

Abstract
Pluripotent stem cells represent one promising source for cell replacement therapy in heart, but differentiating embryonic stem cell-derived cardiomyocytes (ESC-CMs) are highly heterogeneous and show a variety of maturation states. In this study, we employed an ESC clonal line that contains a cardiac-restricted ncx1 promoter-driven puromycin resistance cassette together with a mass culture system to isolate ESC-CMs that display traits characteristic of very immature CMs. The cells display properties of proliferation, CM-restricted markers, reduced mitochondrial mass, and hypoxia-resistance. Following transplantation into rodent hearts, bioluminescence imaging revealed that immature cells, but not more mature CMs, survived for at least one month following injection. These data and comparisons with more mature cells lead us to conclude that immature hypoxia resistant ESC-CMs can be isolated in mass in vitro and, following injection into heart, form grafts that may mediate long-term recovery of global and regional myocardial contractile function following infarction.
1. Introduction
The regenerative capacity of adult mammalian heart is insufficient to fully restore function following myocardial injury or heart failure. In cases of severe cardiac insufficiency, the treatment of choice is cardiac transplantation, but this approach is hampered by both a severe shortage of donor organs and the potential for organ rejection [1, 2]. Cell replacement therapy represents one promising alternative, but it is limited by the availability of transplantable human cardiomyocytes (CMs). To overcome this limitation, cardiovascular researchers have undertaken numerous studies over the past decade to identify cells in animal and human systems with cardiomyogenic potential, including cardiac derivatives isolated at various times of development and noncardiac progenitors derived from bone-marrow, fat, or skeletal muscle [3]. Subsequent preclinical studies have demonstrated that a wide variety of cardiomyogenic stem/progenitor cells can improve the cardiac function after transplantation, but it remains largely unclear whether improved cardiac function occurs through integration and coupling of new CMs with endogenous cells (reviewed by [4, 5]) or conversely through secondary events related to angiogenesis, sparing of endogenous CMs, cell fusion, or anti-inflammatory responses [6–10].
While numerous investigators have described putative stem/progenitor cells with cardiomyogenic potential, myocardial regeneration confirmed through direct functional coupling is limited to a few cell types [11, 12]. Among these, pluripotent stem cells (PSCs), including both embryonic stem (ES) and induced pluripotent stem (iPS) cells, represent the best viable cell source suitable for transplantation therapy [13]. PSCs can be passaged indefinitely and can produce all body cells; consequently, these cells are capable of treating a wide range of debilitating disease states whose underlying pathology involves cell degeneration, death, or acute injury. There are, however, significant challenges to be overcome before these approaches can be applied in the clinic. Constraints to the use of human-derived ESCs for cell therapy include ethical barriers and potential immunogenicity of ESC-progeny [14, 15]. These concerns may be overcome by iPS cells, which are generated in vitro via transcription factor-mediated reprogramming [16–18], but iPS cells also suffer from interline heterogeneity and incomplete epigenetic remodeling [19–21]. Although generation of sufficient numbers of cells for therapeutic intervention is feasible, isolation of a desired cell type in pure form, a systematic analysis of cell delivery systems, and determination of which cell type may be necessary to correct a specific pathology remain unanswered questions [14].
To deal with cell purity issues, several groups reported isolation of ES cell-derived CMs (ESC-CMs) with cardiac-restricted gene promoters (nkx2.5, cardiac α-mhc, mlc2v, cardiac α-actin, ncx1) from mouse, however, each has limitations. Some like nkx2.5 are not restricted to heart, whereas others like α-mhc are only weakly expressed in the primary myocardium before becoming restricted to the atrial regions throughout the remainder of embryonic and early fetal development [22, 23]. In adult rodents, the mlc2v promoter is restricted to ventricle, but early in development, its expression is present in anterior (atrial and atrioventricular) portions of the heart tube, and at later stages, in the caval myocardium [23, 24]. The cardiac α-actin gene promoter is expressed both in cardiac and skeletal muscle during embryogenesis [23]. Moreover, the distal upstream region of the ncx1 promoter is cardiac-restricted during embryonic and fetal development, but it does not distinguish among chamber-specific types of heart cells [25]. Functionally, ESC-CMs are most typical of embryonic or fetal stages of development [23, 26], and no one has successfully generated ESC-CMs in vitro with characteristics of adult-derived ventricular CMs. Moreover, none of these promoters have been employed in conjunction with stage-specific analyses to determine which cell type and maturation state may be necessary to correct a specific pathology. The goal of such a study would be to relate stage-specific CMs with their suitability to regenerate damaged heart muscle.
In the current study, we have begun to test the hypothesis that very immature but committed CMs may be suitable for cardiac regenerative medicine. We have taken advantage of murine ESCs containing a cardiac restricted ncx1 promoter-driven puromycin-resistance gene that eliminates noncardiac cells by antibiotic treatment and permits the isolation of highly enriched and early-staged ESC-CMs [27, 28]. Developmentally, this promoter is active in proliferating myocardium, and we previously showed that it could be employed to isolate proliferative cells using a monolayer culture and that late-stage nonproliferating cells could be generated for therapeutic interventions. In this pilot study designed to determine if early dividing CMs could be isolated from a mass culture system in therapeutically relevant quantities, we present our initial in vitro findings and data following injection of these cells into the mouse heart. These data indicate that early-staged ESC-CMs can be isolated in therapeutic quantities, are hypoxic-resistant and proliferative competent and potentially suitable to treat myocardial infarctions.
