

Abstract
The Caenorhabditis elegans caudal homolog, pal-1, is required for neurogenesis in the male tail. We show that expression of pal-1 in the postembryonic neuroblast cell V6 can be initiated by two alternate pathways. One pathway, acting in wild type, requires a regulatory element in the fifth pal-1 intron. The other pathway, independent of this element, is normally repressed by the newly identified gene sop-1, which encodes a homolog of the mammalian Mediator complex protein TRAP230. In sop-1 mutants, pal-1 is activated by a pathway that is stimulated by bar-1/β-catenin, a component of the Wnt signal transduction pathway. The results support a physiological role of the Mediator complex in conveying regulatory signals to the transcriptional apparatus.
Keywords: Transcription, homeobox gene, development, cell fate, regulation
The properties of differentiated cells are determined by the sets of transcription factors they express and lineage ancestry. The male rays in Caenorhabditis elegans are a model for studying how a transcription factor cascade in a postembryonic cell lineage leads to specification of a defined set of differentiated cells at a particular site in the adult body (Emmons 1999). The transcription factor cascade leading to rays involves several homologs of well-known cell-fate specifying regulatory transcription factors, notably the Hox transcription factors and a caudal homolog (Chow and Emmons 1994; Salser and Kenyon 1996; Hunter et al. 1999). Genes for these transcription factors are turned on and off at specific times during the late embryonic and postembryonic ray cell lineages, defining multiple alternate cell fates necessary for wild-type ray development (Salser and Kenyon 1996; Fereira et al. 1999; Hunter et al. 1999). Here we take advantage of a genetic approach to identify an element of the mechanism that governs the accurate expression of this cascade.
Regulation of gene expression occurs primarily through control of the several steps of the transcription initiation pathway (Kornberg and Lorch 1999; Struhl 1999). This pathway involves opening of the promoter region by chromatin modification and remodeling, followed by recruitment of the polymerase complex and relevant cofactor complexes, and finally initiation of elongation. Each of these steps is a potential target of regulatory factors. Recruitment of holoenzyme and communication of regulatory signals to the core polymerase are thought to occur in part via a multiprotein complex termed the Mediator complex, which includes the targets for several DNA-binding transcription factors (see Björklund et al. 1999). Like other metazoans, C. elegans contains one or more complexes containing homologs of yeast Mediator proteins (Kwon et al. 1999). The C. elegans genome encodes homologs of several additional components present in human complexes but not found in yeast (Ito et al. 1999).
Transcription factor cascades leading to rays can be considered to begin during late embryogenesis in three bilateral pairs of postembryonic neuroectoblasts. The ray transcriptional program in one of these pairs of neuroectoblasts, V6, begins with expression of pal-1, the C. elegans ortholog of the conserved homeodomain transcription factor caudal (Mlodzik and Gehring 1987; Waring and Kenyon 1991). Expression of pal-1 in V6 in late embryogenesis results in the expression of mab-5, a Hox gene of the Antennapedia paralog family (Costa et al. 1988; Hunter et al. 1999). Later in the cell lineage, mab-5 directly or indirectly turns on egl-5 and lin-32 (Wrischnik and Kenyon 1997; Ferreira et al. 1999). egl-5 is a second gene of the C. elegans Hox family, most closely related to the Abdominal-B paralog family (Wang et al. 1993). lin-32 is a bHLH transcription factor related to Drosophila atonal that is proneural in function, initiating the ray sublineage and neurogenesis (Zhao and Emmons 1995). mab-5 and egl-5, in addition to regulating lin-32, define ray-specific properties, such as morphogenetic identity and neurotransmitter expression (Chow and Emmons 1994; Salser and Kenyon 1996; Lints and Emmons 1999).
Accurate expression of this cascade is crucial to the development of the wild-type ray pattern. To define additional components governing its expression, we studied regulation of the first gene, the caudal homolog pal-1. We identified a cis-regulatory region within a pal-1 intron that is necessary for expression of pal-1 in V6. Taking an approach similar to that used previously in yeast to identify components of the transcriptional regulatory apparatus (Carlson 1997), we carried out a suppressor screen for mutations that restore ray development in a strain carrying a mutation in this cis-regulatory region. The screen resulted in the isolation of mutations that restored pal-1 activity in V6.
This strategy allowed us to identify a Mediator component, encoded by the gene sop-1 (suppressor of pal-1), which plays a role in determining which of two alternate activation pathways will express pal-1. SOP-1 corresponds to the C. elegans homolog of a large Mediator protein, TRAP230, identified by in vitro studies of human transcription complexes (Ito et al. 1999). Loss of SOP-1 function releases pal-1 expression from the requirement for the cis regulatory region by allowing pal-1 activation via a different pathway. This alternate pathway involves the function of β-catenin, a conserved DNA-binding component of the Wnt signal transduction pathway. Thus, as it does in yeast and in cell-free transcription systems, the Mediator complex in C. elegans appears to function in regulation of holoenzyme by DNA-binding factors, and to play a role in ensuring the stringency and specificity of their action. SOP-1/TRAP230 is not found in yeast. Thus its role may be in mediating developmental signals.
Results
A lineage-specific cis regulatory element lies in a pal-1 intron
The C. elegans caudal homolog pal-1 plays an essential role to define cell fates in posterior cell lineages during both early and late embryogenesis (Hunter and Kenyon 1996). In the latter part of embryogenesis, pal-1 is also expressed in a bilateral pair of seam cells, the V6 cells, that are descendants of the anterior blastomere of the two-cell embryo. Expression of pal-1 in V6 results in generation of a cell lineage leading to the postembryonic development of male rays (Fig. 1A,B) (Waring and Kenyon 1990, 1991; Hunter et al. 1999).
Figure 1.

sop-1 mutations suppress pal-1(e2091). (A) Lateral view of an L1 larva showing the row of seam cells, V1–V6 and T, on one side. The Wnt ligands EGL-20 and LIN-44 are expressed, respectively, in the rectal epithelium and tail tip epithelium as shown (Herman et al. 1995; Whangbo and Kenyan 1999). (B) Wild-type adult male tail, ventral view, Nomarski photomicrograph. Rays 2–6 are derived from V6, which gives rise to the postembryonic cell lineage shown (larval stages are shown on the vertical scale) (Sulston and Horvitz 1977). (C) pal-1(e2091) mutant. V6 adopts a fate similar to anterior seam cells V1–V4 in wild type, producing alae, longitudinal cuticular ridges, instead of rays. Lineages of V5 and T, leading respectively to ray 1 and rays 7–9, are unaffected. Ray 1 usually fails to migrate posteriorly into the fan region and forms a papilla on the side of the body. (D) pal-1(e2091); sop-1(bx92). V6 produces five normal rays. (E) In pal-1(e2091); sop-1(bx107) double mutants, 13.5% of sides have ray 3 and 4 fused (ray 4 appears to adopt a ray 3-like identity).
The viable pal-1(e2091) mutation results in the specific loss of pal-1 expression in V6, while leaving unperturbed pal-1's essential embryonic functions (Hunter et al. 1999). Absence of pal-1 in V6 results in failure to generate the V6 rays (rays 2–6) in the adult male (Fig. 1C). In pal-1(e2091), descendant cells of V6 divide following a cell lineage similar to the lineages of anterior seam cells, and instead of rays, produce alae, cuticular ridges running along the sides of adult animals from the head through most of the body (Pal phenotype, for posterior alae) (Fig. 1C; Waring and Kenyon 1990).
To determine the basis for the loss of pal-1 expression in V6, we sought to identify the e2091 mutation. Earlier attempts to identify this mutation were unsuccessful, and showed that the nucleotide sequences of the exons and exon/intron boundaries were wild type (Hunter et al. 1999; data not shown). e2091 was therefore presumed to affect a regulatory sequence. To localize the e2091 mutation, we carried out complementation rescue experiments with chimeric genes constructed with segments derived from wild-type and e2091 mutant DNA (Fig. 2A). We found that mutant DNA derived from the 5′ flanking genomic region fused to wild-type coding sequence could rescue the ray defect in pal-1(e2091), but that mutant DNA covering the exons, introns, and 3′ flanking genomic region driven by wild-type 5′ flanking sequence could not. We sequenced the downstream fragment from pal-1(e2091) and found two point mutations in the last intron (Fig. 2B), and no other changes.
