

Abstract
The stabilization of β-catenin is a key regulatory step during cell fate changes and transformations to tumor cells. Several interacting proteins, including Axin, APC, and the protein kinase GSK-3β are implicated in regulating β-catenin phosphorylation and its subsequent degradation. Wnt signaling stabilizes β-catenin, but it was not clear whether and how Wnt signaling regulates the β-catenin complex. Here we show that Axin is dephosphorylated in response to Wnt signaling. The dephosphorylated Axin binds β-catenin less efficiently than the phosphorylated form. Thus, Wnt signaling lowers Axin’s affinity for β-catenin, thereby disengaging β-catenin from the degradation machinery.
Keywords: Axin complex, β-catenin, phosphorylation, Wnt signaling
The stabilization of the β-catenin protein is a key regulatory step during cell fate changes and transformations to tumor cells (for review, see Cadigan and Nusse 1997; Brown and Moon 1998; Wodarz and Nusse 1998; Polakis 1999). Several proteins, including the serine/threonine kinase GSK-3β, the protein product of the Adenomatous polyposis coli (APC) gene, and Axin are implicated in the control of β-catenin protein levels. Signaling by Wnt proteins regulates the stability of β-catenin. β-Catenin levels, in turn, mediate expression of many Wnt target genes by modulating transcription in concert with the Lef-1/Tcf family of transcription factors.
Axin, which directly associates with β-catenin, GSK-3β, and APC, was implicated in negatively regulating the Wnt signaling pathway (Zeng et al. 1997; Behrens et al. 1998; Hart et al. 1998; Ikeda et al. 1998; Itoh et al. 1998; Kishida et al. 1998; Nakamura et al. 1998; Sakanaka et al. 1998; Yamamoto et al. 1998, 1999; Kishida et al. 1999). Overexpressed Axin destabilizes β-catenin and blocks the axis-duplicating activity of XWnt-8 in Xenopus embryos (Zeng et al. 1997; Behrens et al. 1998; Yamamoto et al. 1998) and Wnt-induced accumulation of β-catenin in cell culture (Kishida et al. 1999).
The stability of β-catenin is controlled by phosphorylation, most likely by GSK-3β (Yost et al. 1996; Aberle et al. 1997; Morin et al. 1997; Rubinfeld et al. 1997), which targets β-catenin to the ubiquitin/proteasome pathway (Aberle et al. 1997; Maniatis 1999). Axin is likely to have an indispensable role in the phosphorylation of β-catenin by GSK-3β, as it is able to bind both proteins simultaneously and may thereby bring them into close proximity. β-Catenin is a much better substrate for GSK-3β when Axin is present than when it is absent (Ikeda et al. 1998). It has not been clear, however, whether or how Wnt regulates the interactions among Axin, β-catenin, and GSK-3β. Here we show that Axin is a component of the Wnt signaling pathway as it is dephosphorylated in response to Wnt, and this dephosphorylated Axin binds β-catenin less efficiently than phosphorylated Axin.
Results and Discussion
We examined the effect of Wnt signaling on the endogenous Axin protein in a cell culture system. For detection of Axin, we generated antibodies to various domains of Axin. All antibodies recognize a set of proteins that migrate with an apparent molecular mass of 120–140 kD (Fig. 1; data not shown).
Figure 1.

Wnt signaling modifies Axin. (A) Western analysis of Axin and β-catenin protein from C57MG cells treated with either Wnt-1 or Wnt-3A. (B) Axin and β-catenin protein from L cells treated with Wnt-3A. C57MG and L cells were stimulated with control (−) or Wnt-3A (+)-conditioned medium for 2 hr prior to lysis. C57MG cells containing a tet-repressible Wnt-1 transgene were incubated in tet-containing medium to repress Wnt-1 expression (−) or in tet-free medium for 12 hr to induce Wnt-1 expression (+). (C) Time course of Axin protein modification in response to Wnt-3A. C57MG cells were stimulated with control (−) or Wnt-3A (+)-conditioned medium for the indicated times and immunoblotted with anti-Axin antibody.
