

Abstract
Hemizygosity for the retinoblastoma gene RB in man strongly predisposes to retinoblastoma. In the mouse, however, Rb hemizygosity leaves the retina normal, whereas in Rb−/− chimeras pRb-deficient retinoblasts undergo apoptosis. To test whether concomitant inactivation of the Rb-related gene p107 is required to unleash the oncogenic potential of pRb deficiency in the mouse retina, we inactivated both Rb and p107 by homologous recombination in embryonic stem cells and generated chimeric mice. Retinoblastomas were found in five out of seven adult pRb/p107-deficient chimeras. The retinal tumors showed amacrine cell differentiation, and therefore originated from cells committed to the inner but not the outer nuclear layer. Retinal lesions were already observed at embryonic day 17.5. At this stage, the primitive nuclear layer exhibited severe dysplasia, including rosette-like arrangements, and apoptosis. These findings provide formal proof for the role of loss of Rb in retinoblastoma development in the mouse and the first in vivo evidence that p107 can exert a tumor suppressor function.
Keywords: Retinoblastoma, apoptosis, Rb, p107, tumor suppressor gene, chimeric mice
Hereditary retinoblastoma, a childhood tumor of the eye, has served as a paradigm for studies concerning the role of tumor suppressor genes in cancer predisposition. The rate-limiting step in the initiation of both the hereditary and sporadic form of this tumor is loss of function of the retinoblastoma gene RB in the developing retina. Inheritance of one mutant RB allele not only predisposes to retinoblastoma (90%) early in life but also to osteosarcomas (2%) later on (Draper et al. 1986; Friend et al. 1987). In addition, loss of function of RB has been frequently found in lung, breast, and bladder carcinomas (Harbour et al. 1988; Lee et al. 1988; Horowitz et al. 1990). Also, upstream regulators of pRB are repeatedly found mutated (p16 and CDK4) or overexpressed (cyclin D1) in human tumors (Hall and Peters 1996). Thus, deregulation of pRB function appears to be a common event in the development of many tumor types.
pRB plays an important role during the G1 phase of the cell cycle, when cells are responsive to extracellular positive and negative proliferation signals (Sherr 1994). pRB functions in a pathway that transduces such signals to the cell nucleus modulating the activity of, for example, E2F transcription factors. In G1, the transactivating potential of these proteins is suppressed by their association with hypophosphorylated pRB. The E2Fs are released upon phosphorylation of pRB by cyclin D-dependent kinases whose activity depends on mitogenic stimuli. After passing a restriction point, pRB stays in the hyperphosphorylated, inactive conformation throughout the autonomous program that carries the cell through the remaining of G1, S, and G2 phases of the cell cycle (Weinberg 1995). In both sequence and function pRB is closely related to two other nuclear phosphoproteins, p107 and p130 (Ewen et al. 1991; Hannon et al. 1993; Li et al. 1993; Mayol et al. 1993). Extensive structural homology is found in their so called pocket domain, the binding site for many viral oncoproteins, including adenovirus E1A, simian virus 40 large T antigen, and human papillomavirus E7 (DeCaprio et al. 1988; Whyte et al. 1988; Dyson et al. 1989). Like pRB, p107 and p130 may also act as negative regulators of cell proliferation through interaction with E2F transcription factors (Zhu et al. 1993; Claudio et al. 1994; Qin et al. 1995). However, different pRB family proteins associate with different E2Fs at different times during the cell cycle (Bernards 1997). In mice, the Rb gene family members share a wide expression pattern, with high and overlapping expression of Rb and p107 in embryonic liver and CNS (Jiang et al. 1997).
Retinoblastomas have not been described to occur spontaneously in species other than man. In addition to loss of RB, a limited number of karyotypic rearrangements with unknown functional significance have been found in retinal tumors (Kusnetsova et al. 1982; Squire et al. 1984). In the mouse, hemizygosity for Rb does not lead to retinoblastoma. Instead, Rb+/−mice succumb to pituitary gland tumors from 6–8 months on. Rb−/− embryos show severe defects in central neurogenesis, fetal liver erythropoiesis, lens development, and myogenesis, and die around days 12–15 of gestation when the developing retina appears normal (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992; Morgenbesser et al. 1994; Robanus Maandag et al. 1994; Williams et al. 1994b; Zacksenhaus et al. 1996). However, evidence for a function of pRb at later stages of retinal development has come from the analysis of chimeric Rb−/− mice. Apoptosis was observed in the developing retina beyond day 16 of gestation and the number of Rb−/− cells in the adult retina was significantly reduced (Robanus Maandag et al. 1994). These observations suggest that in the mouse loss of Rb during development of the retina results in cell death rather than enhanced cell proliferation. Therefore, additional mutations may be required to unleash the oncogenic potential of pRb deficiency in mouse retinoblasts. This hypothesis is supported by the analyses of various transgenic and knock-out mouse lines. Retinoblastomas develop in transgenic mice with retina-specific expression [using the human interphotoreceptor retinoid-binding protein (IRBP) promoter] of SV40 Tag or HPV-16 E7, the latter exclusively in a p53−/− background (Al-Ubaidi et al. 1992; Howes et al. 1994). These results suggest a requirement for multiple inactivations that possibly include one or more of the pocket proteins and p53. However, which specific proteins need to be inactivated has not been answered and the oncoproteins may elicit other oncogenic alterations as well. In addition, the use of specific promoters to drive SV40 Tag or HPV-16 E7 limits inactivation to those cells in which the oncoproteins are expressed. As a consequence, analyses have remained restricted to a subset of cells within a specific differentiation window characterized by expression of IRBP. Knockout mouse models lack these limitations of the transgenic mouse models. Mice have been generated with (various combinations of) inactivated candidate genes that may be required for retinoblastoma development. Rb+/−;p107−/− mice do not show any altered tumor predisposition when compared with Rb+/− mice but develop multiple dysplastic lesions of the retina that are absent in Rb+/− and p107−/− mice (Lee et al. 1996). In addition, retinal dysplasias have been observed in 40% of the Rb+/−;p53−/− mice as well as pinealoblastomas that show loss of heterozygosity for Rb (Williams et al. 1994a).
The lethality of pRb/p107-deficient embryos at day 11.5 of gestation precludes to study the effect of concomitant pRb and p107 deficiency at later stages of development and during adult life (Lee et al. 1996). To circumvent this problem, we investigated the tumorigenic and developmental potential of Rb−/−;p107−/− cells in the retina of chimeric mice generated with Rb−/−; p107−/− embryonic stem cells. We report here that loss of function of both Rb and p107 in murine retinoblasts leads to retinoblastoma originating from cells committed to the amacrine cell compartment of the inner nuclear layer but not from those committed to the outer nuclear layer of the retina.
Results
Generation of mutant Rb;p107 chimeras
The p107 gene was inactivated in murine embryonic stem (ES) cells by homologous recombination. Because p107 is expressed in ES cells (Fig. 1B), a targeting vector was constructed with the promoterless IRESβgeo cassette (Mountford et al. 1994) inserted behind codon 145 of p107 (Fig. 1A). Homologous recombination of the p107 targeting vector with mouse genomic DNA was predicted to produce a p107 null allele caused by an in-frame termination codon in the IRES sequence. On introduction of the p107 targeting vector into strain 129/Ola-derived ES cells homologous recombinants were obtained with a frequency of 65%. One of these clones carried IRESβgeo in both p107 alleles giving a p107−/− ES cell line. To confirm full inactivation of p107, the level of p107 protein was examined in extracts of ES cells. Whereas p107 could be readily detected in wild-type ES cells, no p107 protein was detected in p107−/− ES cells by Western blot analysis using the anti-p107 rabbit antibody C-18 (Fig. 1B).
Figure 1.