Figure 2.

The pal-1(e2091) mutation is a point mutation within a conserved intronic sequence. (A) Complementation rescue experiments defining the gene segment containing the mutation. Two genomic DNA fragments were amplified from either wild type or pal-1(e2091) as shown, ligated at the NcoI site in either homologous or heterologous combinations, and injected into pal-1(e2091); him-5 hermaphrodites. Only the 3′ fragment from pal-1(e2091) failed to support rescue. Nucleotide positions are in cosmid C38D4. Likewise, the genomic-cDNA hybrid construct pSC16, which lacks the last intron of pal-1, failed to support rescue. (B) The two sequence differences between pal-1(e2091) and wild type. The sequence of the pal-1(e2091) coding region, including all introns, plus 1 kb of 3′ UTR was determined. One of the two mutations was reverted in the intragenic revertant pal-1(e2091 bx89), thus identifying T24264→C as the pal-1(e2091) mutation. (C) Alignment of part of the last intronic sequence of C. elegans and C. briggsae pal-1, showing the conserved region containing the pal-1(e2091) mutation.
In our screen for pal-1(e2091) suppressors, described below, we recovered a dominant suppressor mutation (bx89) that was linked tightly to e2091. We sequenced the last intron from this presumptive intragenic revertant and found that one of the two point mutations present in pal-1(e2091) was reverted to wild type (Fig. 2B). On this basis, we conclude that this mutation, T(24264)→C, is the e2091 mutation.
A point mutation in an intron might block gene function for one of two reasons. It might act at the level of DNA and affect the function of a cis-acting transcriptional regulatory element, or it might act at the level of RNA and affect splicing or some other aspect of RNA function. According to the first model, the intron is necessary for gene expression, whereas under the second model, if the mutation blocks splicing, a gene lacking the intron altogether would be functional. To distinguish between these two models, we tested whether a pal-1 transgene lacking the last intron could rescue pal-1(e2091). pSC16 is a genomic DNA–cDNA hybrid constructed by joining genomic DNA up to the penultimate pal-1 exon to cDNA sequence (L. Edgar and W.B. Wood, pers. comm.). pSC16 is capable of rescuing the pal-1 null embryonic lethal phenotype (L. Edgar and W.B. Wood, pers. comm.), but we found that it cannot rescue the V6 ray loss phenotype of pal-1(e2091) (Fig. 2A). This result supports the identification of T(24264)→C as the e2091 mutation and indicates that the last intron is necessary for gene function, suggesting that it contains a lineage-specific regulatory element required for expression of pal-1 in V6. An alternative hypothesis is that a sequence within the intron acts at the level of RNA and is required for one or more steps in RNA processing or function other than simple removal of the intron. This possibility seems unlikely in view of the nature of the suppressor mutations discussed below, which affect a protein thought to act in transcription initiation.
If the last intron of pal-1 contained an essential cis-regulatory element, we expected this element to be conserved in closely related nematodes. We therefore sequenced the corresponding intron of pal-1 of Caenorhabditis briggsae. We found the C. briggsae pal-1 homolog contained an intron of similar length (C. briggsae: 1469 nucleotides, C. elegans: 1446 nucleotides) at the same position within the coding region as found in C. elegans. The sequence of the C. briggsae intron was diverged from that of C. elegans with the exception of three conserved blocks of ∼50 nucleotides. In one of these blocks, a 56-nucleotide region identical to C. elegans at 49 positions with one 13 nucleotide gap, the fourth-to-last nucleotide corresponded to the nucleotide mutated in pal-1(e2091) (Fig. 2C). We hypothesize that this mutated sequence corresponds to the binding site for a transcription factor necessary for expression of pal-1 in V6.
Suppressor mutations in sop-1 restore pal-1 activity to V6
To identify genes acting to regulate the pal-1-initiated transcription factor cascade leading to rays, we isolated suppressor mutations that reverted the V6 ray loss phenotype of pal-1(e2091) (see Materials and Methods). The X-linked gene sop-1 was identified by four recessive suppressor mutations (Fig. 1D,E; Table 1, lines 3–6). These suppressor mutations were specific to the function of pal-1 in generation of rays, and did not suppress the embryonic lethal phenotypes of a pal-1(0) mutation (data not shown). When the pal-1(e2091) mutation was crossed out, none of the sop-1 alleles had any obvious mutant phenotype.
Table 1.
V6 rays in various genetic backgrounds
|
|
Genotype
|
Wild-type V6 rays (%)
|
Total sides
|
|---|---|---|---|
| 1 | Wild-type male | 100 | >1000 |
| 2 | pal-1(e2091) | 4 | 214 |
| 3 | pal-1(e2091); sop-1(bx92) | 94 | 2470 |
| 4 | pal-1(e2091); sop-1(bx93) | 96 | 262 |
| 5 | pal-1(e2091); sop-1(bx103) | 62 | 412 |
| 6 | pal-1(e2091); sop-1(bx107) | 87 | 426 |
| 7 | pal-1(e2091); sop-1 (RNAi) | 97 | 226 |
| 8 | pal-1(ct224); sDp3 | 96.4 | 792 |
| 9 | pal-1(ct224); sop-1(bx92); sDp3 | 95.7 | 956 |
| 10 | pal-1(e2091) mab-5(e1239); sop-1(bx92) | 0a | 456 |
| 11 | pal-1(e2091); sop-1(bx92) bar-1(ga80) | 48 | 434 |
| 12 | bar-1(ga80) | 100 | 50 |
| 13 | sop-1(bx92) bar-1(ga80) | 100 | 50 |
| 14 | pal-1(e2091); egl-20(n585); sop-1(bx92) | 91 | 444 |
Percent of sides with a full set of normal V6 rays (rays 2–6). Strains with sDp3 were grown at 25°C.
2.8% of male sides (13/456) have 1–2 V6 rays.
sop-1 mutations appeared to restore rays by reactivating pal-1 activity in the V6 lineage. We gained evidence in support of this conclusion by showing that sop-1 mutations could not suppress ray loss due to pal-1(ct224), a null mutation consisting of a 4.2-kb deletion extending from within exon 1 to the end of pal-1. Because sop-1 mutations do not suppress the embryonic lethal phenotype of pal-1(ct224), it was necessary to carry out this experiment in genetic mosaic animals. To generate mosaics in which pal-1 activity was absent from the V6 lineage but present elsewhere, we employed the free duplication sDp3, a chromosomal fragment carrying a wild-type pal-1 gene copy. In ∼4% of pal-1(ct224); sDp3 animals, sDp3 is lost in the V6 lineage, resulting in a Pal phenotype (Table 1, line 8) (Hunter et al. 1999). We found that the frequency of such mosaic Pal males was unaffected by introduction of sop-1(bx92) (Table 1, line 9). This indicates that ray loss in V6 lineages lacking pal-1 activity is not suppressed by sop-1(bx92). We conclude that suppression requires pal-1 activity, and does not occur via a pal-1-independent bypass pathway.
One function of pal-1 is to activate the Hox gene mab-5, which is not expressed in V6 in pal-1(e2091) (Salser and Kenyon 1996; Hunter et al. 1999). If the effect of sop-1 suppressor mutations was to reactivate the normal pathway of ray development initiated by pal-1, then generation of rays in pal-1(e2091); sop-1 should require mab-5 gene function. We found that this was the case (Table 1, line 10).
sop-1 mutations might result in pal-1-dependent activation of mab-5 in a pal-1(e2091) background by restoring or raising the level of pal-1 activity in V6, or by making mab-5 sensitive to a small amount of pal-1 gene function still supplied by the pal-1(e2091) allele. The following observation argues in favor of the former, that the effect of sop-1 mutation is to elevate the level of pal-1 activity in V6. Salser and Kenyon (1996) found that although pal-1 functioned to turn on mab-5 expression in V6, mab-5 activity alone in the absence of a wild-type pal-1 allele was insufficient to generate a wild-type V6 lineage and normal V6 rays. This was shown by experiments in which MAB-5 was supplied from a heat-shock transgene. In a mab-5(−) pal-1(e2091) double mutant, MAB-5 supplied from a heat-shock transgene was not effective in restoring rays, but was effective in a mab-5(−) pal-1(+) background (Salser and Kenyon 1996). Therefore, pal-1 has a function in addition to activation of mab-5. Mutation in sop-1, which fully restores rays in pal-1(e2091), must result in the provision of this additional activity, as well as in activation of mab-5. The simplest hypothesis is that both effects are brought about by elevation of pal-1 expression in V6.