Stimulation of cells with the Wnt protein was accomplished in two ways: (1) We used the mammary epithelial cell line C57MG into which a tetracycline (tet)-repressible Wnt-1 transgene was introduced (Korinek et al. 1998); and (2) we used conditioned medium from cells overexpressing and secreting Wnt-3A. The Wnt-3A protein in this conditioned medium leads to the elevation of cytoplasmic β-catenin levels in C57MG cells, a Wnt-specific effect (Fig. 1A; Shibamoto et al. 1998).
Activation of the Wnt signaling pathway either by induction of Wnt-1 expression in C57MG cells (Fig. 1A) or by stimulation of C57MG or mouse L cells with Wnt-3A-conditioned medium (Fig. 1A,B) modified the Axin protein, evidenced by an increase in its mobility on gels. As both Wnt-1 induction and Wnt-3A stimulation elicit the same effect on the Axin protein and β-catenin levels, the remaining experiments were performed using Wnt-3A-conditioned medium.
Treating cells with conditioned medium allowed us to perform a kinetic analysis of Axin modification. The change in mobility of Axin in response to Wnt-3A is time-dependent, first becoming visible at 15 min, and nearing completion within 4 hr (Fig. 1C). This is the same time course as observed for β-catenin accumulation in response to Wnt-3A (data not shown). Prolonged stimulation of cells with Wnt-3A leads to an overall decrease in the levels of the Axin protein (240-min time point in Fig. 1C; data not shown). Wnt-induced degradation of Axin has been reported previously (Yamamoto et al. 1999); however, as this effect occurs subsequent to the mobility shift, we did not investigate this aspect of Wnt signaling on Axin any further.
As changes in the mobility of proteins are frequently due to changes in their phosphorylation state, we investigated whether the shift in mobility of Axin in response to Wnt signaling was due to dephosphorylation. Treatment of immunoprecipitated Axin with protein phosphatase 2A (PP2A) leads to a mobility shift in Axin similar to that seen when cells are stimulated with Wnt-3A (Fig. 2A). Phosphatase 1 (PP1) does not shift the mobility of Axin. Furthermore, treatment of cells with okadaic acid, a potent inhibitor of PP2A, blocks the dephosphorylation of Axin in response to Wnt-3A (Fig. 2B). These data demonstrate that in unstimulated cells Axin is phosphorylated and it is dephosphorylated in response to Wnt signaling.
Figure 2.

The Axin protein is phosphorylated. (A) PP2A, but not PP1, dephosphorylates the Axin protein in vitro. Axin protein was immunoprecipitated from cell extracts of C57MG cells treated with control (−) or Wnt-3A (+)-conditioned medium. The immune complexes were treated with phosphatase buffer only, PP2A, or PP1 and immunoblotted with anti-Axin antibody. (B) The phosphatase inhibitor okadaic acid inhibits the dephosphorylation of Axin. Cell lysates were prepared from C57MG cells treated with control (−) or Wnt-3A (+)-conditioned medium and with or without 400 nm okadaic acid and immunoblotted with anti-Axin antibody. (C) Lithium, an inhibitor of GSK-3β, promotes the dephosphorylation of Axin. Cell lysates were prepared from C57MG cells first treated with or without 25 mm LiCl for 4 hr and treated with control (−) or Wnt3A (+)-conditioned medium for an additional 2 hr.
At present, we have no evidence that Wnt signaling regulates this phosphatase activity. Based on inhibition experiments in vitro by microcystin (data not shown) and in vivo by okadaic acid (Fig. 2B), this phosphatase could be PP2A, which can dephosphorylate Axin efficiently in vitro (Figs. 2A and 4A, below) and has been shown to bind to Axin (Hsu et al. 1999). Recently, Seeling et al. (1999) showed that overexpression of the PP2A regulatory subunit B56 reduces β-catenin levels and inhibited transcription of β-catenin target genes in mammalian cells and Xenopus embryo explants. Although these workers primarily entertain the possibility that B56 may direct the dephosphorylation of specific Wnt signaling components, it is also possible that overexpression of this PP2A regulatory subunit prevents dephosphorylation of certain signaling components such as Axin.
