

Abstract
AU-rich RNA-destabilizing elements (AREs) have become a paradigm for studying cytoplasmic mRNA turnover in mammalian cells. Though many RNA-binding proteins have been shown to bind to AREs in vitro, trans-acting factors that participate in the in vivo destabilization of cytoplasmic RNA by AREs remains unknown. Experiments were performed to investigate the cellular mechanisms and to identify potential trans-acting factors for ARE-directed mRNA decay. These experiments identified hnRNP D, a heterogeneous nuclear ribonucleoprotein (hnRNP) capable of shuttling between the nucleus and cytoplasm, as an RNA destabilizing protein in vivo in ARE-mediated rapid mRNA decay. Our results show that the ARE destabilizing function is dramatically impeded during hemin-induced erythroid differentiation and not in TPA-induced megakaryocytic differentiation of human erythroleukemic K562 cells. A sequestration of hnRNP D into a hemin-induced protein complex, termed hemin-regulated factor or HRF, correlates well with the loss of ARE-destabilizing function in the cytoplasm. Further experiments show that in hemin-treated cells, ectopic expression of hnRNP D restores the rapid decay directed by the ARE. The extent of destabilizing effect varies among the four isoforms of hnRNP D, with p37 and p42 displaying the most profound effect. These results demonstrate a specific cytoplasmic function for hnRNP D as an RNA-destabilizing protein in ARE-mediated decay pathway. These in vivo findings support an emerging idea that shuttling hnRNP proteins have not only a nuclear but also a cytoplasmic function in mRNA metabolism. The data further imply that shuttling hnRNP proteins define, at least in part, the nuclear history of individual mRNAs and thereby influence their cytoplasmic fate.
Keywords: AU-rich element, hnRNP protein, mRNA turnover, tetracycline-regulatory system, hematopoietic differentiation
AU-rich RNA destabilizing elements (AREs) are found in mRNAs encoding proteins with diversified functions and synthesized under a vast variety of physiological conditions (Chen and Shyu 1995), suggesting that AREs are a key player in controlling gene expression post-transcriptionally. The potent RNA-destabilizing ability of AREs coupled with transient transcription of the corresponding genes is a prerequisite for achieving a tight temporal and spatial regulation of a transient expression of mRNA (Treisman 1985; Schiavi et al. 1992; Ross 1995). Whereas AREs are found in many different labile mRNAs, they are most commonly found in the cytokine mRNAs whose half-lives change in cells undergoing a stress response, an immune response, and responding to tissue repair (Caput et al. 1986; Shaw and Kamen 1986; Greenberg and Belasco 1993; Chen and Shyu 1995). It is now clear that the regulation of cytoplasmic mRNA turnover plays a critical role in determining the duration and level of expression of many cytokines. Recently, much has been learned concerning the key sequence features of AREs that are necessary for exerting their destabilizing function (Chen and Shyu 1995; Xu et al. 1997). Increasing reports have also been made on how alterations of certain signaling transduction pathways in lymphoid or myeloid cell lines lead to changes of the stability of cytokine mRNAs via mechanisms that require AREs, for example, IL-2, IL-3, and IL-8 mRNAs (Sirenko et al. 1997; Chen et al. 1998; Ming et al. 1998; R. Winzen, M. Kracht, B. Ritten, A. Wilhelm, C.-Y.A. Chen, A.-B. Shyu, M. Muller, M. Gaestel, K. Resch, and H. Holtmann, in prep.). However, relatively little is known concerning the trans-acting factors that participate in or modulate the ARE-directed rapid mRNA turnover in vivo.
By use of in vitro assays, for example, RNA gel mobility shift and UV cross-linking, many protein factors that are capable of forming an RNA–protein complex with an ARE have been described previously. These include AUF1 (Zhang et al. 1993), 3-oxoacyl-CoA thiolase (Nanbu et al. 1993), glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) (Nagy and Rigby 1995), hnRNP A1 (Hamilton et al. 1993), hnRNP C (Hamilton et al. 1993), AUH with enoyl-CoA hydratase activity (Nakagawa et al. 1995), and the ELAV family of RNA-binding proteins (for review, see Antic and Keene 1997). However, the functional consequences and the physiological significance of the observed in vitro RNA–protein interactions in vivo remain largely unknown. Recent in vivo and in vitro evidence has shown that the ELAV family of proteins, in particular HuR, have the ability to inactivate the RNA-destabilizing function of AREs (Jain et al. 1997; Fan and Steitz 1998; Levy et al. 1998; Peng et al. 1998; Ford et al. 1999). Nevertheless, trans-acting factors that participate in mRNA destabilization by AREs in vivo have not been identified, although tristetraprolin (TTP) has been suggested recently to play such a role in the turnover of TNFα mRNA, which bears a potent ARE in its 3′ UTR (untranslated region) (Carballo et al. 1998).
Among all of the ARE-binding proteins, AUF1 is of particular interest because there is a large body of correlative evidence for a functional role of AUF1 in ARE-mediated decay (Buzby et al. 1996; DeMaria and Brewer 1996; Pende et al. 1996; Lafon et al. 1998). It was originally copurified with the fractions of a protein purification that display ARE-binding activity and enhance in vitro decay of c-myc mRNA with cell lysates made from human erythroleukemia K562 cells (Brewer and Ross 1989; Brewer 1991). Nonetheless, it remains to be proven that AUF1 has an in vivo function in the ARE-mediated mRNA decay, especially given the observation that AUF1 is mainly present in the nucleus. Following the cDNA cloning for AUF1, it was realized that the AUF1 is a known member of hnRNP proteins, namely, the hnRNP D (Zhang et al. 1993; Kajita et al. 1995). Subsequent cloning and characterization of the genomic clones indicate that hnRNP D/AUF1 (hnRNP D, thereof) gene is transcribed into a pre-mRNA that undergoes alternative pre-mRNA splicing to give rise to four different protein isoforms with apparent molecular masses of 37, 40, 42, and 45 kD (Wagner et al. 1998). The observation that hnRNP D may be able to shuttle between the nucleus and cytoplasm suggests that it has a function in the cytoplasm (Piñol-Roma and Dreyfuss 1991, 1992; Dreyfuss et al. 1993).
As an effort to identify the destabilizing protein factor(s) and further delineate how ARE function may be regulated, we have chosen to investigate whether and how ARE function may be controlled during hematopoietic differentiation with a human K562 erythroleukemic cell line as a model system. Proliferating K562 cells can undergo megakaryocytic differentiation when stimulated with the phorbol ester (TPA), whereas hemin promotes erythroid differentiation of these cells (Rutherford et al. 1979, 1981; Alitalo 1990). To begin to investigate the regulation of ARE-mediated mRNA decay in K562 cells undergoing cell differentiation, we have developed recently a new transcriptional pulsing strategy by introducing a tetracycline-based promoter system, the so-called Tet-off system, into the K562 cell line (Xu et al. 1998). By use of this new strategy, it is possible to carry out time-course experiments to monitor kinetics of mRNA decay under a vast variety of physiological conditions of cells without using transcription inhibitors (Loflin et al. 1999).
Here, we report that hnRNP D can function in vivo as an RNA-destabilizing factor in the ARE-mediated decay pathway. We show that ectopic expression of the p37 and p42 isoforms releases the inhibition of ARE-mediated rapid decay of cytoplasmic RNA as a result of hemin-induced erythroid differentiation of K562 cells, whereas the p40 and p45 partially restore the rapid decay of the ARE-containing transcripts. Thus, different isoforms of hnRNP D appear to display differential destabilizing effects. The hemin-induced RNA stabilization effect is likely specific for hemin-induced erythroid differentiation as the phorbol ester, TPA, that induces megakaryocytic differentiation of K562 cells, but does not elicit any significant change in ARE function. Our results suggest that the stabilization of ARE-containing mRNA by hemin is accomplished via a mechanism that causes the specific assembly of a protein complex, which we have termed hemin-regulated factor or HRF involving hnRNP D proteins, on the ARE.
Results
Experimental approach
In the past we have used the serum-inducible c-fos promoter system and NIH-3T3 cells to study the ARE-mediated mRNA turnover (Chen et al. 1994; Chen and Shyu 1994; Shyu et al. 1996). However, the c-fos promoter is not able to respond to serum stimulation effectively for a transient burst of mRNA synthesis in K562 cells (unpublished observation). In an effort to address how ARE-destabilizing function may be modulated in proliferating K562 cells or in K562 cells induced to differentiate, we used a new transcriptional pulsing strategy that used the Tet-regulatory promoter system, to monitor kinetics or rates of mRNA turnover (Xu et al. 1998; Loflin et al. 1999). Briefly, a stable K562 cell transfectant, designated K562 III-2, was established that constitutively expressed tTA, a trans-activator that, in the absence of tetracycline, recognizes and activates transcription from genes bearing the tetracycline-regulatory promoter. Prior to the test of candidate AREs for their ability to function as RNA-destabilizing elements in K562 cells, we first characterized the decay of a reporter transcript, the rabbit β-globin mRNA, whose expression is under the control of the Tet-regulatory promoter. These experiments were performed in proliferating cells and in cells undergoing erythroid differentiation by hemin or megakaryocytic differentiation by TPA.
Following Tet-controlled transcriptional pulse, a transient burst of the reporter mRNA synthesis from the Tet-regulatory promoter was achieved. The results demonstrated that neither hemin nor TPA treatment of K562 cells changes the stability of β-globin mRNA (Fig. 1A). β-Globin mRNA appeared in the cytoplasm as a tight band and remained stable for the first 16 hr in all three cell states. During the 16-hr period the poly(A) tail was gradually shortened to around 60 nucleotides in length before the β-globin mRNA was quickly degraded. Poly(A) shortening appeared to precede the decay of the RNA body, supporting that shortening of the poly(A) tail is a prerequisite for degrading the stable β-globin mRNA in K562 cells. The anticipated effects of hemin and TPA on cell differentiation were confirmed by hybridizing RNA blots for respective transferrin receptor (TfR) and for PDGF β mRNA expression (Alitalo et al. 1987, 1988; Miyamoto et al. 1990). The results showed that hemin down-regulated TfR mRNA expression and TPA induced the megakaryocyte-related expression of PDGF β mRNA (data not shown). Taken together, these experiments demonstrated that our model system for controlling the differentiation and transcriptional states of K562 cells was sufficient for studying the in vivo mechanism and regulation of mRNA decay during hematopoietic differentiation.
Figure 1.