To gain further evidence for reactivation of pal-1(e2091) in V6 by sop-1 mutations, we studied MAB-5 and PAL-1 expression in V6 by means of antibody staining. As shown previously, in wild type, MAB-5 could be detected in three lateral epidermal cells in late embryogenesis, identified as P9/10, P11/12, and V6 (Fig. 3A) (Cowing and Kenyon 1992; Hunter et al. 1999). In pal-1(e2091), only two cells were stained, consistent with the loss of PAL-1 expression in V6 (Fig. 3B) (Hunter et al. 1999). In pal-1(e2091); sop-1(bx92), three MAB-5-positive cells were visible (n = 4/4) (Fig. 3C). PAL-1 staining was weak and difficult to localize to individual cells, but was consistent with expression in the cell identified as V6 (Fig. 3C). Therefore, results of antibody staining supported the conclusion that mab-5 is reactivated in V6, and were consistent with elevation of PAL-1 level in this cell.
Figure 3.
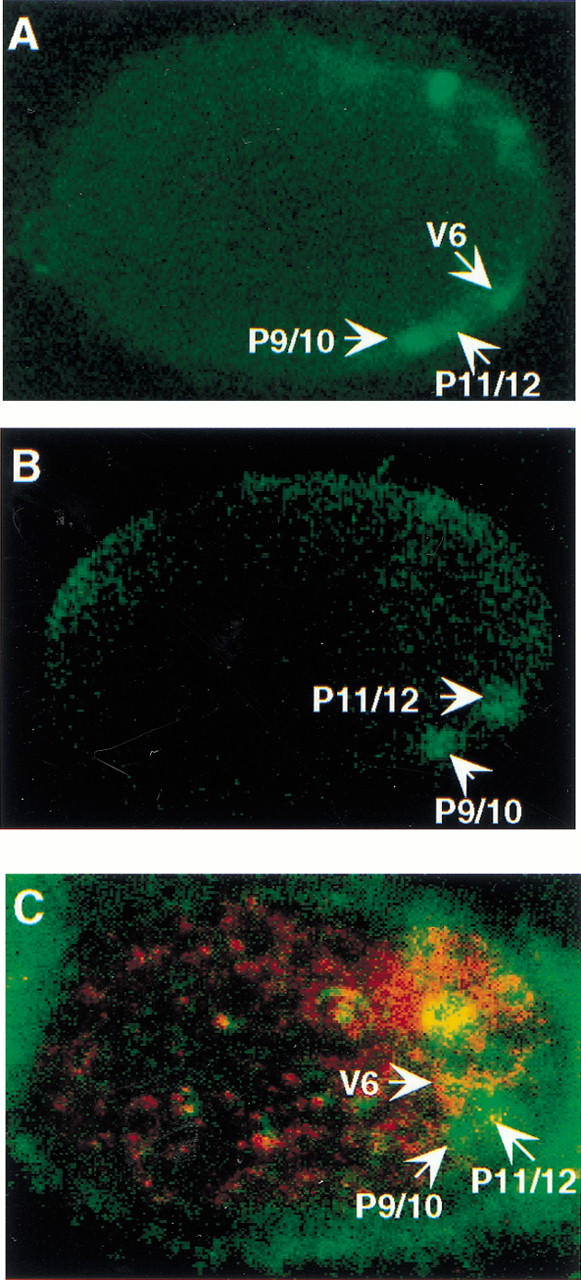
Expression of MAB-5::LACZ and PAL-1 in embryos (confocal fluorescence photomicrograph; anti-LACZ, green; anti-PAL-1, red). (A) MAB-5::LACZ in wild-type embryo. (B) MAB-5::LACZ in pal-1(e2091). In A and B PAL-1 staining was weak and is not shown. (C) MAB-5::LACZ and PAL-1 in pal-1(e2091); sop-1(bx92). Weak overlap (yellow) is detectable in the cell identified by position as V6. The strongly expressing cell above the arrowhead pointing to V6 is in a different plane of focus.
sop-1 encodes a homolog of a component of the human transcription Mediator complex
We identified genomic DNA encoding the sop-1 locus by complementation rescue (Fig. 4A). Rescuing activity was localized to a fragment containing a single predicted gene, F47A4.2, comprising 19 exons encoding a protein of 3498 amino acids (Fig. 4B,C). We confirmed most of the predicted intron/exon boundaries by sequencing cDNAs (see Materials and Methods). To gain further evidence that the large predicted gene was a single functional locus, we introduced frameshift mutations into the amino-terminal region and also constructed amino-terminal and carboxy-terminal deletions. Each of these alterations abolished or severely reduced rescuing activity (Fig. 4B). Finally, reporter genes constructed by in-frame insertion of green fluorescent protein (GFP) respectively into the fourth and seventeenth exons had similar expression patterns, consistent with the assignment of these exons to a single transcriptional unit (Fig. 5).
Figure 4.

sop-1 encodes a TRAP-230 homolog. (A) Map position of sop-1 in the genetic interval defined by the cloned genes dpy-6 and egl-15. The cosmids shown were tested for rescue. Cosmid F47A4 restored the Pal phenotype in pal-1(e2091); sop-1(bx92) mutants. (B) The structure of sop-1 (top). The exons and introns predicted by GeneFinder are shown. The structure from exon 9 to end was confirmed by sequencing cDNAs (see Materials and Methods). The numbers in parenthesis are the positions of the mutations in F47A4. bx103 changes an invariant intron donor CA:gt to CA:at. (Bottom) Rescuing activity of various genomic regions (see Materials and Methods). (C) The predicted amino acid sequence of SOP-1. The carboxy-terminal Q-rich domain is highlighted. The two LxxLL motifs are highlighted in red and underlined. The three marked Q residues correspond to the positions of the mutations in bx93, bx92, and bx107. (D) Schematic of similarity between SOP-1 and TRAP-230.
Figure 5.

SOP-1 is nuclear and expressed ubiquitously. (A) The structure of sop-1::gfp reporter genes. EM#291 and EM#292 have in-frame insertions of gfp and are functional. EM#290 includes an SV40 nuclear localization signal. (B–D) Expression of EM#292 (EM#291 was similar). (B) Ubiquitous expressed in the early embryo. (C) Expression during late embryogenesis showing some nuclei with punctate localization. (D) Punctate expression in neurons in the head of an L4 larva. (E,F) Expression of EM#290. (E) Expression in developing vulval cells. (F) Expression in gut nuclei and neurons in the male posterior region.
sop-1 encodes a homolog of human TRAP230, a component of the transcriptional mediator complex (Fig. 4D) (Ito et al. 1999). SOP-1 and TRAP230 are ∼40% similar and 22% identical in five regions spanning the coding sequence. Both proteins have a glutamine-rich region at the carboxy-terminal end (798 amino acids long and 31% glutamine in SOP-1, 127 amino acids long and 67% glutamine in TRAP230). SOP-1 also contains two ligand-dependent nuclear hormone receptor recognition motifs (LXXLL), as does TRAP230. Apart from the glutamine-rich region and the LXXLL motifs, SOP-1 and TRAP230 are novel proteins containing no other recognizable sequence motifs. SOP-1 is the only predicted TRAP230 homolog in the C. elegans genome.
The similarity of SOP-1 to a known component of RNA polymerase II holoenzyme argues strongly that relief of the pal-1(e2091) mutational block by sop-1 mutation occurs at the level of pal-1 transcriptional initiation, and not at a later step involving RNA processing, transport, or expression. Suppression at the level of transcriptional initiation is consistent with the conclusion that the point mutation in the fifth intron in pal-1(e2091) prevents pal-1 transcription and not with models that postulate this mutation affects RNA processing or other posttranscriptional event.