2. Materials and Methods
2.1. Production of Highly Enriched ESC-CMs
Murine R1 ESCs (clone syNP4) that stably express a puromycin resistant gene cassette under the cardiac-specific promoter of sodium calcium exchanger (ncx1) were employed in this study. Differentiation of ESCs to CMs was performed as previously described, except where differences were noted in the text [27, 28]. To track these cells following injection into mice hearts, syNP4 cells were electroporated with an expression cassette containing a mouse EF1α promoter-luciferase construct and a hygromycin resistance cassette. Clonal lines containing this construct with stable expression of luciferase were isolated using standard protocols. Differentiation of ESC-CMs and their purity and maturation were confirmed using RT-PCR with primers specific for α-SMA, cardiac calsequestrin, and atrial natriuretic factor (ANF) [27, 28] and by immunological techniques using antibodies against α-smooth muscle actin (α-SMA, Biomeda, Foster City, CA), sarcomeric α-actinin (Sigma, St. Louis, MO), cTnT (Millipore, Billerica, MA), p21CIP (ab7960), p27KIP1 (ab7961), and phospho-Ser10-Histone H3 (ab5176) (Abcam, Cambridge, MA). CMs in the S phase of the cell cycle were identified by 5-Bromodeoxyuridine (BrdU) (Sigma) incorporation into DNA as described previously [27].
2.2. Flow Cytometry
DNA content was determined from ice cold 70% ethanol fixed cells using a BD-Canto II after staining with propidium iodide (PI, Sigma). Cell cycle compartments were deconvoluted from single-parameter DNA histograms of 10,000 to 50,000 cells, and DNA cell cycle analysis was performed as previously described [27, 29]. Cell viability based on mitochondrial membrane potentials and mitochondrial content were determined using mitotracker dyes according to the manufacturer's instructions. Live versus dead (apoptotic) cells were analyzed on a flow cytometer using 488 nm excitation after labeling the cells using a JC-1 mitochondrial membrane potential assay kit (Invitrogen, Carlsbad, CA). In this assay, the cationic dye JC-1 spontaneously forms complexes known as J-aggregates that have intense red fluorescence. In apoptotic or unhealthy cells with a low membrane potential, JC-1 remains in the monomeric form and shows only green fluorescence. Mitochondria content was also estimated by staining cells with MitoTracker Green FM (MTG), a mitochondrial-selective green fluorescent dye (Invitrogen, Carlsbad, CA). This probe preferentially accumulates in mitochondria regardless of mitochondrial membrane potential and is useful for determining mitochondrial mass. Cells were incubated in 200 micromolar concentrations of the MTG probe for 30 minutes followed by flow cytometry, according to the manufacturer's instructions.
2.3. Hypoxia and Nutrient Deprivation In Vitro
To induce hypoxia, Day 10 and Day 18 ESC-CM cultures in either glucose- and serum-free Dulbecco's modified Eagle's medium (DMEM) or DMEM supplemented with 5.5 mM glucose and 10% heat-inactivated fetal bovine serum for 24 or 48 hours were transferred into a humidified hypoxic incubator maintained at 37°C with 5% CO2, 0.5% O2, and the balance in N2 gas. Following incubation, culture media was immediately collected to quantify total Lactate Dehydrogenase (LDH, Sigma), an indicator of cell death and injury. Cells were also harvested to measure viability via trypan blue exclusion by two observers blind to experimental conditions.
2.4. In Vivo Studies
Surgical Procedures: all animal protocols were approved by the local Institutional Animal Care and Use Committee of the University of Pennsylvania. Surgical procedures were performed on 8–10 athymic nude mice (NCI) as we have described previously [30–32]. Briefly, animals were anesthetized using an induction dose of 3% isoflurane. Mice were intubated, ventilated, and anesthesia maintained with inhaled isoflurane (1% to 2%). An aseptic left thoracotomy was performed and the pericardium was opened. Mice then received a single mid-anterior intramyocardial injection of 1-2 × 106 ESC-CMs in 50 μL of fibrin glue. The developmental stage of the ESC-CMs is indicated in the text.
In Vivo Bioluminescent Imaging (BLI): BLI was performed on days 1, 3, 7, 14, 21, and 30 on IVIS Lumina (Caliper LifeSciences, Hopkinton MA) as we have shown previously [33, 34]. Mice were maintained under isoflurane (1% mixed with oxygen) via a nose cone. Luciferin (250 mg/kg) was injected i.p. and continuous BLI was performed for 15 min after injection. BLI signal was standardized for exposure time and quantified in units of maximum photons per second per square centimeter per steradian (p/s/cm2/sr). The area with the greatest signal intensity was used for region of interest (ROI) analysis at each time point.
2.5. Statistical Analysis
Data are presented as mean ± standard deviation. Student's t-test was used to compare two groups, and significance was considered for P values of < 0.05.
3. Results
To test the ability of committed but proliferating ESC-CM to form grafts in the heart, we had to demonstrate that therapeutically relevant quantities of very immature cells could be generated in vitro. For this, we took advantage of an ESC line that was manipulated to express the puromycin resistance (PacR) cassette under control of the cardiac-restricted portion of the ncx1 promoter. Using the syNP4 clone and the classic hanging drop technique, we generated CMs from >70–80% of the embryoid bodies (EBs). The addition of ascorbic acid at day 2 of cultivation improves this efficiency such that >90% of the EBs contain contracting cells. As shown in Figure 1(a), following collagenase treatment, the cells plate as a pseudo-monolayer characterized by clusters of beating ESC-CMs found throughout the plate; however, the total number of CMs is relatively low. When selected by puromycin (2.5 μg/mL), CMs can be isolated that are ~98% pure based on α-actinin or cTnT labeling in vitro and by flow cytometric analysis of cTnT labeled cells [27]. Here, we also show that cells isolated at early time points and selected for at least 48 hours are transiently proliferative. As previously described, cell cycle attributes can be assayed by BrdU incorporation, Ser10 phosphorylation of histone H3, and by increasing cell numbers on the plate (Figure 1(a)). However, within 8–10 days of plating and selection (Day 7 + 8–10), the number of cells in G2/M decreases (P < 0.05), histone H3 phosphorylation at Ser10 is no longer observed, and bi-nucleation increases (not shown). Here, we also report that cell markers associated with contact inhibition (proteins—p21CIP and p27KIP1) are upregulated (Figure 1(b)) with time of differentiation, which together with the cell cycle data, suggest that these cells rapidly undergo a maturation process associated with the loss of proliferative capacity. Transcripts for α-smooth muscle actin, a marker of very early cardiac differentiation, are however retained in these cells (Figure 1(b)).