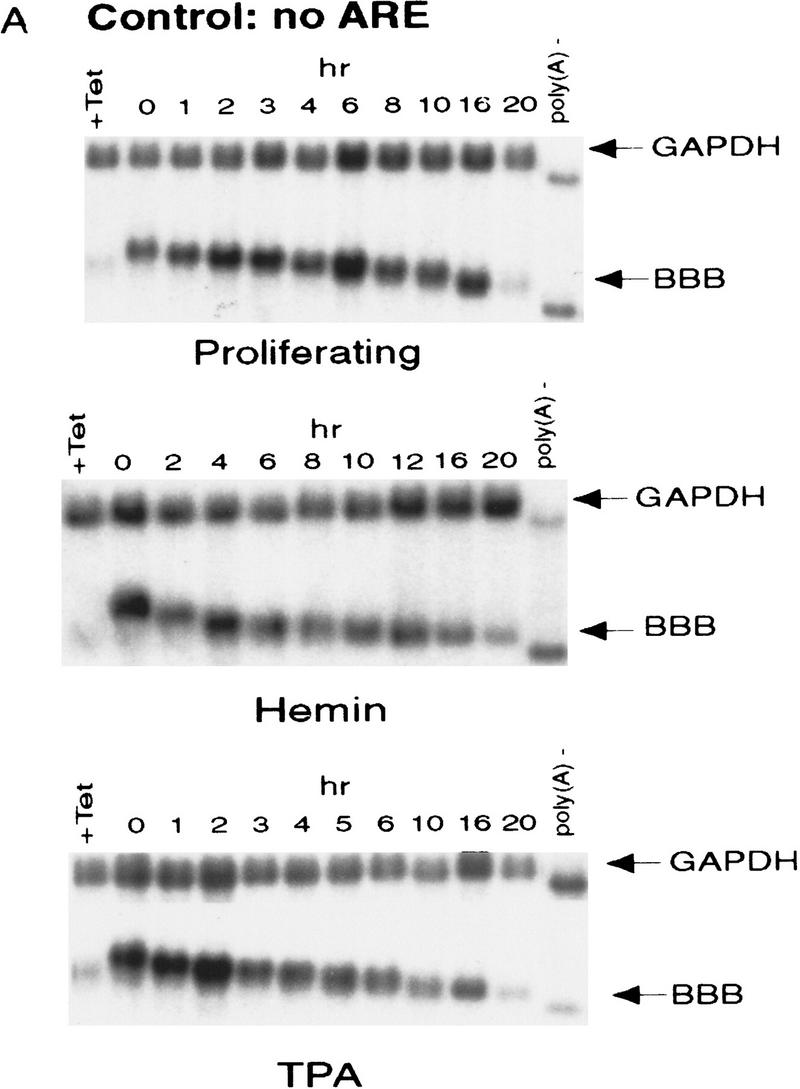
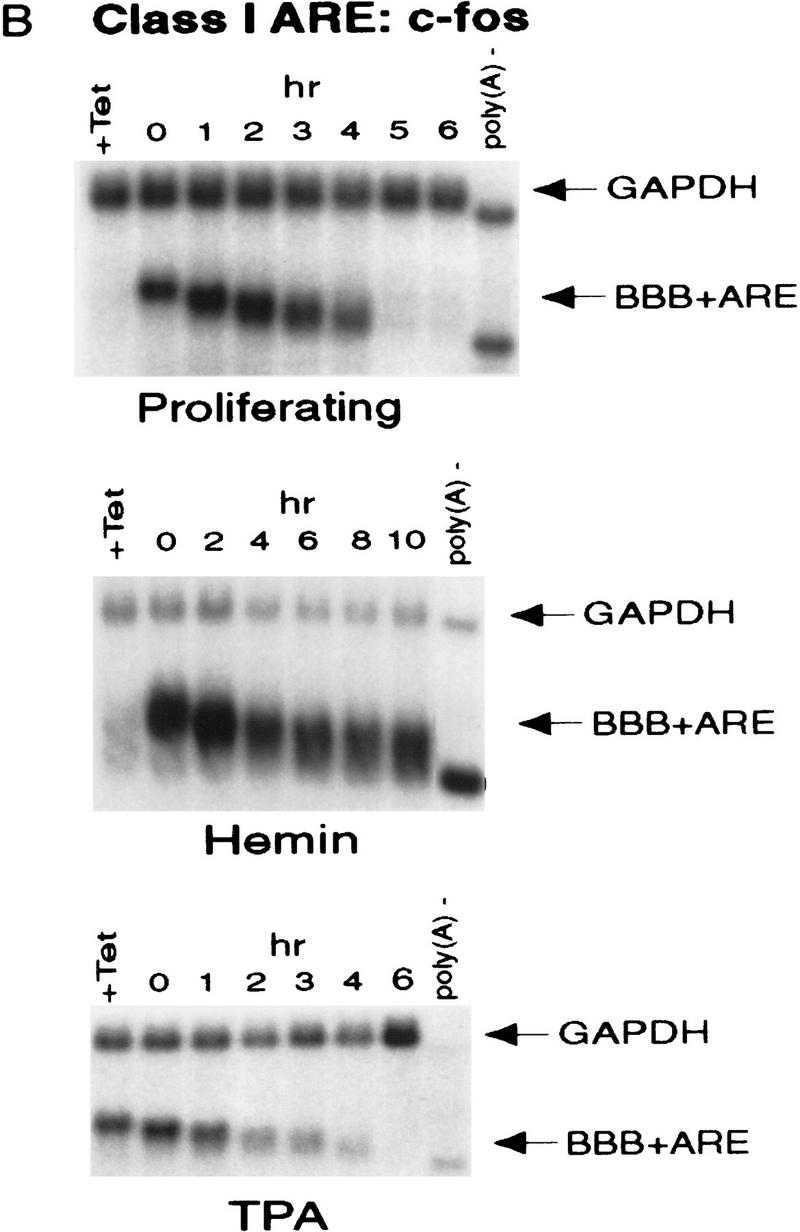
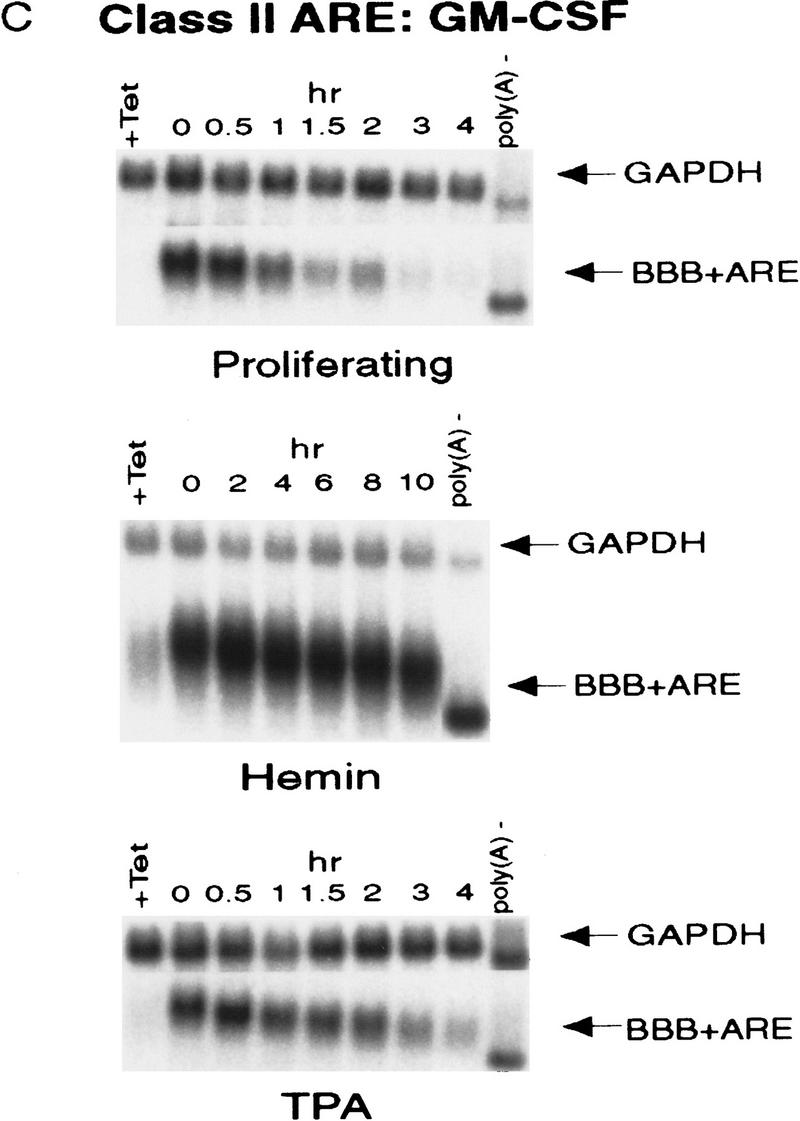
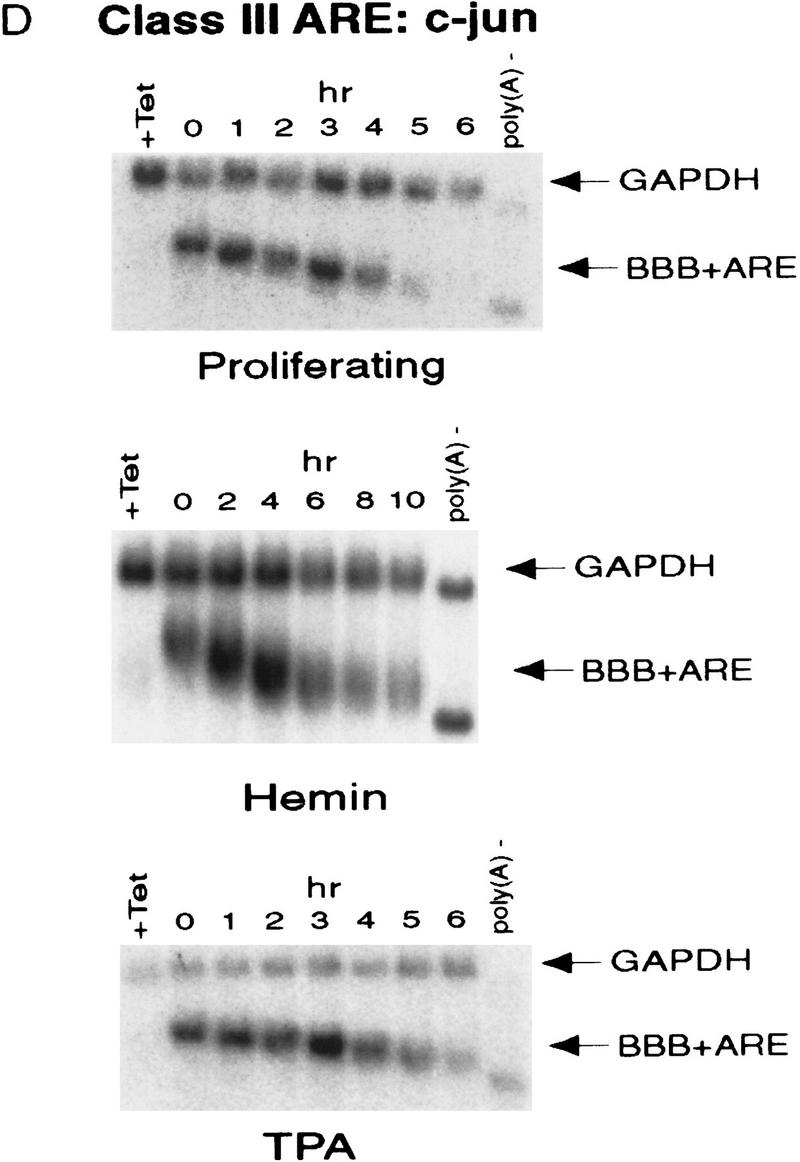
ARE-mediated RNA destabilization in proliferating K562 cells is inhibited during hemin-induced erythroid differentiation. K562 III-2 cells were electroporated with pTetBBB (A) or pTetBBB+ARE bearing various 3′-UTR ARE’s (BD). Cells were kept in medium with 45 ng/ml tetracycline for 22 hr followed by treatment with no drug (proliferating), with 50 μm hemin (erythrocytic differentiation), or with 20 nm TPA (megakaryocytic differentiation) for an additional 24 hr in the presence of 45 ng/ml of tetracycline (+Tet). After transcriptional pulsing (see Materials and Methods), cytoplasmic RNA was isolated immediately for the zero time point or 500 ng/ml Tet was added for the various time intervals as indicated before cytoplasmic RNA was extracted. RNA samples were analyzed by Northern blotting. Poly(A)− RNA was prepared in vitro by treating the zero time point sample with oligo(dT) and RNase H. (BBB) β-Globin mRNA; (BBB+ARE) β-globin mRNA bearing an ARE; (GAPDH) glyceraldehyde-3-phosphate dehydrogenase mRNA served as an internal control.