Targeted disruption of p107. (A) Restriction map of the wild-type p107 allele around codon 145 at the EcoRV site and DNA targeting construct. Black boxes indicate determined exons. Upon homologous recombination a fusion transcript is generated of p107, truncated behind codon 145, with IRESβgeo. Probe A and probe B detect modifications at p107. (B) Western blot analysis of p107 in lysates prepared from p107+/+ and p107−/− ES cells. Positions of molecular mass standards and p107 are indicated.
Subsequently, in this p107−/− ES cell line both alleles of the retinoblastoma gene Rb were inactivated by two rounds of homologous recombination with the isogenic targeting vectors 129Rb-hyg (Te Riele et al. 1992) and 129Rb-his. These vectors carry the hygromycin and histidinol resistance genes, respectively, inserted into exon 19 of Rb, which on homologous recombination lead to Rb null alleles (Clarke et al. 1992).
In an attempt to investigate the effect of combined loss of Rb, p107, and p53 in the retina, we also introduced into Rb−/−;p107−/− ES cells the dominant-negative p53 mutant minigene p53DD (Shaulian et al. 1992) driven by the 1.3-kb human interphotoreceptor retinoid-binding protein (hIRBP) promoter fragment (Liou et al. 1990). p53DD has been shown to elicit a biological effect corresponding to genetic loss of p53 including a reduction in apoptosis and acceleration of tumorigenesis (Bowman et al. 1996). We expected Rb−/−;p107−/−;hIRBPp53DD chimeras to mimic hIRBP-E7;p53−/− transgenic mice, which developed retinoblastoma (Howes et al. 1994). Multiple copies of hIRBPp53DD were introduced into Rb−/−;p107−/− ES cells by coelectroporation with the selection marker PGKpur.
Rb+/−;p107−/−, Rb−/−;p107−/−, and Rb−/−;p107−/−;hIRBPp53DD ES cell clones were verified for the correct karyotype and injected into C57BL/6 blastocysts to generate chimeras. The level of pigmentation in the retinal pigment epithelium (RPE) served as a rough indication for the extent of chimerism of the eye (nonpigmented areas result from ES cell contribution).
Poor chimerism in Rb−/−;p107−/− chimeras
Chimeric Rb+/−;p107−/− mice were readily obtained (28/62 births) and were able to transmit ES cell-derived alleles through the germ line. In 6/56 chimeric eyes, the retina showed some dysplasia. For example, Figure 2A shows a small lesion in the transition region of the inner nuclear to the outer plexiform layer. In contrast, Rb−/−;p107−/− chimeras were obtained with low efficiency (7/56 births) and only when a low number of ES cells (4–6) per blastocyst was injected. In general, the ES cell contribution in the tissue samples of these animals was reduced twofold with respect to that of Rb−/− chimeras (Robanus Maandag et al. 1994) (not shown).
Figure 2.
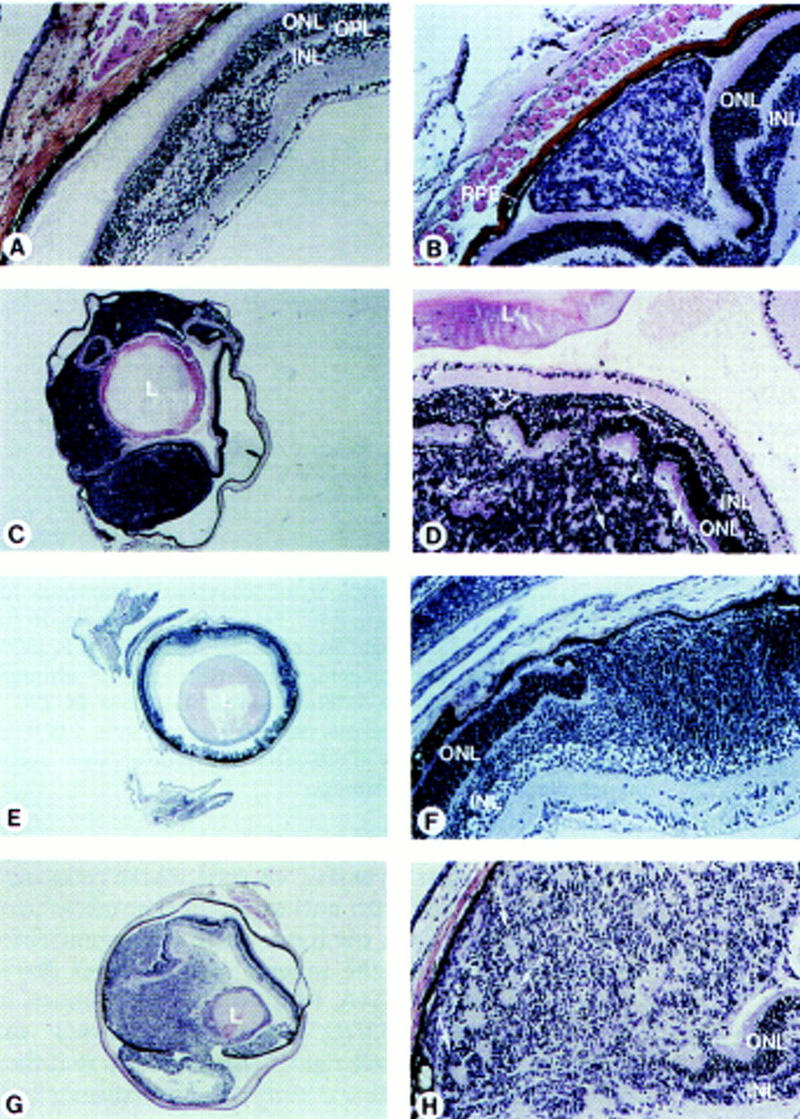
Retinoblastoma in chimeric Rb−/−; p107−/−(;hIRBPp53DD) mice. Histological sections of eyes, stained with hematoxilyn-eosin. (A) Dysplasia in the transit region of the inner nuclear to the outer plexiform retinal layer of a chimeric Rb+/−;p107−/− adult mouse (5 months). (B) Developing retinal tumor consisting of inner-nuclear-layer-like cells growing between the outer nuclear layer and the RPE in a chimeric Rb−/−;p107−/− mouse (1 month). (C) Large retinoblastoma in a chimeric Rb−/−;p107−/− mouse (3.5 months), with (D) rosettes consisting of rearranged photoreceptor cells (open arrows) and tumor cells (solid arrows). (E) Malignant retinoblastoma, with (F) nodular growth of the inner nuclear layer disrupting the outer nuclear layer in a young chimeric Rb−/−;p107−/−; hIRBPp53DD mouse (P15). (G) Large retinoblasto ma in a chimeric Rb−/−;p107−/−;hIRBPp53DD mouse (2.5 months), with (H) rosette-like structures (arrows). (RPE) Retinal pigment epithelium; (ONL) outer nuclear layer consisting of photoreceptor cells; (OPL) outer plexiform layer; (INL) inner nuclear layer; (L) lens. Magnification (C,E,G) 25×; (A,B,D,F,H) 200×.
Retinoblastoma in Rb−/−;p107−/− chimeras
Retinoblastomas were found in five of seven Rb−/−; p107−/−(;hIRBPp53DD) chimeras: Two of six eyes in Rb−/−;p107−/− and four of eight eyes (one bilateral case) in Rb−/−;p107−/−;hIRBPp53DD chimeras. Histological examination of the eyes of Rb−/−;p107−/− chimeras revealed in a 1-month-old chimera a developing retinal tumor between the photoreceptor layer and the RPE, consisting of inner-nuclear-layer-like cells (Fig. 2B). In a chimera of 3.5 months, one of the eyes contained a large tumor process. Microscopically, this appeared to be a retinoblastoma that had invaded into the anterior eye chamber (Fig. 2C,D). The tumor cells often formed small irregular circles. Also, invasion of tumor cells into the outer nuclear layer apparently induced the rearrangement of normal photoreceptor cells into rosettes (Fig. 2D). In a chimeric Rb−/−;p107−/−;hIRBPp53DD mouse of postnatal day 15 (P15) we observed a malignant nodular growth of inner-nuclear-layer-like cells at multiple regions (Fig. 2E,F); similar to the tumor in Figure 2B, the tumor cells tended to invade between the photoreceptor layer and RPE (Fig. 2F). We found three large tumors in chimeric Rb−/−;p107−/−;hIRBPp53DD mice of 2.5 (Fig. 2G,H) and 4 months. In the four large tumors, 3–10 mitotic figures were counted per high power field with a 40× objective (not shown). Thus, both types of chimeras developed retinoblastoma with similar incidence (although the numbers were small), inner-nuclear-layer-like appearance, and tendency to grow between the photoreceptor layer and RPE.
