

Abstract
An assessment of several widely used exchange--correlation potentials in computing charge-transfer integrals is performed. In particular, we employ the recently proposed Coulomb-attenuated model which was proven by other authors to improve upon conventional functionals in the case of charge-transfer excitations. For further validation, two distinct approaches to compute the property in question are compared for a phthalocyanine dimer.
Keywords: Charge-transfer integral, Density functional theory, Long-range corrected functionals, Organic electronics, Phthalocyanine
Introduction
The charge–transfer integral is an essential parameter in several theoretical models describing charge carrier transport in organic materials [1–7]. It is very often assumed that charge carrier mobility is proportional to the square of the charge–transfer integral (J) which describes the transport of a charge between adjacent molecular sites. An inherent issue of practical computations of charge–transfer integrals represents the choice of an approach to solve the Schrödinger equation. Currently, the DFT framework is commonly used to model charge transport in organic materials [3, 8–14]. Certain exchange–correlation potentials are recognized to predict accurately geometries of molecules and shapes of molecular orbitals. However, it is well known, that wrong asymptotic behavior of conventional functionals create a real problem in calculations of some properties, especially for molecular complexes [15, 16]. A recent systematic study of Peach and co–workers may serve as an illustrative example [17]. The authors showed that conventional exchange–correlation functionals have difficulties with reliable description of excitation energies to charge–transfer states in molecules and molecular complexes. The charge–transfer integral (J) involves orbitals localized on the two adjacent sites. For this reason, its evaluation might also present a challenge for the conventional exchange–correlation potentials commonly used nowadays. The primary goal of this study is to shed some light on this issue by employing recently proposed long–range corrected density functional theory (hereafter denoted as LRC–DFT) to compute charge–transfer integrals. The LRC–DFT is still being extensively tested primarily with an eye toward electric dipole (hyper)polarizabilities, linear and nonlinear optical spectra [17–29]. Here, we use two LRC functionals, namely LC-BLYP [30] and CAM–B3LYP [31] together with their conventional counterparts. The LRC functionals employ the Ewald split of 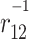 operator which, in the case of the CAM-B3LYP functional, takes the following form [31]:
operator which, in the case of the CAM-B3LYP functional, takes the following form [31]:
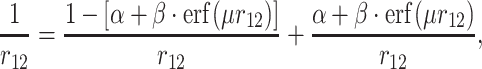 |
1 |
where the first and the second term on the rhs of the equation account for the short– and the long–range interactions, respectively, and α and β are constants. Here, we use the models based on the above equation with μ = 0.33 for CAM–B3LYP and μ = 0.47 for LC–BLYP [32].
As a model system to evaluate the performance of conventional exchange–correlation potentials in computing charge–transfer integrals we have chosen metal–free phthalocyanine dimer. Phthalocyanines are often considered as conductive materials with potential applications in organic electronics [33–36]. In crystalline phase phthalocyanine molecules usually form regular columns and liquid crystals composed of phthalocyanines are promising materials for organic electronics [37]. The liquid crystals in question are usually built from flat aromatic phthalocyanine center and aliphatic side groups. Likewise, aromatic core of molecules in liquid crystal state form regular columns with molecules in stacked conformations and the fastest charge transport is observed inside a column with much smaller probability of charge transport between columns. The charge–transfer integral between monomers in dimer can be used to describe charge transport inside of column composed of phthalocyanine molecules and as a first approximation of charge–transfer in phthalocyanine based liquid crystals. In this work only charge–transfer integrals between highest occupied molecular orbitals (HOMOs) of adjacent monomers are considered. This represents the charge–transfer integral related to the transport of positive charge carrier (hole transport).
Computational details
The initial structure of the dimer was composed of two phthalocyanine monomers in stacked conformation with rotation axis passing through the centers of mass of monomers and perpendicular to the plane of monomer. The structure of the phthalocyanine monomer was optimized at the B3LYP/6-31G(d) level of theory and was not reoptimized in dimer. The configurational space was spanned by a set of three parameters, namely intermolecular distance, twist angle around the symmetry axis and lateral slide of one of the monomers in two directions (see Fig. 1). What follows is a brief description of techniques used to compute charge–transfer integrals which are defined as:
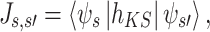 |
2 |
where h KS is the Kohn–Sham Hamiltonian, Ψs , Ψs′ denote wave functions of the charge carrier localized on the sites s and s′, respectively. In many organic systems there is non–zero spatial overlap between orbitals of the molecular sites. To account for this effect in calculations of charge–transfer integrals, the effective charge–transfer integral (J eff) may be introduced [4]:
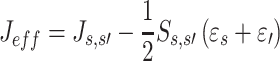 |
3 |
S s,s′ denotes overlap integral of orbitals s and s′; εs and εs′ stand for energies of the sites s and s′, respectively, and hereafter will be referred to as site energies.