Figure 1.
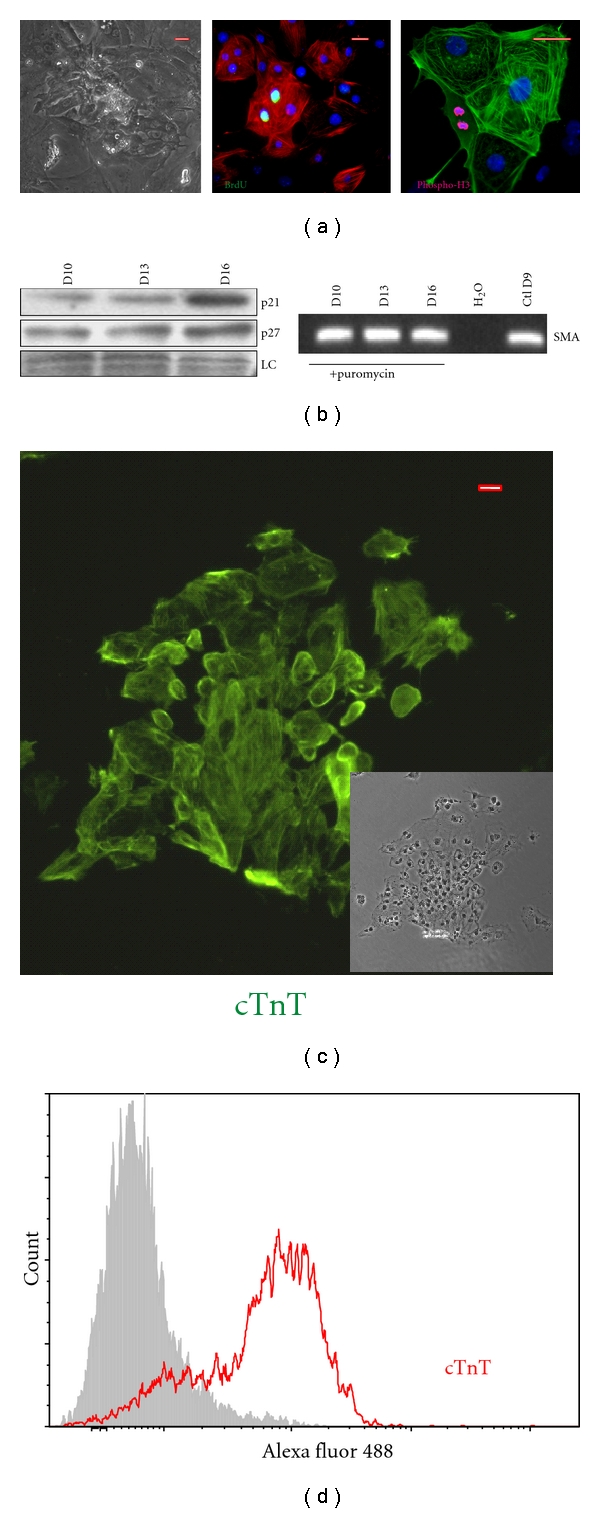
Characterization of syNP4 ES cell derived-CMs. (a) Traits of ESC-CM generated using the hanging drop technique. Following differentiation and plating, puromycin-resistant cells aggregate in clusters and show spontaneous contractions. Day 8-9 CMs (one or two days after plating) readily incorporate BrdU (green) and dividing cells can be readily identified by the use of an antibody against a phosphorylated form of histone H3. (b) As a function of differentiation time, syNP4 derived-CMs begin to express p21CIP and p27KIP1 markers of contact inhibition and cell cycle checkpoint control, but the cells also continue to express transcripts to α-smooth muscle actin (α-SMA), a marker of immature cardiomyocytes. (c) Using the mass culture system, plated puromycin-resistant syNP4-derived CMs cluster similarly to that described in 1A above (see inset). Cells were immunostained with an antibody against cardiac TnT (cTnT), demonstrating that these clusters consist of CMs. (d) By flow cytometry, the purity of the cells has been determined to range from 75 to 92% (n = 5), depending on the cultivation and selection conditions. LC: loading control. Size markers = 50 μm.
When the syNP4 clone is cultured in a spinner flask system for the production of CMs, a number of experimental changes are required to achieve high yields of CMs [28]. The system requires EB formation and suspension for 8-9 days with media changes on a daily basis. At day 6, the addition of BMP2 improves the yield of CMs, and selection with puromycin begins at day 9. The partially selected EBs are then collagenase dissociated and cultivated as a monolayer culture. This method routinely produces ~35 million ESC-CM in a 16-17 day cycle after initial seeding of 25 million ESC, and we have been able to reproduce these results here (see Figure 1(c)). Consistent with our previous report, CM enrichment at day 14 of differentiation estimated by TnT staining was >85% when manually counted under microscope but ranging from 75 to 92% by flow cytometry (Figure 1(d)). The cultures are however heterogeneous, and the presence of α-smooth muscle actin protein is present in a subset of these cells [28].