To investigate whether the tumors were ES cell-derived, tumor cells were scraped from formalin-fixed, paraffin-embedded tissue sections, DNAs were isolated and subjected to simple sequence repeat (SSR) analysis. PCR amplification of a polymorphic SSR marker on chromosome 2 (D2mit94) showed that the four large tumors that could be tested in this way were of ES cell origin (Fig. 3). These results indicate that loss of function of both Rb and p107 leads to retinoblastoma in the mouse.
Figure 3.
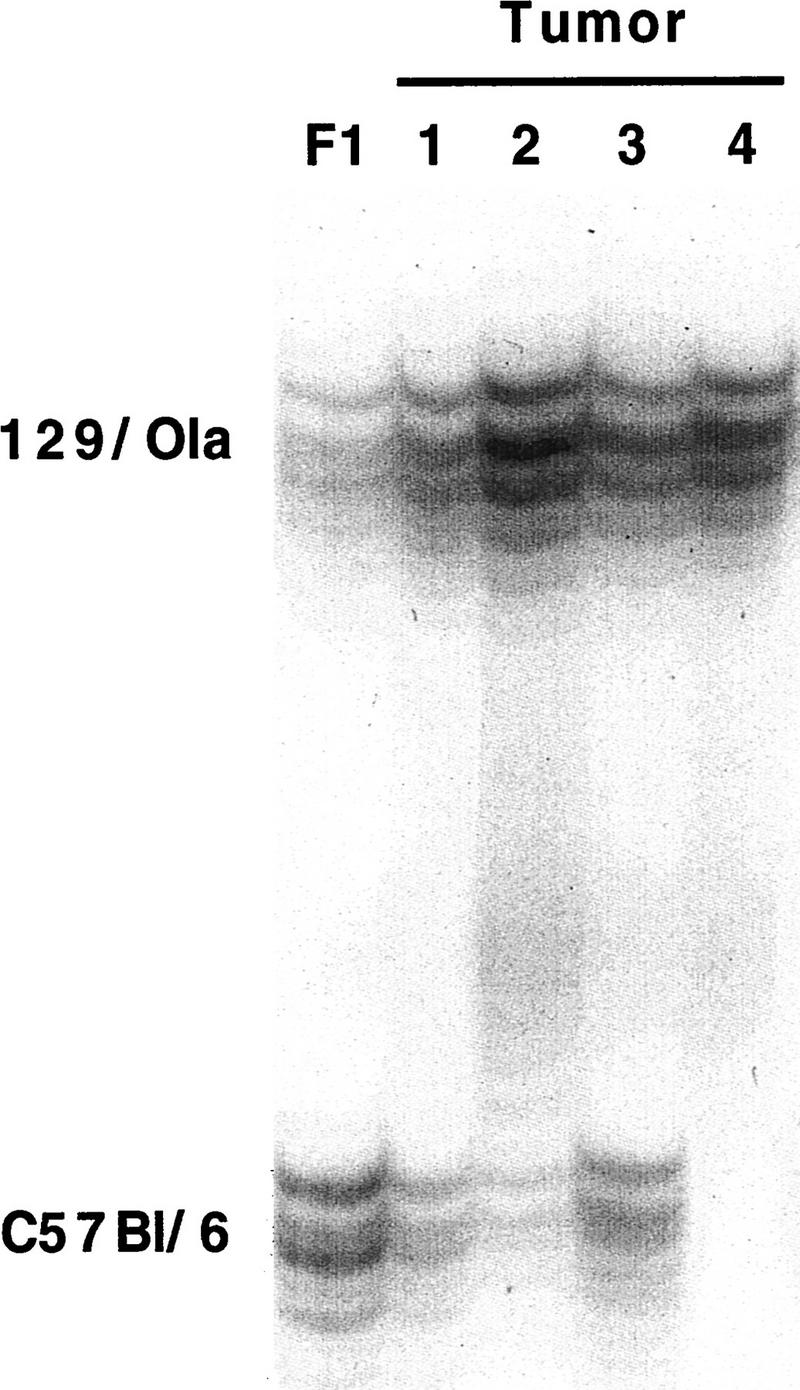
Retinoblastomas are derived from Rb−/−;p107−/− cells. PCR products of a simple sequence repeat polymorphic for 129/Ola (190 nucleotides) and C57Bl/6 (160 nucleotides) in tumor DNAs. (Lane 1) Control liver of 129/Ola:C57Bl/6 F1 (50% 129/Ola). (Lane 2) Retinoblastoma in a Rb−/−;p107−/− chimera (82% 129/Ola). (Lanes 3–5) Three retinoblastomas in Rb−/−; p107−/−;hIRBPp53DD chimeras (96%, 79%, and 99% 129/Ola, respectively). Percentages of 129/Ola contribution were determined using the PhosphorImager.
Retinoblastoma has characteristics of the inner nuclear layer
Our results suggested that the hIRBPp53DD transgene had not contributed to tumorigenesis. To address this point, we further characterized the tumors by immunohistochemistry. Staining with an anti-p53 antibody of the retina of a control hIRBPp53DD transgenic mouse identified a compartment of non-IRBP-expressing cells in the inner nuclear layer (Fig. 4B, left). These cells were positively identified with anti-syntaxin that labels neuronal amacrine cells in the inner nuclear layer and their synaptic processes in the inner plexiform layer (Barnstable et al. 1985) (Fig. 4A, left). None of the tumors in Rb−/−;p107−/−;hIRBPp53DD chimeras expressed the p53DD transgene (Fig. 4B, right). This indicates that they did not express IRBP, as was confirmed by staining with an anti-IRBP antibody recognizing the outer segments of photoreceptor cells (Carter-Dawson et al. 1986) (Fig. 4C). Instead, all retinal tumors showed extensive positive staining with anti-syntaxin (Fig. 4A, right). Furthermore, the tumors were positive with an antibody against neuron-specific enolase (NSE), labeling all neuronal cells of the retina (Schmechel et al. 1978) (Fig. 4E), but negative for the antibody against 200-kD neurofilament protein (NF200kd) which identifies the nerve fibers of the ganglion cells and axonless horizontal cells (Drager et al. 1984) (Fig. 4F). Some staining in the tumors was found with an antibody against glial fibrillary acidic protein (GFAP) (Fig. 4D), which recognizes glial cells, that is, retinal astrocytes in the ganglion cell layer and shows some positivity in the outer plexiform layer (Bjorklund et al. 1985). Double staining with anti-syntaxin and anti-GFAP showed that the cell bodies labeled by anti-syntaxin did not coincide with those labeled by anti-GFAP, indicating that the tumors contained two different cell types, neuronal amacrine and glial cells (Fig. 4G,H). The abnormally high GFAP staining in the adjacent retina may be indicative of reactive Müller cells, which express increased levels of GFAP under pathogenic conditions (Eisenfeld et al. 1984) (Fig. 4I; see also Fig. 4D, right).
Figure 4.
