
- 9.Legrand P, Romero EA, Cohen E, Bolard J. Effects of aggregation and solvent on the toxicity of AmB to human erythrocytes. Antimicrob Agents Chemother. 1992;36:2518–2522. doi: 10.1128/aac.36.11.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brajtburg J, Bolard J. Carrier effects on biological activity of AmB. Clin Microbiol Rev. 1996;9:512–31. doi: 10.1128/cmr.9.4.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawabata M, Onda M, Mita T. Effect of aggregation of AmB on lysophosphatidylcholine micelles as related to its complex formation with cholesterol or ergosterol. J Biochem. 2001;129:725–732. doi: 10.1093/oxfordjournals.jbchem.a002912. [DOI] [PubMed] [Google Scholar]
- 12.Olson JA, Adler-Moore JP, Jensen GM, Schwartz J, Dignani MC, Proffitt RT. Comparison of the physicochemical, antifungal and toxic properties of two liposomal AmB products. Antimicrob Agents Chemother. 2008;52:259–268. doi: 10.1128/AAC.00870-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sundar S, Mehta H, Suresh AV, Singh SP, Rai M, Murray HW. AmB treatment for Indian visceral leishmaniasis: conventional versus lipid formulations. Clin Infect Dis. 2004;38:377–383. doi: 10.1086/380971. [DOI] [PubMed] [Google Scholar]
- 14.Sundar S, Chakravarty J, Agarwal D, Rai M, Murray HW. Single-dose liposomal AmB for visceral leishmaniasis in India. N Engl J Med. 2010;362:504–512. doi: 10.1056/NEJMoa0903627. [DOI] [PubMed] [Google Scholar]
- 15.Murray HW, Brooks EB, DeVecchio JL, Heinzel FP. Immuno-enhancement combined with AmB as treatment for experimental visceral leishmaniasis. Antimicrob Agents Chemother. 2003;47:2513–2517. doi: 10.1128/AAC.47.8.2513-2517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to L. major in mice. Nat Rev Immunol. 2002;2:845–858. doi: 10.1038/nri933. [DOI] [PubMed] [Google Scholar]
- 17.Stanley AC, Engwerda CR. Balancing immunity and pathology in visceral leishmaniasis. Immunol Cell Biol. 2007;85:138–147. doi: 10.1038/sj.icb7100011. [DOI] [PubMed] [Google Scholar]
- 18.Noben-Trauth N, Kropf P, Müller I. Susceptibility to L. major infection in IL-4 deficient mice. Science. 1996;271:987–990. doi: 10.1126/science.271.5251.987. [DOI] [PubMed] [Google Scholar]
- 19.Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4+CD25-Foxp3-Th1 cells are the source of IL-10 mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med. 2007;204:285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, et al. The role of IL-10 in the persistence of L. major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med. 2001;194:1497–1506. doi: 10.1084/jem.194.10.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray HW, Lau CM, Mauze S, Freeman S, Moreira AL, Kaplan G, et al. L-10 in experimental visceral leishmaniasis and IL-10 receptor blockade as immunotherapy. Infect Immun. 2002;70:6284–6293. doi: 10.1128/IAI.70.11.6284-6293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagase H, Jones KM, Anderson CF, Noben-Trauth N. Despite increased CD4+Foxp3+ cells within the infection site, BALB/c IL-4 receptor-deficient mice reveal CD4+Foxp3- T-cells as a source of IL-10 in L. major susceptibility. J Immunol. 2007;179:2435–2444. doi: 10.4049/jimmunol.179.4.2435. [DOI] [PubMed] [Google Scholar]
- 23.Zelikin AN, Price AD, Stadler B. Polymethacrylic acid polymer hydrogel capsules: drug carriers, sub-compartmentalized microreactors, artificial organelles. Small. 2010;6:2201–2207. doi: 10.1002/smll.201000765. [DOI] [PubMed] [Google Scholar]
- 24.Mandel M, Leyte JC, Stadhouder MG. The conformational transition of polymethacrylic acid in solution. J Phys Chem. 1967;71:603–612. [Google Scholar]
- 25.Ikawa T, Abe K, Honda K, Tsuchida E. Interpolymer complex between polyethylene oxide and polycarboxylic acid. J Polymer Science. 1975;13:1505–1514. [Google Scholar]