To isolate immature and potentially proliferative and hypoxic resistant cells, we employed the same mass culture system, except that differentiating syNP4 ESC-CMs were dissociated at day 8 by collagenase treatment and immediately treated with puromycin for 24 to 48 hours prior to analysis. For all experiments, Day 9–11 cells were compared with Day 14–16 or Day 21 cells. At the earlier time point, the overall yield of CMs is decreased significantly, but when expanded ~10 million or more cells can be routinely isolated. The cells plated as a pseudo-monolayer, and most clusters showed asynchronous automaticity. Similar to the later staged cells described earlier, the ESC-CMs were heterogeneous and positive for α-SMA and α-actinin staining (Figure 2(a)). The degree of maturation of ESC-CM as a function of differentiation time is characterized by altered gene expression. More specifically, the ratio of genes encoding ANF (early marker) and calsequestrin (late marker) decreases with increased time of maturation (P < 0.05). Calsequestrin is undetectable by RT-PCR in very early Day 9-10 cells, prior to being upregulated in Day 14–16 cells.
Figure 2.
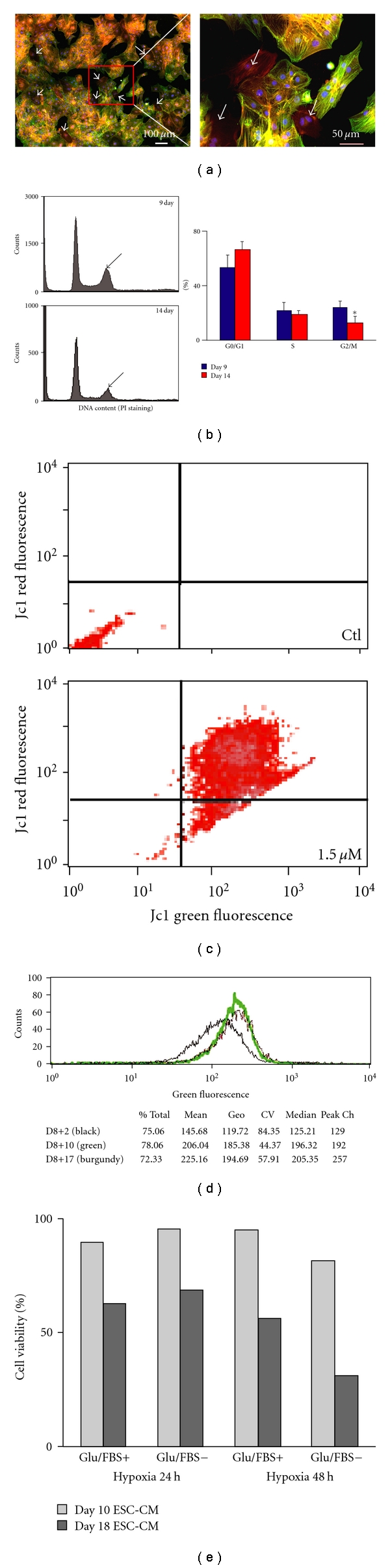
Characterization of immature syNP4 ES cell derived-CMs. (a) The purity and maturity of ESC-CMs at Day 11 were estimated by costaining with anti-α-SMA (red) and anti-α-actinin (sarcomeric) (green) antibodies. All cells express α-SMA, while 90 ± 2% cells are α-actinin positive. The right figure is a high magnification of the box in the left one. Arrows mark cells with α-SMA positive and α-actinin negative staining. (b) The DNA content of puromycin-selected ESC-CMs generated by mass culture was determined following PI staining. In these experiments (n = 3), the histogram indicates that Day 9 cells contain a higher percentage of G2/M phase cells than Day 14 cells. The number of G0/G1 cells also tends to increase with time of differentiation (P > 0.05). (c) Relative to controls, immature ESC-CMs incubated with 1.5 μM Jc1 had a strong increase in red fluorescence, which based on membrane potentials is indicative of live cells. Moreover, lack of change in membrane potential in either cell population indicates that the data shown from the MTG assay is directly comparable. (d) Typical results from MTG-stained cells assessed by flow cytometry. The green fluorescence signal is directly proportional to the mitochondrial content. As shown in the figure and table, the mitochondrial content increases with time of differentiation. (e) Cell viability of ESC-CMs cultivated under hypoxic and nutrient deprivation conditions. Data are presented as a percentage of control values.
At selected time points, we measured DNA content on propidium iodide- (PI, Sigma) stained nuclei from puromycin selected ESC-CMs. Consistent with our previous results with hanging drop derived ESC-CMs, the DNA content of PI labeled cells showed an increase in the number of G0/G1 cells and a decrease in G2/M phase cells between days 9 and 14 of differentiation (Figure 2(b)). In contrast to our previous report, no significant change in the percentages of S phase cells was observed during this time period. The changes in G1 versus G2/M phase cells is, however, consistent with the isolation of a proliferating and consequently less mature cell population than those isolated at Days 14–16.
