

Abstract
SMAD proteins mediate signals from receptor serine–threonine kinases (RSKs) of the TGF-β superfamily. We demonstrate here that HGF and EGF, which signal through RTKs, can also mediate SMAD-dependent reporter gene activation and induce rapid phosphorylation of endogenous SMAD proteins by kinase(s) downstream of MEK1. HGF induces phosphorylation and nuclear translocation of epitope-tagged Smad2 and a mutation that blocks TGF-β signaling also blocks HGF signal transduction. Smad2 may thus act as a common positive effector of TGF-β- and HGF-induced signals and serve to modulate cross talk between RTK and RSK signaling pathways.
Keywords: SMAD, TGF-β, signal transduction, protein phosphorylation, HGF, receptor kinases
Receptor complexes of the TGF-β family consist of a ligand-binding type II receptor that, following ligand activation, binds to and transphosphorylates the signal transducing type I receptor (Massagué 1996; Heldin et al. 1997). Experiments in Drosophila and Caenorhabditis elegans identified a family of proteins called SMADs, which are mediators of signals emanating from receptor serine–threonine kinases (RSKs). Studies in mammalian cells have shown that SMAD proteins bind to and are phosphorylated by type I receptors, associate with Smad4, and then translocate to the nucleus and act as cofactors in transcriptional complexes (Chen et al. 1997; Heldin et al. 1997; Kim et al. 1997; Liu et al. 1997; Yingling et al. 1997). SMAD–receptor interactions are specific, that is, Smad1 interacts with type I bone morphogenetic protein (BMP) receptors, and Smad2 binds to the type I TGF-β receptor (Macias-Silva et al. 1996; Kretzschmar et al. 1997b).
In contrast to TGF-β family ligands, hepatocyte growth factor (HGF) and epidermal growth factor (EGF) signal their responses through transmembrane receptor tyrosine kinases (RTKs) (Pawson 1995; Cantley and Songyang 1997). Multiple signaling pathways have been identified that originate from these receptors, the most prominent being the Ras pathway, which leads to phosphorylation and activation of the serine–threonine kinase MAPK, which in turn activates several transcription factors. Although some data suggest a cell-specific activation of Ras signaling by TGF-β (Yan et al. 1994; Hartsough et al. 1996) and a MEKK family member, TAK-1, has been implicated in signaling from TGF-β receptors (Yamaguchi et al. 1995), generalized activation of common pathways by RTKs and RSKs has not been demonstrated.
TGF-β can act synergistically with ligands signaling through RTKs in many developmental and biological systems, suggesting that certain intermediates in their signaling pathways might be shared. TGF-β was originally identified for its ability to transform normal rat kidney (NRK) fibroblasts in vitro, an effect that was dependent on the presence of EGF (Roberts et al. 1983). HGF and TGF-β both strongly up-regulate the extracellular protease inhibitors plasminogen activator inhibitor-1 (PAI-1; Keski-Oja et al. 1988; Wojta et al. 1994) and tissue inhibitor of metalloproteinases-3 (P. Castagnino and D. Bottaro, in prep.). TGF-β can also potentiate scatter of epithelial cells induced by HGF or EGF (Stolz and Michalopoulos 1997). Similarly, although BMPs oppose the actions of fibroblast growth factor (FGF) in limb bud development (Niswander and Martin 1993), TGF-β or activin can act synergistically with FGF in heart formation (Lough et al. 1996), chondrogenesis (Frenz et al. 1994), and myogenesis (Stern et al. 1997). The recent demonstration of inhibition of Smad1 signaling by RTKs (Kretzschmar et al. 1997a) suggests that SMAD proteins may play a pivotal role in mediating cross talk between the RSK and RTKs. Here, we sought to determine if SMAD proteins could mediate positive responses from both RTKs and RSKs. We demonstrate that either HGF or EGF can stimulate phosphorylation of endogenous SMAD proteins and that the SMAD signaling pathway, particularly Smad2, plays a role in transmitting activating signals from the RTKs.
Results and Discussion
Smad4 is structurally and functionally unique among the SMADs, acting as an essential component downstream of TGF-β, activin, and BMP receptors (Lagna et al. 1996; de Caestecker et al. 1997; de Winter et al. 1997; Zhang et al. 1997). In Smad4 homozygous null MDA-MB-468 cells, introduction of Smad4 is required to elicit a TGF-β-induced response from the reporter construct 3TP–Lux (Fig. 1A), which contains 3 TPA-responsive elements and a small portion of the PAI-1 promoter. We sought to determine if Smad4 was also necessary for induction of 3TP–Lux activity following activation of RTKs. In MDA-MB-468 cells, HGF did not induce 3TP–Lux in the absence of Smad4; however, cotransfection of Smad4 restored a response to HGF (Fig. 1A). EGF induction of 3TP–Lux activity was also Smad4-dependent, but a moderate increase in luciferase activity in the absence of Smad4 suggests that a SMAD-independent pathway also contributes to EGF-induced 3TP–Lux activity. To rule out a role for autocrine or paracrine TGF-β in inducing SMAD-dependent reporter activity, cells were treated with HGF or EGF and both total and active TGF-β in the culture medium were measured. No active TGF-β was detected in the conditioned medium, and total TGF-β concentrations did not change following HGF treatment (from 3.6 ng/ml acid-activated TGF-β in untreated controls to 2.5 ng/ml in HGF-treated samples), suggesting no additional autocrine or paracrine stimulation. EGF, however, induced a twofold increase in acid-activatable TGF-β in the culture medium after 24 hr of treatment (from 3.6 ng/ml untreated to 7.1 ng/ml treated).
Figure 1.