Fig. 1.
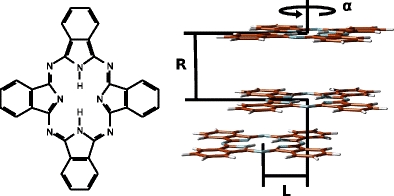
The schematic representation of three geometrical parameters used to describe the structure of phthalocyanine dimer: α denotes twist angle, R is intermolecular distance and L stands for lateral slide
Equation 2 can be directly used to compute the charge–transfer integral. In doing so, we express the Hamiltonian of the dimer (h KS) in the basis of molecular orbitals of the monomers [3, 38]:
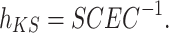 |
4 |
In order to compute the charge–transfer integral one needs to determine the eigenvalues for a dimer (E), the eigenvectors in the basis of atomic orbitals (AO) for a dimer (C
AO), the spatial overlap integrals in AO representation for a dimer (S
AO ) and eigenvectors for monomers in AO representation 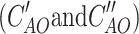 .The eigenvector matrix for a dimer and spatial overlap matrix was transformed from AO representation to molecular orbital representation of monomers as follows:
.The eigenvector matrix for a dimer and spatial overlap matrix was transformed from AO representation to molecular orbital representation of monomers as follows:
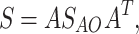 |
5 |
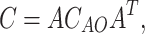 |
6 |
where A denotes transformation matrix which is diagonal block matrix with monomer eigenvector matrices 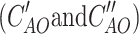 on the diagonal and A
T is transposed transformation matrix. The off–diagonal elements of h
KS matrix in monomer orbital basis represent charge–transfer integrals.
on the diagonal and A
T is transposed transformation matrix. The off–diagonal elements of h
KS matrix in monomer orbital basis represent charge–transfer integrals.
Another approach to compute the charge–transfer integrals, even more frequently used than the one described above, is the method which introduces energy splitting in dimer [39–41]. In this approach, the eigenvalue of sth molecular orbital of the Hamiltonian for dimer is given by:
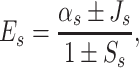 |
7 |
where αs is the s–th site energy integral, J s and S s are the charge–transfer and the overlap integral between orbitals denoted by index s of two molecules forming dimer, respectively. It is assumed that the mixing between s–th orbital and the other molecular orbitals is not significant. The splitting in energy of s–th molecular orbital of monomer in a dimer can be written as:
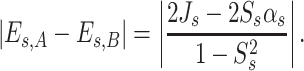 |
8 |
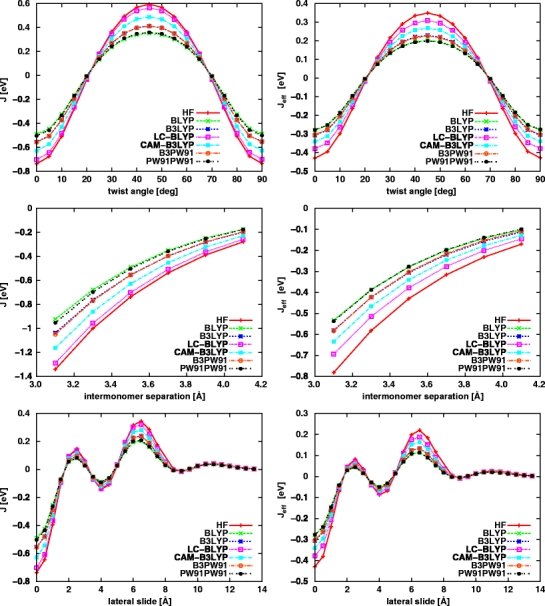
In Eq. (8) E s,A and E s,B denote eigenvalues for s–th molecular orbital of molecules A and B, respectively. J s , αs , S s stand for the charge–transfer integral, site energy and overlap integral of s–th molecular orbital of molecules A and B, respectively. Since we consider a dimer composed of two identical monomers, it is further assumed that αs is the same for monomers A and B. For a system composed of two nonequivalent molecules, expressions for the eigenvalue E s and the charge transfer integral J s take more complicated form [41]. Usually, it is also assumed that spatial overlap integral is equal zero (S s = 0), which is reasonable assumption considering charge transport between two organic molecules. In organic materials spatial overlap between orbitals of neighboring molecules are usually <<1. Thus, Eq. (8) can be rewritten as:
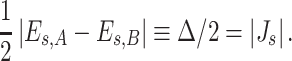 |
9 |
In the present study we compute charge–transfer integrals using both above described methods. Therefore, once the splitting in energy is known, the charge-transfer integral J can be calculated according to this relation.