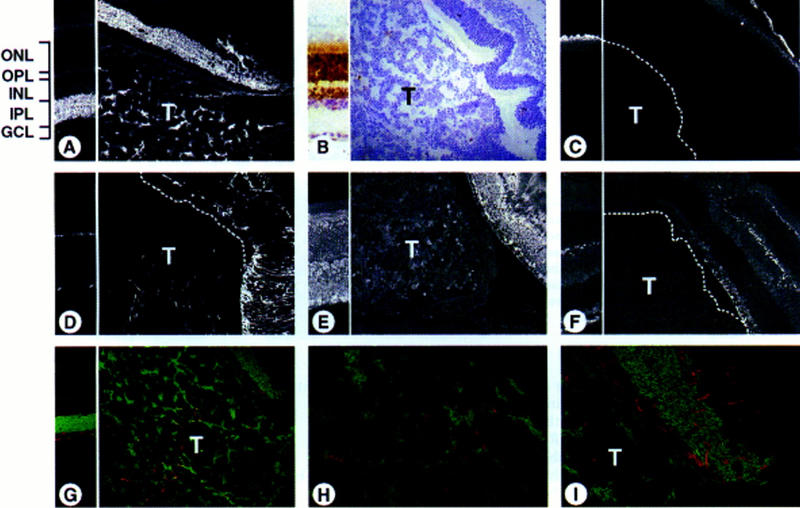
Retinoblastomas show immunocharacteristics of amacrine and glial cells. Sections of a normal retina (transgenic hIRBPp53DD mouse, left parts) and a tumor-containing eye (chimeric Rb−/−;p107−/−;hIRBPp53DD mouse, right parts), immunostained with various antibodies using FITC, Texas Red, and DAB. (ONL) Outer nuclear layer containing photoreceptor (rod and cone) cells; (OPL) outer plexiform layer; (INL) inner nuclear layer containing horizontal, bipolar, Müller, and amacrine cells; (IPL) inner plexiform layer; (GCL) ganglion cell layer containing ganglion cells and astrocytes. (A) Extensive positive staining of the tumor with anti-syntaxin that labels amacrine cells in the INL and their synaptic processes in the IPL (FITC). (B) Whereas the control retina shows positive staining of the ONL and the outer half of the INL with anti-p53, the tumor is negative (DAB). Note the nuclear staining caused by the nuclear localization signal in p53DD. (C) No tumor staining with anti-IRBP that recognizes the outer segments of photoreceptor cells (FITC). (D) Tumor area with cells positive for anti-GFAP that labels glial cells (astrocytes in the GCL) and shows some positivity in the OPL (FITC). (E) Positive staining of the tumor with anti-NSE that immunoreacts with all neuronal cells (FITC). (F) No tumor staining with anti-NF200kd that labels the nerve fibers of ganglion cells in the GCL and axonless horizontal cells in the INL (FITC). (G–I) No double-stained (yellow) cells with anti-syntaxin (FITC) and anti-GFAP (Texas Red) in the tumor (H), or in the adjacent retina (I) showing a staining pattern suggestive of reactive Müller cells spanning across the retina. Dotted lines mark the border of the tumor (T). Magnification (A–G) 200×; (H,I) 600×.
Taken together, these results show that the tumors largely originated from cells committed to the amacrine cell compartment of the inner nuclear layer.
The majority of Rb−/−;p107−/− retinoblasts do not form tumors but undergo apoptosis
During normal retinal development, the non-IRBP-expressing amacrine and ganglion cell compartments are formed prenatally, the Müller cell compartment postnatally. Also prenatally, IRBP-expressing cells begin to appear which will ultimately constitute the outer nuclear layer (photoreceptor cell compartment) and the outer part of the inner nuclear layer (bipolar and horizontal cell compartment) (Young 1985; Al-Ubaidi et al. 1992). Our results suggest that the tumors originate prenatally from a retinoblast population committed to form amacrine cells, but not from the population committed to form IRBP-expressing cells (Duke-Elder and Cook 1963). To identify early stages of tumor development and to study the fate of pRb/p107-deficient IRBP-expressing cells, we analyzed chimeric eyes at various stages of retinal development.
Whereas only a limited number of chimeras survived into adulthood, at day 17.5 of gestation chimeric Rb−/−; p107−/−(;hIRBPp53DD) embryos were present at normal frequency and often showed a high contribution of ES cells to the RPE. At this stage of development, 11/23 chimeric eyes already showed severe dysplasia, rosette-like arrangements and many pyknotic nuclei, indicative of apoptotic cell death (Fig. 5B–D), this in contrast to 6/6 normal chimeric Rb+/−;p107−/− retinas of the same age (Fig. 5A). Clear anti-p53DD antibody staining in the ventricular layer of the developing retina identified Rb−/−; p107−/−;hIRBPp53DD retinoblasts that had apparently reached the differentiation stage of IRBP-expressing cells (Fig. 6B). These cells were virtually absent at P15, even at retinal regions that were highly chimeric as deduced from the presence of malignant nodular growths of the inner nuclear layer (Fig. 6C, see also Fig. 4B). Apparently, pRb/p107-deficient cells can contribute to the IRBP-expressing compartment, but are excluded from the retina between E17.5 and P15. Indeed, apoptotic cell death in chimeric Rb−/−;p107−/− retinas could be detected as early as day 17.5 of gestation and continued at least until P11 (Fig. 7B,D).
Figure 5.

Severe retinal abnormalities in chimeric Rb−/−;p107−/− embryos. Histological sections of eyes, stained with hematoxilyn–eosin. (A) Normal retina of a chimeric Rb+/−;p107−/− embryo (E17.5). (B) Dysplastic retina of a chimeric Rb−/−;p107−/− embryo (E17.5), with rosettes (C), and an increased number of pyknotic nuclei (D) indicative of apoptosis. Magnification (A,B) 100×; (C) 200×; (D) 400×.
Figure 6.
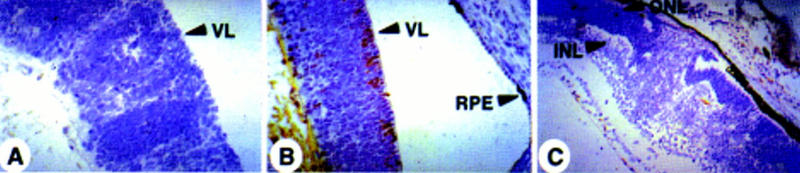
IRBP-expressing Rb−/−;p107−/− cells disappear from the retina before P15. Immunohistochemical staining for p53DD protein with pAb421 plus counterstaining with hematoxilyn. (A) No staining in the dysplastic retina of a chimeric Rb−/−;p107−/− embryo (E17.5). (B) Retina of a chimeric Rb−/−;p107−/−;hIRBPp53DD embryo (E17.5) with positive cells in the ventricular layer (arrows). (C) No staining in the retina with malignant nodular growth of a young chimeric Rb−/−;p107−/−;hIRBPp53DD mouse (P15). (RPE) Retinal pigment epithelium; (VL) ventricular layer; (ONL) outer nuclear layer; (INL) inner nuclear layer. Magnification (A,B) 400×; (C) 200×.
Figure 7.