Stabilization of ARE-containing mRNAs during hemin-induced erythroid differentiation
Next, we set out to address whether the RNA-destabilizing function of all three classes of AREs (I, II, and III) observed in proliferating cells are modulated in hemin-induced erythrocytic differentiation and/or in TPA-induced megakaryocytic differentiation of K562 cells. We first examined the decay of the reporter β-globin mRNA bearing an ARE in K562-III cells undergoing erythroid differentiation. The β-globin mRNA bearing one of the following ARE representatives (pTet-BBB+ARE) was tested individually, including c-fos and c-myc AREs for class I, GM-CSF, and TNFα AREs for class II, and c-jun ARE for class III (Chen and Shyu 1995). Transiently transfected cells were treated with hemin for 24 hr to induce erythroid differentiation. A short burst of mRNA synthesis was then induced by manipulating Tet concentrations in the medium, and the time course experiments were performed to measure mRNA deadenylation and decay. Remarkably, there was a dramatic stabilization of transcripts bearing all three classes of AREs in hemin-treated cells when compared with proliferating cells (Figs. 1 and 2). mRNAs bearing class I and class II AREs remained at the same level over the time course, whereas the class III-containing mRNA decreased slightly by approximately twofold after 10 hr. It appeared that the deadenylation step was not affected. Instead, hemin impaired the decay of the RNA body following poly(A) shortening. In contrast to the hemin treatment, TPA treatment for 24 hr did not cause any significant change of the stability of mRNAs bearing all three classes of AREs. All three classes of AREs remained capable of directing rapid degradation of β-globin mRNA, although the transcript bearing class II AREs (GM-CSF and TNF-α) were slightly retarded compared with proliferating cells (Figs. 1 and 2). These results indicate that the pathway for ARE-mediated mRNA decay in K562 cells is impeded by hemin treatment.
Figure 2.




Kinetic analysis of the decay of β-globin mRNA with or without an ARE. Northern blot analysis depicted in the legend to Fig. 1 was analyzed on a digital scanner. The corresponding signals for β-globin mRNA were normalized against the corresponding signals observed for GAPDH. The identities of the AREs tested are indicated at the top of each graph. Symbols correspond to the following treatments: (open oval) proliferating; (solid triangle) 20 nm TPA; (solid oval) 50 μm hemin.
Formation of a specific cytoplasmic ARE/protein super complex in response to hemin-induced erythroid differentiation
To begin to elucidate the mechanism underlying this dramatic stabilization of transcripts bearing AREs by hemin, we sought to identify trans-acting factor(s) that may be involved. We chose to focus on the c-fos ARE in subsequent experiments because it was the best-characterized ARE. Gel mobility shift assays were carried out using 32P-labeled c-fos ARE as the RNA substrate. c-fos ARE probe was incubated with cytoplasmic lysates individually prepared from proliferating, hemin-treated, or TPA-treated K562 cells. Figure 3A showed that when proliferating lysate was used, two major and several minor RNA–protein complexes were detected. However, when cytoplasmic lysate prepared from hemin-treated cells was used, a significant reduction of nearly all of the complexes was observed, which concomitantly resulted in the appearance of a slow-migrating super complex. In contrast, the patterns of mobility shift were nearly identical between proliferating cells and TPA-treated cells with a couple of minor bands loosing their intensity in TPA-treated cells. To demonstrate that formation of the hemin-induced complex was ARE-specific, competition experiments were performed. Figure 3B showed that increasing amounts of unlabeled homologous c-fos ARE could specifically and readily abolish the formation of hemin-induced super complex, whereas increasing amounts of the nonspecific unlabeled β-globin RNA had little effect. Together, these experiments suggest the existence of a functional correlation between hemin-induced inactivation of ARE-mediated decay and the formation of a hemin-induced ARE–protein super complex.
Figure 3.

Formation of a specific cytoplasmic ARE/protein super complex in response to hemin-induced erythroid differentiation. (A) Cytoplasmic lysates prepared from proliferating cells (Pr), from cells treated with 20 nm TPA for 24 hr (TPA), or from cells treated with 50 μm hemin for 24 hr were incubated for 15 min with the fos ARE RNA substrate labeled with [32P]UTP. The samples were then treated with RNase T1 for 20 min and the RNA–protein complexes were separated on a nondenaturing 6% polyacrylamide gel. (B) Competition analysis was carried out by incubating cytoplasmic lysate from hemin-treated cells with [32P]UTP-labeled fos–ARE RNA substrate in the presence of increasing amounts of nonlabeled fos ARE RNA (specific) or a β-globin coding region RNA (nonspecific) at the various molar excess as indicated.
The hemin-induced stabilization of ARE-containing mRNA is time dependent and correlates with the time-dependent formation of the hemin-induced super complex
To seek further evidence to support the aforementioned correlation, we examined the time frame in which hemin was able to induce the super complex formation and whether this complex formation coincided with the loss of ARE-destabilizing function. Cytoplasmic lysates were prepared from either proliferating K562 cells or cells treated with hemin for 3, 6, 9, 12, 16, and 24 hr. In the parallel experiments, decay of BBB+AREfos mRNA was also monitored in K562 cells pretreated with 50 μm hemin for 3, 9, 12, 16, and 24 hr. Gel mobility shift assays showed the first appearance of the super complex at ∼9 hr of hemin treatment with an increase in intensity between 12 and 24 hr of hemin treatment (Fig. 4A). This gradual and time-dependent appearance of the hemin-induced super complex coincided with a time-dependent inactivation of ARE destabilizing function. It took ∼9 hr of hemin treatment to begin to observe some RNA stabilization effect. A nearly complete stabilization effect by hemin was seen when cells were treated with hemin for 16 or 24 hr (Fig. 4B). Therefore, a correlation can be established between the time-dependent formation of the hemin-induced ARE–protein super complex and the time-dependent loss of ARE function. Taken together, the above experiments suggest a potential role for this hemin-induced ARE–protein super complex, which we termed hemin-regulated factor or HRF, in down-regulating ARE destabilizing function.
Figure 4.
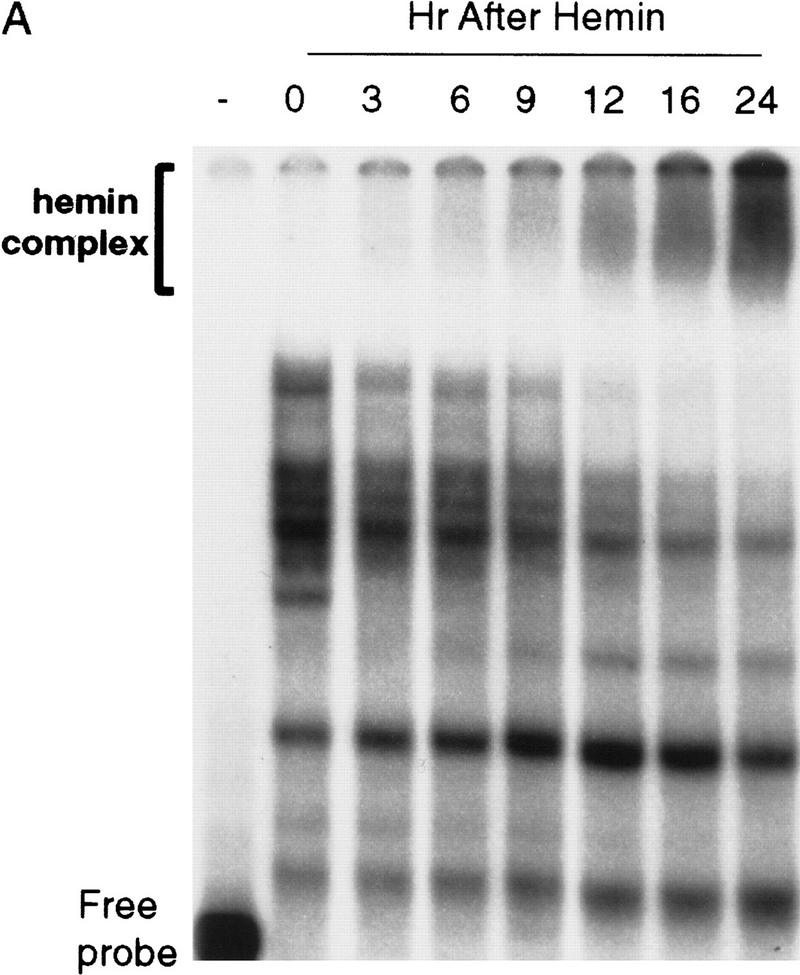

The time frame for hemin-induced super complex formation correlates with that of hemin-induced stabilization of the ARE-containing mRNA. (A) Gel mobility shift analyses of cytoplasmic lysates prepared from K562 cells treated with 50 μm hemin for the various time intervals indicated. Cytoplasmic lysates were incubated with a [32P]UTP-labeled fos–ARE RNA, after which RNase T1 digestion was performed. Complexes were then analyzed by electrophoresis through a 6% polyacrylamide nondenaturing gel. (B) Kinetic analysis of Northern blots preformed on K562 III-2 cells transfected with pTetBBB+AREfos and treated with 50 μm hemin for various time intervals. The results were normalized and plotted as described in the legend to Fig. 2 and are depicted as no hemin (open oval), 3 hr hemin treatment (solid oval), 9 hr hemin treatment (open triangle), 12 hr hemin treatment (solid triangle), 16 hr hemin treatment (open square) and 24 hr hemin treatment (solid square).