Mitochondria content/activity as a function of differentiation time was estimated in the ESC-CMs by staining with fluorescent dyes and analysis by flow cytometry (Figures 2(c) and 2(d)). The cells were initially analyzed by flow cytometer to assess live versus apoptotic cells based on mitochondrial membrane potentials. As shown in Figure 2(c), JC1 fluorescence indicates that the vast majority (>90%) of Day 8 + 3 cells are alive (based on red versus green fluorescence) following selection. Mitochondria content as a function of differentiation time was also estimated by staining ESC-CMs with MTG, a mitochondrial-selective green fluorescent dye. ESC-CMs appear relatively rich in mitochondria, but importantly, the mitochondria content increases as a function of differentiation time. The fluorescence signals at Days 10, 18, and 25 of differentiation are 145 ± 84, 206 ± 44, and 225 ± 57, respectively (Figure 2(d)). This result shows that the content of mitochondria in cells isolated at Day 10 is less than that isolated at Day 18 or 25; and consequently, these cells should be less sensitive to changes in oxygen content.
To test this possibility, we induced hypoxia and nutrient depletion in these cells. Under hypoxic and glucose and serum-deprived (double stress) conditions, early stage ESC-CMs, which were isolated at Day 10, survived remarkably better than those mature ESC-CMs isolated at Day 18 (Figure 2(e)). In these experiments, over 80% of Day 10 ESC-CMs were viable, which is on average 36% greater than Day 18 cells at 48 hrs. Consistently, under double stress conditions, Day 18 ESC-CMs released much more total LDH into the media than Day 10 cells. In fact, within 24 hours the increase in LDH release from Day 18 cells was twofold greater and at 48 hours threefold greater than that observed from Day 10 cells.
Finally, to monitor survival of CMs grafted in the heart, a subclone of syNP4 cells that stably express firefly luciferase (Fluc) was established following selection with hygromycin. Here, only one Fluc-expression sub-clone was employed for in vivo analyses. In these experiments, 1-2 million ESC-CMs (Day 8 + 3) selected by puromycin and expressing Fluc were injected into the mid ventricular wall of a normal (noninfarcted) heart in nude mice. Bioluminescent imaging (BLI) was performed at days 1, 3, 7, 14, 21, and 30 post transplantation, and the signal was quantified from an ROI as described previously [35, 36]. As shown in (Figures 3(a) and 3(b)), BLI signal from very immature ESC-CMs was detectable for at least 30 days, whereas the signal from more mature cells was lost within one week of injection. The signal intensity observed in Figure 3(a) falls substantially during the first week, achieves a plateau from day 7 to 21, and then declines further by 1 month. Perhaps more importantly, Fluc signal was only detected in the thoracic region during the entire experiments, indicating that injected cells were retained primarily at the site of injection. At least within the detection limits of the method, the “wash out” cells either did not take up residence elsewhere or did not survive in noncardiac (e.g., liver) locations [37]. Finally, these mice were examined for teratoma formation to ensure against false Fluc signals due to tumor formation. In these animals, no teratoma formation was observed in either experimental group one month following injection (n = 2), confirming that early and proliferating CMs are not tumorgenic.
Figure 3.
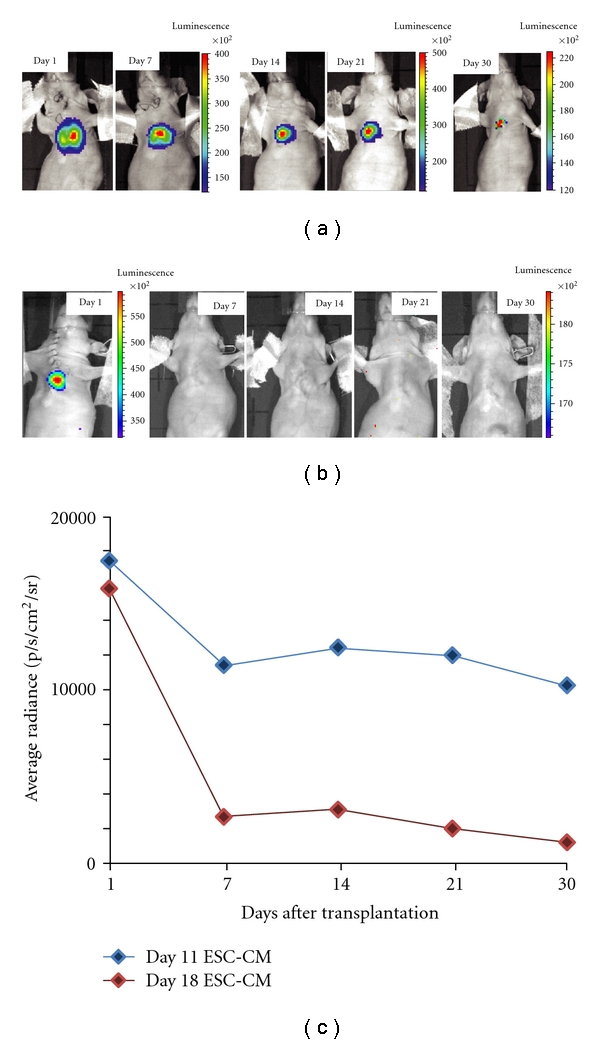
Image analysis of immature syNP4-derived cardiomyocytes injected into heart. (a, b) Representative longitudinal bioluminescence imaging (BLI) of athymic nude mice following intramyocardial transplantation of 2 × 106 syNP4-derived CMs stably expressing a firefly luciferase reporter construct. The images shown in (a) involved CMs isolated at day 11 in culture, while those shown in (b) involved CMs isolated at day 18. (c) Analysis of BLI data from two sets of ESC-CMs shows a robust cardiac signal on day1, which decreases substantially in the first week, achieves a plateau from days 7 to 21, and then declines at 1 month, whereas those cells, which are slightly more mature, have minimal signals within 7 days of injection.