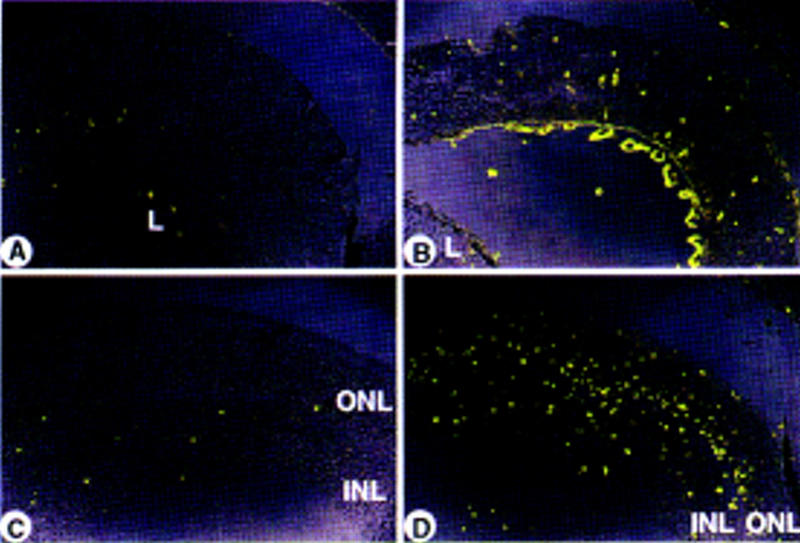
Abnormal apoptosis during retinal development of chimeric Rb−/−;p107−/− mice. FITC staining of apoptotic retinal cells (TUNEL assay). (A) Normal level of apoptosis in chimeric Rb+/−;p107−/−;hIRBPp53DD embryo (E17.5). (B) Increased level of apoptosis in chimeric Rb−/−;p107−/− embryo (E17.5). (C) Normal level of apoptosis in wild-type C57Bl/6 neonate (P11). (D) Massive apoptosis in chimeric Rb−/−;p107−/− neonate (P11). (ONL) Outer nuclear layer; (INL) inner nuclear layer; (L) lens. Magnification (A–D) 200×.
These results allow us to draw two conclusions. Firstly, oncogenic transformation of pRb/p107-deficient retinoblasts occurs as early as day 17.5 of gestation and involves cells committed to the non-IRBP-expressing amacrine cell compartment of the inner nuclear layer. Secondly, pRb/p107-deficient cells can contribute to the IRBP-expressing compartment, but these cells do not grow out to retinoblastoma. Instead, they undergo apoptosis before P15, likely at the stage of differentiation to mature bipolar, horizontal, and photoreceptor cells.
Discussion
In contrast to the situation in humans, mice hemizygous for the retinoblastoma gene Rb do not develop retinoblastoma, but pituitary gland tumors. Also, chimeric Rb−/−mice do not form retinoblastomas but, instead, undergo retinal apoptosis during development. Apparently, the murine retina is better protected against tumorigenesis than the human retina. For example, in the mouse, additional mutations may be required for the oncogenic transformation of pRb-deficient retinoblasts. Indications for this have come from studies in transgenic mice in which concomitant inactivation of pocket proteins and p53 by overexpression of HPV-16 E7 (in a p53 null background) or SV40 Tag led to development of retinoblastoma. However, these transgenic mouse models do not accurately specify the mutational requirements for retinal tumorigenesis. On the other hand, the use of knockout mice carrying specific (combinations of) mutations is limited by the early death of pRb-deficient embryos. Therefore, we generated chimeric mice using ES cells carrying disruptions in both Rb and p107. This approach permitted us to address the question whether loss of p107 unleashes the expected oncogenic potential of loss of Rb in retinoblasts.
Our results clearly demonstrate that this is the case: In the mouse, loss of function of both Rb and its close relative p107 leads to retinoblastoma. Although it is generally believed that loss of pRb function is the key event in retinoblastoma development in the various transgenic mouse models, proof for this has been lacking. Here we have provided formal evidence for the role of loss of Rb in the development of retinoblastomas in mice. Moreover, our results indicate that p107 can act as a tumor suppressor gene in the mouse. p107 exerts its tumor suppressor function in a conditional fashion, that is, it suppresses tumorous outgrowth of pRb-deficient retinoblasts. In the presence of wild-type pRb activity, hetero- or homozygosity for mutant p107 by itself does not lead to tumorigenesis (Lee et al. 1996). Thus, p107 and pRb may functionally overlap, as was previously suggested by their structural homology and capacity to block the cell cycle in vitro (Zhu et al. 1993; Beijersbergen et al. 1995). Our results provide the first demonstration of functional synergism of pRb and p107 in controlling proliferation in vivo. It remains unclear why this safeguard mechanism does not operate in human retinal cells. It is possible that p107, in contrast to the situation in the mouse, is not adequately expressed in the human retina. Alternatively, murine p107 may respond to upstream regulators of pRb, whereas human p107 may not.
The cell of origin of human retinoblastoma is a moot point (Tsokos et al. 1986; Nork et al. 1995). Many believe it is a primitive multipotential cell, but differentiation toward amacrine cells has only sporadically been found (Albert et al. 1974; Tarkkanen et al. 1984; Tsokos et al. 1986). Others suggest it is a cell capable of bipotential differentiation into photoreceptor and glial cells. However, proof for the neoplastic nature of glial cells in human retinoblastoma is lacking. Also, the retinoblastoma cell type in the transgenic mouse models remained largely undefined, although oncogenic transformation was directed to the IRBP-expressing cell compartment. In our model system, immunohistochemistry of the tumors revealed two distinct cell types: Non-IRBP-expressing neuronal amacrine cells (majority) and glial cells (minority). This may indicate that the tumors originate from a primitive retinoblast with bipotential differentiation capacity into amacrine cells and glial cells. The relatively modest GFAP staining in the tumors may also represent nontumorigenic, reactive Müller cells that increased GFAP expression under pathogenic conditions (Eisenfeld et al. 1984). Such cells may support the malignant outgrowth of amacrine-like tumor cells. The in situ identification of individual b−/−;p107−/− cells in the chimeras by a marker will further address this issue. Formally, we cannot fully exclude the possibility that the tumors in our system had originated from IRBP-expressing precursors that were destined to form the photoreceptor cell layer, but had lost this capacity (and IRBP expression) through oncogenic transformation and acquired amacrine cell differentiation.
Although the retinal tumors arise at a high frequency in chimeric Rb−/−;p107−/−(;hIRBPp53DD) mice, additional mutations may be required. First, some but not all retinas that were chimeric in the RPE formed retinoblastomas. Second, Rb+/−;p107−/− mice (Lee et al. 1996) and chimeric Rb+/−;p107−/− mice (our data) often showed regions of retinal dysplasia but never developed a malignant tumor. Third, the tumors apparently arose from developmental defects that occurred as early as embryonic day 17.5. At this stage, the primitive nuclear layer showed severe dysplasia but also extensive apoptosis. This result suggests that in addition to loss of Rb and p107, a genetic alteration counteracting apoptotic cell death is required for development of retinal tumors. We cannot exclude, however, that apoptotic cell death only included the IRBP-expressing pRb/p107-deficient compartment of the retina (see below). In line with this, we could not obtain evidence for involvement of p53 mutations in retinoblastoma development. Single-strand conformation polymorphism and sequence analyses of p53 exons 5–8 in DNA of the large Rb−/−;p107−/− tumor did not reveal a p53 mutation (E. Robanus-Maandag and A. Berns, unpubl.). Moreover, none of the tumors immunoreacted with the anti-p53 antibody.
IRBP-expressing retinoblastomas did not develop in our system. IRBP-expressing pRb/p107-deficient cells were present in the ventricular layer of the embryonic retina, however, these cells underwent massive apoptosis and had completely disappeared from the developing retina by postnatal day 15. Death of IRBP-expressing pRb/p107-deficient cells is in agreement with the observed retinal degeneration in the hIRBP-E7 transgenic mice (Howes et al. 1994). These authors showed that hIRBP-driven expression of E7 could give rise to retinoblastoma exclusively in a p53−/− background, suggesting that p53 counteracted apoptosis of cells that lacked Rb (and p107) function. Therefore, the absence of p53DD-expressing retinoblastomas in our chimeric Rb−/−;p107−/−; hIRBPp53DD mice was unexpected. It is possible that the wild-type p53 was not fully inactivated by p53DD. Alternatively, other as yet unknown oncogenic alterations may be required for the development of outer nuclear layer tumors. In the hIRBP-E7;p53−/− transgenic mice, genetic instability throughout all stages of development may have provided the required mutation(s) [note that in hIRBP-E7;p53+/− transgenic mice no retinal tumors were found (Howes et al. 1994)]. In conclusion, our observations show that Rb−/−;p107−/− retinoblasts committed to the non-IRBP-expressing inner nuclear layer have the potential to form tumors at high incidence, whereas IRBP-expressing Rb−/−;p107−/− retinoblasts do not. Because both types of retinoblasts originate from the same Rb−/−;p107−/− retinal stem cell, different, and possibly fewer, mutations may be required for the development of inner nuclear layer tumors than for the development of outer nuclear layer tumors.