We localized the four sop-1 mutations by transformation rescue experiments employing combinations of gene subregions derived from wild-type and mutant DNA and sequenced subregions that could not support rescue (see Materials and Methods). In bx103, the weakest allele, an invariant residue necessary for splice donor activity at the start of the seventh intron is mutated from G to A (Fig. 4B). In bx92, bx93, and bx107, CAG glutamine codons are mutated to TAG amber stop codons (Fig. 4B). In bx93, the strongest allele, the terminal one-third of the protein, including the entire glutamine-rich domain, is predicted to be truncated (950 amino acids). bx92 and bx107 lie downstream of bx93 within the glutamine-rich region and are predicted to truncate respectively 333 and 299 carboxy-terminal amino acids. Among the three stop codon mutations, the degree of suppression increases with increasing protein truncation (Table 1, lines 3,4,6). However, on microinjection into pal-1(e2091); sop-1(bx92), DNA from even the strongest allele, bx93, gave up to 20% rescue (see Materials and Methods). Therefore all of these mutants may retain some gene activity.
To determine the expression pattern of sop-1, we constructed three GFP reporters (Fig. 5A). EM#290 contains 2.2 kb of upstream genomic sequence plus the first four SOP-1 exons joined in-frame to GFP. EM#291 and EM#292 are functional reporters that have GFP inserted in-frame into an amino-terminal and a carboxy-terminal exon, respectively. During embryogenesis, all three reporters are expressed in all cells starting after the 8-cell stage of embryogenesis (Fig. 5B). Fluorescence is nuclear, and for EM#291 and EM#292 but not EM#290, beginning in late embryogenesis, it is punctate (Fig. 5C). In postembryonic development and in adult animals, EM#291 and EM#292 are expressed mainly in neurons and fluorescence is punctate (Fig. 5D), whereas EM#290 is expressed widely (Fig. 5E,F). Expression of these reporters in V6 during embryogenesis was observed and is consistent with cell-autonomous function of sop-1 in regulation of pal-1 expression. Nuclear localization is consistent with a function in transcription.
Decrease of sop-1 gene function results in suppression of pal-1(e2091)
To gain information regarding the effect of pal-1(e2091) suppressor mutations on sop-1 gene activity, we used the RNAi (RNA interference) technique to reduce or eliminate gene function (Fire et al. 1998). We asked whether reduction of sop-1 activity resulted in suppression of pal-1(e2091). Double-stranded RNA covering the ninth exon of sop-1 was microinjected into the syncytial gonad of adult pal-1(e2091) hermaphrodites. Progeny embryos were scored for viability and adult males were scored for suppression of the Pal ray-loss phenotype. No loss of viability was observed, and the Pal phenotype of males was suppressed (Table 1, line 7; Materials and Methods). This result indicates that suppression of pal-1(e2091) results from decrease of sop-1 gene function, rather than from some special property of the four suppressor mutations. This is consistent with the recessive nature of these mutations. The experiment does not indicate whether or not sop-1 is an essential gene, because we do not know whether gene function was eliminated completely in every cell by the RNAi technique.
Wild-type sop-1 blocks a bypass pathway stimulated by β-catenin
Previous studies had suggested that pal-1(e2091) could be suppressed by activation of the Wnt signal transduction pathway (Hunter et al. 1999). We therefore investigated whether the Wnt pathway was responsible for suppression of pal-1(e2091) in a sop-1 mutant background. The known components of the Wnt pathway active in the male tail are shown in Figure 6. Evidence for involvement of this pathway in ray development came from studies of pry-1. In a pry-1 mutant, the ray developmental program is activated in anterior seam cells, which as a result generate anterior rays (pry stands for polyray) (Maloof et al. 1999). Activation of the ray pathway results from the ectopic expression of mab-5 and requires the function of bar-1/β-catenin but not of the EGL-20 ligand or the LIN-17 receptor. It was therefore proposed that pry-1 acts downstream of the receptor but upstream of bar-1/β-catenin to block inappropriate activation of the Wnt signal transduction pathway.
Figure 6.

The known components of the Wnt pathway in the male seam.
Mutations in pry-1 suppress pal-1(e2091) (Hunter et al. 1999). By inference it was concluded that pal-1(e2091) could be suppressed by activation of the Wnt signal transduction pathway in V6. We therefore tested the effect of a bar-1 mutation on ray development in pal-1(e2091); sop-1. The number of rays in pal-1(e2091); sop-1(bx92) was significantly reduced by introduction of bar-1(ga80) (Table 1, line 11). bar-1(ga80) had an effect only in the presence of both the pal-1(e2091) mutation and a sop-1 mutation. bar-1(ga80) did not reduce the number of rays in pal-1(+); sop-1(+) (Hunter et al. 1999; Table 1, line 12). Therefore bar-1 is normally not required for expression of pal-1 or any other gene in the ray developmental pathway. Nor did bar-1(ga80) reduce the number of rays in pal-1(+); sop-1(bx92) (Table 1, line 13). Therefore, a sop-1 mutation does not make pal-1 or any other gene sensitive to bar-1 function.
The requirement for bar-1 only in a pal-1(e2091); sop-1 mutant provides evidence that sop-1 mutations allow pal-1 activation via a bypass pathway, rather than via reactivation of the normal pathway requiring the fifth pal-1 intron. It is unlikely that sop-1 mutations restore binding of the putative intronic factor, because the activation pathway involving this factor does not require bar-1. Rather, sop-1 mutations activate a different pathway with a different genetic requirement. In wild type, by blocking the pathway stimulated by bar-1, sop-1(+) enforces the requirement for the intronic cis-regulatory element.
Development of rays in pal-1(e2091); sop-1 was not affected by introduction of a mutation in egl-20 (Table 1, line 14). We were unable to test the effect of the ligand gene lin-44 or the receptor gene lin-17 because mutations in these genes are pleiotropic and have severe effects on tail development. lin-44 might act redundantly with egl-20 to activate the Wnt pathway in the tail, explaining why there was no effect of an egl-20 mutation. Or sop-1 mutants could activate the Wnt signal transduction pathway in a ligand-independent manner similar to the action of pry-1 mutants.
sop-1 prevents activation of the Hox gene egl-5 by bar-1
We asked whether sop-1 acted at additional steps of the ray transcription factor cascade to prevent gene activation by the Wnt signal transduction pathway. One of the later steps of the cascade is activation of the Hox gene egl-5 in V6.ppp (Ferreira et al. 1999). Normally, activation of egl-5 is dependent on the function of mab-5. However, we found that in sop-1 mutants, egl-5 was activated weakly in the absence of mab-5 gene function. Whereas in mab-5(e1239), a null allele, there are no V6 rays (rays 2–6) or V5 rays (ray 1), and alae extend through the region where the V-rays normally form (Kenyon 1986), in mab-5(e1239); sop-1(bx92) mutants, although there were no normal sets of V6 rays, in 13% of male sides alae stopped short of the ray domain and in these cases an average of 1.5 rays were generated (n = 205 sides).This effect of sop-1 required egl-5 activity, because in a mab-5(e1239) egl-5(n486); sop-1(bx92) triple mutant, alae always extended into the ray domain and there were no V rays (n = 132 sides).
To determine whether activation of egl-5 was stimulated by the Wnt signal transduction pathway, we introduced bar-1(ga80). In this background in all male sides alae extended into the tail region and no rays were generated (n = 205). Thus as it does for pal-1, sop-1 enforces regulatory stringency on egl-5. By preventing activation of egl-5 by bar-1, sop-1 makes egl-5 expression completely dependent on mab-5.
sop-1 mutations do not elevate gene expression generally
The foregoing results showing activation of pal-1 and egl-5 by sop-1 mutations might be explained if sop-1 mutations had a global effect on transcription, possibly resulting in a general elevation of gene expression. To determine whether sop-1 mutations nonselectively elevated the expression of all genes, we tested the effect of sop-1 mutations on a known expression-sensitive locus unrelated to ray development. Loss-of-function mutations of lin-15 cause a hermaphrodite multivulva (Muv) phenotype. The percentage of Muv animals in the hypomorphic background lin-15(n765ts) at 20°C is sensitive to changes in the level of global gene expression (Villeneuve and Meyer 1987). We found that the percent of lin-15(n765ts) Muv animals (>99%) was not affected by introduction of sop-1(bx92) [n = 424 for lin-15(n765ts) sop-1(+); n = 576 for lin-15(n765ts) sop-1(bx92)]. Therefore sop-1 mutations do not appear to elevate levels of gene expression generally, arguing that they act selectively on the regulation of only certain genes.