4. Discussion
During myocardial infarction, heart muscle is damaged by ischemia due to a loss of blood flow and a lack of oxygen. Some if not most of the CMs in the affected area may survive brief and transient episodes of ischemia; however, the longer the injury without treatment the more likely the probability of significant cell death. This occurs in part to hypoxic stresses, mitochondrial dysfunction, pH changes, and the accumulation of reactive oxygen species. If however the ischemic episode is severe and is followed by reperfusion as typically occurs following many coronary occlusions, CMs die in mass due to the opening of the membrane permeability transition pore of the mitochondria. A spreading wave of reactive oxygen species causes further damage. An inflammatory response is mounted, and the process of left ventricular remodeling begins shortly thereafter. This latter process involves expansion of the infarcted area, ventricular dilatation, extensive fibrosis, and thinning of the ventricular wall [38–41].
For cell-based therapies designed to treat heart failure to become reality, problems associated with immunogenicity, cell differentiation, and isolation, and delivery systems must be overcome. While there is general agreement that ventricular defects should be treated with ventricular cells, the stage of cell maturation is also an area of active debate. This is particularly true with regards to the ability of transplanted cells to tolerate ischemic conditions. Cell replacement therapy of a myocardial infarction, particularly after extensive remodeling, is going to require the introduction of cells into poorly vascularized and therefore hypoxic conditions. Under these conditions, mature CMs are unlikely to survive; however, immature CMs with innate hypoxic resistance should survive to repopulate damaged tissue. Consistent with this view, Reinecke et al. have shown that highly differentiated adult CMs do not form viable grafts, which makes adult hearts unsuitable as a source of donor cells [42]. Moreover, in animal models, the vast majority of neonatal CMs die within one week of grafting; however, a modest improvement in survival is seen following grafting into vascularized tissue [43]. Reinecke and coworkers also documented that grafted fetal CMs survive and proliferate in the impaired myocardium as well as in the normal myocardium [42, 44]. Significant effort has therefore been made to find CMs that have the ability to improve left ventricular function in damaged heart, and currently it appears that fetal CMs are the best cell candidates for myocardial repair.
For ethical reasons, fetal tissue sources cannot be used to treat human heart failure; however, the derivation of CMs from embryonic or adult stem cells with embryonic or fetal-like characteristics represents one promising alternative [14, 26, 45, 46]. These cells show a wide range of functional attributes in part due to the state of maturation. Some researchers have therefore advocated the use of more mature stem cell-derived CMs that contain a developed contractile apparatus and can integrate through gap junctions and intercalated disks with host myocardium. When functionally coupled, these cells should work to increase force generation and decrease the stresses on surviving CMs [47]. Alternatively, we and others have argued for the use of progenitor cells or very immature but committed CMs that have the capacity to integrate and mature in vivo. These immature cells should ultimately form work-producing cells that couple with endogenous cells. While there are merits to both arguments, the appropriate cell type is likely to be disorder (e.g., myocardial infarction, hypertrophic or dilated cardiomyopathy) specific as well as location dependent; and to date, no research efforts have been designed to address this critical issue.
Here, we report the isolation of therapeutic quantities of ESC-CMs that are functionally immature based on automaticity, cell cycle analysis, hypoxia resistance, lower mitochondrial mass, and the presence of markers typical of immature embryonic or fetal heart cells. Because the puromycin resistant cells are all CMs, based on previous electrophysiological analyses and immunostaining, we conclude that these cells are fully committed CMs, albeit nonchamber specific [27]. Fijnvandraat et al. previously reported that Day 7 to 13 ESC-CMs corresponded most closely to E8.75–9 mouse heart cells based on a variety of chamber-specific markers and electrophysiological properties [26]. The ESC-CMs analyzed by patch clamp in that study all showed automaticity, similar to what we observed here. While contraction is observed in cells present in older cultures (Day 14–21), it is not cell autonomous. Instead, synchronous contractions are driven by the presence of pacemaker-like cells [45], which has led investigators to conclude that later-staged cells were most analogous to fetal or even neonatal stages of development. Based on these published findings, the presence of smooth muscle actin, time of differentiation, and the dynamic proliferative capacity, we conclude that the immature ESC-CMs described here correspond most closely to cells present in mouse primary heart tube stage prior to E8.75–9.
Moreover, we present the first evidence to indicate that these cells can in fact be introduced into myocardium, and that the cells survive for at least one month in mouse heart. Moreover, these cells survive preferentially to those isolated at slightly later times of differentiation. We further demonstrate that the purified ESC-CMs take up residence in the myocardium without teratoma formation, at least during the time-period analyzed, and that Fluc signals can be followed as a function of time to test therapeutic potentials. Together with our recent publication utilizing more mature cells [28], the development of this model system means that we can begin addressing the possibility that committed and proliferating CMs will be therapeutically superior to more mature cells in the treatment of myocardial infarction.
5. Conclusions
In conclusion, we present data showing that the mass culture system employed here can be adapted to generate immature proliferating CMs tolerant of hypoxia. With the development of this system, ESC-CMs at defined differentiation stages can now be tested in vivo to determine their value for treatment of myocardial infarctions. Techniques like cell surface capturing technologies [48] will however be required to isolate pure ventricular populations, but once accomplished, we predict that the results will be analogous to those generated with fetal, neonatal, and adult ventricular cells isolated directly from heart. Early stage-specific CMs should lead to increased cell survival and regeneration of damaged heart, and once confirmed in mouse, this approach could become a powerful strategy for treating human cardiac disease states and injuries.