Functional loss of Rb in murine retinoblasts has been generally believed as essential for the development of retinoblastoma analogous to the situation in man. However, the absence of retinoblastoma in Rb+/− and chimeric Rb−/− mice indicated that besides loss of function of Rb additional mutations are required to induce tumorigenesis in the murine retina. Our data unequivocally demonstrate that the inactivation of both Rb and the closely related gene p107 leads to oncogenic transformation of cells committed to the amacrine cell compartment of the inner nuclear layer but not of cells committed to other retinal compartments. Thus, p107 can act as a tumor suppressor gene in the mouse. Finally, our results illustrate that the generation of chimeric mice with ES cells carrying multiple gene lesions is a valuable tool to assess the role of these genes in development and tumorigenesis.
Materials and methods
Marker plasmids
For the disruption of multiple genes in a single cell line, we generated two new selectable markers: PGKpur and PGKhis. The Streptomyces alboniger puromycin phosphotransferase gene pur (Lacalle et al. 1989; kindly provided by A. Jimenez) was provided with a Kozak consensus sequence and inserted between the PGK promoter and poly(A) sequences (McBurney et al. 1991), giving PGKpur. The Salmonella typhimurium histidinol dehydrogenase gene his (Hartman and Mulligan 1988) was inserted between the PGK promoter and poly(A) sequences, giving PGKhis.
Generation of DNA fragments for electroporation
For construction of the p107 targeting vector (129p107–IRESβgeo), a 129-derived genomic clone covering a portion of the mouse p107 gene was isolated using the human p107 cDNA (kindly provided by M. Ewen and D. Livingston, Dana-Farber Cancer Institute, Boston, MA) as a probe. An exon was identified within the genomic clone by a combination of Southern blot and sequence analysis. Into a unique EcoRV site within this exon, IRESβgeo [derived from the plasmid pGT1.8IRES-βgeo (Mountford et al. 1994), kindly provided by A. Smith] was inserted, resulting in a fusion transcript containing p107 codons 1–145 and IRESβgeo. The targeting vector was linearized before electroporation into ES cells (Fig. 1A). Two comparable isogenic targeting vectors for the Rb locus were used: 129Rb–hyg (Te Riele et al. 1992) and 129Rb–his, carrying instead of PGKhyg the 2.2-kb PGKhis fragment. Both markers were inserted into the BglII site of exon 19.
For the construction of hIRBPp53DD the plasmid pSPp53DD (Shaulian et al. 1992; kindly provided by M. Oren) was digested with BamHI, filled in with Klenow polymerase, and digested with EcoRI. The resulting 800-bp p53DD cDNA fragment, containing amino acids 1–13 and 302–390, was ligated into the EcoRV–EcoRI-digested plasmid containing the 1.3-kb hIRBP promoter fragment, kindly provided by G. Liou (Medical College of Georgia, Augusta). phIRBPp53DD was linearized with BamHI, pPGKpur with XhoI.
Generation of mutant ES cell clones and chimeric mice
The E14 ES cell line, derived from 129/Ola and kindly provided by M. Hooper (Western General Hospital, Edinburgh, UK), was subcloned. Subclone IB10 and its derivatives were grown on feeder layers of γ-irradiated murine embryonic fibroblasts in Glasgow modified Eagle medium supplemented with 10% fetal calf serum, 1× nonessential amino acids, 1 mm sodium pyruvate, 2 mm l-glutamine, 0.1 mm 2-mercaptoethanol, and 1000 U/ml of ESGRO-LIF. During selection the ES cells were cultured in BRL-conditioned medium (Hooper et al. 1987).
129p107-IRESβgeo was introduced into IB10 ES cells and G418-resistant cells were selected as described (Te Riele et al. 1992). Southern blot analysis with the 5′ probe A and 3′ probe B, located as in Figure 1A, of EcoRI-digested DNA from selected clones showed in case of a homologous recombinant both a band of 20 kb of the nonmodified p107 locus, and a band of 3.4 and 16.6 kb of the modified p107 locus, respectively. In addition, one p107−/− ES cell clone was obtained that had inserted the promoterless IRESβgeo in both p107 alleles. Subsequently, in this p107−/− ES cell clone targeting at the Rb locus was performed with 129Rb-hyg as described (Te Riele et al. 1992). In a resulting Rb+/−;p107−/− clone the second wild-type Rb allele was targeted with 129Rb-his. One day after electroporation, ES cells were selected for resistance to 1.5–2.5 mm histidinol for 7 days. Southern blot analysis with the 5′ Rb probe B (Te Riele et al. 1992) of EcoRI-digested DNA from selected clones showed a 7.2-kb band in case of a correctly recombined 129Rb-his fragment.
The 5.0-kb hIRBPp53DD fragment was coelectroporated with the 4.3-kb PGKpur fragment into Rb−/−;p107−/− ES cells in a molar ratio of 10:1. One day after electroporation, ES cells were selected for resistance to 1.8 μg/ml puromycin for 7 days. A 450-bp EcoRV–XbaI fragment of hIRBPp53DD was used as probe in the Southern blot analysis of EcoRV-digested DNA from the selected clones. A 5-kb band indicated a head-to-tail integration of hIRBPp53DD.
Selected ES cell clones were verified for the correct karyotype (>12/15 metaphase chromosome spreads with 40 chromosomes). Chimeric mice were generated by injection of 4–12 ES cells into C57Bl/6 blastocyst stage embryos.
Western blot analysis
ES cells (3 × 106) were resuspended in 50 μl of 2× Laemmli sample buffer. The lysates were boiled for 10 min and, after centrifugation for 2 min, 30% of the supernatant was loaded on a 10% SDS–polyacrylamide gel. After resolution, the gel was transferred to a Protran membrane (Schleicher & Schüll) by electroblotting. For the antibody incubation with anti-p107, performed in 5% Blotto dissolved in TBST (Tris-buffered saline; 0.1% Tween-20), the polyclonal rabbit anti-human antibody C-18 was used that recognizes amino acids 1052–1068 of p107 (Santa Cruz Biotechnology). Subsequently, the membrane was incubated with goat anti-rabbit horseradish peroxidase-labeled antibody. Antigen–antibody complexes were detected by enhanced chemoluminescence (ECL; Amersham).
Generation of transgenic hIRBPp53DD mice
The 2.1-kb ClaI–BamHI fragment of phIRBPp53DD was microinjected into FVB zygotes. Southern blot analysis of EcoRV-digested DNA from tail biopsies was performed as described (Laird et al. 1991) using the 450-bp EcoRV–XbaI fragment of phIRBPp53DD as probe.
ES cell contribution in (tumor) tissues
DNA was isolated from tissue samples as described by Laird et al. (1991). The extent of chimerism was determined by detection of the Rb wild-type and mutated EcoRI fragments with probe A on Southern blots as described before (Te Riele et al. 1992) using the PhosphorImager. Cells of the tumor areas in the unstained 10 μm tissue sections were scraped off with a scalpel from the plain glass slides and transferred to 0.5 ml of xylene to dissolve the paraffin for 5 min. One volume of 100% ethanol was mixed with the supernatants and after 5 min the tissues were pelleted, dried at 55°C, and incubated in 50 mm Tris (pH 8.5), 1 mm EDTA, 0.5% Tween-20, and 200 μg/ml proteinase K overnight at 55°C and for 10 min at 95°C. To determine the percentage of ES cell-derived cells in the tumors, simple sequence repeat analyses were performed on the DNA solutions with the primer set D2mit94 (Mouse MapPairs, Research Genetics, Huntsville, AL) as described (Dietrich et al. 1992).