Calculations were performed with the aid of several exchange–correlation potentials using different basis sets, including Dunning’s correlation consistent cc-pVDZ basis set [42] as well as recently proposed Jensen’s basis set [43]. The results of calculations presented in this work were carried out using the GAUSSIAN 09 program [32].
Results and discussion
Among several factors responsible for accuracy of results of computations of charge–transfer integrals one should not overlook the choice of the basis set. Most quantum–chemistry programs use the Gaussian functions, introduced by Boys in the mid 1950s, to approximate a wave function. Huang and Kertesz recently made an attempt to analyze the charge–transfer integrals using various basis sets and proved that basis set limit might be reached quite rapidly for the property in question [40]. It should be underlined that the smooth convergence of charge–transfer integrals with the basis set size is observed for effective charge–transfer integrals or in the case of negligible spatial overlap. Since the phthalocyanine complexes studied in the present work do not fulfill the latter criteria, we find it interesting to explore this topic more deeply by including the spatial overlap in computations of charge–transfer integrals. In Fig. 2a the values of spatial overlap integrals, charge–transfer integrals as well as effective charge–transfer integrals for different intermonomer separations are presented. Indeed, as seen in the figure, the values of charge–transfer integrals are quite insensitive to the basis set, provided the spatial overlap is taken into account.
Fig. 2.
(a) Spatial overlap integrals (S), charge–transfer integrals (J) and effective charge–transfer integrals (J eff) computed using the B3LYP functional. (b) Dependence of the spatial overlap on the geometrical parameters calculated using various exchange–correlation potentials and the HF method. The cc–pVDZ basis set was used
The values of the spatial overlap for different geometrical parameters calculated with the aid of different exchange–correlation potentials as well as using the Hartree–Fock method are presented in Fig. 2b. As seen in the figure, the values of the spatial overlap calculated with use of different DFT functionals are comparable for all considered geometrical parameters. The values of spatial overlap integral calculated using the HF wavefunction seem to be overestimated and differ substantially from the values determined within the DFT framework.
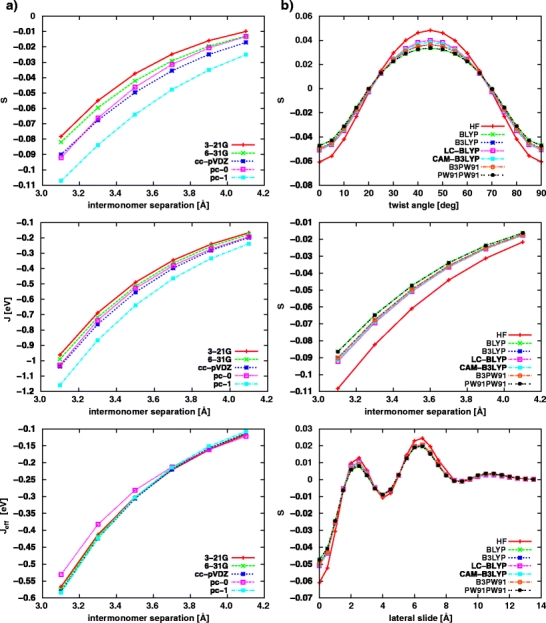
As seen in Fig. 3 the choice of exchange–correlation potential is quite important as far as the magnitude of charge–transfer integral (J) and effective charge–transfer integral (J eff) is concerned. The commonly used conventional exchange–correlation potentials such as BLYP or B3LYP predict much smaller values of charge–transfer integrals than the HF method. The PW91 functional, suggested as the best choice for computations of charge–transfer integrals in π–conjugated systems in stacked configurations [3, 41], gives comparable results to those determined with the aid of the BLYP and the B3LYP potentials. The values of charge–transfer integrals predicted by long–range corrected functionals, namely CAM–B3LYP and LC-BLYP, lie between HF and conventional DFT results (see Fig. 3). It was shown by Peach and co-workers that long–range corrected functionals improve substantially upon their traditional counterparts as far as excitation energies to charge–transfer states are concerned [17]. It is a particularly notable observation for the Coulomb–attenuated model (CAM-B3LYP). Since both quantities in principle might be similar in nature, the LRC potentials should give better results also in the case of charge–transfer integrals. For this reason, with a bit of scepticism due to the lack of more solid quantitative basis, we use the CAM-B3LYP potential as a reference. We conclude that conventional DFT functionals underestimate the values of charge–transfer integrals in comparison with their LRC counterparts.