Figure 4.
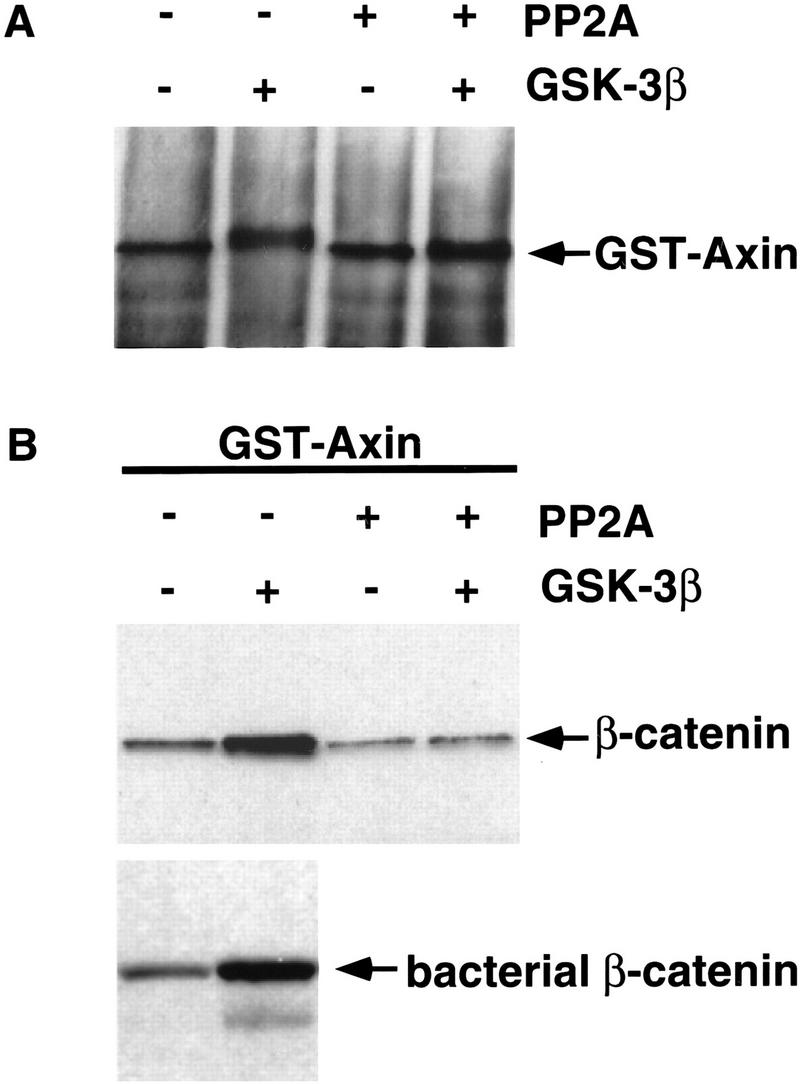
Phosphorylation of Axin with GSK-3β increases Axin’s affinity for β-catenin. (A) Phosphorylation of GST–Axin with GSK-3β and subsequent dephosphorylation with PP2A. GST–Axin immobilized on glutathione–Sepharose beads was treated with (+) or without (−) purified GSK-3β. Phosphorylated or unphosphorylated GST–Axin subsequently was washed to remove the ATP and incubated in the absence (−) or presence (+) of purified PP2A. The GST–Axin protein was visualized by immunoblotting with anti-Axin antibody. (B) GSK-3β phosphorylated Axin binds β-catenin significantly better than unphosphorylated or dephosphorylated GST–Axin. The GSK-3β and PP2A-modified GST–Axin proteins were used to precipitate β-catenin from lysates of L cells treated with Wnt-3A. Phosphorylated and unphosphorylated GST–Axin was also used to precipitate bacterially produced β-catenin protein. Axin-binding proteins were immunoblotted with anti-β-catenin antibody.