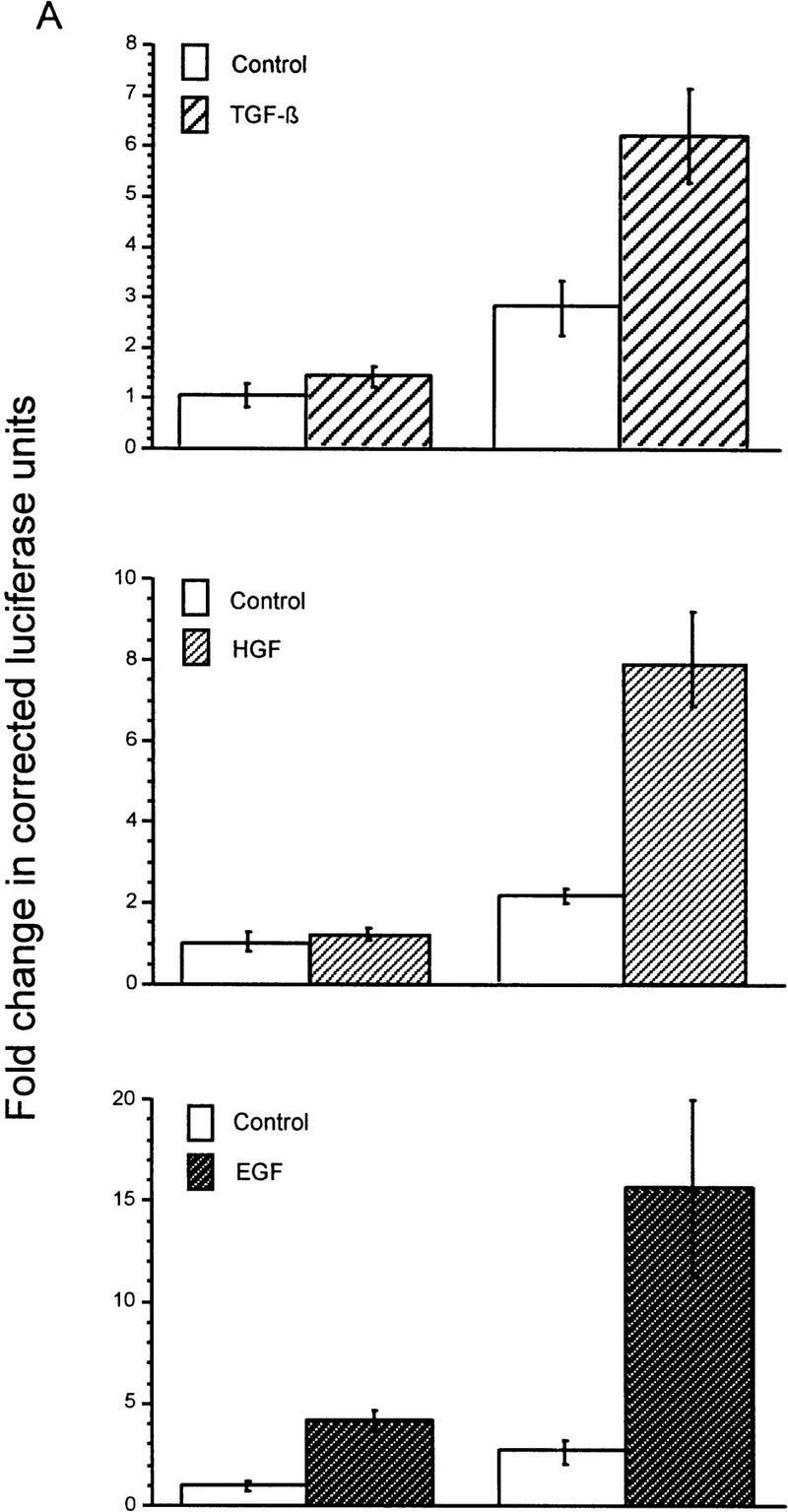


HGF and EGF signaling through SMAD proteins. (A) Smad4 null MDA-MB-468 cells were transiently transfected with the reporter 3TP–Lux and vector (left) or a Smad4 expression construct (right). Cells were treated with TGF-β (3 ng/ml), HGF (200 ng/ml), EGF (30 ng/ml), or untreated. Luciferase activity is in arbitrary units expressed as fold change above the untreated vector control and represents the average and standard error of four experiments. (B) TGF-β neutralizing antibody was added following transfection and before ligand addition. Results are averages of triplicate wells ±s.e.m. (C) Cells were transfected without or with DNTβR-II and treated with increasing amounts TGF-β (10 ng/ml) or HGF (600 ng/ml).
To demonstrate that induction of 3TP–Lux luciferase activity was not caused by TGF-β induction following HGF or EGF treatment, transiently transfected cells were treated with TGF-β neutralizing antibody, which inhibits Smad4-dependent signaling in MDA-MB-468 cells (de Caestecker et al. 1997). Although this antibody clearly eliminated activation by exogenous TGF-β, it had no effect on HGF induction of 3TP–Lux (Fig. 1B). A partial inhibition of EGF-induced signaling was observed, suggesting that some luciferase activity induced by EGF is dependent on up-regulation or activation of TGF-β (M.P. de Caestecker, unpubl.). To further demonstrate the TGF-β independence of the HGF response, cells were transiently transfected with a truncated form of the type II receptor for TGF-β (DNTβR-II) lacking the cytoplasmic domain. This construct effectively repressed induction of cotransfected 3TP–Lux reporter activity by TGF-β, whereas it had no effect on activation induced by HGF (Fig. 1C). These data suggest a role for the SMAD signaling pathway as a positive effector of HGF and EGF signal transduction and demonstrate that this signal is independent of TGF-β and TGF-β receptors.