Histological analysis and immunostaining
Embryos and tissues were fixed in phosphate-buffered formalin, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin according to standard procedures.
For immunohistochemical detection of antigens, the rehydrated tissue sections were boiled for 15 min in citrate buffer at pH 6.0 and cooled down slowly before preincubation with 1% normal goat serum. The following primary antibodies were used: (1) mouse monoclonal anti-human p53 that recognizes amino acids 370–378 (pAb421, Harlow et al. 1981; Oncogene Science); (2) mouse monoclonal anti-rat syntaxin (HPC-1, Sigma Biosciences); (3) rabbit polyclonal anti-bovine IRBP (kindly provided by Yvonne De Kozak, U450 INSERM, Paris, France); (4) rabbit polyclonal anti-cow glial fibrillary acidic protein (GFAP; DAKO); (5) rabbit polyclonal anti-bovine neuron-specific enolase (NSE; Chemicon International); and (6) rabbit polyclonal anti-bovine neurofilament, 200-kD subunit (NF200kd; Sigma).
Expression of the p53DD transgene was detected by the indirect immunoperoxidase assay with DAB substrate as described (Ivanyi et al. 1989). Expression of the endogenous retinal antigens was determined by the indirect immunofluorescence assay with goat anti-mouse or pig anti-rabbit FITC (DAKO) and, in case of double staining, goat anti-rabbit Texas Red (Molecular Probes, Leiden, The Netherlands). Incorporated fluorescein was detected by confocal laser scan microscopy.
In situ detection of apoptosis
TUNEL analyses (Gavrieli et al. 1992) were performed on 8-μm tissue sections as described (In Situ Cell Death Detection kit, Boehringer Mannheim). Incorporated fluorescein was detected by confocal laser scan microscopy.
Acknowledgments
We thank Paul Krimpenfort, Karin van Veen-Buurman, and René Bobeldijk for assistance in zygote and blastocyst injections; Jurjen Bulthuis, Kees de Goeij, Lia Kuijper-Pietersma, and Eva van Muylwijk for histotechnical assistance; Rein Regnerus for tail DNA analysis; Fina van der Ahé, Kwamé Ankama, Nel Bosnie, Halfdan Raasø, Loes Rijswijk, and Auke Zwerver for animal care; Lauran Oomen for assistance with the confocal laser scan microscope; René Bernards, Gabriel Gil-Gómez, and Marc Vooijs for critically reading the manuscript. This work was supported by The Netherlands Organization for Scientific Research (NWO) through a program grant to A.B. (E.R.-M.), the European Community (E.R.-M.), and the Netherlands Cancer Foundation (J.-H.D.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL hriele@nki.nl; FAX 31 205121954.
References
- Albert DM, Lahav M, Lesser R, Craft J. Recent observations regarding retinoblastoma, I: Ultrastructure, tissue culture growth, incidence, and animal models. Trans Ophthalmol Soc UK. 1974;94:909–928. [PubMed] [Google Scholar]
- Al-Ubaidi MR, Font RL, Quiambao AB, Keener MJ, Liou GI, Overbeek PA, Baehr W. Bilateral retinal and brain tumors in transgenic mice expressing simian virus 40 large T antigen under control of the human interphotoreceptor retinoid-binding protein promoter. J Cell Biol. 1992;119:1681–1687. doi: 10.1083/jcb.119.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnstable CJ, Hofstein R, Akagawa K. A marker of early amacrine cell development in rat retina. Brain Res. 1985;20:286–290. doi: 10.1016/0165-3806(85)90116-6. [DOI] [PubMed] [Google Scholar]
- Beijersbergen RL, Carlée L, Kerkhoven RM, Bernards R. Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes & Dev. 1995;9:1340–1353. doi: 10.1101/gad.9.11.1340. [DOI] [PubMed] [Google Scholar]
- Bernards R. E2F: A nodal point in cell cycle regulation. Biochim Biophys Acta. 1997;1333:33–40. doi: 10.1016/s0304-419x(97)00027-9. [DOI] [PubMed] [Google Scholar]
- Bjorklund H, Bignami A, Dahl D. Immunohistochemical demonstration of glial fibrillary acidic protein in normal rat Müller glia and retinal astrocytes. Neurosci Lett. 1985;54:363–368. [PubMed] [Google Scholar]
- Bowman T, Symonds H, Gu L, Yin C, Oren M, Van Dyke T. Tissue-specific inactivation of p53 tumor suppression in the mouse. Genes & Dev. 1996;10:826–835. doi: 10.1101/gad.10.7.826. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson L, Alvarez RA, Fong S-L, Liou GI, Sperling HG, Bridges CDB. Rhodopsin, 11-cis vitamin A, and interstitial retinol-binding protein (IRBP) during retinal development in normal and rd mutant mice. Dev Biol. 1986;116:431–438. doi: 10.1016/0012-1606(86)90144-2. [DOI] [PubMed] [Google Scholar]
- Clarke AR, Robanus Maandag E, van Roon M, van der Lugt NMT, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- Claudio PP, Howard CM, Baldi A, De Luca A, Fu Y, Condorelli G, Sun Y, Colburn N, Calabretta B, Giordano A. p130/pRB2 has growth suppressive properties similar to yet distinctive from those of retinoblastoma family members pRB and p107. Cancer Res. 1994;54:5556–5560. [PubMed] [Google Scholar]
- DeCaprio JA, Ludlow JW, Figge J, Shew J-Y, Huang C-M, Lee W-H, Marsilio E, Paucha E, Livingston DM. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- Dietrich W, Katz H, Lincoln SE, Shin H-S, Friedman J, Dracopoli NC, Lander ES. A genetic map for the mouse suitable for typing intraspecific crosses. Genetics. 1992;131:423–447. doi: 10.1093/genetics/131.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drager UC, Edwards DL, Barnstable CJ. Antibodies against filamentous proteins in discrete cell types of the mouse retina. J Neurosci. 1984;4:2025–2042. doi: 10.1523/JNEUROSCI.04-08-02025.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper GJ, Sanders BM, Kingston JE. Second primary neoplasms in patients with retinoblastoma. Br J Cancer. 1986;53:661–671. doi: 10.1038/bjc.1986.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke-Elder S, Cook C. Embryology. In: Duke-Elder S, editor. System of ophthalmology. London, UK: Kimpton; 1963. pp. 81–109. [Google Scholar]
- Dyson N, Howley PM, Munger K, Harlow E. The human papillomavirus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–936. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- Eisenfeld AJ, Bunt-Milam AH, Sarthy PV. Müller cell expression of glial fibrillary acidic protein after genetic and experimental photoreceptor degeneration in the rat retina. Invest Ophthalmol Vis Sci. 1984;25:1321–1328. [PubMed] [Google Scholar]
- Ewen ME, Xing Y, Lawrence JB, Livingston DM. Molecular cloning, chromosomal mapping, and expression of the cDNA for p107, a retinoblastoma gene product-related protein. Cell. 1991;66:1155–1164. doi: 10.1016/0092-8674(91)90038-z. [DOI] [PubMed] [Google Scholar]