It has been shown previously that the Ser/Thr kinase GSK-3β is partially inhibited by Wnt signaling (Cook et al. 1996) and that lithium (Li+) is a potent in vivo inhibitor of its kinase activity (Klein and Melton 1996; Stambolic et al. 1996; Hedgepeth et al. 1997). Furthermore, Ikeda et al. (1998) have shown that GSK-3β can directly phosphorylate Axin in vitro. To address the possibility that GSK-3β phosphorylates Axin in vivo, we treated cells with Li+ to inhibit GSK-3β kinase activity. This produced an effect similar to treatment with Wnt-3A, which leads to the dephosphorylation of Axin (Fig. 2C) and to the elevation of β-catenin levels in the cytoplasm (data not shown). This observation suggests that Wnt signaling primarily acts to inhibit GSK-3β kinase activity while having little or no effect on the above-mentioned phosphatase activity. As GSK-3β activity is inhibited by Wnt signaling, a constitutive phosphatase activity leads to an overall dephosphorylation of Axin which, in turn, binds β-catenin less efficiently.
We then examined whether Wnt-3A signaling has an effect on the formation of complexes between Axin and its known binding partners. GSK-3β, β-catenin, and APC bind to Axin through distinct domains (Behrens et al. 1998; Hart et al. 1998; Ikeda et al. 1998; Itoh et al. 1998; Kishida et al. 1998; Nakamura et al. 1998; Sakanaka et al. 1989; Yamamoto et al. 1998). Both β-catenin (Fig. 3A) and GSK-3β (data not shown) can be coimmunoprecipitated with Axin antibodies. No dramatic differences in the amount of coimmunoprecipitated β-catenin or GSK-3β in response to Wnt-3A signaling are detected by this approach. Furthermore, we did not detect a striking change in Axin-associated GSK-3β kinase activity in response to Wnt signaling (data not shown).
Figure 3.

In the absence of a Wnt signal, Axin binds β-catenin with higher affinity. (A) β-Catenin coimmunoprecipitates with Axin. Axin protein was immunoprecipitated from L cells treated with control (−) or Wnt-3A (+)-conditioned medium and immunoblotted with either anti-Axin or anti-β-catenin antibodies. (B) Axin from Wnt-3A-stimulated cells binds β-catenin less efficiently. β-Catenin-coated beads were used to retrieve the Axin protein from cell extracts prepared from C57MG cells treated with control (−) or Wnt-3A (+)-conditioned medium in the absence (−) or presence (+) of 25 mm LiCl. The precipitating Axin protein was then visualized by immunoblotting with anti-Axin antibodies. The bracket indicates the position of phosphorylated Axin (Axin-P); the arrowhead indicates the position of un- or under-phosphorylated Axin (Axin).
Despite the failure to detect a change in the amount of complexes formed, one result struck us as peculiar: Wnt-3A signaling significantly up-regulates cytoplasmic levels of β-catenin (Figs. 1A,B and 3A); however, there is no corresponding increase in the amount of β-catenin associated with Axin (Fig. 3A). This suggests two possibilities: (1) Axin levels are limiting so that it is completely saturated with β-catenin prior to Wnt-3A signaling, or (2) Wnt-3A signaling modifies the Axin protein in a way that prevents it from binding β-catenin as effectively. Because Wnt-3A signaling modifies Axin through dephosphorylation we investigated the latter possibility.
To address whether Wnt signaling lowers Axin’s ability to bind β-catenin, we covalently immobilized bacterially produced β-catenin protein on Sepharose beads and used the beads to retrieve Axin from lysates prepared from either unstimulated or Wnt-3A-stimulated cells. The β-catenin-precipitating proteins are then immunoblotted with anti-Axin antibodies. As shown in Figure 3B, Axin from unstimulated cells binds to β-catenin significantly better than Axin from Wnt-3A-stimulated cells, suggesting that Axin in Wnt-3A-stimulated cells binds β-catenin less effectively than does Axin in unstimulated cells. Quantitation of multiple immunoblots revealed that Axin protein levels in the whole cell lysate declined by 11%–16% (also see Fig. 1C), whereas Axin levels in the β-catenin-coated bead precipitations decreased by 75%–80% upon Wnt stimulation. Moreover, the β-catenin-coated beads precipitate the slower migrating phosphorylated Axin (Axin-P) exclusively, and never the underphosphorylated Axin (Axin) (Fig. 3B). Furthermore, treatment of cells with Li+, which leads to dephosphorylation of Axin in vivo (Figs. 2C and 3B, left), also lowers the amounts of Axin binding to the β-catenin-coated beads.