Because Smad4 was found to be essential for reporter gene activation, we sought to determine if endogenous receptor-activated SMAD proteins are phosphorylated following HGF or EGF treatment. In Mv1Lu mink lung epithelial cells treated with TGF-β, endogenous SMAD phosphoproteins of 58 and 54 kD (presumably Smad2 and Smad3) are immunoprecipitated using affinity-purified antibodies (Fig. 2A). In contrast, only a 58-kD band (corresponding to the size of Smad1, Smad2, or Smad5) is recognized in HGF-treated lanes. To rule out activation of TGF-β type I receptors leading to SMAD phosphorylation, we performed the same experiment in R1B cells that lack TβR-I and therefore cannot transmit a TGF-β-induced signal (Laiho et al. 1990). In HGF- and EGF-treated R1B cells, only the 58-kD phosphoprotein is immunoprecipitated (Fig. 2A). As with TGF-β treatment of other cell types (Lechleider et al. 1996; Nakao et al. 1997) phosphorylation is readily evident by 15 min following treatment with HGF or EGF (Fig. 2B,C), but in contrast to TGF-β stimulation, the phosphorylation of SMADs by HGF and EGF is transient, diminishing by 60 min. Differences in the duration of phosphorylation and, hence, activation, may regulate which genes or responses are modulated by SMADs, much like differences in the duration of activation of MEK by different ligands in PC12 cells lead to different phenotypic responses (Traverse et al. 1992). Phosphoamino acid analysis of this band revealed serine phosphorylation only (data not shown). These data show that an endogenous SMAD protein(s) are phosphorylated by a non-TGF-β receptor kinase following stimulation by either HGF or EGF.
Figure 2.





Phosphorylation of endogenous SMAD proteins in mink lung epithelial cells. (A) R1B or Mv1Lu cells were metabolically labeled with [32P]orthophosphate and stimulated with HGF (200 ng/ml) or TGF-β (10 ng/ml) for 30 min. (Arrow) Phosphorylated SMAD. The lower band in the TGF-β-treated lane likely represents Smad3. (B) Metabolically labeled R1B cells were treated with HGF (B) or EGF (C) for the times indicated and analyzed as in A. R1B (D) or Mv1Lu (E) cells were metabolically labeled with [32P]orthophosphate and treated without or with MEK inhibitor PD98059 (20 μm unless indicated) prior to the addition of ligand.
HGF and EGF signal in part through the Ras pathway, which leads to activation of the MEKK Raf and ultimately phosphorylation and activation of p42/p44 MAPK by MEK1. We sought to determine whether the SMAD kinase was in the Ras pathway. Pretreatment of R1B cells with the MEK1 inhibitor PD98059 (Coolican et al. 1997) prior to treatment with HGF or EGF diminished the level of phosphorylation of the immunoprecipitated SMAD protein (Fig. 2D), whereas it had no effect on phosphorylation induced by TGF-β in Mv1Lu cells (Fig. 2E). This indicates that the SMAD kinase(s) activated by HGF and EGF lie downstream of MEK1. Several HGF and EGF responses are also mediated by PI3–kinase activation, which can lead to MAPK activation through Ras-dependent and -independent mechanisms (Rahimi et al. 1996). Treatment of metabolically labeled cells with the PI3–kinase inhibitor wortmannin (Rahimi et al. 1996) prior to treatment with HGF had no effect on SMAD phosphorylation (data not shown), suggesting that MEK1-dependent phosphorylation of SMAD is not regulated by PI3–kinase in this system. These data suggest that the kinase phosphorylating endogenous SMADs in response to HGF or EGF is downstream of MEK1, although the exact location is not determined.