Fig. 3.
The dependence of the charge–transfer integral (J, left side) and the effective charge–transfer integral (J eff, right side) on the geometrical parameters for different DFT functionals using the cc-pVDZ basis set
One of the theories most commonly used for describing charge transport in organic system is the one proposed by Marcus [44, 45]. In this approach, the hopping rate constant between sites i and j is defined as:
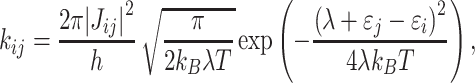 |
10 |
where h, k B and T are Planck constant, Boltzmann constant and temperature, respectively. εi and εj denote energies of the charge carrier localized on the sites i and j and λ stands for reorganization energy [4, 13, 46]. In order to estimate the charge carrier mobility in the system without structural disorder (assuming that each hopping rate is the same) one can use the relation [47, 48]:
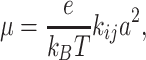 |
11 |
where e is elementary charge, and a denotes distance between molecular sites. For the studied phthalo-cyanine dimers, the internal reorganization energy calculated at the B3LYP/cc–pVDZ level of theory is 0.043 eV, which is similar to the results presented in literature [13, 49, 50]. For the intermolecular distance 3.5 Å, the rotation angle 0 and lateral slide 1.5 Å (this is the structure similar to the crystal structure of the phthalocyanine) the charge–transfer integrals calculated with B3LYP and CAM–B3LYP functional are −0.16 eV and −0.18 eV respectively. The charge carrier mobility values calculated from Eq. (11) for this two charge–transfer integrals are 3.9 cm2 /Vs and 4.9 cm2 /Vs. Thus, one can easily see, that DFT functional has a significant influence on the mobility value. A close look at Fig. 2 leads to the conclusion that for certain areas of conformational space the differences might be even higher.
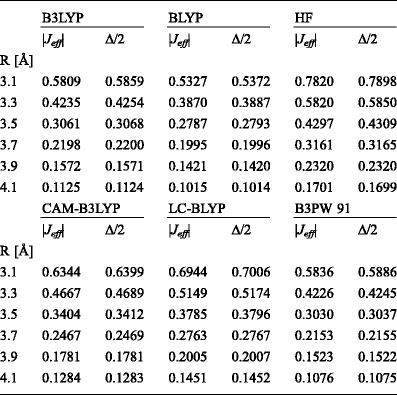
A comparison of the effective charge-transfer integrals calculated using Eq. (3) and the charge-transfer integrals determined from the energy splitting (Eq. (9)) is shown in Table 1. The data show that the differences in the values of charge–transfer integrals calculated based on the two approaches are insignificant and do not exceed a few thousandths of eV. At first glance, it appears that it is sufficient to employ less accurate method, based on the energy splitting in dimer with assumption of zero spatial overlap, to compute J between molecules in π interacting system. However, as it has already been mentioned, it is important to include spatial overlap in calculations of charge–transfer integrals from the definition. Otherwise, the values of J might strongly depend on the size of the basis set used in calculations. The other drawback of the method based on energy splitting in dimer is the lack of information about the sign of charge–transfer integral. However, if the knowledge of the sign is important, it can be subsequently determined from the bonding–antibonding character of the interaction between the corresponding orbitals [51].
Table 1.