- 26.Khutoryanskiy VV. Hydrogen-bonded interpolymer complexes as materials for pharmaceutical applications. Int J Pharmaceutics. 2007;334:15–26. doi: 10.1016/j.ijpharm.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 27.Longo GS, de la Cruz MO, Szleifer I. Molecular theory of weak polyelectrolyte gels: the role of pH and salt concentration. Macromolecules. 2011;44:147–158. [Google Scholar]
- 28.Yessine M-A, Leroux J-C. Membrane-destabilizing polyanions: interaction with lipid bilayers and endosomal escape of biomacromolecules. Adv Drug Del Rev. 2004;56:999–1021. doi: 10.1016/j.addr.2003.10.039. [DOI] [PubMed] [Google Scholar]
- 29.Astier A, Doat B, Ferrer M-J, Benoit G, Fleury J, Rolland A, et al. Enhancement of adriamycin antitumor activity by its binding with an intracellular sustained-release form, polymethacrylate nanospheres, in U-937 cells. Cancer Res. 1988;48:1835–1841. [PubMed] [Google Scholar]
- 30.Murthy N, Robichard JR, Tirrell DA, Stayton PS, Hoffman AS. The design and synthesis of polymers for eukaryotic membrane disruption. J Controlled Release. 1999;61:137–143. doi: 10.1016/s0168-3659(99)00114-5. [DOI] [PubMed] [Google Scholar]
- 31.Boudier A, Aubert-Pouëssel A, Louis-Plence P, Gérardin C, Jorgensen C, Devoisselle JM. The control of dendritic cell maturation by pH-sensitive polyion complex micelles. Biomaterials. 2009;20:233–241. doi: 10.1016/j.biomaterials.2008.09.033. [DOI] [PubMed] [Google Scholar]
- 32.Shaunak S, Thornton M, John S, Teo I, Peers E, Mason P, et al. Reduction of the viral load of HIV-1 after the intraperitoneal administration of dextrin 2-sulphate in patients with AIDS. AIDS. 1998;12:399–409. doi: 10.1097/00002030-199804000-00009. [DOI] [PubMed] [Google Scholar]
- 33.Thornton M, Barkley L, Shaunak S. The anti-Kaposi's sarcoma and anti-angiogenic activity of sulphated dextrins. Antimicrob Agents & Chemotherapy. 1999;43:2528–2533. doi: 10.1128/aac.43.10.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Obeidat WM, Abuznait AH, Sallam AS. Sustained release tablets containing soluble polymethacrylates: comparison with tableted polymethacrylate IPEC polymers. AAPS PharmSciTech. 2010;11:54–63. doi: 10.1208/s12249-009-9348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corware KD, Rogers M, Teo I, Muller I, Shaunak S. An amphotericin B-based drug for treating Leishmania major infection. Trans Royal Soc of Trop Med & Hyg. 2010;104:749–750. doi: 10.1016/j.trstmh.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Espada R, Valdespina S, Alfonso C, Rivas G, Paloma Ballesteros M, Torrado JJ. Effect of aggregation state on the toxicity of different AmB preparations. Int J Pharmaceutics. 2008;361:64–69. doi: 10.1016/j.ijpharm.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 37.Shaunak S, Thomas S, Gianasi E, Godwin A, Jones E, Teo I, et al. Polyvalent dendrimer glucosamine conjugates prevent scar tissue formation. Nat Biotechnol. 2004;22:977–985. doi: 10.1038/nbt995. [DOI] [PubMed] [Google Scholar]
- 38.Locksley RM, Heinzel FP, Sadick MD, Holaday BJ, Gardner KD. Murine cutaneous leishmaniasis susceptibility correlates with differential expansion of helper T cell subsets. Ann Inst Pasteur/Immunol. 1987;138:744–749. doi: 10.1016/s0769-2625(87)80030-2. [DOI] [PubMed] [Google Scholar]
- 39.Katzman SD, Fowell DJ. Pathogen imposed skewing of mouse chemokine and cytokine expression at the infected tissue site. J Clin Invest. 2008;118:801–811. doi: 10.1172/JCI33174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell WJ, Meinardus-Hager G, Neugebauer DC, Sorg C. Differences in the onset of the inflammatory response to cutaneous leishmaniasis in resistant and susceptible mice. J Leucoc Biol. 1992;52:135–142. doi: 10.1002/jlb.52.2.135. [DOI] [PubMed] [Google Scholar]