These results showed a correlation between Axin modification and binding affinity for β-catenin. We then tested directly whether the phosphorylation of Axin by GSK-3β regulates the binding of Axin to β-catenin. We produced Axin protein in bacteria as a GST fusion protein. As shown previously (Ikeda et al. 1998) and in Figure 4A, Axin protein can be phosphorylated in vitro by GSK-3β. This phosphorylation leads to a mobility shift of GST–Axin, similar to the difference in mobility seen between Axin from Wnt-3A-stimulated versus unstimulated cells. The GSK-3β phosphorylations can subsequently be removed by treating the GST–Axin protein with PP2A (Fig. 4A). Protein kinase A and CK2 also phosphorylate Axin in vitro, but these phosphorylations fail to shift the mobility of Axin (data not shown).
We then used this unphosphorylated and GSK-3β-phosphorylated Axin to examine whether it would bind β-catenin differentially. As a source of β-catenin protein we used whole cell lysates from L cells that were stimulated with Wnt-3A and therefore contained high levels of uncomplexed β-catenin protein. Phosphorylated GST–Axin binds β-catenin significantly better than unmodified GST–Axin (Fig. 4B). Furthermore, dephosphorylation of Axin with PP2A reverses these effects so that dephosphorylated Axin binds β-catenin as poorly as does unmodified GST–Axin. GSK-3β-phosphorylated GST–Axin also precipitated bacterially produced β-catenin protein (Fig. 4B), arguing that this interaction between Axin and β-catenin is direct and requires no other proteins and modifications on β-catenin. A similar experiment has been performed previously but did not reveal such a difference (Ikeda et al. 1998)—a discrepancy we do not understand.
These results provide a plausible mechanism for the way in which Wnt signaling regulates interactions between Axin and β-catenin and the subsequent stabilization of β-catenin (Fig. 5). In an unstimulated cell, Axin is phosphorylated by GSK-3β, promoting effective binding to β-catenin. Axin-associated β-catenin is then phosphorylated by GSK-3β and targeted for degradation, which involves APC and the ubiquitin–proteasome pathway (Aberle et al. 1997). Upon reception of a Wnt signal, GSK-3β is inhibited (Cook et al. 1996), and Axin is dephosphorylated by a phosphatase and releases β-catenin.
Figure 5.

A model for the role of Axin in Wnt signal transduction. In an unstimulated cell, GSK-3β is active and phosphorylates Axin, which in turn, recruits β-catenin into the Axin/GSK-3β complex. By virtue of its proximity to GSK-3β, β-catenin is then phosphorylated. Phosphorylated β-catenin is then targeted for degradation. Upon transduction of the Wnt signal through the Frizzled (Fz) receptors to Dishevelled (Dvl), GSK-3β kinase activity is inhibited so that PP2A dephosphorylates Axin. Unphosphorylated Axin, in turn, no longer recruits β-catenin to the complex. Failure of β-catenin to associate with the Axin/GSK-3β complex prevents its phosphorylation by GSK-3β so that it can accumulate to high levels in the cytoplasm and nucleus and activate transcription in concert with the Tcf/Lef-1 family of transcription factors. GSK-3β also phosphorylates APC, which may facilitate β-catenin recruitment into the complex; however, this event has not been shown to be regulated by Wnt signaling.
An additional implication of these results is that the primary target for GSK-3β phosphorylation is Axin. Previous models argued that GSK-3β directly phosphorylates β-catenin and thereby targets it for degradation. However, GSK-3β does not bind directly to β-catenin, and efficient in vitro phosphorylation of β-catenin by GSK-3β requires the presence of Axin, which binds both proteins (Ikeda et al. 1998). Efficient Axin phosphorylation by GSK-3β on the other hand, does not require additional proteins (Fig. 4A; Ikeda et al. 1998). Thus, in a Wnt-stimulated cell, β-catenin fails to be phosphorylated by GSK-3β because it is not recruited into the Axin/GSK-3β complex. It should also be noted that phosphorylation of APC by GSK-3β increases β-catenin binding to APC (Rubinfeld et al. 1996); however, in contrast to Axin, phosphorylation of APC has not been shown to be regulated by Wnt signaling.