To determine the SMAD isoform phosphorylated following HGF treatment, COS-1 and R1B (data not shown) or HepG2 (Fig. 3A; data not shown) cells transiently transfected with epitope-tagged Smad1 or Smad2 were analyzed by metabolic labeling and immunoprecipitation. Both Smad1 and Smad2 were phosphorylated following HGF treatment.
Figure 3.

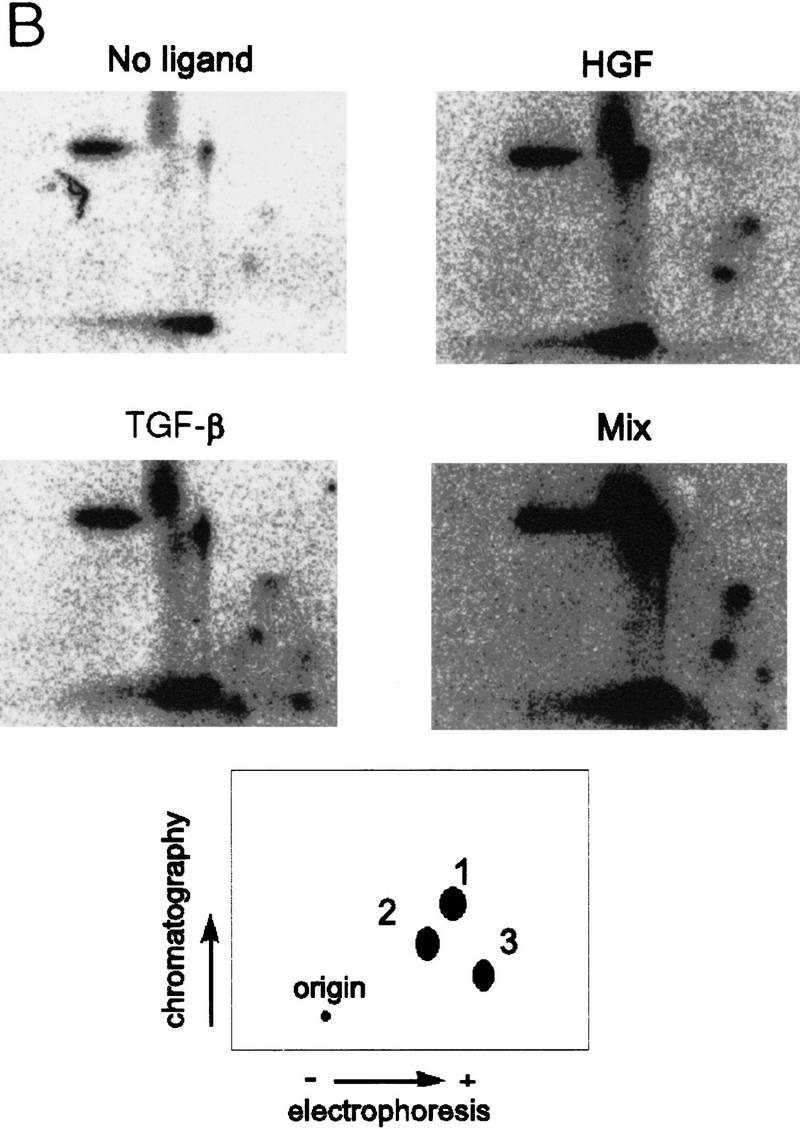
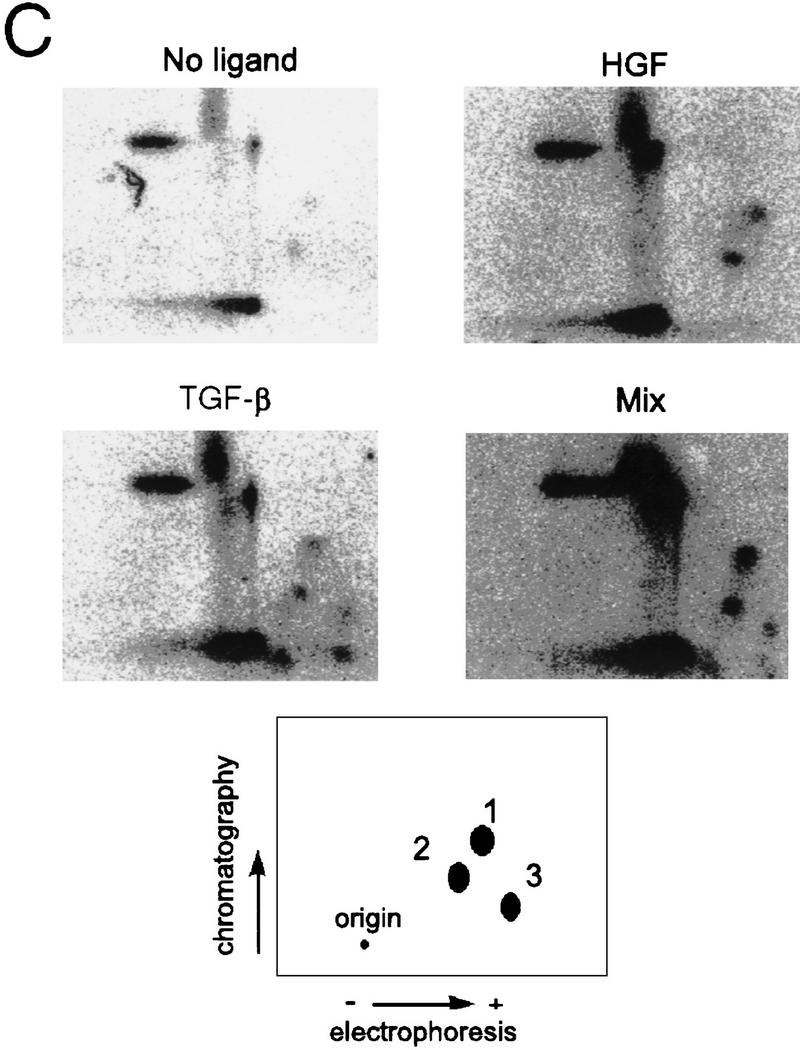
Identification of phosphorylated SMADs. (A) HepG2 cells transiently transfected with the Flag-tagged Smad2 wild-type (W/T) (left) or mutant (3S→A) (right) constructs as indicated. (Top) [32P]orthophosphate labeled; (bottom) an immunoblot of controls transfected in parallel. (B) Metabolically labeled Smad2 immunoprecipitates were analyzed by tryptic digest, treatments are indicated and mixture of the two digests (Mix) is also shown.
Because we show a SMAD-dependent activation of HGF signaling pathways, we sought to determine if HGF treatment could lead to Smad1 or Smad2 phosphorylation on the three highly conserved carboxy-terminal serine residues (SSMS) that lead to SMAD activation. These residues have been shown to be phosphorylated by activated type I receptors, and their phosphorylation is essential for signaling (Macias-Silva et al. 1996; Abdollah et al. 1997; Kretzschmar et al. 1997b; Souchelnytskyi et al. 1997). We used mutants lacking these phosphorylation sites [Smad1(3S>A) and Smad2(3S>A)] to determine if HGF could induce phosphorylation on the same residues. Both Smad1 and Smad1(3S>A) were phosphorylated following HGF addition to transiently transfected HepG2 cells (data not shown). In contrast, although phosphorylation was clearly evident on wild-type Smad2, phosphorylation of the mutant Smad2(3S>A) was not observed with either TGF-β or HGF (Fig. 3A), suggesting that these residues are critical phosphorylation or regulatory sites for both ligands.
Phosphotryptic peptide maps of wild-type Smad2 (Fig. 3C) indicated that HGF induced phosphorylation on a subset of TGF-β-dependent sites (Fig. 3C, spots 1 and 2), with a single additional phosphopeptide appearing following treatment with TGF-β (spot 3). Previously published data (Macias-Silva et al. 1996; Abdollah et al. 1997; Souchelnytskyi et al. 1997) indicate that spot 3 likely represents the SSMS phosphopeptide and that the additional spots represent phosphorylation of other, as yet unidentified, tryptic residues (Souchelnytskyi et al. 1997). As there is no inducible phosphorylation of the Smad2(3S>A) mutant following treatment with either ligand (Fig. 3A), these data indicate that the SSMS motif is necessary to enable phosphorylation of these additional tryptic sites (spots 1 and 2) following treatment with TGF-β or HGF.
To demonstrate that Smad2 could directly transmit an HGF-induced signal, we used a Gal4 fusion reporter system. In this system Gal4–Smad2 is activated by treatment with TGF-β (Hayashi et al. 1997). HGF or TGF-β treatment of HepG2 cells transiently transfected with the Gal4–Smad2 caused a reproducible increase in luciferase activity, indicating that Smad2 was activated (Fig. 4A). Cotransfection with DNTβR-II nearly eliminated the induction by TGF-β but had no effect on HGF-induced activity. Similarly, induction by TGF-β was blocked by TGF-β-neutralizing antibody, but the response to HGF remained unchanged, confirming that the response was not due to activation or secretion of TGF-β (Fig. 4B). To determine if we could block HGF-induced 3TP–Lux reporter gene activation, we used the dominant inhibitor Smad2(3S>A). In R1B cells, transfection of Smad2(3S>A) significantly reduced reporter gene activity induced by HGF, but wild-type Smad2 had no effect (Fig. 4C).