hnRNP D is an ARE-binding component in the hemin regulated complex, HRF
Having established a correlation between the appearance of HRF and the RNA stabilization by hemin, we next asked what RNA-binding protein(s) may be responsible for the ARE-binding activity in the HRF. In the past few years, there have been many reports identifying ARE-binding activity in crude cell extracts; at least 11 of them have been either cloned or found to be identical to known gene products (see Introduction). There is a large body of correlative evidence for a potential role of the hnRNP D or HuR in the ARE-mediated decay. Therefore, we have focused our initial effort on addressing whether HuR or hnRNP D participates in the hemin-induced RNA stabilization. Antibodies raised against HuR or hnRNP D were used to perform antibody supershift assays. Antibodies were added in two ways, either before (Fig. 5, Bf) or after (Fig. 5, Af) RNA substrate was mixed with lysate. Experiments with lysates prepared from proliferating cells (Fig. 5) showed that a significant portion of the ARE–protein complexes were supershifted by anti-AUF1 polyclonal antibody and not by preimmune serum. More importantly, the hemin-induced ARE–protein super complex or HRF was further shifted by the anti-AUF1 antiserum but not by preimmune serum, demonstrating the presence of hnRNP D in HRF (Fig. 5). These experiments also demonstrated that hnRNP D was part of normal ARE–protein complexes detected in proliferating lysate and becomes part of the HRF on prolonged hemin treatment. In contrast, similar supershifts were not observed for the anti-HuR antibody, suggesting that HuR was not part of the ARE–protein complexes (Fig. 5).
Figure 5.

hnRNP D is part of ARE–protein complexes detected in proliferating lysate and becomes an integral part of a hemin super complex induced by hemin. Gel mobility shift assays with a [32P]UTP-labeled fos–ARE probe were performed as described in the legend to Fig. 2. Antibodies against hnRNP D/AUF (α-AUF) or HuR (α-HuR) were added in two ways for antibody super-shift assays, either before (Bf) or after (Af) RNA substrate was mixed with lysate. Cytoplasmic lysates from proliferating K562 III-2 cells (left) or cells treated with 50 μm hemin (right) were as indicated. RNA/lysate/antibody mixtures were then analyzed by nondenaturing 6% polyacrylamide gel electrophoresis. (PI) Preimmune serum.
Hemin induces time-dependent changes of relative distribution of hnRNP D isoforms between the nucleus and cytoplasm
Previously, it has been reported that hnRNP D has four distinct isoforms resulting from alternative RNA splicing (Wagner et al. 1998), termed p37, p40, p42, and p45 (Fig. 6A). Therefore, we asked whether there exists an isoform-specific formation of ARE–protein complexes in both proliferating and hemin-treated cells and whether hemin treatment changes relative distribution of these isoforms between the nucleus and cytoplasm. Cytoplasmic and nuclear extracts were prepared from proliferating, TPA-treated, and hemin-treated cells and subjected to Western blot analysis with a monoclonal antibody, 5B9, to a common epitope present in the RNA-binding domain II of all isoforms of hnRNP D (Kajita et al. 1995). The mAb, when used in the gel mobility supershift assays, yielded results similar to those obtained with polyclonal antibody against AUF1 (data not shown). Figure 6B shows the distinct distribution patterns for the different isoforms present in nuclear and cytoplasmic extracts. In proliferating K562 cells, p45 is found nearly exclusively in the cytoplasm, whereas the other three isoforms are found predominantly in the nucleus. In addition, some p40 can be readily detected in the cytoplasm. In K562 cells treated with hemin for 24 hr, there was a significant increase of p40 and p42 in the cytoplasm (Fig. 6B). It appeared that a modest increase of p37 in the cytoplasm was also detected. The weak signal for p37 in the cytoplasm, which has been seen in other studies (Pende et al. 1996; Kiledjian et al. 1997; Sirenko et al. 1997), makes it difficult to assess its relative distribution between the nucleus and cytoplasm. Little change of distribution between the nucleus and cytoplasm for p45 was detected. In contrast to hemin treatment, TPA treatment of K562 cells did not lead to any significant change of isoform distributions. Collectively, these results provided further evidence to support a functional correlation between the hemin-induced RNA stabilization and the appearance of hnRNP D in the cytoplasm.
Figure 6.



Hemin induces changes of the subcellular localization of hnRNP D isoforms. (A) Schematic diagram of the four hnRNP D isoforms. The two RNA-binding domains (RBD1 and RBD2) are depicted as the central two gray-shaded regions. The four proteins are identical except for a 19-amino-acid exon present at the amino terminus of p40 and p45 (denoted by the filled square region) and a carboxy-terminal 49-amino-acid region present in p42 and p45 (represented by the dotted region). (B) Cytoplasmic (C) and nuclear (N) proteins of K562 III-2 cells from either proliferation cells (Prolf), cells treated with 20 nm TPA for 24 hr (TPA), or cells treated with 50 μm hemin for 24 hr (Hemin) were separated on a 12% SDS–polyacrylamide gel and transferred to nitrocellulose membranes for Western blot analysis. The blots were probed with a monoclonal antibody (5B9) for endogenous hnRNP D and a monoclonal antibody for α-tubulin simultaneously followed by chemilumenescent detection with an anti-mouse IgG antibody. Molecular weights of the four hnRNP D isoforms as well as α-tubulin are indicated. (C) The time frame for hemin-induced changes of localization of hnRNP D isoforms was analyzed with lysates from K562 III-2 cells treated with 50 μm Hemin for the various time intervals indicated. Equal amounts of cytoplasmic and nuclear protein were used for all experiments.
To further substantiate this correlation, we asked if the aforementioned changes of distribution among different hnRNP D isoforms coincided with the time-dependent formation of HFR and RNA stabilization described in Figure 4. To address this issue, cytoplasmic and nuclear lysates prepared from K562 cells treated with hemin for 3, 6, 9, 12, 16, and 24 hr were also analyzed by Western blot analysis (Fig. 6C). The results showed that p42 had the most prominent change. It was detected after 9-hr hemin treatment and increased to reach maximal levels by the 24-hr time point. p40 also increased in the cytoplasm after 9-hr hemin treatment with little change afterwards. In addition, p37 started to appear in the cytoplasm at around 9 hr. There was little change of subcellular distribution of p45. Therefore, these experiments show a hemin-induced and time-dependent increase of three hnRNP D isoforms in the cytoplasm, which coincides with the hemin-induced formation of HRF and RNA stabilization. Taken together, the results suggest a functional involvement of hnRNP D in hemin-induced stabilization of ARE-containing mRNAs.
Ectopic expression of hnRNP D in proliferating K562 cells does not cause stabilization of ARE-containing mRNA
The above lines of evidence establish a correlation between hemin-induced RNA stabilization and hemin-induced changes in the intracellular localization of hnRNP D isoforms. To determine whether hnRNP D plays a causal role in regulating the stability of ARE-containing transcripts, we ectopically overexpressed hnRNP D in proliferating or hemin-treated K562 cells, and examined how hnRNP D affect the decay of ARE-containing mRNAs. One possibility is that hnRNP D could have an RNA stabilization role. Hemin might induce redistribution of hnRNP D from the nucleus to the cytoplasm. Subsequent formation of HRF through hnRNP D on the ARE might then prevent the ARE to act as an RNA destabilizer. A second possibility is that that hnRNP D might function as an RNA-destabilizing factor in proliferating cells. In this scenario, hemin might induce the formation of HRF on top of the hnRNP D/ARE complex in the cytoplasm and thus prevent RNA destabilization. As such, hnRNP D becomes accumulated or trapped in the cytoplasm as a result of HRF formation and can no longer exert its normal destabilization function.
The first possibility is not without a precedent. Recently, we and others have reported that ectopic overexpression of HuR, an ARE-binding protein that is able to shuttle between the nucleus and cytoplasm, leads to its accumulation in the cytoplasm, which in turn causes retardation of ARE-mediated mRNA decay (Fan and Steitz 1998; Peng et al. 1998). Therefore, we first examined whether ectopic overexpression of the p42 isoform in proliferating K562 cells could impede the c-fos ARE-mediated mRNA decay as it showed the most prominent change of subcellular distribution among the four isoforms when cells were treated with hemin for 24 hr (Fig. 6B). To distinguish exogenous from endogenous hnRNP D, myc epitope-tagged sequence was introduced on the amino terminus of each of the four isoforms of hnRNP D. Final hybrid cDNAs were then subcloned into a mammalian overexpression vector (Peng et al. 1998) and were cotransfected with the reporter gene. Decay of the BBB+ARE mRNA was then monitored in the presence of ectopically expressed myc-tagged p42 in proliferating K562 cells. As shown in Figure 7A, no significant change of the stability of BBB+AREfos mRNA can be detected when compared with the control. In a separate set of experiments, we further address whether other isoforms may have the ability to impede the ARE-mediated decay in proliferating cells. As shown in Figure 7B (left), neither p42 nor the other three isoforms, when ectopically overexpressed, could retard the rapid decay of BBB mRNA bearing the c-fos ARE. Western blot analysis of the cytoplasmic lysates (Fig. 7B, right) showed that in all cases myc-tagged hnRNP D isoforms can be readily detected. Thus, unlike HuR, hnRNP D does not impede the ARE-mediated mRNA decay in proliferating K562 cells when ectopically expressed (Peng et al. 1998).
Figure 7.


Ectopic expression of hnRNP D in proliferating K562 cells has little effect on decay of the ARE-containing mRNA. Transfection of K562 III-2 cells, RNA extraction, and RNA blot analysis were carried out as described in the legend to Fig. 1. (A) RNA blots showing decay of the β-globin (BBB) mRNA bearing the fos ARE in the presence (left) or absence (right) of the p42 isoforms in proliferating cells. (B) (Left) Graph showing kinetics of decay of the BBB+AREfos mRNA in proliferating cells overexpressing individual isoforms of hnRNP D. Data collection and analysis are as described in Fig. 2. Symbols are depicted as follows: (Solid oval) Vector alone or control; (solid square) p37; (solid diamond) p40; (x) p42; (+) p45. (Right) Western blot analysis for exogenously expressed myc-tagged hnRNP D was performed as described in the legend to Fig. 6. Cytoplasmic lysates were prepared from proliferating cells transfected with cDNAs for individual isoforms of hnRNP D.