This result also shows that sop-1 differs in its action from another putative Mediator component, sur-2. Loss-of-function mutations in sur-2 suppress the effects of an activated Ras/MAP kinase pathway (Singh and Han 1995). In particular, they suppress the Muv phenotype of lin-15(n765ts). Thus Mediator contains components with selective effects in promoting the actions of different transcriptional regulatory signals.
Discussion
Model for pal-1 regulation
Our results identify a component of the Mediator complex that is required for normal regulation of genes in a postembryonic transcriptional cascade. Expression of the caudal homolog pal-1 in V6 in late embryogenesis initiates a transcription factor cascade that results in generation of rays in the late L4 male. We have shown that pal-1 can be activated in V6 either by a pathway requiring an intronic cis-regulatory element, or by a pathway involving the action of BAR-1/β-catenin. The two pathways appear to initiate the cascade equally well , producing a normal set of rays. SOP-1, a homolog of a component of the human Mediator complex, suppresses the BAR-1-stimulated pathway normally, thus making pal-1 expression dependent on the pathway that utilizes the cis-regulatory element. SOP-1 similarly makes expression of a later gene of the cascade, egl-5, entirely dependent on the transcription factor MAB-5 by suppressing the action of BAR-1/β-catenin on this gene. Thus the mode of regulation of pal-1 and egl-5 are influenced by a component of the Mediator complex.
The data presented here suggest the model for pal-1 activation shown in Figure 7. This model postulates that a transcription factor (labeled I in the figure) binds an intronic enhancer to activate pal-1 transcription (Fig. 7, top panel). The identity of this factor is unknown, as is its role in the transcription complex assembly pathway. It might act early, binding to chromatin-bound DNA to initiate the assembly pathway. Alternatively, it could act at later steps to recruit chromatin-remodeling factors or holoenzyme, or to release the preinitiation complex for transcription. We suggest that because the requirement for this factor can be overcome by mutation in a component of the Mediator, Factor I is more likely to act at one of the later steps of the pathway. We postulate that opening of the chromatin structure and recruitment of holoenzyme is accomplished by one or more other factors (Factor II in Fig. 7) as explained below.
Figure 7.
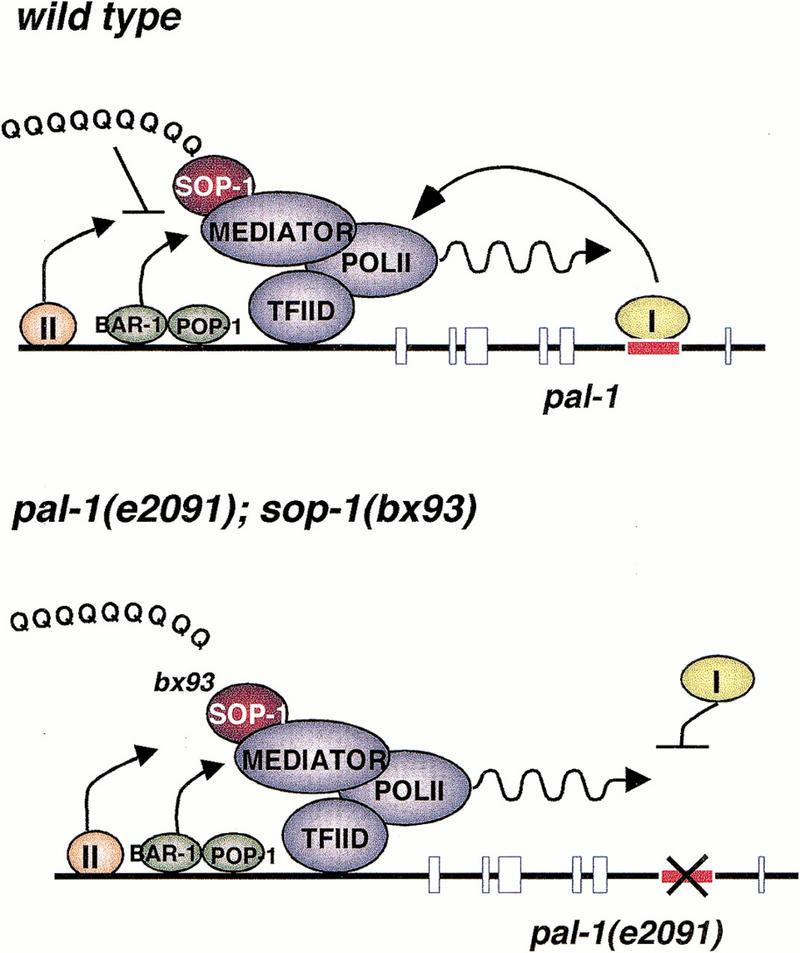
Model for pal-1 regulation. In wild type, the intronic factor I allows a preinitiation complex containing SOP-1 to transcribe pal-1. The holoenzyme, consisting of the Mediator complex and PolII-core polymerase complex are brought to the promoter through the action of other factors, here represented by factor II. Such factors, together with the general A/P specifying factors BAR-1 and POP-1, are prevented by SOP-1 from activating transcription in the absence of factor I. In a pal-1(e2091); sop-1(−) background, the negative effect of SOP-1 is reduced, allowing pal-1 transcription. The bx92, bx93, and bx107 mutations reduce or eliminate the carboxy-terminal glutamaine-rich domain of SOP-1, implicating this domain in the SOP-1-mediated repression.
In a pal-1(e2091) mutant background, the model postulates that the intronic factor cannot effectively bind to the mutated cis-regulatory element (Fig. 7, bottom panel). Under these circumstances, decrease of sop-1 gene function allows pal-1 transcription to be activated by an alternate pathway that is stimulated by BAR-1. Because BAR-1 together with the TCF/LEF family transcription factor POP-1 is likely to act on many genes to convey the anteroposterior position of a cell (Lin et al. 1998), we suggest that BAR-1 acts at a step in the assembly pathway after initiation of the pathway by a pal-1-specific factor. Initiation of the pathway is the function of the postulated Factor II (see Fig. 7). It seems unlikely that DNA binding by factors such as Factor II and BAR-1/POP-1 will be affected by mutation within the Mediator. A mutation in the Mediator would only be expected to affect recruitment of holoenzyme by DNA-bound factors or the function of holoenzyme after recruitment. Therefore, Factor II and BAR-1/POP-1 most likely also bind in wild type, as shown in Figure 7 (top panel). It is for this reason that we suggest that Factor I acts at a later step in the assembly pathway, after Factor II has initiated the assembly process. Although we show BAR-1/POP-1 as acting in the nucleus at the pal-1 promoter, we have no direct evidence for nuclear action of BAR-1. One mode of BAR-1 action appears to be cytoplasmic, involving the LIT-1 kinase (Rocheleau et al. 1999; Shin et al. 1999), and this could be the pathway that is involved in preventing expression of pal-1(e2091).
In addition, the model assumes that the effects of SOP-1 mutations occur at the pal-1 promoter, but it is formally possible that they occur at the promoter of another gene that acts as an alternate pal-1 activator.
C. elegans Mediator complex
Analysis of the C. elegans genome sequence reveals homologs of 11 components of the mammalian and yeast Mediator complexes (Boyer et al. 1999; Ito et al. 1999; Kwon et al. 1999; this work, data not shown). An additional seven components of the mammalian complex (TRAP220, TRAP150, TRAP100, TRAP95, TRAP80, CRSP34, CRSP70) are not represented in the C. elegans genome sequence (Ito et al. 1999; this work). A functional role in developmental gene regulation has been defined for one of the genes conserved between C. elegans and humans, sur-2. Loss-of-function mutations of sur-2 were identified as suppressors of an activated Ras/MAP kinase pathway (Singh and Han 1995). Biochemical studies have shown that hSur-2 (human sur-2) is a binding target of the adenovirus E1A transcription factor, as well as of the Ras/MAP kinase-activated transcription factor Elk-1 (Boyer et al. 1999). However, hSur-2 did not appear to be involved in gene activation by Gal4-VP16, a transcription factor with a transcriptional activator domain of a different class. Therefore SUR-2/hSur-2 appears to be a Mediator target that is involved selectively in gene activation by the Ras/MAP kinase pathway. Two additional selective transcription factor targets have been identified in in vitro studies of human Mediator. TRAP220 is a binding and activation target of TR (thyroid hormone receptor) and p53, whereas TRAP80 is a binding and activation target of VP16 and p53 (Ito et al. 1999).