Figure 4.


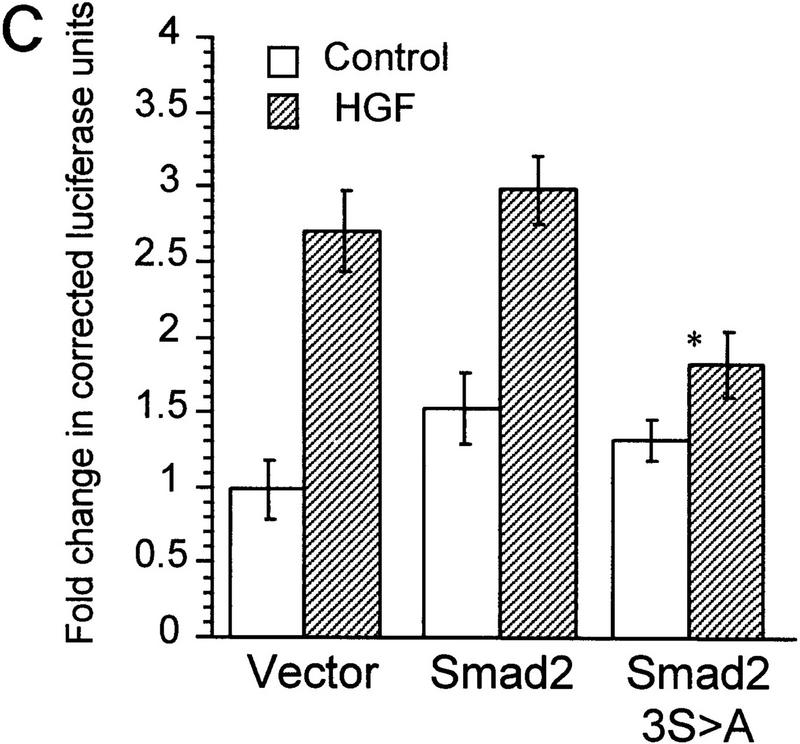
Smad2 mediates HGF-induced transcriptional responses. (A) The Gal4 reporter construct pG5–E1B–Luc was transfected into HepG2 cells with Gal4 alone (−) or Gal4–Smad2 fusion construct (+) and luciferase activity assayed following HGF (400 ng/ml) or TGF-β (0.3 ng/ml) treatment with or without DNTβR-II. Values are arbitrary luciferase units and represent the mean ± s.e.m. of triplicate wells. (B) Cells were transfected with Gal4–Smad2 and treated with TGF-β or HGF as in A either in the absence or presence of TGF-β antibody. (C) R1B cells were transfected with the reporter 3TP–Lux and Smad2 constructs and treated with HGF (400 ng/ml) as indicated. Values represent the mean ±s.e.m. of at least four experiments, with P < .05 for the difference of vector only with HGF vs. Smad2(3S→A) plus HGF (*).
To determine if HGF could induce SMAD nuclear translocation, we transiently transfected HepG2 cells with Flag-tagged Smad2, treated the cells with either TGF-β or HGF, and observed intracellular localization by indirect immunofluorescence microscopy. Smad2 clearly translocated to the nucleus in cells treated with either HGF or TGF-β (Fig. 5), whereas the Smad2(3S>A) mutant showed no increase in nuclear staining with either HGF or TGF-β, as expected. Likewise, neither cotransfection with DNTβR-II nor treatment with TGF-β-neutralizing antibodies affected the magnitude of nuclear translocation induced by HGF but greatly reduced nuclear translocation following TGF-β treatment (data not shown). The magnitude of translocation observed in these cells with HGF treatment was always smaller than that observed following TGF-β stimulation, indicating that the absolute level of Smad2 activation by HGF may not be as great as with TGF-β. Similarly, the magnitude of the maximal response in the Gal4 heterologous activation assay was ∼20-fold higher following TGF-β versus HGF treatment. This suggests a regulatory mechanism for transcriptional activation whereby levels of SMAD activation play a role in determining which subset of target genes are induced.
Figure 5.