HnRNP D functions as an RNA-destabilizing protein in the ARE-mediated mRNA decay
To test the second possibility (see above), the decay of BBB+AREfos mRNA was first examined in hemin-treated cells that ectopically overexpressed the hnRNP D p42. It is possible that ectopically overexpressing p42 in hemin-treated K562 cells could overcome the stabilization effect from HRF. Remarkably, ectopic expression of p42 led to a nearly complete restoration of rapid decay of BBB+AREfos mRNA, whereas decay of BBB+AREfos mRNA in hemin-treated cells with overexpression of vector alone remain impeded (Fig. 8A). To further substantiate this result, decay of BBB+AREGM–CSF mRNA was examined. A similar restoration of the RNA-destabilizing function of GM-CSF ARE in hemin-treated cells was observed (Fig. 8B). From these experiments, we conclude that the hnRNP D p42 isoform has an RNA-destabilizing role in the ARE-mediated decay in the cytoplasm.
Figure 8.



Ectopic expression of hnRNP D in hemin-treated K562 cells restore the rapid decay of ARE-containing transcripts. Transfection of K562III-2 cells, RNA extraction, and RNA blot analysis were carried out as described in the legend to Fig. 1. RNA blots showing decay of the β-globin (BBB) mRNA bearing the fos ARE (A) or the GM-CSF ARE (B) in the presence (left) or absence (right) of the p42 isoforms in K562 cells treated with hemin for 24 hr. (C) (Left) Graph showing kinetics of decay of the BBB+AREfos mRNA in hemin-treated cells overexpressing individual isoforms of hnRNP D. Data collection and analysis are as described in the legend to Fig. 2. Symbols are depicted as follows: (Solid oval) Vector alone; (solid square) p37; (solid diamond) p40; (x) p42; (+) p45. (Right) Western blot analysis for exogenously expressed myc-tagged hnRNP D was performed as described in the legend to Fig. 6. Cytoplasmic lysates were prepared from hemin-treated cells transfected with cDNAs for individual isoforms of hnRNP D.
Next, we asked whether other isoforms were also able to rescue hemin-induced RNA stabilization. The results (Fig. 8C, left) showed that like p42, p37 also exerted a very profound destabilizing effect, whereas p40 and p45 had lesser ability to restore the rapid decay of BBB+AREfos mRNA in hemin-treated cells. The results of Western blot analysis (Fig. 8C, right) showed a readily detectable expression of all myc-tagged isoforms in the cytoplasm. Taken together, these experiments provide further in vivo evidence to substantiate the above conclusion that hnRNP D can function as an RNA-destabilizing protein in the ARE-mediated decay in the cytoplasm. Moreover, they showed that different isoforms vary considerably in their destabilizing effects.
Discussion
In this report we have used a new transcriptional pulsing strategy, which made it possible to monitor kinetics of mRNA decay in human erythroleukemia K562 cells without using transcription inhibitors (Xu et al. 1998; Loflin et al. 1999). Therefore, the new system has offered an opportunity to study the regulation of ARE function during hematopoietic differentiation. We show that when K562 cells were induced by hemin to differentiate down the erythroid lineage, ARE-containing transcripts are dramatically stabilized. In contrast, when proliferating K562 cells were induced by TPA to undergo megakaryocytic differentiation, there was little change of stability of all three classes of ARE-containing transcripts. The stabilization engendered by hemin was not due to a blockade in deadenylation, as shown by our experiments examining poly(A) tail loss after shut-off of transcription. It appears that decay of the body of poly(A) shortened mRNA is profoundly impeded. It is striking that this loss of rapid RNA turnover in hemin-treated cells can be significantly rescued by ectopically overexpressing hnRNP D. This rescue appears to be more profound for p37 and p42, than for p40 or p45 (see below). Our findings provide, to our knowledge, the first in vivo evidence that identifies a destabilizing protein for the ARE-mediated mRNA turnover. Furthermore, they also demonstrate a cytoplasmic role for hnRNP D as a destabilizing protein, thus extending the roles played by hnRNP proteins from the nucleus to the cytoplasm.
Diversified roles and functions in RNA biogenesis and transcription have been described for hnRNP proteins in the nucleus (Dreyfuss et al. 1993, 1996). The findings that hnRNP proteins, including hnRNP A1, D, and K, shuttle between the nucleus and the cytoplasm have raised the possibility that shuttling hnRNP proteins may be involved in the export of mature mRNAs out of the nucleus with shuttling signals like the M9 or the KNS (Weighardt et al. 1996; Izaurralde et al. 1997; Michael et al. 1997; Siomi et al. 1997). Moreover, they also have led to the idea that RNA-binding proteins that initially bind RNA in the nucleus, such as shuttling hnRNP proteins, can influence the fate of mRNA in the cytoplasm (Piñol-Roma and Dreyfuss 1992; Dreyfuss et al. 1996). Several recent reports have lent support for such cytoplasmic functions for hnRNP proteins. For example, hnRNP K has been shown to be part of a cytoplasmic complex that is formed on a 3′ UTR element termed DICE of 15-lipoxygenase mRNA and regulates its translation during erythroid differentiation (Ostareck et al. 1997). HnRNP L and I both have been implicated in the control of cap-independent translation of various RNAs bearing an internal ribosomal entry site (Svitkin et al. 1996; Visa et al. 1996; Hahm et al. 1998). HnRNP A1, which is able to interact with AU-rich sequences, has been shown to be associated with cytoplasmic poly(A)+ mRNAs and is found to be present abundantly in the cytoplasm of human T lymphocytes (Hamilton et al. 1997). HnRNP D has been shown recently to be an integral component of a cytoplasmic complex, termed mRNA-stabilizing α-complex that is formed on a CU-rich region in the 3′ UTR of the α-globin mRNA (Kiledjian et al. 1991). Although the α-complex is necessary for maintaining the stability of α-globin mRNA in the cytoplasm during erythroid differentiation (Kiledjian et al. 1995; Wang and Liebhaber 1996), the role played by hnRNP D remains unclear. It will be important to test directly whether and how hnRNP D may participate in the regulation of α-globin mRNA stability in vivo. Nevertheless, our study identifies a new role for shuttling hnRNP proteins in the cytoplasm as an RNA-destabilizing protein.
How do cells achieve differential regulation of ARE-destabilizing function by different isoforms of hnRNP D? Our results show that p37 and p42 are more capable of releasing hemin-induced stabilization of transcripts bearing an ARE than p40 and p45 (Fig. 8). These properties correlate with the in vitro RNA-binding affinities displayed by the four isoforms, p37>p42>p45>p40 (DeMaria and Brewer 1996; DeMaria et al. 1997). Recent studies showed that several distinct structural determinants in hnRNP D are required for high-affinity binding to AREs. HnRNP D and A1 share a common structural backbone that consists of two canonical RRMs followed by carboxy-terminal RGG motifs (Zhang et al. 1993; Kajita et al. 1995). Although the two RRMs are necessary for RNA binding, they are not sufficient for high affinity for AREs (DeMaria et al. 1997). Both an alanine-rich region of the amino terminus and a short glutamine-rich region in the carboxyl terminus are required to achieve the highest affinity binding of hnRNP D to an ARE. It is interesting to note that unlike p40 and p45, both p37 and p42 lack a small peptide (19 amino acids) insertion at the amino terminus that may have interfered with the participation of the amino-terminal alanine-rich region in the ARE binding (DeMaria and Brewer 1996; DeMaria et al. 1997). This may explain the diminished affinity of p40 and p45 for AREs. Another possibility is that each isoform may elicit interaction with distinct proteins to form different complexes that help to fulfill multiple roles in the cytoplasm, for example, in the ARE-mediated decay or in the stabilization of α-globin mRNA.
Given the observations that the carboxy-terminal RGG-containing domain of hnRNP can be methylated, glycosylated, and phosphorylated (e.g., Soulard et al. 1993; Pype et al. 1994; Bosser et al. 1995; Shen et al. 1998; Valentini et al. 1999), one could imagine that through various extents of post-translational modifications among the isoforms of hnRNP D, a change of isoforms-binding specificity and affinity for a given substrate may be achieved. Therefore, differential post-translational modifications of different isoforms of hnRNP D could offer more versatility to its function. For example, they can enhance, reduce, or eliminate the binding specificity or affinity for AREs by altering the nucleocytoplasmic distribution of hnRNP D or by altering their interactions with auxiliary protein factors that are necessary for substrate recognition and binding. Whereas the exact relationship between post-translational modifications and changes of functionality of hnRNP D isoforms await further experimentation, several studies suggest the existence of such a functional correlation. It is clear that shuttling hnRNP proteins, including A1, D, F/H and K, exhibit differential distribution in a cell-type dependent and tissue-specific manner (Faura et al. 1995; Kamma et al. 1995). Moreover, the distribution pattern of an individual hnRNP protein varies during proliferation or differentiation. It has been shown that arginine methylation of RGG motifs facilitates the nuclear export of hnRNP proteins (Shen et al. 1998). Therefore, it will be interesting to determine the functional consequence in the ARE-mediated decay by mutating these sites in hnRNP D, which are potential targets for post-translational modifications.
One of the critical issues remaining to be addressed is the nature of the hemin-induced mRNA stabilization in K562 cells. The lack of any immediate stabilization effect by hemin suggests the RNA stabilization is related to the prolonged treatment of K562 cells with hemin. The time-frame for hemin to exhibit profound RNA-stabilization effect coincides with the time required for differentiation of K562 cells further toward erythrocytes (Baliga et al. 1993; Nakajima et al. 1997). In addition, hemin is known to induce stress response in K562 cells that leads to some significant changes of gene expression (Theodorakis et al. 1989; Sistonen et al. 1992). These observations suggest that hemin-induced RNA stabilization may be related to erythroid differentiation, physiological changes as a result of prolonged stress, or both. Preliminary experiments with various pharmacological drugs that are capable of inducing further erythroid differentiation do not show the mRNA stabilization effect (unpublished data). Several lines of evidence we have gathered indicate that the hemin-induced stabilization we described here may be a consequence of stress response induced by prolonged hemin treatment (unpublished data). We have identified, along with hnRNP D, heat-shock proteins in HRF, and have shown that a reductant, when added to the culture medium, can abrogate the stabilization effect by hemin. If the hemin-induced stabilization is related to stress response, the questions become if stress-activated signaling pathways, for example, p38 map kinase and JUK kinase pathways, might be involved and if hemin as a whole or iron or protoporphyrin alone is sufficient for the effect. It is interesting to note that several recent reports have shown that either signal pathway can regulate the ARE-destabilizing function, depending on the systems and the nature of stress evoked (Sirenko et al. 1997; Chen et al. 1998; Ming et al. 1998).