Our results suggest that SOP-1/TRAP230 may be a Mediator target of pathways regulating transcriptional response to the Wnt pathway. Because sop-1 appears to block action of the Wnt signal transduction pathway, and because it is expressed widely, its effect may have to be relieved wherever the Wnt pathway acts. In this role, SOP-1 may act as an integrator of multiple signaling pathways impinging on the nucleus. One pathway known to act in concert with the Wnt signal transduction pathway in C. elegans is the Ras/MAP kinase pathway. Ras/MAP kinase and Wnt/bar-1/β-catenin act together to activate the Hox gene lin-39 in vulval development (Eisenmann et al. 1998), and the Hox gene egl-5 in the P12 neuroectoblast cell (Jiang and Sternberg 1998). The Ras/MAP kinase pathway does not appear to be involved in development of the rays, and as noted above, sop-1, unlike sur-2, does not suppress an activated ras pathway. Therefore, sop-1 may integrate one or more non-Ras/MAP kinase pathway with the Wnt pathway.
The glutamine-rich domain of human TRAP230 functions as a transcriptional activation domain (Ito et al. 1999). This is in contrast to the results reported here in which the glutamine-rich domain appears to function to repress transcription. Mutation of human TRAP230 affecting the glutamine-rich region results in an elevated susceptibility to mental retardation in males (Philibert et al. 1998). Our results suggest that altered cell fate specification within the central nervous system, consequent on misexpression of such key cell-fate specification genes as caudal and Hox genes, could be the cause of this condition.
Activation of ray development by the Wnt pathway
The pathway that sop-1 integrates with the Wnt pathway may be a signaling pathway regulated by cell contacts. Expression of the ray developmental program is regulated by the contacts that seam cells make with their neighbors. Normally, rays develop from only the three most posterior cells in two bilateral rows of nine seam cells, and not from seam cells in the anterior part of the body (Sulston and Horvitz 1977). If, however, anterior seam cells lose contact with their neighbors, they can undergo a cell fate transformation and generate rays (Sulston and White 1980; Austin and Kenyon 1994; Hunter et al. 1999). A similar effect of neighbor-cell contact is seen in pal-1(e2091). In a pal-1(e2091) background, where rays are not generated by V6, the ray developmental program is activated in V6 if the posterior neighbor of V6 is ablated (Waring and Kenyon 1990). Activation of ray development following such neighbor-cell ablation, like activation by mutation of sop-1, is stimulated by the function of bar-1 (Hunter et al. 1999). Thus, it is possible that the same pathway is activated both by loss of cell contact and by sop-1 mutation. As we have shown, in a normal cellular context, bar-1 is inhibited by sop-1. After cell ablation, SOP-1 action could be blocked by an inhibitory signal generated as a result of loss of cell contact.
Unlike the alternate pathway in a sop-1 background, the alternate pathway activated by loss of cell contact, both in the anterior seam in wild-type and in V6 in pal-1(e2091), requires the Wnt ligand EGL-20 (Hunter et al. 1999). One possible explanation for the absence of EGL-20-dependence of the sop-1 pathway is that EGL-20 and LIN-44, the two Wnt ligands known to be expressed in the tail, normally act redundantly on V6. After T-cell ablation, access of LIN-44 to V6 might be blocked by cell debris or damage, thus giving rise to an artifactual dependence on EGL-20 in this experiment. Alternatively, activation of bar-1 in sop-1 mutants may be ligand independent. In this case, one effect of the normal context of cellular contacts may be to activate BAR-1/β-catenin.
Materials and methods
Nematodes
Strain maintenance, mutagenesis, and genetic analysis followed Brenner (1974); nematodes were grown at 20°C unless otherwise noted. Most strains carried the him-5(e1490) mutation, which gives a high frequency of males in selfing populations. The following additional mutations or strains were used (see Hodgkin 1997): LGII: tra-2(q276)/unc-4(e120) dpy-10(e128); LGIII: pal-1(e2091), pal-1(ct224), mab-5(e1239); LGIV: egl-20(n585); LGX: dpy-6(e14), egl-15(n484), bar-1(ga80). Rearrangement: sDP3, a free duplication covering the left portion of LGIII including pal-1.
Identification of pal-1(e2091)
To localize the mutation in pal-1(e2091) either to the 5′ flanking promoter region or to the coding region, introns, and 3′ flanking region, we amplified the pal-1 gene into two overlapping DNA fragments with the following primers (nucleotide numbers are from cosmid C38D4, GenBank accession no. Z46241): 5′ fragment—(31080–31055) 5′-ACCTGGTCGATACACTCAACGTGTGC-3′; (26622–26641) 5′-AATGAGACAGGTCGGCAAGG-3′; 3′ fragment—(28157–28131) 5′-GGAGCACCTGTTGATGCTGCAAAGGAG-3′; (21737–21762) 5′-GTCTCGAAGCGGCTCAAAATGTGGAG-3′ (Fig. 2A). Both fragments were amplified from either wild-type or pal-1(e2091) worms and digested with NcoI, which cut at position 28093. The 2.9-kb 5′ fragment (31080–28093) and 6.4-kb 3′ fragment (28093–21737) were gel-purified and ligated, and injected into pal-1(e2091) animals together with pRF4, which carries the dominant rol-6 (su1006) marker (Mello et al. 1991). F1 and F2 Rol males were scored for ray phenotype. The 3′ fragment from pal-1(e2091) failed to support rescue, showing that the mutation was downstream of most of the promoter region (Fig. 2A). Therefore, genomic DNA from pal-1(e2091) including all of the exons, introns, and ∼1 kb of 3′ UTR was amplified and sequenced. The only differences from wild type were the two point mutations in intron 5 shown in Figure 2B.
Sequence of intron 5 from C. briggsae
The sequence of C. briggsae pal-1 cDNA was kindly provided by C. Hunter (Harvard University, Cambridge, MA). Primers lying within regions corresponding to the fifth and sixth C. elegans exons (5′-TCGGCTTTCATAACTTCGGATCG-3′, 5′-GTGCTTCAGACATCTTAGAGTCG-3′) were used to amplify C. briggsae genomic DNA and the amplification product was sequenced.
Isolation and characterization of bx89, bx92, bx93, bx103, and bx107
sop-1 mutations were isolated as genetic suppressors of the V6 ray loss phenotype of pal-1(e2091). F2 or F3 male progeny of EMS-treated pal-1(e2091); him-5(e1490) hermaphrodites were screened for the presence of V6 rays; mutations were recovered from sibling hermaphrodites. Approximately 4000 haploid genomes were screened, and 19 suppressor mutations in >10 genes were recovered, including the intragenic revertant bx89.
bx89 is an intragenic revertant of pal-1(e2091) by the following evidence. First, it is a fully penetrant dominant suppressor. Second, it mapped close to pal-1. Third, one of the two sequence differences between pal-1(e2091) and wild-type (T24264C) is reverted in pal-1(e2091 bx89) (Fig. 2B).
sop-1 mutants are rescued by maternal gene function: All male progeny from tra-2/unc-4 dpy-10; pal-1(e2091); sop-1/+ hermaphrodites are Pal, whereas 25% are expected to be of genotype tra-2; pal-1(e2091); sop-1 and hence suppressed.
bx92, bx93, bx103, and bx107 were mapped by three-factor crosses to a single region on LG X between the cloned genes dpy-6 and egl-15. Complementation tests were performed as follows. tra-2; pal-1(e2091); sop-1(a) males were mated to dpy-10 unc-4; pal-1(e2091); him-5; sop-1(b) hermaphrodites. Non-Dpy nonUnc hermaphrodite progeny were picked [genotype tra-2/dpy-10 unc-4; pal-1; him-5/+; sop-1(a)/sop(b)] and allowed to self. F2 males (homozygous for tra-2) were scored. Because sop-1 is rescued maternally, complementation should have resulted in nearly 100% Pal males, but <50% were observed. (For unknown reasons, the penetrance of sop-1 suppression in tra-2 mutant males is lower than in him-5 males.)