The absolute values of effective charge transfer integrals (|J eff|, given in eV) computed with the aid of Eqs. (3)–(6) and the charge transfer integrals calculated using energy splitting in dimer approach (|∆/2|). The cc-pVDZ basis set was employed in all calculations. R is the intermolecular distance
Conclusions
The primary aim of the present study was to evaluate the performance of commonly employed conventional exchange–correlation potentials that are used to compute charge–transfer integrals. In doing so, we apply the recently proposed Coulomb–attenuated model as a reference as this approach is proven to be very successful in predicting excitation energies to charge–transfer states. It is shown that for certain areas of conformational space in phthalocyanine dimer the differences in values of charge-transfer integrals between the conventional schemes and the CAM-B3LYP functional in values of charge–transfer integrals might be quite significant. The same is revealed for triphenylene dimer [52]. As a result, the values of charge carrier mobilities estimated using Marcus formula might differ by 20% and more. Likewise, theoretical predictions of peaks intensity in electro-absorption spectrum of molecular crystals and molecular aggregates [53, 54] might be determined to a large extent by the accuracy of charge-transfer integrals (Kulig W, Petelenz P, (2010). Private communication). We have also confirmed the findings reported by other authors [41] that the size of the basis set used in calculations of charge–transfer integrals plays only a minor role provided the spatial overlap is included in the theoretical model.
Acknowledgments
This work was supported by computational grants from Wroclaw Center for Networking and Supercom- puting (WCSS) and ACK Cyfronet. Work in the USA was supported by the HRD-0833178 grant. One of the authors (RZ) would like to acknowledge support from a grant from Iceland, Liechtenstein and Norway through the EEA Financial Mechanism - Scholarship and Training Fund. Financial support from Wroclaw University of Technology and the Czech Science Foundation (Project No. P205/10/2280) and the European Commission through the Human Potential Programme (Marie-Curie RTN BIMORE, Project No. MRTN-CT-2006-035859) is also acknowledged.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
An erratum to this article can be found at http://dx.doi.org/10.1007/s00894-011-1008-5
References
- 1.Brédas JL, Calbert JP, da Silva Filho DA, Cornil J. PNAS. 2002;99(9):5804. doi: 10.1073/pnas.092143399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Nan G, Yang X, Peng Q, Li Q, Shuai Z. Chem Soc Rev. 2010;39:423. doi: 10.1039/b816406c. [DOI] [PubMed] [Google Scholar]
- 3.Senthilkumar K, Grozema FC, Bickelhaupt FM, Siebbeles LDA. J Chem Phys. 2003;119:9809. doi: 10.1063/1.1615476. [DOI] [PubMed] [Google Scholar]
- 4.Newton MD. Chem Rev. 1991;91:767. doi: 10.1021/cr00005a007. [DOI] [Google Scholar]
- 5.Cheung DL, Troisi A. Phys Chem Chem Phys. 2008;10:5941. doi: 10.1039/b807750a. [DOI] [PubMed] [Google Scholar]
- 6.Bassler H. Phys Stat Sol (b) 1993;175:15. doi: 10.1002/pssb.2221750102. [DOI] [Google Scholar]