Finally, how may hnRNP D be involved in ARE-mediated mRNA destabilization in proliferating cells and how does hemin abolish its function? It is possible that hnRNP D may be first assembled on the ARE while the message is still in the nucleus. Once mature ARE-bearing mRNP is formed, hnRNP D may then function to escort this ARE-containing mRNP complex to the cytoplasm (Müller et al. 1992). On reaching the cytoplasm, hnRNP D may gain new functions. For example, it may direct the mRNA to a specific location, release it for degradation, and then return to the nucleus. The other possibility is that it may be required to elicit an assembly of a decay complex, through protein–protein interactions, on the ARE for RNA degradation. In this latter scenario, hnRNP D may play an active and direct role in RNA decay. Once RNA is degraded, it is released from the complex and then returned to the nucleus. We further hypothesize that when a cell is treated with hemin, it induces an assembly of the HRF on the ARE through interaction with hnRNP D in the cytoplasm and thus interferes with the RNA destabilizing function of hnRNP D, leading to mRNA stabilization. This is consistent with our observations that hnRNP D is part of the HRF and that hnRNP D still retains its ARE-binding ability when associated with other factors to form HRF (Fig. 5). Because ectopic expression of hnRNP D alone is sufficient to release the hemin-induced RNA stabilization, it is tempting to speculate that excess and free hnRNP D in the cytoplasm compete with the HRF and replace its binding at the ARE to resume rapid mRNA decay by AREs. It is interesting to note that deadenylation rates appear not to be affected in hemin-induced cells. Given the observations that in ARE-mediated decays deadenylation always precedes the decay of the RNA body, it is possible that the hemin-induced complex does not abrogate the stimulatory effect of the ARE on deadenylation but does impede its stimulatory effect on the decay of the RNA body following poly(A) removal. This would imply that hnRNP D participates in the second step of the ARE-mediated mRNA decay.
In summary, our studies have identified the first destabilizing protein in the ARE-mediated decay pathway in vivo. The system we describe here will offer an opportunity to conduct in vivo dissection and characterization of structural features of hnRNP D proteins that are functionally important for the ARE-mediated decay pathway and more importantly its regulation. An elucidation of a potential role of stress-activated signaling pathways in this case may provide further insight into mechanisms underlying the regulation of ARE function during cell growth, differentiation, and immune response. Moreover, the formation of HRF and its correlation with the loss of ARE-destabilizing ability suggests an experimental basis for identifying other protein factors that also participate in the ARE-mediated mRNA decay in vivo. Characterization of the protein composition of HRF should shed light on this process. Given that a wide variety of changes in physiological conditions lead to changes of the stability of transcripts bearing an ARE, our studies suggest that hnRNP D has a general and key role in mediating or manifesting those physiological responses via AREs at the level of message stability.
Materials and methods
Plasmid constructions
When necessary, DNA with 5′- or 3′-protruding ends was treated with Klenow fragment or T4 DNA polymerase to make ends blunt. The construction of plasmids pTet-BBB and pTet-BBB+ARE has been described previously (Xu et al. 1998). Briefly, plasmid pBBB(Stu) was created by first using site directed mutagenesis to insert a StuI site at a position immediately upstream of the transcription start site of rabbit β-globin gene in plasmid pBBB. pBBB(Stu) was then digested with StuI (fill in) and KpnI (fill in) and the fragment containing the β-globin sequence was subcloned between the EcoRI (fill in) and NotI (fill in) sites of plasmid pTet-splice (GIBCO-BRL) to create pTet-BBB. Fragments containing various AREs were synthesized by PCR as described previously (Chen and Shyu 1994; Xu et al. 1997), with various pBBB+ARE plasmids as templates. These ARE fragments were flanked by a BamHI site at the 5′ end and a BglII site at the 3′ end. Following BamHI and BglII digestion, the fragment was inserted into the unique BglII site in pTet-BBB to create pTet-BBB+ARE. To construct pSVmyc45, which expresses myc-tagged hnRNP D isoform p45, a p45 cDNA fragment was first prepared by NdeI and EcoRI digestions of plasmid pET-21(c)-cDx9-His6 (kindly provided by F. Ishikawa, Department of Bioengineering, Tokyo Institute of Technology, Yokohama, Japan), blunt ended, and then inserted into the SalI (blunt-ended) site of plasmid pMyc-over (Peng et al. 1998). Plasmid pMyc-over is a pCAT3 (Promega) derivative in which the CAT-coding sequence has been replaced by a sequence encoding the myc epitope tag. The His epitope tag present in the carboxyl terminus of p45 was subsequently removed by replacing the XcmI–EcoRI fragment spanning the His-tag region with a XcmI–ApaI (blunt-ended) fragment of the plasmid pEx10x-cDx9, which is kindly provided by F. Ishikawa (Kajita et al. 1995). To construct pSVmyc42, a 711-bp PCR product amplified from a plasmid carrying a full-length AUF1-p37 cDNA (Kiledjian et al. 1997), kindly provided by Gary Brewer (Wake Forest University School of Medicine, Winston, Salem, NC), was first subcloned into pKS Bluscript (Stratagene). A SalI fragment spanning the PCR-amplified hnRNP D/AUF1 cDNA was then subcloned into the SalI site of pTet-Myc-over (Peng et al. 1998) to create pTetMycAUF1. A StuI fragment of pSVmyc45 containing the 5′ portion of p45 cDNA was then replaced by a corresponding StuI fragment of pTetMycAUF1 to generate pSVmyc42. Plasmid pSVmyc40 was constructed by removing the BstXI fragment from pSVmyc45 that carries the 49-amino-acid insertion at the carboxyl terminus of p45 isoform. Plasmid pSVmyc37 was created by replacing the StuI fragment of pSVmyc40 containing the 5′ portion of p40 cDNA with a corresponding StuI fragment from pTetMycAUF1. Plasmid pT7β3′C was constructed by first excising a unique 150-bp BamHI–BglII fragment from the 608-bp PCR-amplified rabbit β-globin cDNA and then inserting it into the unique BamHI site of plasmid T3–T7α18 (GIBCO-BRL).
Analysis of mRNA decay and deadenylation
Establishment of a stable K562 cell transfectant III-2 harboring the gene for tTA (tetracycline-controlled transcriptional activator) was described previously (Xu et al. 1998; Loflin et al. 1999). Cell culture, DNA transfection, isolation of total cytoplasmic RNA, and Northern blot analysis were conducted as described previously (Xu et al. 1998; Loflin et al. 1999). Briefly, cells were cultured at 37°C, 8% CO2 in RPMI1640 (GIBCO-BRL) with 10% fetal bovine serum (FBS) in the presence of tetracycline (500 ng/ml) to a density of 106 cells/ml, harvested, washed with PBS, and resuspended in RPMI at a density of 3 × 107 cells/0.4 ml before transfection cells. Cells (3 × 107) were transfected by electroporation in a 2-mm gap cuvette with 30 μg of pTetBBB or pTetBBB+ARE and 90 μg of pMyc-over or its derivatives carrying hnRNP D cDNA (Loflin et al. 1999). After electroporation, cells were maintained in RPMI/10%FBS containing 45 ng/ml of tetracycline (Sigma) for ∼16 hr. When needed, cells were treated with 50 μm hemin (Sigma) or 20 nm TPA (LC Laboratories, Boston, MA) for various time intervals as indicated in each experiment (see Figures). Cells were then transferred to fresh medium without Tet for 3–4 hr to resume transcription, followed by the addition of 500 ng/ml Tet to block further transcription. Total cytoplasmic RNA was isolated at various time intervals after the addition of Tet (500 ng/ml), and Northern blot analysis was performed with a probe spanning 60 bp of the rabbit β-globin 3′-UTR for detection of the transfected β-globin message without (BBB) or with an ARE (BBB+ARE). A 300-bp fragment spanning the second exon of GAPDH was used for detection of endogenous GAPDH, which was used as an internal control. The quantitation of data was obtained by scanning the radioactive blots with an imager (Packard). All experiments described in this manuscript have been performed in duplicate or triplicate.
Preparation of K562 cytoplasmic and nuclear extracts
Cytoplasmic and nuclear lysates were prepared as described previously (Peng et al. 1998). Briefly, cytoplasmic lysates were prepared from K562 cells by lysis at 4°C in a lysis/extraction buffer containing 10 mm HEPES (pH 7.6), 3 mm MgCl2, 40 mm KCl, 2 mm DTT, 5% glycerol, 0.5% NP-40, 8 μg/ml aprotinin, 8 μg/ml leupeptin, and 100 μg/ml PMSF. Nuclei were removed by centrifugation (1250g) at 4°C for 5 min. Nuclei were then resuspended and spun down in the lysis/extraction buffer twice to avoid contamination of cytoplasmic proteins. Nuclear extracts were prepared from the pelleted nuclei in nuclear extraction buffer containing 10 mm HEPES (pH 7.9), 0.1 mm EGTA, 1.5 mm MgCl2, 420 mm NaCl, 0.5 mm DTT, 0.5 mm PMSF, and 25% glycerol. After incubation on ice for 20 min, cellular debris was removed by centrifugation (1250g) at 4°C for 5 min. Protein concentration was analyzed by the BCA protein assay reagent (Pierce).