Antibodies and immunostaining
Simultaneous immunohistochemical staining of PAL-1 and MAB-5 followed the method of Hunter et al. (1999). MAB-5 was detected with mouse anti-β-GAL antibody (40-1a, J.R. Sanes, Johns Hopkins University, Baltimore, MD) in a strain (EM512) of genotype pal-1(e2091); sop-1(bx92); muIs3. muIS3 is an integrated transgenic array consisting of mab-5::lacZ and the transformation marker pRF4 (Cowing and Kenyon 1992). Secondary antibody was labeled with FITC. Rabbit anti-PAL-1 antibodies were kindly provided by C. Hunter and were detected by means of secondary antibody labeled with Cy3.
Cloning of sop-1
sop-1 was cloned by complementation rescue. Cosmids from the genetic region of sop-1 (A. Coulson, Sanger Center, Cambridge, UK) were injected into pal-1(e2091); sop-1(bx92) hermaphrodites at a concentration of 20–50 ng/μl (Fig. 4A). pRF4 was coinjected at a concentration of 100–200 ng/μl. F2 Rol males were scored for presence of V6 rays. Cosmid F47A4 restored the ray loss phenotype of the unsuppressed pal-1 background. Strong rescuing activity (86% of sides with Pal phenotype compared with 6% in uninjected) was localized to a 14-kb HpaI–XhoI fragment (6081–20759) (Fig. 4B).
sop-1 gene structure from exon 9 to the end was confirmed by sequencing cDNA ESTs yk266a1, yk542c11, yk495a10, and yk492f6 kindly provided by Y. Kohara (MIG, Japan). These cDNA ESTs cover the region 14967–7040.
Because of its large size, sop-1 was cloned in two fragments: the XhoI–EagI DNA fragment of F47A4 (20759–13973) was cloned into pBluescript (EM#293); the HpaI–BglII DNA fragment of F47A4 (6081–17057) was cloned into EcoRV–BamHI sites of pBR322 (EM#294). To reconstruct the gene, the 8.4-kb EagI–PpuMI fragment of EM#293 (20759–15306) and the 10.3-kb BglI–PpuMI fragment of EM#294 (6081–15306) were gel purified, ligated, and injected.
The 5′ upstream sequence and the first 35 amino acids were deleted by cleaving EM#293 with EcoRV and PpuMI. The resulting 3.5-kb fragment (18837–15306) was gel purified, ligated to PpuMI-cleaved EM#294, and tested for rescuing activity. Similarly, a carboxy-terminal deletion was constructed by cleaving EM#294 with BamHI and PpuMI. The resulting 7.8-kb fragment, which lacked the 3′ UTR plus the 41 carboxy-terminal amino acids (7494–15306) was gel purified, ligated to PpuMI-cleaved EM#293, and tested for rescue. Frameshift mutations were introduced into the amino terminal region by filling in with Klenow enzyme either the XbaI site (18089) (EM#295) or the NcoI site (16480) (EM#296) of EM#293 and religating the blunt ends. The resulting fragments were cleaved with PpuMI, ligated to PpuMI-cleaved EM#294, and tested for rescue.
Mapping the location of mutations in bx92, bx93, bx103, and bx107
To molecularly identify the sop-1 mutations, they were first localized to regions of the sop-1 gene, as follows. sop-1 was amplified from mutants in two overlapping fragments. Fragment 1, ∼6 kb, covered the amino-terminal portion of the gene [primers: 5′-GGCGTTTCAGATCAACGAGAACCG-3′ (20834–20811), 5′-AGCAATCTTGTCGTCAACTGCCTTC-3′ (14541–14565)], while fragment 2, ∼10 kb, covered the carboxy-terminal portion [primers: 5′-TCTCAAGCATGGTGCAGTACATGG-3′ (16005–15982), 5′-GCTCAAGCGTTATCTTGATGACGC-3′ (6041–6064)]. Each fragment was digested with PpuMI and ligated as appropriate with either the wild-type 10.4-kb BglI– PpuMI fragment of EM#294 or the wild-type 8.4-kb EagI–PpuMI fragment of EM#293 and tested for rescuing activity. PCR amplification products that failed to support rescue were directly sequenced.
RNAi experiments
RNAi experiments were performed as described by Fire et al. (1998). The template DNA fragment, covering the ninth exon of sop-1 (15724–14416), was amplified with the following primer pair: 5′-CACTAGAATTAACCCTCACTAAAGGGTCGTCGGAAATATACATTCGCAGCAGG-3′, 5′-CACTAGTAATACGACTCACTATAGGGGATCATTCCGTGCGACTTACATCGGC-3′. The amplification product was transcribed using MEGAscript T3 and T7 kit (Ambion). About 200 ng/μl dsRNA was injected into pal-1(e2091); him-5 or him-5 hermaphrodites. Eggs laid between 4 to 24 hr after injection were collected at 12 hr intervals.
sop-1 reporter genes
EM#290 contained 2.2 kb of sop-1 5′-upstream region and the first four exons. It was constructed by joining the XbaI fragment of F47A4 (21136–18089) in-frame into the XbaI site of pPD95.67, which contains an SV40 nuclear localization signal, gfp coding sequence, and unc-54 3′ UTR (for structure of gfp vectors, see Fire laboratory vector kit). EM#291 contained a gfp in-frame fusion at the carboxy-terminal NotI site (8918). It was constructed by inserting the 1-kb NotI fragment of pPD103.87 into the NotI site of EM#294. EM#292 contained a gfp in-frame insertion at the amino-terminal XbaI site (18089). It was constructed by inserting the 1-kb XbaI fragment of pPD102.33 into the XbaI site of EM#293. Before transformation, EM#291 and EM#292 were cleaved with PpuMI and ligated to PpuMI-cleaved EM#293 or EM#294, respectively.
Acknowledgments
We thank C. Hunter for anti-PAL-1 antibody, the C. briggsae pal-1 cDNA sequence, and for insightful discussions. We thank L. Edgar for pal-1(ct224), and pSC16. We are grateful for helpful discussions and comments on the manuscript provided by G. Kalpana, G. Prelich, Y. Cang, and members of the Emmons laboratory. Some nematode strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources. This work was supported by a grant from the National Institutes of Health (R01 GM39353). S.W.E. is the Siegfried Ullmann Professor of Molecular Genetics.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL emmons@aecom.yu.edu; FAX (718) 430-8778.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.814700.