- 41.Titus RG, Marchand M, Boon T, Louis JA. A limiting dilution assay for quantifying L. major in tissues of infected mice. Parasite Immunol. 1985;7:545–555. doi: 10.1111/j.1365-3024.1985.tb00098.x. [DOI] [PubMed] [Google Scholar]
- 42.Tacchini-Cottier F, Zweifel C, Belkaid Y, Mukankundiya C, Vasei M, Launois P, et al. An immuno-modulatory function for neutrophils during the induction of a CD4+ Th2 response in BALB/c mice infected with L. major. J Immunol. 2000;165:2628–2636. doi: 10.4049/jimmunol.165.5.2628. [DOI] [PubMed] [Google Scholar]
- 43.Odo MEY, Cuce LC, Odo LM, Natrielli A. Action of sodium deoxycholate on subcutaneous human tissue: local and systemic effects. Dermatol Surg. 2007;33:178–189. doi: 10.1111/j.1524-4725.2006.33036.x. [DOI] [PubMed] [Google Scholar]
- 44.Schuller-Petrovic S, Wolkart G, Hofler G, Neuhold N, Freisinger F, Brunner F. Tissue toxic effects of phosphatidylcholine/deoxycholate after subcutaneous injection for fat dissolution in rats and a human volunteer. Dermatol Surg. 2008;34:529–543. doi: 10.1111/j.1524-4725.2008.34128.x. [DOI] [PubMed] [Google Scholar]
- 45.Oussoren C, Eling WMC, Crommelin DJA, Storm G, Zuidema J. The influence of the route of administration and liposome composition on the potential of liposomes to protect tissue against local toxicity of two antitumor drugs. Biochimica et Biophysica Acta. 1998;1369:159–172. doi: 10.1016/s0005-2736(97)00221-6. [DOI] [PubMed] [Google Scholar]
- 46.Kadir F, Eling VM, Abrahams D, Zuidema J, Crommelin DJ. Tissue reaction after intramuscular injection of liposomes in mice. Int J Clin Pharmacol Ther Toxicol. 1992;30:374–382. [PubMed] [Google Scholar]
- 47.Corware K. PhD thesis. Imperial College London; 2010. Modifying AmB for the treatment of cutaneous leishmaniasis. [Google Scholar]
- 48.Harris D. PhD thesis. University of London; 2006. A homopolymer complex of AmB for the pharmacological treatment and immunotherapy of leishmaniasis. [Google Scholar]
- 49.Bell WJ, Meinardus-Hager G, Neugebauer DC, Sorg C. Differences in the onset of the inflammatory response to cutaneous leishmaniasis in resistant and susceptible mice. J Leucoc Biol. 1992;52:135–142. doi: 10.1002/jlb.52.2.135. [DOI] [PubMed] [Google Scholar]
- 50.Iezzi G, Frölich A, Ernst B, Ampenberger F, Saeland S, Glaichenhaus N, et al. Lymph node resident rather than skin derived dendritic cells initiate specific T cell responses after L. major infection. J Immunol. 2006;177:1250–1256. doi: 10.4049/jimmunol.177.2.1250. [DOI] [PubMed] [Google Scholar]
- 51.Katzman SD, Fowell DJ. Pathogen imposed skewing of mouse chemokine and cytokine expression at the infected tissue site. J Clin Invest. 2008;118:801–811. doi: 10.1172/JCI33174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cangussú SD, de Souza CC, Campos CF, Vieira LQ, Afonson LCC, Arantes RM. Histopathology of L. major infection: revisiting L. major histopathology in the ear dermis infection model. Mem Inst Oswaldo Cruz. 2009;104:918–922. doi: 10.1590/s0074-02762009000600017. [DOI] [PubMed] [Google Scholar]
- 53.Murray HW, Delph-Etienne S. Roles of endogenous IFN-γ and macrophage microbicidal mechanisms in host response to chemotherapy in experimental visceral leishmaniasis. Infect Immun. 68:288–293. doi: 10.1128/iai.68.1.288-293.2000. 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murphy ML, Wille U, Villegas EN, Hunter CA, Farrell JP. IL-10 mediates susceptibility to L. donovani infection. Eur J Immunol. 2001;31:2848–2856. doi: 10.1002/1521-4141(2001010)31:10<2848::aid-immu2848>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 55.Merigan TC, Finkelstein MS. Interferon stimulating and in vivo antiviral effects of various synthetic anionic polymers. Virology. 1968;35:363–374. doi: 10.1016/0042-6822(68)90215-8. [DOI] [PubMed] [Google Scholar]
- 56.Otterlei M, Ostgaard K, Skjåk-Braek G, Smidsrød O, Soon-Shiong P, Espevik T. Induction of cytokine production from human monocytes stimulated with alginate. J Immunother. doi: 10.1097/00002371-199108000-00007. [DOI] [PubMed] [Google Scholar]