Gel mobility shift assay and supershift analysis
Preparation of RNA probes by in vitro transcription and analysis of RNA–protein interaction were described previously (You et al. 1992). Transcription reactions were performed according to Promega instructions, with T3 or T7 RNA polymerase. Labeled RNA transcripts were produced by inclusion of [α-32P]UTP (DuPont, 800 Ci/mmole) in the reactions. The c-fos ARE probe is a sense transcript synthesized from HindIII-linearized pT3ARE (You et al. 1992) with T3 polymerase. The β-globin RNA used as a nonspecific competitor in Figure 3 is an antisense transcript spanning the 150-nucleotide 3′ portion of the protein coding region of the rabbit β-globin mRNA and is synthesized from BamHI-linearized pT7β3′C with T7 polymerase. Cytoplasmic lysate (6 μg of protein) and 32P-labeled RNA (1 ng) were incubated at room temperature for 15 min in a buffer containing 10 mm HEPES (pH 7.6), 3 mm MgCl2, 40 mm KCl, 2 mm DTT, 5% glycerol, and 0.5% NP-40. Heparin (5 μg/ml, final concentration) and yeast total RNA (200 μg/ml, final concentration) were added to reduce nonspecific binding. The final volume of each reaction was 10 μl. Subsequently, unbound RNA was digested for 20 min by 0.6 units of RNase T1 (Calbiochem) at room temperature. RNA–protein complexes were resolved on 6% nondenaturing polyacrylamide gels. Gel mobility supershift analysis was performed by the addition of antibody into the binding reaction (final volume, 10 μl; 1:20 dilution for antibody to AUF1, and 1:10 dilution for antibody to HuR), which was preincubated for 15 min at room temperature to allow RNA–protein interactions to occur, and the antibody–RNA–protein mixture was then incubated at room temperature for another 15 min. The RNA–protein–antibody complexes were resolved on 6% nondenaturing polyacrylamide gels. The purified monoclonal antibody against the myc-tag (0.1 μg of IgG/μl) was purchased from Calbiochem. The polyclonal antibody to AUF1 was kindly provided by G. Brewer (Pende et al. 1996; Kiledjian et al. 1997; Sirenko et al. 1997) and the antibody to human HuR was a gift from H. Furneaux (Memorial Sloan Kettering Cancer Center, NY) and was raised against the peptide sequence spanning the first 13 amino acids of human HuR (Peng et al. 1998).
Western blotting analysis
Cytoplasmic and nuclear lysates were resolved on a 12% SDS–polyacrylamide gel and analyzed by Western blotting with an ECL Western-blotting kit (Amersham). The blots were probed with specific antibodies as described in the legends to the figures. The purified monoclonal antibody against α-tubulin (DM1A) was purchased from Sigma and was used at a 1:20,000 dilution. The antibody to myc tag was as described above and was used at a 1:100 dilution. The monoclonal antibody to hnRNP D (5B9) was kindly provided by G. Dreyfuss (Howard Hughes Medical Institute, University of Pennsylvania School of Medicine, Philadelphia, PA) and was used at 1:10,000 dilution (Faura et al. 1995; Kamma et al. 1995; Kiledjian et al. 1997).
Acknowledgments
We thank M. Blackburn, G. Cote, and M. Wilkinson for critical reading of the manuscript and their valuable comments, N. Xu for technical assistance and discussion, G. Brewer and G. Dreyfuss for the antibodies against hnRNP D/AUF1, G. Brewer, F. Ishikawa, and M. Kiledjian for the AUF1 and hnRNP D cDNA clones. This work was supported by a grant from National Institutes of Health (RO1 GM 46454) and in part by a grant from the Council of Tobacco Research-USA, Inc. A.-B.S. is the recipient of an American Heart Association Established Investigator Award.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL abshyu@bmb.med.uth.tmc.edu; FAX (713) 500-0652.
References
- Alitalo R. Induced differentiation of K562 leukemia cells: A model for studies of gene expression in early megakaryoblasts. Leuk Res. 1990;14:501–514. doi: 10.1016/0145-2126(90)90002-q. [DOI] [PubMed] [Google Scholar]
- Alitalo R, Andersson LC, Betsholtz C, Nilsson K, Westermark B, Heldin CH, Alitalo K. Induction of platelet-derived growth factor gene expression during megakaryoblastic and monocytic differentiation of human leukemia cell lines. EMBO J. 1987;6:1213–1218. doi: 10.1002/j.1460-2075.1987.tb02356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alitalo R, Mäkelä TP, Koskinen P, Andersson LC, Alitalo K. Enhanced expression of transforming growth factor beta during megakaryoblastic differentiation of K562 leukemia cells. Blood. 1988;71:899–906. [PubMed] [Google Scholar]
- Antic D, Keene JD. Insights from model systems-Embryonic lethal abnormal visual RNA-binding proteins involved in growth, differentiation, and posttranscriptional gene expression. Am J Hum Genet. 1997;61:273–278. doi: 10.1086/514866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliga BS, Mankad M, Shah AK, Mankad VN. Mechanism of differentiation of human erythroleukaemic cell line K562 by hemin. Cell Prolif. 1993;26:519–529. doi: 10.1111/j.1365-2184.1993.tb00030.x. [DOI] [PubMed] [Google Scholar]
- Bosser R, Faura M, Serratosa J, Renau-Piqueras J, Pruschy M, Bachs O. Phosphorylation of rat liver heterogeneous nuclear ribonucleoproteins A2 and C can be modulated by calmodulin. Mol Cell Biol. 1995;15:661–670. doi: 10.1128/mcb.15.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer G. An A + U-rich element RNA-binding factor regulates c-myc mRNA stability in vitro. Mol Cell Biol. 1991;11:2460–2466. doi: 10.1128/mcb.11.5.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer G, Ross J. Regulation of c-myc mRNA stability in vitro by a labile destabilizer with an essential nucleic acid component. Mol Cell Biol. 1989;9:1996–2006. doi: 10.1128/mcb.9.5.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzby JS, Lee SM, Van Winkle P, DeMaria CT, Brewer G, Cairo MS. Increased granulocyte-macrophage colony-stimulating factor mRNA instability in cord versus adult mononuclear cells is translation-dependent and associated with increased levels of A + U-rich element binding factor. Blood. 1996;88:2889–2897. [PubMed] [Google Scholar]
- Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci. 1986;83:1670–1674. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. Selective degradation of early-response-gene mRNAs: Functional analyses of sequence features of the AU-rich elements. Mol Cell Biol. 1994;14:8471–8482. doi: 10.1128/mcb.14.12.8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- Chen CY, Chen TM, Shyu AB. Interplay of two functionally and structurally distinct domains of the c-fos AU-rich element specifies its mRNA-destabilizing function. Mol Cell Biol. 1994;14:416–426. doi: 10.1128/mcb.14.1.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Del Gatto-Konczak F, Wu Z, Karin M. Stabilization of interleukin-2 mRNA by the c-Jun NH2-terminal kinase pathway. Science. 1998;280:1945–1949. doi: 10.1126/science.280.5371.1945. [DOI] [PubMed] [Google Scholar]
- DeMaria CT, Brewer G. AUF1 binding affinity to A + U-rich elements correlates with rapid mRNA degradation. J Biol Chem. 1996;271:12179–12184. doi: 10.1074/jbc.271.21.12179. [DOI] [PubMed] [Google Scholar]
- DeMaria CT, Sun Y, Long L, Wagner BJ, Brewer G. Structural determinants in AUF1 required for high affinity binding to A + U-rich elements. J Biol Chem. 1997;272:27635–27643. doi: 10.1074/jbc.272.44.27635. [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Matunis MJ, Piñol-Roma S, Burd CG. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Hentze M, Lamond AI. From transcript to protein. Cell. 1996;85:963–972. doi: 10.1016/s0092-8674(00)81298-2. [DOI] [PubMed] [Google Scholar]
- Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998;17:3448–3460. doi: 10.1093/emboj/17.12.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faura M, Renau-Piqueras J, Bachs O, Bosser R. Differential distribution of heterogeneous nuclear ribonucleoproteins in rat tissues. Biochem Biophys Res Comm. 1995;217:554–560. doi: 10.1006/bbrc.1995.2811. [DOI] [PubMed] [Google Scholar]
- Ford LP, Watson J, Keene JD, Wilusz J. ELAV proteins stabilize deadenylated intermediates in a novel in vitro mRNA deadenylation/degradation system. Genes & Dev. 1999;13:188–201. doi: 10.1101/gad.13.2.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Belasco JG. Control of the decay of labile protooncogene and cytokine mRNAs. In: Belasco JG, Brawerman G, editors. Control of messenger RNA stability. San Diego, CA: Academic Press; 1993. pp. 199–218. [Google Scholar]
- Hahm B, Kim YK, Kim JH, Kim TY, Jang SK. Heterogeneous nuclear ribonucleoprotein L interacts with the 3′ border of the internal ribosomal entry site of hepatitis C virus. J Virol. 1998;72:8782–8788. doi: 10.1128/jvi.72.11.8782-8788.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BJ, Nagy E, Malter JS, Arrick BA, Rigby WF. Association of heterogeneous nuclear ribonucleoprotein A1 and C proteins with reiterated AUUUA sequences. J Biol Chem. 1993;268:8881–8887. [PubMed] [Google Scholar]
- Hamilton BJ, Burns CM, Nichols RC, Rigby WFC. Modulation of AUUUA response element binding by heterogeneous nuclear ribonucleoprotein A1 in human T lymphocytes. The roles of cytoplasmic location, transcription, and phosphorylation. J Biol Chem. 1997;272:28732–28741. doi: 10.1074/jbc.272.45.28732. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Jarmolowski A, Beisel C, Mattaj IW, Dreyfuss G, Fischer U. A role for the M9 transport signal of hnRNP A1 in mRNA nuclear export. J Cell Biol. 1997;137:27–35. doi: 10.1083/jcb.137.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RG, Andrews LG, McGowan KM, Pekala PH, Keene JD. Ectopic expression of Hel-N1, an RNA-binding protein, increases glucose transporter (GLUT1) expression in 3T3-L1 adipocytes. Mol Cell Biol. 1997;17:954–962. doi: 10.1128/mcb.17.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajita Y, Nakayama J, Aizawa M, Ishikawa F. The UUAG-specific RNA binding protein, heterogeneous nuclear ribonucleoprotein D0. Common modular structure and binding properties of the 2xRBD-Gly family. J Biol Chem. 1995;270:22167–22175. doi: 10.1074/jbc.270.38.22167. [DOI] [PubMed] [Google Scholar]
- Kamma H, Portman DS, Dreyfuss G. Cell type-specific expression of hnRNP proteins. Exp Cell Res. 1995;221:187–196. doi: 10.1006/excr.1995.1366. [DOI] [PubMed] [Google Scholar]