References
- Austin J, Kenyon C. Cell contact regulates neuroblast formation in the Caenorhabditis elegans lateral epidermis. Development. 1994;120:313–324. doi: 10.1242/dev.120.2.313. [DOI] [PubMed] [Google Scholar]
- Björklund S, Almouzni G, Davidson I, Nightingale KP, Weiss K. Global transcription regulators of eukaryotes. Cell. 1999;96:759–767. doi: 10.1016/s0092-8674(00)80586-3. [DOI] [PubMed] [Google Scholar]
- Boyer TG, Martin MED, Lees E, Ricciardi RP, Berk AJ. Mammalian Srb/Mediator complex is targeted by adenovirus E1A protein. Nature. 1999;399:276–279. doi: 10.1038/20466. [DOI] [PubMed] [Google Scholar]
- Brenner S. The Genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M. Genetics of transcriptional regulation in yeast: Connections to the RNA polymerase II CTD. Annu Rev Cell Dev Biol. 1997;13:1–23. doi: 10.1146/annurev.cellbio.13.1.1. [DOI] [PubMed] [Google Scholar]
- Chow KL, Emmons SW. HOM-C/Hox genes and four interacting loci determine the morphogenetic properties of single cells in the nematode male tail. Development. 1994;120:2579–2593. doi: 10.1242/dev.120.9.2579. [DOI] [PubMed] [Google Scholar]
- Costa M, Weir M, Coulson A, Sulston J, Kenyon C. Posterior pattern formation in C. elegans involves position-specific expression of a gene containing a homeobox. Cell. 1988;55:747–756. doi: 10.1016/0092-8674(88)90131-6. [DOI] [PubMed] [Google Scholar]
- Cowing D, Kenyon C. Expression of the homeotic gene mab-5 during Caenorhabditis elegans embryogenesis. Development. 1992;116:481–490. doi: 10.1242/dev.116.2.481. [DOI] [PubMed] [Google Scholar]
- Eisenmann DM, Maloof JN, Simske JS, Kenyon C, Kim SK. The β-catenin homolog BAR-1 and LET-60 Ras coordinately regulate the Hox gene lin-39 during Caenorhabditis elegans vulval development. Development. 1998;125:3667–3680. doi: 10.1242/dev.125.18.3667. [DOI] [PubMed] [Google Scholar]
- Emmons SW. Cell fate determination in Caenorhabditis elegans ray development. In: Moody SA, editor. Cell lineage and fate determination. San Diego, CA: Academic Press; 1999. pp. 139–155. [Google Scholar]
- Ferreira HB, Zhang Y, Zhao C, Emmons SW. Patterning of Caenorhabditis elegans posterior structures by the Abdominal-B homolog, egl-5. Dev Biol. 1999;207:215–228. doi: 10.1006/dbio.1998.9124. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Herman MA, Vassilieva LL, Horvitz HR, Shaw JE, Herman RK. The C. elegans gene lin-44, which controls the polarity of certain asymmetric cell divisions, encodes a Wnt protein and acts cell nonautonomously. Cell. 1995;83:101–110. doi: 10.1016/0092-8674(95)90238-4. [DOI] [PubMed] [Google Scholar]
- Hodgkin J. Appendix 1, Genetics. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 881–1047. [Google Scholar]
- Hunter CP, Kenyon C. Spatial and temporal controls target pal-1 blastomere-specification activity to a single blastomere lineage in C. elegans embryos. Cell. 1996;87:217–226. doi: 10.1016/s0092-8674(00)81340-9. [DOI] [PubMed] [Google Scholar]
- Hunter CP, Harris JM, Maloof JN, Kenyon C. Hox gene expression in a single Caenorhabditis elegans cell is regulated by a caudal homolog and intercellular signals that inhibit Wnt signaling. Development. 1999;126:805–814. doi: 10.1242/dev.126.4.805. [DOI] [PubMed] [Google Scholar]
- Ito M, Yuan CX, Malik S, Gu W, Fondell JD, Yamamura S, Fu ZY, Zhang X, Qin J, Roeder RG. Identity between TRAP and SMCC complexes indicates novel pathways for the function of nuclear receptors and diverse mammalian activators. Mol Cell. 1999;3:361–370. doi: 10.1016/s1097-2765(00)80463-3. [DOI] [PubMed] [Google Scholar]
- Jiang LI, Sternberg PW. Interactions of EGF, Wnt, and HOM-C genes specify the P12 neuroectoblast fate in C. elegans. Development. 1998;125:2337–2347. doi: 10.1242/dev.125.12.2337. [DOI] [PubMed] [Google Scholar]
- Kenyon C. A gene involved in the development of the posterior body region of C. elegans. Cell. 1986;46:477–487. doi: 10.1016/0092-8674(86)90668-9. [DOI] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- Kwon JY, Park JM, Gim BS, Han SJ, Lee J, Kim Y. Caenorhabditis elegans Mediator complexes are required for develomental-specific transcriptional activation. Proc Natl Acad Sci. 1999;96:14990–14995. doi: 10.1073/pnas.96.26.14990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Hill RJ, Priess JR. POP-1 and anterior-posterior fate decisions in C. elegans embryos. Cell. 1998;92:229–239. doi: 10.1016/s0092-8674(00)80917-4. [DOI] [PubMed] [Google Scholar]
- Lints R, Emmons SW. Patterning of dopaminergic neurotransmitter identity among Caenorhabditis elegans ray sensory neurons by a TGFβ family signaling pathway and a Hox gene. Development. 1999;126:5819–5831. doi: 10.1242/dev.126.24.5819. [DOI] [PubMed] [Google Scholar]
- Maloof JN, Whangbo J, Harris JM, Jongeward GD, Kenyon C. A Wnt signaling pathway controls Hox gene expression and neuroblast migration in C. elegans. Development. 1999;126:37–49. doi: 10.1242/dev.126.1.37. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlodzik M, Gehring WJ. Expression of the caudal gene in the germ line of Drosophila: Formation of an RNA and protein gradient during early embryogenesis. Cell. 1987;48:465–478. doi: 10.1016/0092-8674(87)90197-8. [DOI] [PubMed] [Google Scholar]
- Philibert RA, King BH, Winfield S, Cook EH, Lee YH, Stubblefield B, Damschroder-Williams P, Dea C, Palotie A, Tengstrom C, et al. Association of an X-chromosome dodecamer insertional variant allele with mental retardation. Mol Psychiatry. 1998;3:303–309. doi: 10.1038/sj.mp.4000442. [DOI] [PubMed] [Google Scholar]
- Rocheleau CE, Yasuda J, Shin TH, Lin R, Sawa H, Okano H, Priess JR, Davis RJ, Mello CC. WRM-1 activates the LIT-1 protein kinase to transduce anterior/posterior polarity signals in C. elegans. Cell. 1999;97:717–726. doi: 10.1016/s0092-8674(00)80784-9. [DOI] [PubMed] [Google Scholar]
- Salser SJ, Kenyon C. A C. elegans Hox gene switches on, off, on and off again to regulate proliferation, differentiation and morphogenesis. Development. 1996;122:1651–1661. doi: 10.1242/dev.122.5.1651. [DOI] [PubMed] [Google Scholar]
- Shin TH, Yasuda J, Rocheleau CE, Lin R, Soto M, Bei Y, Davis RJ, Mello CC. MOM-4, a MAP kinase kinase kinase-related protein, activates WRM-1/LIT-1 kinase to transduce anterior/posterior polarity signals in C. elegans. Mol Cell. 1999;4:275–280. doi: 10.1016/s1097-2765(00)80375-5. [DOI] [PubMed] [Google Scholar]
- Singh N, Han M. sur-2, a novel gene, functions late in the let-60 ras-mediated signaling pathway during Caenorhabditis elegans vulval induction. Genes & Dev. 1995;9:2251–2265. doi: 10.1101/gad.9.18.2251. [DOI] [PubMed] [Google Scholar]
- Struhl K. Fundamentally different logic of gene regulation in eukaryotes and prokaryotes. Cell. 1999;98:1–4. doi: 10.1016/S0092-8674(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode Caenorhabditis elegans. Dev Biol. 1977;56:111–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Sulston JE, White JG. Regulation and cell autonomy during postembryonic development of Caenorhabditis elegans. Dev Biol. 1980;78:577–597. doi: 10.1016/0012-1606(80)90353-x. [DOI] [PubMed] [Google Scholar]
- Villeneuve AM, Meyer BJ. sdc-1: A link between sex determination and dosage compensation in C. elegans. Cell. 1987;48:25–37. doi: 10.1016/0092-8674(87)90352-7. [DOI] [PubMed] [Google Scholar]
- Wang BB, Müller-Immergluck MM, Austin J, Robinson NT, Chisholm A, Kenyon C. A homeotic gene cluster patterns the anteroposterior body axis of C. elegans. Cell. 1993;74:29–42. doi: 10.1016/0092-8674(93)90292-x. [DOI] [PubMed] [Google Scholar]
- Waring DA, Kenyon C. Selective silencing of cell communication influences anteroposterior pattern formation in C. elegans. Cell. 1990;60:123–131. doi: 10.1016/0092-8674(90)90722-q. [DOI] [PubMed] [Google Scholar]
- ————— Regulation of cellular responsiveness to inductive signals in the developing C. elegans nervous system. Nature. 1991;350:712–715. doi: 10.1038/350712a0. [DOI] [PubMed] [Google Scholar]
- Whangbo J, Kenyon C. A Wnt signaling system that specifies two patterns of cell migration in C. elegans. Mol Cell. 1999;4:851–858. doi: 10.1016/s1097-2765(00)80394-9. [DOI] [PubMed] [Google Scholar]
- Wrischnik LA, Kenyon CJ. The role of lin-22, a hairy/enhancer of split homolog, in patterning the peripheral nervous system of C. elegans. Development. 1997;124:2875–2888. doi: 10.1242/dev.124.15.2875. [DOI] [PubMed] [Google Scholar]
- Zhao C, Emmons SW. A transcription factor controlling development of peripheral sense organs in C. elegans. Nature. 1995;373:74–78. doi: 10.1038/373074a0. [DOI] [PubMed] [Google Scholar]