Acknowledgments
The research was supported by NIH Grants R21EB-2473 (RZ), 4R00HL094798-03 (RLG), R01-HL081185 (RZ), R01-HL081185S1 (RZ) the Innovation Center at the Medical College of Wisconsin (RLG), and the Intramural Research Program (IRP) of the NIH, National Institute on Aging (KRB, RPW). The authors also thank Dr. M. Celeste Simon for helping with hypoxic studies.
References
- 1.Boheler KR, Fiszman MY. Can exogenous stem cells be used in transplantation? Cells Tissues Organs. 1999;165(3-4):237–245. doi: 10.1159/000016684. [DOI] [PubMed] [Google Scholar]
- 2.Li RA, Moore JC, Tarasova YS, Boheler KR. Human embryonic stem cell-derived cardiomyocytes: therapeutic potentials and limitations. Journal of Stem Cells. 2006;1(2):109–124. [Google Scholar]
- 3.Perino MG, Yamanaka S, Li J, Wobus AM, Boheler KR. Cardiomyogenic stem and progenitor cell plasticity and the dissection of cardiopoiesis. Journal of Molecular and Cellular Cardiology. 2008;45(4):475–494. doi: 10.1016/j.yjmcc.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wollert KC, Drexler H. Cell-based therapy for heart failure. Current Opinion in Cardiology. 2006;21(3):234–239. doi: 10.1097/01.hco.0000221586.94490.d2. [DOI] [PubMed] [Google Scholar]
- 5.Rubart M, Field LJ. Stem cell differentiation: cardiac repair. Cells Tissues Organs. 2008;188(1-2):202–211. doi: 10.1159/000112846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murry CE, Soonpaa MH, Reinecke H, et al. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature. 2004;428(6983):664–668. doi: 10.1038/nature02446. [DOI] [PubMed] [Google Scholar]
- 7.Balsam LB, Wagers AJ, Christensen JL, Kofidis T, Weissmann IL, Robbins RC. Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature. 2004;428(6983):668–673. doi: 10.1038/nature02460. [DOI] [PubMed] [Google Scholar]
- 8.Ying QL, Nichols J, Evans EP, Smith AG. Changing potency by spontaneous fusion. Nature. 2002;416(6880):545–548. doi: 10.1038/nature729. [DOI] [PubMed] [Google Scholar]
- 9.Terada N, Hamazaki T, Oka M, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416(6880):542–545. doi: 10.1038/nature730. [DOI] [PubMed] [Google Scholar]
- 10.Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, et al. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425(6961):968–973. doi: 10.1038/nature02069. [DOI] [PubMed] [Google Scholar]
- 11.Rubart M, Field LJ. Cell-based approaches for cardiac repair. Annals of the New York Academy of Sciences. 2006;1080:34–48. doi: 10.1196/annals.1380.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubart M, Field LJ. Cardiac regeneration: repopulating the heart. Annual Review of Physiology. 2006;68:29–49. doi: 10.1146/annurev.physiol.68.040104.124530. [DOI] [PubMed] [Google Scholar]
- 13.Boheler KR. Pluripotency of human embryonic and induced pluripotent stem cells for cardiac and vascular regeneration. Thrombosis and Haemostasis. 2010;104(1):23–29. doi: 10.1160/TH09-07-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wobus AM, Boheler KR. Embryonic stem cells: prospects for developmental biology and cell therapy. Physiological Reviews. 2005;85(2):635–678. doi: 10.1152/physrev.00054.2003. [DOI] [PubMed] [Google Scholar]
- 15.Nussbaum J, Minami E, Laflamme MA, et al. Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. FASEB Journal. 2007;21(7):1345–1357. doi: 10.1096/fj.06-6769com. [DOI] [PubMed] [Google Scholar]
- 16.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 18.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 19.Yamanaka S. A Fresh Look at iPS Cells. Cell. 2009;137(1):13–17. doi: 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 20.Daley GQ, Lensch MW, Jaenisch R, Meissner A, Plath K, Yamanaka S. Broader implications of defining standards for the pluripotency of iPSCs. Cell Stem Cell. 2009;4(3):200–201. doi: 10.1016/j.stem.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu SM, Fujiwara Y, Cibulsky SM, et al. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell. 2006;127(6):1137–1150. doi: 10.1016/j.cell.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 23.Franco D, Lamers WH, Moorman AFM. Patterns of expression in the developing myocardium: towards a morphologically integrated transcriptional model. Cardiovascular Research. 1998;38(1):25–53. doi: 10.1016/s0008-6363(97)00321-0. [DOI] [PubMed] [Google Scholar]
- 24.Christoffels VM, Habets PE, Franco D, et al. Chamber formation and morphogenesis in the developing mammalian heart. Developmental Biology. 2000;223(2):266–278. doi: 10.1006/dbio.2000.9753. [DOI] [PubMed] [Google Scholar]
- 25.Koban MU, Brugh SA, Riordon DR, et al. A distant upstream region of the rat multipartite Na+—Ca2+ exchanger NCX1 gene promoter is sufficient to confer cardiac-specific expression. Mechanisms of Development. 2001;109(2):267–279. doi: 10.1016/s0925-4773(01)00548-2. [DOI] [PubMed] [Google Scholar]
- 26.Fijnvandraat AC, van Ginneken ACG, de Boer PAJ, et al. Cardiomyocytes derived from embryonic stem cells resemble cardiomyocytes of the embryonic heart tube. Cardiovascular Research. 2003;58(2):399–409. doi: 10.1016/s0008-6363(03)00282-7. [DOI] [PubMed] [Google Scholar]