- Kiledjian M, Wang X, Liebhaber SA. Identification of two KH domain proteins in the alpha-globin mRNP stability complex. EMBO J. 1995;14:4357–4364. doi: 10.1002/j.1460-2075.1995.tb00110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiledjian M, DeMaria CT, Brewer G, Novick K. Identification of AUF1 (heterogeneous nuclear ribonucleoprotein D) as a component of the alpha-globin mRNA stability complex. Mol Cell Biol. 1997;17:4870–4876. doi: 10.1128/mcb.17.8.4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon I, Carballes F, Brewer G, Poiret M, Morello D. Developmental expression of AUF1 and HuR, two c-myc mRNA binding proteins. Oncogene. 1998;16:3413–3421. doi: 10.1038/sj.onc.1201895. [DOI] [PubMed] [Google Scholar]
- Levy NS, Chung S, Furneaux H, Levy AP. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998;273:6417–6423. doi: 10.1074/jbc.273.11.6417. [DOI] [PubMed] [Google Scholar]
- Loflin TL, Chen C-YA, Xu N, Shyu A-B. Transcriptional pulsing approaches for analysis of mRNA turnover in mammalian cells. Methods: Companion Methods Enzymol. 1999;17:11–20. doi: 10.1006/meth.1998.0702. [DOI] [PubMed] [Google Scholar]
- Michael WM, Eder PS, Dreyfuss G. The K nuclear shuttling domain: A novel signal for nuclear import and nuclear export in the hnRNP K protein. EMBO J. 1997;16:3587–3598. doi: 10.1093/emboj/16.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming XF, Kaiser M, Moroni C. c-jun N-terminal kinase is involved in AUUUA-mediated interleukin-3 mRNA turnover in mast cells. EMBO J. 1998;17:6039–6048. doi: 10.1093/emboj/17.20.6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Kosaka M, Eto Y, Shibai H, Saito S. Effect of erythroid differentiation factor on erythroid differentiation and proliferation of K-562 cells. Biochem Biophys Res Comm. 1990;168:1149–1156. doi: 10.1016/0006-291x(90)91149-m. [DOI] [PubMed] [Google Scholar]
- Müller WE, Slor H, Pfeifer K, Hühn P, Bek A, Orsulic S, Ushijima H, Schröder HC. Association of AUUUA-binding protein with A+U-rich mRNA during nucleo-cytoplasmic transport. J Mol Biol. 1992;226:721–733. doi: 10.1016/0022-2836(92)90628-w. [DOI] [PubMed] [Google Scholar]
- Nagy E, Rigby WF. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold) J Biol Chem. 1995;270:2755–2763. doi: 10.1074/jbc.270.6.2755. [DOI] [PubMed] [Google Scholar]
- Nakagawa J, Waldner H, Meyer-Monard S, Hofsteenge J, Jeno P, Moroni C. AUH, a gene encoding an AU-specific RNA binding protein with intrinsic enoyl-CoA hydratase activity. Proc Natl Acad Sci. 1995;92:2051–2055. doi: 10.1073/pnas.92.6.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima O, Iwasaki S, Hashimoto Y. Hemin-induced erythroid differentiation of human myeloleukemia K562 cell line and its modification by bioresponse modifiers. Cell Mol Biol (Noisy-le-grand) 1997;43:115–134. [PubMed] [Google Scholar]
- Nanbu R, Kubo T, Hashimoto T, Natori S. Purification of an AU-rich RNA binding protein from Sarcophaga peregrina (flesh fly) and its identification as a Thiolase. J Biochem (Tokyo) 1993;114:432–437. doi: 10.1093/oxfordjournals.jbchem.a124193. [DOI] [PubMed] [Google Scholar]
- Ostareck DH, Ostareck-Lederer A, Wilm M, Thiele BJ, Mann M, Hentze MW. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell. 1997;89:597–606. doi: 10.1016/s0092-8674(00)80241-x. [DOI] [PubMed] [Google Scholar]
- Pende A, Tremmel KD, DeMaria CT, Blaxall BC, Minobe WA, Sherman JA, Bisognano JD, Bristow MR, Brewer G, Port J. Regulation of the mRNA-binding protein AUF1 by activation of the beta-adrenergic receptor signal transduction pathway. J Biol Chem. 1996;271:8493–8501. doi: 10.1074/jbc.271.14.8493. [DOI] [PubMed] [Google Scholar]
- Peng S-P, Chen C-Y, Xu N, Shyu A-B. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 1998;17:3461–3470. doi: 10.1093/emboj/17.12.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñol-Roma S, Dreyfuss G. Transcription-dependent and transcription-independent nuclear transport of hnRNP proteins. Science. 1991;253:312–314. doi: 10.1126/science.1857966. [DOI] [PubMed] [Google Scholar]
- ————— Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature. 1992;355:730–732. doi: 10.1038/355730a0. [DOI] [PubMed] [Google Scholar]
- Pype S, Slegers H, Moens L, Merlevede W, Goris J. Tyrosine phosphorylation of a M(r) 38,000 A/B-type hnRNP protein selectively modulates its RNA binding. J Biol Chem. 1994;269:31457–31465. [PubMed] [Google Scholar]
- Ross J. mRNA stability in mammalian cells. Microbiol Rev. 1995;59:423–450. doi: 10.1128/mr.59.3.423-450.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford TR, Clegg JB, Weatherall DJ. K562 human leukaemic cells synthesize embryonic haemoglobin in response to haemin. Nature. 1979;280:164–165. doi: 10.1038/280164a0. [DOI] [PubMed] [Google Scholar]
- Rutherford T, Clegg JB, Higgs DR, Jones RW, Thompson J, Weatherall DJ. Embryonic erythroid differentiation in the human leukemic cell line K562. Proc Natl Acad Sci. 1981;78:348–352. doi: 10.1073/pnas.78.1.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavi SC, Belasco JG, Greenberg ME. Regulation of proto-oncogene mRNA stability. Biochim Biophys Acta. 1992;1114:95–106. doi: 10.1016/0304-419x(92)90009-n. [DOI] [PubMed] [Google Scholar]
- Shaw G, Kamen R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell. 1986;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- Shen EC, Henry MF, Weiss VH, Valentini SR, Silver PA, Lee MS. Arginine methylation facilitates the nuclear export of hnRNP proteins. Genes & Dev. 1998;12:679–691. doi: 10.1101/gad.12.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu A-B, Garcia-Sanz JA, Mullner E. Analysis of mRNA decay in mammalian cells. In: Lefkovits I, editor. The immunology methods manual. London, UK: Academic Press; 1996. pp. 450–456. [Google Scholar]
- Siomi MC, Eder PS, Kataoka N, Wan L, Liu Q, Dreyfuss G. Transportin-mediated nuclear import of heterogeneous nuclear RNP proteins. J Cell Biol. 1997;138:1181–1192. doi: 10.1083/jcb.138.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirenko OI, Lofquist AK, DeMaria CT, Morris JS, Brewer G, Haskill JS. Adhesion-dependent regulation of an A+U-rich element-binding activity associated with AUF1. Mol Cell Biol. 1997;17:3898–3906. doi: 10.1128/mcb.17.7.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistonen L, Sarge KD, Phillips B, Abravaya K, Morimoto RI. Activation of heat shock factor 2 during hemin-induced differentiation of human erythroleukemia cells. Mol Cell Biol. 1992;12:4104–4111. doi: 10.1128/mcb.12.9.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulard M, Della Valle V, Siomi MC, Piñol-Roma S, Codogno P, Bauvy C, Bellini M, Lacroix JC, Monod G, Dreyfuss G, et al. hnRNP G: Sequence and characterization of a glycosylated RNA-binding protein. Nucleic Acids Res. 1993;21:4210–4217. doi: 10.1093/nar/21.18.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin YV, Ovchinnikov LP, Dreyfuss G, Sonenberg N. General RNA binding proteins render translation cap dependent. EMBO J. 1996;15:7147–7155. [PMC free article] [PubMed] [Google Scholar]
- Theodorakis NG, Zand DJ, Kotzbauer PT, Williams GT, Morimoto RI. Hemin-induced transcriptional activation of the HSP70 gene during erythroid maturation in K562 cells is due to a heat shock factor-mediated stress response. Mol Cell Biol. 1989;9:3166–3173. doi: 10.1128/mcb.9.8.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman R. Transient accumulation of c-fos RNA following serum stimulation requires a conserved 5′ element and c-fos 3′ sequences. Cell. 1985;42:889–902. doi: 10.1016/0092-8674(85)90285-5. [DOI] [PubMed] [Google Scholar]
- Valentini SR, Weiss VH, Silver PA. Arginine methylation and binding of Hrp1p to the efficiency element for mRNA 3′-end formation. RNA. 1999;5:272–280. doi: 10.1017/s1355838299981633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visa N, Alzhanova-Ericsson AT, Sun X, Kiseleva E, Björkroth B, Wurtz T, Daneholt B. A pre-mRNA-binding protein accompanies the RNA from the gene through the nuclear pores and into polysomes. Cell. 1996;84:253–264. doi: 10.1016/s0092-8674(00)80980-0. [DOI] [PubMed] [Google Scholar]
- Wagner BJ, DeMaria CT, Sun Y, Wilson GM, Brewer G. Structure and genomic organization of the human AUF1 gene: Alternative pre-mRNA splicing generates four protein isoforms. Genomics. 1998;48:195–202. doi: 10.1006/geno.1997.5142. [DOI] [PubMed] [Google Scholar]
- Wang X, Liebhaber SA. Complementary change in cis determinants and trans factors in the evolution of an mRNP stability complex. EMBO J. 1996;15:5040–5051. [PMC free article] [PubMed] [Google Scholar]
- Weighardt F, Biamonti G, Riva S. The roles of heterogeneous nuclear ribonucleoproteins (hnRNP) in RNA metabolism. BioEssays. 1996;18:747–756. doi: 10.1002/bies.950180910. [DOI] [PubMed] [Google Scholar]
- Winzen R, Kafert S, Preiss B, Mylius-Spencker HA, Resch K, Holtmann H. Interaction between the mRNA of the 55-kDa tumor necrosis factor receptor and cellular proteins. Possible involvement in post-transcriptional regulation of receptor expression. J Biol Chem. 1996;271:13461–13467. doi: 10.1074/jbc.271.23.13461. [DOI] [PubMed] [Google Scholar]
- Xu N, Chen C-YA, Shyu A-B. Modulation of the fate of cytoplasmic mRNA by AU-rich elements: Key sequence features controlling mRNA deadenylation and decay. Mol Cell Biol. 1997;17:4611–4621. doi: 10.1128/mcb.17.8.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Loflin P, Chen C-YA, Shyu A-B. A broader role for AU-rich element-mediated mRNA turnover revealed by a new transcriptional pulse strategy. Nucleic Acids Res. 1998;26:558–565. doi: 10.1093/nar/26.2.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y, Chen CY, Shyu AB. U-rich sequence-binding proteins (URBPs) interacting with a 20-nucleotide U-rich sequence in the 3′ untranslated region of c-fos mRNA may be involved in the first step of c-fos mRNA degradation. Mol Cell Biol. 1992;12:2931–2940. doi: 10.1128/mcb.12.7.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wagner BJ, Ehrenman K, Schaefer AW, DeMaria CT, Crater D, DeHaven K, Long L, Brewer G. Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol Cell Biol. 1993;13:7652–7665. doi: 10.1128/mcb.13.12.7652. [DOI] [PMC free article] [PubMed] [Google Scholar]