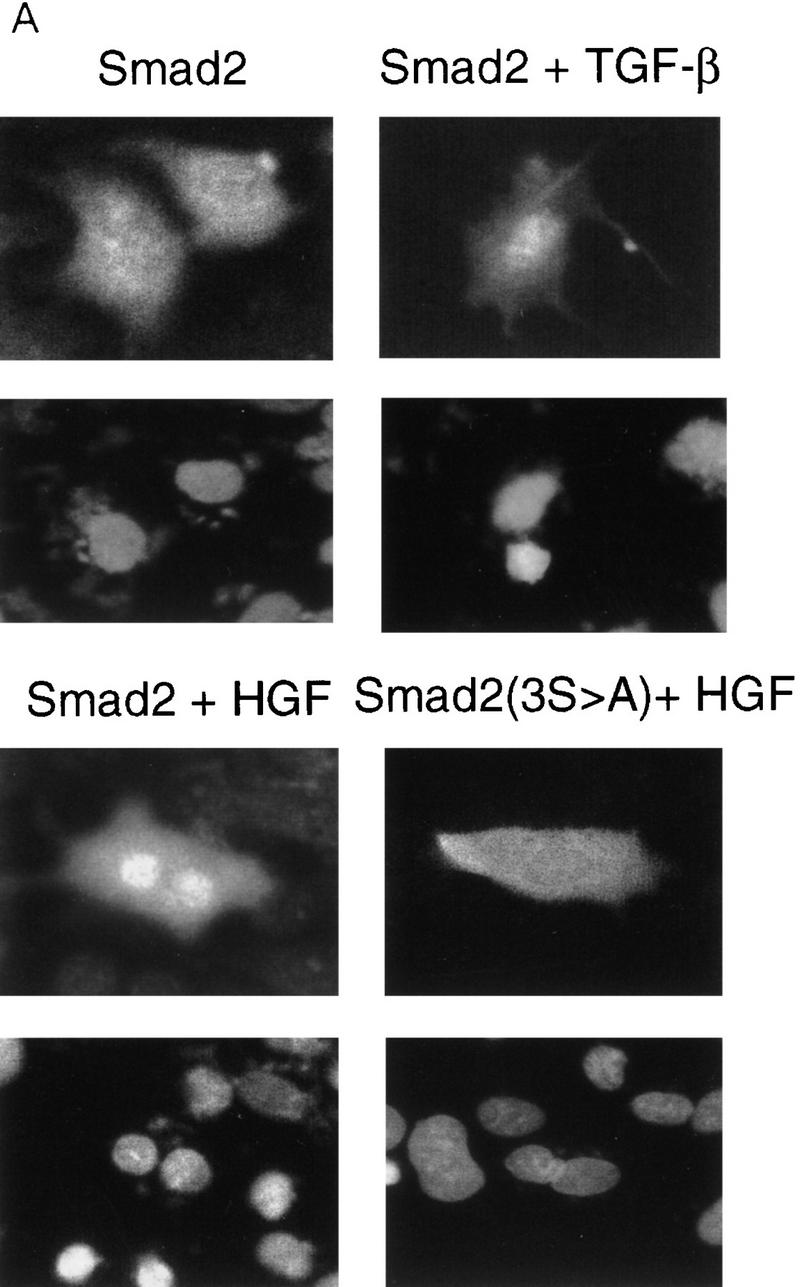

Smad2 indirect immunofluorescence microscopy. (A) HepG2 cells were transiently transfected with Flag-tagged Smad2 or Smad2 (3S→A) and treated as indicated, and Smad2 localization visualized by indirect immunofluorescence (top). (Bottom) DAPI staining of the same field. (B) Graphic representation of percent positive nuclei following ligand treatment. Cotransfection of DNTβR-II or treatment with TRF-β blocking antibodies had no effent on HGF nuclear translocation.
These data suggest that SMAD proteins can signal responses from both TGF-β family RSKs and HGF and EGF. The initial observation that Smad4 is a necessary component of the signaling pathway leading to activation of the reporter 3TP–Lux by HGF or EGF is somewhat surprising, as the three potential AP-1 binding sites were presumed to be activated independent of SMAD pathways. This suggests that SMAD proteins may be more general partners in transcriptional activation than previously thought, or that activation of these sites may be context specific.
The identity of the Smad1 and Smad2 kinase(s) remains to be determined definitively. ERK2 can phosphorylate Smad1 in the middle-linker region in vitro, but whether this occurs in vivo is not yet known (Kretzschmar et al. 1997a). The corresponding sites are not present in Smad2. Inhibition of SMAD phosphorylation by PD98059 suggests that the SMAD kinase activated by HGF lies downstream of MEK1, but whether it is likewise downstream of ERK1 or ERK2 remains to be determined. Similarily, the TGF-β type I receptor is not responsible for Smad2 phosphorylation by HGF or EGF, as this phosphorylation is evident in R1B cells, which lack type I receptors. We cannot rule out that other TGF-β family members or receptors might mediate the results observed.
Mutant phosphorylation site studies and tryptic phosphopeptide mapping of Smad2 indicates that treatment with HGF induces phosphorylation of a subset of sites (Fig. 3B, spots 1 and 2) that are also phosphorylated following treatment with TGF-β. Phosphorylation of these sites is sufficient to enable nuclear translocation and transcriptional activation of Smad2 following treatment with HGF. Our findings that phosphorylation of Smad2 by HGF or TGF-β is dependent on the SSMS are compatible with the functional data demonstrating dominant inhibitory effects of the Smad2(3S>A) mutant on HGF and TGF-β signaling. Phosphorylation of spots 1 and 2 is likely mediated by a kinase other than TβR-I and suggests that regulation of Smad2 signaling is more complex than identified previously.
Our data suggest a model where some of the transcriptional effects of HGF, and likely EGF and other ligands, are mediated by Smad2. The magnitude of the response was consistently lower following treatment with HGF as compared to TGF-β, suggesting that Smad2 activation by HGF may serve to activate only a subset of genes responsive to this protein. This supports the observations that some, but not all, of the biological activities observed with these two ligands overlap. Taken together with the recent observations about Smad1 phosphorylation, these data suggest a complex set of regulatory mechanisms. Smad1 activation by HGF likely inhibits Smad1 function, leading to prevention of BMP-regulated responses (Kretzschmar et al. 1997a). However, HGF activates Smad2, which may act synergistically or additively with signals emanating from TGF-β receptors to cause specific responses. This cross talk between RTK and RSK signaling pathways has important implications in development, normal physiology, and pathology. These observations provide an initial framework for understanding the complex interplay of signaling cascades that are generated from both classes of receptors.
Materials and methods
Cell lines and constructs
MDA-MB-468, Cos-1, HepG2, Mv1Lu, and Mv1Lu clone R1B (a gift from Dr. J. Massagué, Memorial Sloan-Kettering, New York, NY) were maintained in Dulbecco’s modified Eagle medium with 10% FBS and antibiotics. Cells were transfected with the constructs indicated using LT-2 (PanVera Corp.), Lipofectamine (Life Technologies), or Superfect (Qiagen) according to the manufacturers instructions. Wild-type and mutant forms of Smad1 and Smad2 were generated by PCR as necessary and cloned into the appropriate vectors. DNTβR-II was created by cloning the extracellular and transmembrane domains (amino acids 1–191) of human TβR-II into pCDNA3.