Materials and methods
Plasmids and antibodies
The pGEX-β-catenin plasmid used for the production of GST–β-catenin was kindly provided by Rolf Kemler (Max-Planck Institute for Immunology, Freiburg, Germany). GST fusion proteins for Axin antibody production were expressed from the pGEX4T-1 vector (Pharmacia). The GST fusion to full-length Axin was expressed from pGEX4T-2 into which a 2.5-kb StuI–NotI Axin cDNA, including a carboxy-terminal myc tag, was cloned (starting at position 43 from the first methionine and extending to the end of the published Axin sequence).
Antisera to Axin were obtained from rats and rabbits by immunization with bacterially produced GST fusion to either the amino or carboxyl termini of mouse Axin. Antisera were affinity purified against the fusion proteins to which they were raised. All antisera recognized a similar set of bands at ∼120–140 kD. Mouse monoclonal anti-β-catenin antibody was purchased from Transduction Laboratories.
Cell culture
Culture conditions and stimulation protocols of C57MG cells with a tet-repressible Wnt-1 transgene are described elsewhere (Korinek et al. 1998). L cells were cultured in DMEM, 10% FBS, and penicillin/streptomycin. Preparation of Wnt-3A and control conditioned medium (CM) and Wnt-3A stimulations were performed as described (Shibamoto et al. 1998). Unless specified otherwise, cells were treated with Wnt-3A or control CM for 2 hr. When indicated, cells are incubated at the same time with 400 nm okadaic acid (Calbiochem) or for 4 hr with 25 mm LiCl.
Preparation of cell lysates, immunoprecipitations, phosphatase treatment, and immunoblotting
Cells were washed with PBS and lysed on a plate on ice for 20 min in lysis buffer (1% Triton X-100, 150 mm NaCl, 50 mm Tris-HCl at pH 8) supplemented with protease inhibitors (1 mm Pefabloc, 0.5 μg/ml leupeptin, 1 μg/ml pepstatin A) and phosphatase inhibitors (0.4 μm microcystin-LR, 1 mm vanadate). Addition of microcystin in the lysis buffer is essential to recover the fully phosphorylated form of Axin in unstimulated cells. Insoluble proteins were pelleted at 20,000g, and protein concentrations were determined using the Bio-Rad protein assay dye reagent. For visualization of β-catenin accumulation in C57MG cells (Fig. 1A), cells were lysed into hypotonic buffer as described (Shibamoto et al. 1998). Twenty micrograms of total protein per lane was resolved by SDS-PAGE after boiling for 5 min in sample buffer (50 mm Tris-HCl at pH 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, Bromophenol blue). For immunoprecipitation, 200 μg of total protein was incubated overnight at 4°C with 1 μl of affinity-purified rabbit anti-Axin antibody and 10 μl of a 1:1 slurry of protein A–Sepharose. Immune complexes were washed four times with lysis buffer, boiled in SDS-PAGE sample buffer, and resolved by SDS-PAGE. For phosphatase treatment, the immune complexes prior to boiling were incubated in phosphatase reaction buffer (lysis buffer with 0.1% β-mercaptoethanol, 1 mm EGTA, 0.5 mg/ml BSA, and protease inhibitors) and incubated with 1 unit of PP1 (Promega) or 1 unit of PP2A (Upstate Biotechnologies, Inc.) for 1 hr at 30°C. Proteins resolved by SDS-PAGE were transferred to nitrocellulose, and blots were incubated in blocking buffer (3% nonfat dry milk, 1% BSA in 20 mm Tris-HCl, 150 mm NaCl, 0.2% Tween 20 at pH 8) and incubated overnight at 4°C with anti-Axin antibody at 1:1000 or anti-β-catenin antibody at 1:500 in blocking buffer. Proteins were detected using HRP-conjugated secondary antibodies (Bio-Rad) with ECL Western blot detection reagents (Amersham).