- 7.Toman P, Nespurek S, Bartkowiak W. Mater Sci Pol. 2009;27:797. [Google Scholar]
- 8.Hreha RD, George CP, Haldi A, Domercq B, Malagoli M, Barlow S, Brédas J-L, Kippelen B, Marder SR. Adv Funct Mater. 2003;13:967. doi: 10.1002/adfm.200304464. [DOI] [Google Scholar]
- 9.Yang B, Kim S-K, Xu H, Park Y-I, Zhang H, Gu C, Shen F, Wang C, Liu D, Liu X, Hanif M, Tang S, Li W, Li F, Shen J, Park J-W, Ma Y. Chem Phys Chem. 2008;9:2601. doi: 10.1002/cphc.200800513. [DOI] [PubMed] [Google Scholar]
- 10.Gao H, Zhang H, Mo R, Sun S, Su Z-M, Wang Y. Synth Met. 2009;159:1767. doi: 10.1016/j.synthmet.2009.05.023. [DOI] [Google Scholar]
- 11.Zbiri M, Johnson MR, Kearley GJ, Mulder FM. Theor Chem Acc. 2010;125:445. doi: 10.1007/s00214-009-0559-3. [DOI] [Google Scholar]
- 12.Delgado MCR, Kim EG, da DA, Filho S, Brédas JL. J Am Chem Soc. 2010;132:3375. doi: 10.1021/ja908173x. [DOI] [PubMed] [Google Scholar]
- 13.Lee C, Sohlberg K. Chem Phys. 2010;367:7. doi: 10.1016/j.chemphys.2009.10.005. [DOI] [Google Scholar]
- 14.Chen S, Ma J. J Comp Chem. 2009;30:1959. doi: 10.1002/jcc.21194. [DOI] [PubMed] [Google Scholar]
- 15.Baerends EJ, Gritsenko OV. J Phys Chem A. 1997;101:5383. doi: 10.1021/jp9703768. [DOI] [Google Scholar]
- 16.Tozer DJ, Amos RD, Handy NC, Roos BO, Serrano-Andres L. Mol Phys. 1999;97:859. doi: 10.1080/00268979909482888. [DOI] [Google Scholar]
- 17.Peach MJG, Benfield P, Helgaker T, Tozer DJ. J Chem Phys. 2008;128:044118. doi: 10.1063/1.2831900. [DOI] [PubMed] [Google Scholar]
- 18.Kamiya M, Sekino H, Tsuneda T, Hirao K. J Chem Phys. 2005;122:234111. doi: 10.1063/1.1935514. [DOI] [PubMed] [Google Scholar]
- 19.Kirtman B, Bonness S, Ramirez-Solis A, Champagne B, Matsumoto H, Sekino H. J Chem Phys. 2008;128:114108. doi: 10.1063/1.2885051. [DOI] [PubMed] [Google Scholar]
- 20.Jacquemin D, Perpéte EA, Scalmani G, Frisch MJ, Kobayashi R, Adamo C. J Chem Phys. 2007;126:144105. doi: 10.1063/1.2715573. [DOI] [PubMed] [Google Scholar]
- 21.Loboda O, Zaleśny R, Avramopoulos A, Luis JM, Kirtman B, Tagmatarchis N, Reis H, Papadopoulos MG. J Phys Chem A. 2009;113:1159. doi: 10.1021/jp808234x. [DOI] [PubMed] [Google Scholar]
- 22.Jacquemin D, Perpète EA, Medved M, Scalmani G, Frisch MJ, Kobayashi R, Adamo C. J Chem Phys. 2007;126:191108. doi: 10.1063/1.2741246. [DOI] [PubMed] [Google Scholar]
- 23.Hammond JR, Kowalski K. J Chem Phys. 2009;130:194108. doi: 10.1063/1.3134744. [DOI] [PubMed] [Google Scholar]
- 24.Limacher PA, Mikkelsen KV, Lüthi HP. J Chem Phys. 2009;130:194114. doi: 10.1063/1.3139023. [DOI] [PubMed] [Google Scholar]
- 25.Zaleśny R, Wójcik G, Mossakowska I, Bartkowiak W, Avramopoulos A, Papadopoulos MG. J Mol Struct THEOCHEM. 2009;901:46. doi: 10.1016/j.theochem.2009.04.011. [DOI] [Google Scholar]
- 26.Casida ME, Salahub DR. J Chem Phys. 2000;113:8918. doi: 10.1063/1.1319649. [DOI] [Google Scholar]
- 27.Cai ZL, Crossley MJ, Reimers JR, Kobayashi R, Amos RD. J Phys Chem B. 2006;110:15624. doi: 10.1021/jp063376t. [DOI] [PubMed] [Google Scholar]
- 28.Silva D, Krawczyk P, Bartkowiak W, Mendonca CR. J Chem Phys. 2009;131:244516. doi: 10.1063/1.3271239. [DOI] [PubMed] [Google Scholar]