- 27.Yamanaka S, Zahanich I, Wersto RP, Boheler KR. Enhanced proliferation of monolayer cultures of embryonic stem (ES) cell-derived cardiomyocytes following acute loss of retinoblastoma. PLoS ONE. 2008;3(12) doi: 10.1371/journal.pone.0003896. Article ID e3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiao H, Zhang H, Yamanaka S, et al. Long-term improvement in postinfarct left ventricular global and regional contractile function is mediated by embryonic stem cell-derived cardiomyocytes. Circulation: Cardiovascular Imaging. 2011;4(1):33–41. doi: 10.1161/CIRCIMAGING.110.957431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarasov KV, Tarasova YS, Tam WL, et al. B-MYB is essential for normal cell cycle progression and chromosomal stability of embryonic stem cells. PLoS ONE. 2008;3(6) doi: 10.1371/journal.pone.0002478. Article ID e2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou R, Thomas DH, Qiao H, et al. In vivo detection of stem cells grafted in infarcted rat myocardium. Journal of Nuclear Medicine. 2005;46(5):816–822. [PMC free article] [PubMed] [Google Scholar]
- 31.Qiao H, Surti S, Choi SR, et al. Death and proliferation time course of stem cells transplanted in the myocardium. Molecular Imaging and Biology. 2009;11(6):408–414. doi: 10.1007/s11307-009-0222-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiao H, Zhang H, Zheng Y, et al. Embryonic stem cell grafting in normal and infarcted myocardium: serial assessment with MR imaging and PET dual detection. Radiology. 2009;250(3):821–829. doi: 10.1148/radiol.2503080205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gruber PJ, Li Z, Li H, et al. In vivo imaging of MLC2v-luciferase, a cardiac-specific reporter gene expression in mice. Academic Radiology. 2004;11(9):1022–1028. doi: 10.1016/j.acra.2004.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Qiao H, Bakken A, et al. Utility of dual-modality bioluminescence and MRI in monitoring stem cell survival and impact on post myocardial infarct remodeling. Academic Radiology. 2011;18(1):3–12. doi: 10.1016/j.acra.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gruber PJ, Li Z, Li H, et al. In vivo imaging of MLC2v-luciferase, a cardiac-specific reporter gene expression in mice. Academic Radiology. 2004;11(9):1022–1028. doi: 10.1016/j.acra.2004.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Qiao H, Bakken A, et al. Utility of dual-modality bioluminescence and MRI in monitoring stem cell survival and impact on post myocardial infarct remodeling. Academic Radiology. 2011;18(1):3–12. doi: 10.1016/j.acra.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao F, Lin S, Xie X, et al. In vivo visualization of embryonic stem cell survival, proliferation, and migration after cardiac delivery. Circulation. 2006;113(7):1005–1014. doi: 10.1161/CIRCULATIONAHA.105.588954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frantz S, Bauersachs J, Ertl G. Post-infarct remodelling: contribution of wound healing and inflammation. Cardiovascular Research. 2009;81(3):474–481. doi: 10.1093/cvr/cvn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silvestre JS, Mallat Z, Tedgui A, Lévy BI. Post-ischaemic neovascularization and inflammation. Cardiovascular Research. 2008;78(2):242–249. doi: 10.1093/cvr/cvn027. [DOI] [PubMed] [Google Scholar]
- 40.Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovascular Research. 2006;72(2):210–219. doi: 10.1016/j.cardiores.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 41.Ertl G, Frantz S. Healing after myocardial infarction. Cardiovascular Research. 2005;66(1):22–32. doi: 10.1016/j.cardiores.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 42.Reinecke H, Zhang M, Bartosek T, Murry CE. Survival, integration, and differentiation of cardiomyocyte grafts: a study in normal and injured rat hearts. Circulation. 1999;100(2):193–202. doi: 10.1161/01.cir.100.2.193. [DOI] [PubMed] [Google Scholar]
- 43.Zhang M, Methot D, Poppa V, Fujio Y, Walsh K, Murry CE. Cardiomyocyte grafting for cardiac repair: graft cell death and anti-death strategies. Journal of Molecular and Cellular Cardiology. 2001;33(5):907–921. doi: 10.1006/jmcc.2001.1367. [DOI] [PubMed] [Google Scholar]
- 44.Murry CE, Whitney ML, Reinecke H. Muscle cell grafting for the treatment and prevention of heart failure. Journal of Cardiac Failure. 2002;8(supplement 6):S532–S541. doi: 10.1054/jcaf.2002.129268. [DOI] [PubMed] [Google Scholar]
- 45.Boheler KR, Czyz J, Tweedie D, Yang HT, Anisimov SV, Wobus AM. Differentiation of pluripotent embryonic stem cells into cardiomyocytes. Circulation Research. 2002;91(3):189–201. doi: 10.1161/01.res.0000027865.61704.32. [DOI] [PubMed] [Google Scholar]
- 46.Fijnvandraat AC, van Ginneken ACG, Schumacher CA, et al. Cardiomyocytes purified from differentiated embryonic stem cells exhibit characteristics of early chamber myocardium. Journal of Molecular and Cellular Cardiology. 2003;35(12):1461–1472. doi: 10.1016/j.yjmcc.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 47.Musunuru K, Domian IJ, Chien KR. Stem cell models of cardiac development and disease. Annual Review of Cell and Developmental Biology. 2010;26:667–687. doi: 10.1146/annurev-cellbio-100109-103948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gundry RL, Boheler KR, van Eyk JE, Wollscheid B. A novel role for proteomics in the discovery of cell-surface markers on stem cells: scratching the surface. Proteomics—Clinical Applications. 2008;2(6):892–903. doi: 10.1002/prca.200780122. [DOI] [PMC free article] [PubMed] [Google Scholar]