Luciferase assays
For 3TP–Lux reporter assays, MDA-MB-468 cells were transfected with 3TP–Lux and pTK–β-gal with or without pCDNA3–Smad4 as indicated. Twenty-four hours after transfection, cells were switched to 0.2% serum for 16 hr and then treated with the biologically active form of HGF, NK-1 (Cioce et al. 1996) (referred to as HGF throughout the text), EGF, or TGF-β for 20 hr. Cells were lysed and luciferase and β-gal activity determined as described (de Caestecker et al. 1997). For dominant inhibitory experiments, R1B cells were transfected and treated as above with the indicated constructs. For Gal4 reporter assay experiments, Gal4–SMAD fusion constructs were transfected with the Gal4 reporter pG5–E1B into HepG2 cells, and luciferase activity was determined. Antibody blocking experiments used 50 μg/ml of a TGF-β inhibitory antibody (ID11, Genzyme) that blocks all three mammalian isoforms added 1 hr prior to ligand addition. All luciferase values are corrected for transfection efficiency with β-galactosidase. Statistical significance was determined by two-tailed paired t-test.
Metabolic labeling
Mv1Lu or R1B cells were labeled for 3 hr with [32P]orthophosphate, treated with ligand for 20 min or as indicated, and lysates immunoprecipitated using the pan-specific anti-SMAD antibody 367 as described (Lechleider et al. 1996). COS-1, R1B, or HepG2 cells were transiently transfected with the indicated constructs and split into equal aliquots before labeling with [32P]orthophosphate. Labeled lysates were immunoprecipitated with the monoclonal anti-Flag antibody M5 (Kodak). The MEK1 inhibitor PD98059 (NE Biolabs) or DMSO vehicle was added to the indicated lanes 1 hr prior to ligand addition. Immunoprecipitated phosphoproteins were separated by denaturing gel electrophoresis and detected by autoradiography. Equal loading was determined by immunoblotting using the M5 antibody following the manufacturers directions. Phosphopeptide mapping was performed as described (Macias-Silva et al. 1996) with electrophoresis in pH 1.9 buffer. All phosphorylation experiments were repeated at least twice with similar results.
Indirect immunofluorescence
HepG2 cells were transiently transfected in chamber slides, serum starved overnight, and treated for 2 hr with the indicated ligand. Following treatment, cells were fixed in 2% paraformaldehyde, permeablized in ice-cold methanol, and incubated with M5 antibody overnight at 4°C, followed by incubation with goat anti-mouse FITC secondary antibody and mounting in medium containing DAPI. Cells were visualized with a Zeiss immunofluorescence microscope. Percent nuclear localization represents 100 cells counted at high power by a trained observer blinded as to the construct transfected and the treatment. Cells shown are representative, and quantitation is representative of two experiments.
TGF-β bioassay
MDA-MB-468 cells were cultured in 0.2% serum with or without ligand. Supernatants were collected after 24 hr of treatment and the amount of TGF-β determined by bioassay as described (Abe et al. 1994). TGF-β values were determined before and after acid activation of supernatants using concentrated HCl. Values represent the average of three determinations.
Acknowledgments
We thank L. Attisano, S.-J. Kim, J. Massagué, M. Ptashne, L. Wakefield, and J. Wrana for DNA constructs and S.G. Choi, Y. Yi, and Y.S. Kim for oligonucleotide synthesis. N. Roche performed the TGF-β bioassay, and C. Eng and J. Ryan provided expert technical assistance. We thank S. Byers, E. Van Obberghen-Schilling, and the members of the Signal Transduction Unit for helpful advice. M.P.D is the recipient of a Wellcome Trust Advanced Training Fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lechleir@dce41.nci.nih.gov; FAX (301) 496-8395.
References
- Abdollah S, Macias-Silva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL. TβR-I phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J Biol Chem. 1997;272:27678–27685. doi: 10.1074/jbc.272.44.27678. [DOI] [PubMed] [Google Scholar]
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Songyang Z. Specificity in protein-tyrosine kinase signaling. Adv Second Messenger Phosphoprotein Res. 1997;31:41–48. doi: 10.1016/s1040-7952(97)80007-9. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Cioce V, Csaky KG, Chan AML, Bottaro DP, Taylor WG, Jensen R, Aaronson SA, Rubin JS. Hepatocyte growth factor (HGF)/NK1 is a naturally occurring HGF/scatter factor variant with partial agonist/antagonist activity. J Biol Chem. 1996;271:13110–13115. doi: 10.1074/jbc.271.22.13110. [DOI] [PubMed] [Google Scholar]
- Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997;272:6653–6662. doi: 10.1074/jbc.272.10.6653. [DOI] [PubMed] [Google Scholar]
- de Caestecker MP, Hemmati P, Larisch-Bloch S, Ajmera R, Roberts AB, Lechleider RJ. Characterization of functional domains within Smad4/DPC4. J Biol Chem. 1997;272:13690–13696. doi: 10.1074/jbc.272.21.13690. [DOI] [PubMed] [Google Scholar]
- de Winter JP, Roelen BA, ten Dijke P, van der Burg B, van den Eijnden-van Raaij AJ. DPC4 (SMAD4) mediates transforming growth factor-beta1 (TGF-β1) induced growth inhibition and transcriptional response in breast tumour cells. Oncogene. 1997;14:1891–1899. doi: 10.1038/sj.onc.1201017. [DOI] [PubMed] [Google Scholar]
- Frenz DA, Liu W, Williams JD, Hatcher V, Galinovic-Schwartz V, Flanders KC, Van de Water TR. Induction of chondrogenesis: Requirement for synergistic interaction of basic fibroblast growth factor and transforming growth factor-β. Development. 1994;120:415–424. doi: 10.1242/dev.120.2.415. [DOI] [PubMed] [Google Scholar]