- 57.Flo TH, Ryan L, Latz E, Takeuchi O, Monks BG, Lien E, et al. Involvement of TLR2 and TLR4 in cell activation by mannuronic acid polymers. J Biol Chem. 2002;277:35489–35495. doi: 10.1074/jbc.M201366200. [DOI] [PubMed] [Google Scholar]
- 58.Otterlei M, Ostgaard K, Skjåk-Braek G, Smidsrød O, Soon-Shiong P, Espevik T. Induction of cytokine production from human monocytes stimulated with alginate. J Immunother. 1991;10:286–291. doi: 10.1097/00002371-199108000-00007. [DOI] [PubMed] [Google Scholar]
- 59.Flo TH, Ryan L, Latz E, Takeuchi O, Monks BG, Lien E, et al. Involvement of TLR2 and TLR4 in cell activation by mannuronic acid polymers. J Biol Chem. 2002;277:35489–35495. doi: 10.1074/jbc.M201366200. [DOI] [PubMed] [Google Scholar]
- 60.Cohen Stuart MA, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, et al. Emerging applications of stimuli-responsive polymer materials. Nature Materials. 2010;9:101–113. doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- 61.Moore K, Matlashewski G. Intracellular infection by L. donovani inhibits macrophage apoptosis. J Immunol. 1994;152:2930–2937. [PubMed] [Google Scholar]
- 62.Carrera L, Gazzinelli RT, Badolato R, Hieny S, Muller W, Kuhn R, et al. Leishmania promastigotes selectively inhibit IL-12 induction in bone marrow derived macrophages from susceptible and resistant mice. J Exp Med. 2006;183:515–526. doi: 10.1084/jem.183.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Belkaid Y, Butcher B, Sacks DL. Analysis of cytokine production by inflammatory mouse macrophages at the single-cell level: selective impairment of IL-12 induction in Leishmania infected cells. Eur J Immunol. 1998;28:1389–1400. doi: 10.1002/(SICI)1521-4141(199804)28:04<1389::AID-IMMU1389>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 64.Buates S, Matlashewski G. General suppression of macrophage gene expression during L. donovani infection. J Immunol. 2001;166:3416–34122. doi: 10.4049/jimmunol.166.5.3416. [DOI] [PubMed] [Google Scholar]
- 65.Lee N, Bertholet S, Debrabant A, Muller J, Duncan R, Nakhasi HL, et al. Programmed cell death in the unicellular protozoan parasite Leishmania. Cell Death Differ. 2002;9:53–64. doi: 10.1038/sj.cdd.4400952. [DOI] [PubMed] [Google Scholar]
- 66.Hondowicz B, Scott P. Influence of parasite load on the ability of Th1 T-cells to control L. major infection. Infect Immun. 2002;70:498–503. doi: 10.1128/IAI.70.2.498-503.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kropf P, Freudenberg MA, Modolell M, Price HP, Herath S, Antoniazi S, et al. TLR4 contributes to efficient control of infection with the protozoan parasite L. major. Infect Immun. 2004;72:1920–1928. doi: 10.1128/IAI.72.4.1920-1928.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lecoeur H, Buffet P, Morizot G, Goyard S, Guigon G, Milon G, et al. Optimization of topical therapy for L. major localized cutaneous leishmaniasis using a reliable C57BL/6 model. PLoS Negl Trop Dis. 2007;1(2):e34. doi: 10.1371/journal.pntd.0000034. doi:10.1371/journal.pntd.0000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee JW, Park J-H, Prausnitz MR. Dissolving microneedles for transdermal drug delivery. Biomaterials. 2008;29:2113–2124. doi: 10.1016/j.biomaterials.2007.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sullivan SP, Murthy N, Prausnitz MR. Minimally invasive protein delivery with rapidly dissolving polymer microneedles. Adv Mater. 2008;20:933–938. doi: 10.1002/adma.200701205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JW, Choi SO, Felner EI, Prausnitz MR. Dissolving microneedle patch for transdermal delivery of human growth hormone. Small. 2011;7:531–539. doi: 10.1002/smll.201001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.