- 29.Rostov IV, Amos RD, Kobayashi R, Scalmani G, Frisch MJ. J Phys Chem B. 2010;114:5547. doi: 10.1021/jp911329g. [DOI] [PubMed] [Google Scholar]
- 30.Iikura H, Tsuneda T, Yanai T, Hirao K. J Chem Phys. 2001;115:3540. doi: 10.1063/1.1383587. [DOI] [Google Scholar]
- 31.Yanai T, Tew DP, Handy NC. Chem Phys Lett. 2004;393:51. doi: 10.1016/j.cplett.2004.06.011. [DOI] [Google Scholar]
- 32.Frisch MJ et al (2009) Gaussian 09 Revision A.1. Gaussian Inc. Wallingford CT
- 33.Facchetti A. Mater Today. 2007;10(3):28. doi: 10.1016/S1369-7021(07)70017-2. [DOI] [Google Scholar]
- 34.Newman CR, Frisbie CD, da Silva Filho DA, Brédas JL, Ewbank PC, Mann KR. Chem Mater. 2004;16:4436. doi: 10.1021/cm049391x. [DOI] [Google Scholar]
- 35.Toman P, Nespurek S, Yakushi K. J Porphyr Phthalocyanines. 2002;6:556. doi: 10.1142/S1088424602000695. [DOI] [Google Scholar]
- 36.Shirota Y, Kageyama H. Chem Rev. 2007;107:953. doi: 10.1021/cr050143+. [DOI] [PubMed] [Google Scholar]
- 37.Sergeyev S, Pisula W, Geerts YH. Chem Soc Rev. 2007;36:1902. doi: 10.1039/b417320c. [DOI] [PubMed] [Google Scholar]
- 38.Mikolajczyk MM, Toman P, Bartkowiak W. Chem Phys Lett. 2010;485:253. doi: 10.1016/j.cplett.2009.12.048. [DOI] [Google Scholar]
- 39.Cornil J, Beljonne D, Calbert JP, Brédas JL. Adv Mater. 2001;13(14):1053. doi: 10.1002/1521-4095(200107)13:14<1053::AID-ADMA1053>3.0.CO;2-7. [DOI] [Google Scholar]
- 40.Huang J, Kertesz M. Chem Phys Lett. 2004;390:110. doi: 10.1016/j.cplett.2004.03.141. [DOI] [Google Scholar]
- 41.Huang J, Kertesz M. J Chem Phys. 2005;122:234707. doi: 10.1063/1.1925611. [DOI] [PubMed] [Google Scholar]
- 42.Dunning TH. J Chem Phys. 1989;90:1007. doi: 10.1063/1.456153. [DOI] [Google Scholar]
- 43.Jensen F. J Chem Phys. 2001;115:9113. doi: 10.1063/1.1413524. [DOI] [Google Scholar]
- 44.Marcus RA. Rev Mod Phys. 1993;65:599. doi: 10.1103/RevModPhys.65.599. [DOI] [Google Scholar]
- 45.Barbara PF, Meyer TJ, Ratner MA. J Phys Chem. 1996;100:13148. doi: 10.1021/jp9605663. [DOI] [Google Scholar]
- 46.Hutchison GR, Ratner MA, Marks TJ. J Am Chem Soc. 2005;127:2339. doi: 10.1021/ja0461421. [DOI] [PubMed] [Google Scholar]
- 47.Grozema FC, Siebbeles LDA. Int Rev Phys Chem. 2008;27:87. doi: 10.1080/01442350701782776. [DOI] [Google Scholar]
- 48.Berlin YA, Grozema FC, Siebbeles LDA, Ratner MA. J Phys Chem C. 2008;112:10988. doi: 10.1021/jp801646g. [DOI] [Google Scholar]
- 49.Tant J, Geerts YH, Lehmann M, Cupere VD, Zucchi G, Laursen BW, Bjornholm T, Lemaur V, Marcq V, Burquel A, Hennebicq E, Gardebien F, Viville P, Beljonne D, Lazzaroni R, Cornil J. J Chem Phys B. 2005;109:20315. doi: 10.1021/jp054778o. [DOI] [PubMed] [Google Scholar]
- 50.Chang YC, Chao I. J Phys Chem Lett. 2010;1:116. doi: 10.1021/jz900042x. [DOI] [Google Scholar]
- 51.Seo D, Hoffmann R. Theor Chem Acc. 1999;102:23. [Google Scholar]
- 52.Mikołajczyk M, Zaleśny R, Czyżnikowska Z, Bartkowiak W, Toman P, Leszczynski J (2009). Unpublished results [DOI] [PMC free article] [PubMed]
- 53.Stradomska A, Petelenz P. Mol Phys. 2006;104:2063. doi: 10.1080/00268970600655485. [DOI] [Google Scholar]
- 54.Petelenz P. Org Electron. 2004;5:115. doi: 10.1016/j.orgel.2003.12.003. [DOI] [Google Scholar]