- Hartsough MT, Frey RS, Zipfel PA, Buard A, Cook SJ, McCormick F, Mulder KM. Altered transforming growth factor signaling in epithelial cells when ras activation is blocked. J Biol Chem. 1996;271:22368–22375. doi: 10.1074/jbc.271.37.22368. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone Jr MA, Wrana JL, Falb D. The MAD-related protein Smad7 associates with the TGF-β receptor and functions as an antagonist of TGF-β signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Keski-Oja J, Raghow R, Sawdey M, Loskutoff DJ, Postlethwaite AE, Kang AH, Moses HL. Regulation of mRNAs for type-1 plasminogen activator inhibitor, fibronectin, and type I procollagen by transforming growth factor-β. Divergent responses in lung fibroblasts and carcinoma cells. J Biol Chem. 1988;263:3111–3115. [PubMed] [Google Scholar]
- Kim J, Johnson K, Chen HJ, Carroll S, Laughon A. Drosophila Mad binds to DNA and directly mediates activation of vestigial by Decapentaplegic. Nature. 1997;388:304–308. doi: 10.1038/40906. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Massagué J. Opposing BMP and EGF signalling pathways converge on the TGF-β family mediator Smad1. Nature. 1997a;389:618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Liu F, Hata A, Doody J, Massagué J. The TGF-β family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes & Dev. 1997b;11:984–995. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- Lagna G, Hata A, Hemmati-Brivanlou A, Massagué J. Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature. 1996;383:832–836. doi: 10.1038/383832a0. [DOI] [PubMed] [Google Scholar]
- Laiho M, Weis MB, Massagué J. Concomitant loss of transforming growth factor (TGF)-β receptor types I and II in TGF-β-resistant cell mutants implicates both receptor types in signal transduction. J Biol Chem. 1990;265:18518–18524. [PubMed] [Google Scholar]
- Lechleider RJ, de Caestecker MP, Dehejia A, Polymeropoulos MH, Roberts AB. Serine phosphorylation, chromosomal localization, and transforming growth factor-β signal transduction by human bsp-1. J Biol Chem. 1996;271:17617–17620. doi: 10.1074/jbc.271.30.17617. [DOI] [PubMed] [Google Scholar]
- Liu F, Hata A, Baker JC, Doody J, Carcamo J, Harland RM, Massagué J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature. 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- Liu F, Pouponnot C, Massagué J. Dual role of the Smad4/DPC4 tumor suppressor in TGF-β-inducible transcriptional complexes. Genes & Dev. 1997;11:3157–3167. doi: 10.1101/gad.11.23.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lough J, Barron M, Brogley M, Sugi Y, Bolender DL, Zhu X. Combined BMP-2 and FGF-4, but neither factor alone, induces cardiogenesis in non-precardiac embryonic mesoderm. Dev Biol. 1996;178:198–202. doi: 10.1006/dbio.1996.0211. [DOI] [PubMed] [Google Scholar]
- Macias-Silva M, Abdollah S, Hoodless PA, Pirone R, Attisano L, Wrana JL. MADR2 is a substrate of the TGFβ receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell. 1996;87:1215–1224. doi: 10.1016/s0092-8674(00)81817-6. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGFβ signaling: Receptors, transducers, and Mad proteins. Cell. 1996;85:947–950. doi: 10.1016/s0092-8674(00)81296-9. [DOI] [PubMed] [Google Scholar]
- Nakao A, Roijer E, Imamura T, Souchelnytskyi S, Stenman G, Heldin CH, ten Dijke P. Identification of Smad2, a human Mad-related protein in the transforming growth factor β signaling pathway. J Biol Chem. 1997;272:2896–2900. doi: 10.1074/jbc.272.5.2896. [DOI] [PubMed] [Google Scholar]
- Niswander L, Martin GR. FGF-4 and BMP-2 have opposite effects on limb growth. Nature. 1993;361:68–71. doi: 10.1038/361068a0. [DOI] [PubMed] [Google Scholar]
- Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- Rahimi N, Tremblay E, Elliott B. Phosphatidylinositol 3-kinase activity is required for hepatocyte growth factor-induced mitogenic signals in epithelial cells. J Biol Chem. 1996;271:24850–24855. doi: 10.1074/jbc.271.40.24850. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Frolik CA, Anzano MA, Sporn MB. Transforming growth factors from neoplastic and nonneoplastic tissues. Fed Proc. 1983;42:2621–2626. [PubMed] [Google Scholar]
- Souchelnytskyi S, Tamaki K, Engstrom U, Wernstedt C, ten Dijke P, Heldin CH. Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-β signaling. J Biol Chem. 1997;272:28107–28115. doi: 10.1074/jbc.272.44.28107. [DOI] [PubMed] [Google Scholar]
- Stern HM, Lin-Jones J, Hauschka SD. Synergistic interactions between bFGF and a TGF-β family member may mediate myogenic signals from the neural tube. Development. 1997;124:3511–3523. doi: 10.1242/dev.124.18.3511. [DOI] [PubMed] [Google Scholar]
- Stolz DB, Michalopoulos GK. Synergistic enhancement of EGF, but not HGF, stimulated hepatocyte motility by TGF-β 1 in vitro. J Cell Physiol. 1997;170:57–68. doi: 10.1002/(SICI)1097-4652(199701)170:1<57::AID-JCP7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Traverse S, Gomez N, Paterson H, Marshall C, Cohen P. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J. 1992;288:351–355. doi: 10.1042/bj2880351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojta J, Nakamura T, Fabry A, Hufnagl P, Beckmann R, McGrath K, Binder BR. Hepatocyte growth factor stimulates expression of plasminogen activator inhibitor type 1 and tissue factor in HepG2 cells. Blood. 1994;84:151–157. [PubMed] [Google Scholar]
- Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Isao O, Ueno N, Yaniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science. 1995;270:2008–2112. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- Yan Z, Winawer S, Friedman E. Two different signal transduction pathways can be activated by transforming growth factor β 1 in epithelial cells. J Biol Chem. 1994;269:13231–13237. [PubMed] [Google Scholar]
- Yingling JM, Datto MB, Wong C, Frederick JP, Liberati NT, Wang XF. Tumor suppressor Smad4 is a transforming growth factor beta-inducible DNA binding protein. Mol Cell Biol. 1997;17:7019–7028. doi: 10.1128/mcb.17.12.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Musci T, Derynck R. The tumor suppressor Smad4/DPC 4 as a central mediator of Smad function. Curr Biol. 1997;7:270–276. doi: 10.1016/s0960-9822(06)00123-0. [DOI] [PubMed] [Google Scholar]