- Friend SH, Horowitz JM, Gerber MR, Wang X-F, Bogenmann E, Li FP, Weinberg RA. Deletions of a DNA sequence in retinoblastomas and mesenchymal tumors: Organization of the sequence and its encoded protein. Proc Natl Acad Sci. 1987;84:9059–9063. doi: 10.1073/pnas.84.24.9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- Hannon GJ, Demetrick D, Beach D. Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes & Dev. 1993;7:2378–2391. doi: 10.1101/gad.7.12a.2378. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Lai S-L, Whang-Peng J, Gazdar AF, Minna JD, Kaye FJ. Abnormalities in structure and expression of the human retinoblastoma gene in SCLC. Science. 1988;241:353–357. doi: 10.1126/science.2838909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E, Crawford LV, Pim DC, Williamson NM. Monoclonal antibodies specific for simian virus 40 tumor antigens. J Virol. 1981;39:861–869. doi: 10.1128/jvi.39.3.861-869.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman S, Mulligan RC. Two dominant-acting selectable markers for gene transfer studies in mammalian cells. Proc Natl Acad Sci. 1988;85:8047–8051. doi: 10.1073/pnas.85.21.8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper M, Hardy K, Handyside A, Hunter S, Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- Horowitz JM, Park S-H, Bogenmann E, Cheng J-C, Yandell DW, Kaye FJ, Minna JD, Drya TP, Weinberg RA. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc Natl Acad Sci. 1990;87:2775–2779. doi: 10.1073/pnas.87.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes KA, Ransom N, Papermaster DS, Lasudry JGH, Albert DM, Windle JJ. Apoptosis or retinoblasoma: Alternative fates of photoreceptors expressing the HPV-16 E7 gene in the presence or absence of p53. Genes & Dev. 1994;8:1300–1310. doi: 10.1101/gad.8.11.1300. [DOI] [PubMed] [Google Scholar]
- Ivanyi D, Ansink A, Groeneveld E, Hageman PC, Mooi WJ, Heintz APM. New monoclonal antibodies recognizing epidermal differentiation-associated keratins in formalin-fixed, paraffin-embedded tissue. Keratin 10 expression in carcinoma of the vulva. J Pathol. 1989;159:7–12. doi: 10.1002/path.1711590105. [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Zacksenhaus E, Gallie BL, Phillips RA. The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene. 1997;14:1789–1797. doi: 10.1038/sj.onc.1201014. [DOI] [PubMed] [Google Scholar]
- Kusnetsova LE, Prigogina EL, Pogosianz HE, Belkina BM. Similar chromosomal abnormalities in several retinoblastomas. Hum Genet. 1982;61:201–204. doi: 10.1007/BF00296442. [DOI] [PubMed] [Google Scholar]
- Lacalle RA, Pulido D, Vara J, Zalacaín M, Jiménez A. Molecular analysis of the pac gene encoding a puromycin N-acetyl transferase from Streptomyces alboniger. Gene. 1989;79:375–380. doi: 10.1016/0378-1119(89)90220-5. [DOI] [PubMed] [Google Scholar]
- Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EY-HP, To H, Shew J-Y, Bookstein R, Scully P, Lee W-H. Inactivation of the retinoblastoma susceptibility gene in human breast cancers. Science. 1988;241:218–221. doi: 10.1126/science.3388033. [DOI] [PubMed] [Google Scholar]
- Lee EY-HP, Chang C-Y, Hu N, Wang Y-C, Lai C-C, Herrup K, Lee W-H, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Lee M-H, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, Harlow E, Jacks T. Targeted disruption of p107: Functional overlap between p107 and Rb. Genes & Dev. 1996;10:1621–1632. doi: 10.1101/gad.10.13.1621. [DOI] [PubMed] [Google Scholar]
- Li Y, Graham C, Lacy S, Duncan AMV, Whyte P. The adenovirus E1A-associated 130-kD protein is encoded by a member of the retinoblastoma gene family and physically interacts with cyclins A and E. Genes & Dev. 1993;7:2366–2377. doi: 10.1101/gad.7.12a.2366. [DOI] [PubMed] [Google Scholar]
- Liou GI, Geng L, Al-Ubaidi MR, Matragoon S, Hanten G, Baehr W, Overbeek PA. Tissue-specific expression in transgenic mice directed by the 5′-flanking sequences of the human gene encoding interphotoreceptor retinoid-binding protein. J Biol Chem. 1990;265:8373–8376. [PubMed] [Google Scholar]
- Mayol X, Grana X, Baldi A, Sang N, Hu Q, Giordano A. Cloning of a new member of the retinoblastoma gene family (pRb2) which binds to the E1A transforming domain. Oncogene. 1993;8:2561–2566. [PubMed] [Google Scholar]
- McBurney MW, Sutherland LC, Adra CN, Leclair B, Rudnicki MA, Jardine K. The mouse Pgk-1 gene promoter contains an upstream activator sequence. Nucleic Acids Res. 1991;19:5755–5761. doi: 10.1093/nar/19.20.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenbesser SD, Williams BO, Jacks T, DePinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature. 1994;371:72–74. doi: 10.1038/371072a0. [DOI] [PubMed] [Google Scholar]
- Mountford P, Zevnik B, Düwel A, Nichols J, Li M, Dani C, Robertson M, Chambers I, Smith A. Dicistronic targeting constructs: Reporters and modifiers of mammalian gene expression. Proc Natl Acad Sci. 1994;91:4303–4307. doi: 10.1073/pnas.91.10.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nork TM, Schwartz TL, Doshi HM, Millecchia LL. Retinoblastoma: Cell of origin. Arch Ophthalmol. 1995;113:791–802. doi: 10.1001/archopht.1995.01100060117046. [DOI] [PubMed] [Google Scholar]
- Qin X-Q, Livingston DM, Ewen M, Sellers WR, Arany Z, Kaelin Jr WG. The transcription factor E2F-1 is a downstream target of RB action. Mol Cell Biol. 1995;15:742–755. doi: 10.1128/mcb.15.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robanus Maandag EC, van der Valk M, Vlaar M, Feltkamp C, O’Brien J, van Roon M, van der Lugt N, Berns A, te Riele H. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 1994;13:4260–4268. doi: 10.1002/j.1460-2075.1994.tb06746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmechel D, Marangos PJ, Brightman M. Neuron-specific enolase is a molecular marker for peripheral and central neuroendocrine cells. Nature. 1978;276:834–836. doi: 10.1038/276834a0. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Zauberman A, Ginsberg D, Oren M. Identification of a minimal transforming domain of p53: Negative dominance through abrogation of sequence-specific DNA binding. Mol Cell Biol. 1992;12:5581–5592. doi: 10.1128/mcb.12.12.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. G1 phase progression: Cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- Squire J, Phillips RA, Boyce S, Godbout R, Rogers B, Gallie BL. Isochromosome 6p, a unique chromosomal abnormality in retinoblastoma: Verification by standard staining techniques, new densitometric methods, and somatic cell hybridization. Hum Genet. 1984;66:46–53. doi: 10.1007/BF00275185. [DOI] [PubMed] [Google Scholar]
- Tarkkanen A, Tervo T, Tervo K, Panula P. Immunohistochemical evidence for preproenkephalin A synthesis in human retinoblastoma. Invest Ophthalmol Vis Sci. 1984;25:1210–1212. [PubMed] [Google Scholar]
- Te Riele H, Robanus Maandag E, Berns A. Highly efficient gene targeting in embryonic stem cells through homologous recombination with isogenic DNA constructs. Proc Natl Acad Sci. 1992;89:5128–5132. doi: 10.1073/pnas.89.11.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokos M, Kyritsis AP, Chader GJ, Triche TJ. Differentiation of human retinoblastoma in vitro into cell types with characteristics observed in embryonal or mature retina. Am J Pathol. 1986;123:542–552. [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, Harlow E. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988;334:124–129. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nature Genet. 1994a;7:480–484. doi: 10.1038/ng0894-480. [DOI] [PubMed] [Google Scholar]
- Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, Jacks T. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J. 1994b;13:4251–4259. doi: 10.1002/j.1460-2075.1994.tb06745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes & Dev. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]
- Zhu L, van den Heuvel S, Helin K, Fattaey A, Ewen M, Livingston D, Dyson N, Harlow E. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes & Dev. 1993;7:1111–1125. doi: 10.1101/gad.7.7a.1111. [DOI] [PubMed] [Google Scholar]