Production of β-catenin beads and Axin precipitation
GST–β-catenin protein was expressed in Escherichia coli strain BL-21 (Stratagene) and purified with glutathione–Sepharose CL4B beads as described by the vendor (Pharmacia). The immobilized β-catenin was released from the beads by cleavage in PBS containing 2 U/ml Thrombin (Pharmacia) for 2 hr at room temperature. The β-catenin protein was then either used in precipitation experiments with GST–Axin fusion proteins or covalently immobilized to Sepharose beads using the AminoLink Plus Immobilization kit (Pierce, 0.2 mg of purified β-catenin/ml of Sepharose beads). Twenty microliters of a 1:1 slurry of immobilized β-catenin (β-catenin-coated beads) was incubated for 15 hr at 4°C with 500 μg of whole cell extracts of C57MG cells treated or not treated with Wnt-3A-conditioned medium and treated or not treated with 25 mm LiCl. The beads were washed three times with lysis buffer, boiled in loading dye, and resolved by SDS-PAGE.
Modification of GST–Axin and β-catenin precipitation
GST–Axin protein was expressed in E. coli strain BL-21 (Stratagene) and purified with glutathione–Sepharose CL4B beads. Twenty-five microliters of a 1:1 slurry carrying ∼1 μg of GST–Axin/glutathione–Sepharose complexes were washed extensively with lysis buffer and then treated as follows: (1) washed once with kinase buffer (50 mm Tris-HCl, 10 mm MgCl2, 0.2 mm ATP, 1 mm DTT at pH 8), and incubated in the presence or absence of 5 units of GSK-3β (New England Biolabs) in 50 μl total volume of kinase buffer for 1 hr at 37°C; and (2) washed three times with lysis buffer, once with phosphatase buffer (50 mm Tris-HCl, 0.1% β-mercaptoethanol, 0.1 mg/ml BSA at pH 7.5) and incubated in the presence or absence of 1.0 unit of PP2A in 50 μl total volume of phosphatase buffer for 6 hr at 30°C. These modified GST–Axin proteins were then incubated for 15 hr at 4°C with 400 μg of total lysate prepared from L cells treated for 2 hr with Wnt-3A-conditioned medium or purified β-catenin protein (30 ng in 200 μl). The beads were washed four times with lysis buffer, boiled in loading dye, and resolved by SDS-PAGE.
Acknowledgments
We are grateful to William Weis, James Nelson, James Ferrell, Catriona Logan, and Rob Howes for critically reading the manuscript. The L cells producing Wnt-3A protein were kindly provided by Dr. S. Takada. This work was supported by the Howard Hughes Medical Institute (K.W. and R.N) and in part by The Mochida Memorial Foundation for Medical and Pharmaceutical Research (S.S).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rnusse@cmgm.stanford.edu; FAX (650) 723-1399.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. β-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3β. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Brown JD, Moon RT. Wnt signaling: Why is everything so negative? Curr Opin Cell Biol. 1998;10:182–187. doi: 10.1016/s0955-0674(98)80140-3. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R. Wnt signaling: A common theme in animal development. Genes & Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Cook D, Fry MJ, Hughes K, Sumathipala R, Woodgett JR, Dale TC. Wingless inactivates glycogen synthase kinase-3 via an intracellular signalling pathway which involves a protein kinase c. EMBO J. 1996;15:4526–4536. [PMC free article] [PubMed] [Google Scholar]
- Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P. Downregulation of β-catenin by human Axin and its association with the APC tumor suppressor, β-catenin and GSK-3β. Curr Biol. 1998;8:573–581. doi: 10.1016/s0960-9822(98)70226-x. [DOI] [PubMed] [Google Scholar]
- Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VMY, Klein PS. Activation of the wnt signaling pathway: A molecular mechanism for lithium action. Dev Biol. 1997;185:82–91. doi: 10.1006/dbio.1997.8552. [DOI] [PubMed] [Google Scholar]
- Hsu W, Zeng L, Costantini F. Identification of a domain of Axin that binds to the serine/threonine protein phosphatase 2A and a self-binding domain. J Biol Chem. 1999;274:3439–3445. doi: 10.1074/jbc.274.6.3439. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Krupnik VE, Sokol SY. Axis determination in Xenopus involves biochemical interactions of axin, glycogen synthase kinase 3 and β-catenin. Curr Biol. 1998;8:591–594. doi: 10.1016/s0960-9822(98)70229-5. [DOI] [PubMed] [Google Scholar]
- Kishida M, Koyama S, Kishida S, Matsubara K, Nakashima S, Higano K, Takada R, Takada S, Kikuchi A. Axin prevents Wnt-3a-induced accumulation of β-catenin. Oncogene. 1999;18:979–985. doi: 10.1038/sj.onc.1202388. [DOI] [PubMed] [Google Scholar]
- Kishida S, Yamamoto H, Ikeda S, Kishida M, Sakamoto I, Koyama S, Kikuchi A. Axin, a negative regulator of the wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of β-catenin. J Biol Chem. 1998;273:10823–10826. doi: 10.1074/jbc.273.18.10823. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Willert K, Molenaar M, Roose J, Wagenaar G, Markman M, Lamers W, Destree O, Clevers H. Two members of the Tcf family implicated in Wnt/β-catenin signaling during embryogenesis in the mouse. Mol Cell Biol. 1998;18:1248–1256. doi: 10.1128/mcb.18.3.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T. A ubiquitin ligase complex essential for the NF-κB, Wnt/Wingless, and Hedgehog signaling pathways. Genes & Dev. 1999;13:505–510. doi: 10.1101/gad.13.5.505. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Hamada F, Ishidate T, Anai K, Kawahara K, Toyoshima K, Akiyama T. Axin, an inhibitor of the Wnt signalling pathway, interacts with β-catenin, GSK-3β and APC and reduces the β-catenin level. Genes Cells. 1998;3:395–403. doi: 10.1046/j.1365-2443.1998.00198.x. [DOI] [PubMed] [Google Scholar]
- Polakis P. The oncogenic activation of β-catenin. Curr Opin Genet Dev. 1999;9:15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK-3β to the APC–β-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- Sakanaka C, Weiss JB, Williams LT. Bridging of β-catenin and glycogen synthase kinase-3β by axin and inhibition of β-catenin-mediated transcription. Proc Natl Acad Sci. 1998;95:3020–3023. doi: 10.1073/pnas.95.6.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeling JM, Miller JR, Gil R, Moon RT, White R, Virshup DM. Regulation of β-catenin signaling by the B56 subunit of protein phosphatase 2A. Science. 1999;283:2089–2091. doi: 10.1126/science.283.5410.2089. [DOI] [PubMed] [Google Scholar]
- Shibamoto S, Higano K, Takada R, Ito F, Takeichi M, Takada S. Cytoskeletal reorganization by soluble Wnt-3A protein signalling. Genes Cells. 1998;3:659–670. doi: 10.1046/j.1365-2443.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics Wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Wodarz A, Nusse R. Mechanisms of Wnt signalling in development. Annu Rev Cell Biol. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Kishida S, Uochi T, Ikeda S, Koyama S, Asashima M, Kikuchi A. Axil, a member of the Axin family, interacts with both glycogen synthase kinase-3β and β-catenin and inhibits axis formation of Xenopus embryos. Mol Cell Biol. 1998;18:2867–2875. doi: 10.1128/mcb.18.5.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, Kikuchi A. Phosphorylation of Axin, a Wnt signal negative regulator, by glycogen synthase kinase-3β regulates its stability. J Biol Chem. 1999;274:10681–10684. doi: 10.1074/jbc.274.16.10681. [DOI] [PubMed] [Google Scholar]
- Yost C, Torres M, Miller J, Huang E, Kimelman D, Moon R. The axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes & Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL, III, Lee JJ, Tilghman SM, Gumbiner BM, Costantini F. The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell. 1997;90:181–192. doi: 10.1016/s0092-8674(00)80324-4. [DOI] [PubMed] [Google Scholar]