

Abstract
The 9-cis-retinoic acid receptors (RXRα, RXRβ, and RXRγ) are nuclear receptors that play key roles in multiple hormone-signaling pathways. Biochemical data indicate that, in the absence of ligand, RXR can exist as an inactive tetramer and that its dissociation, induced by ligand, is important for receptor activation. In this article we report the inactivated tetramer structures of the RXRα ligand-binding domain (LBD), either in the absence of or in the presence of a nonactivating ligand. These structures reveal that the RXR LBD tetramer forms a compact, disc-shaped complex, consisting of two symmetric dimers that are packed along helices 3 and 11. In each monomer, the AF-2 helix protrudes away from the core domain and spans into the coactivator binding site in the adjacent monomer of the symmetric dimer. In this configuration, the AF-2 helix physically excludes the binding of coactivators and suggests an autorepression mechanism that is mediated by the AF-2 helix within the tetramer. The RXR–tetramer interface is assembled from amino acids that are conserved across several closely related receptors, including the HNF4s and COUP transcription factors, and may therefore provide a model for understanding structure and regulation of this subfamily of nuclear receptors.
Keywords: Crystal structure, nuclear receptors, RXR LBD, tetramer
The three retinoid X receptors (RXRα, RXRβ, and RXRγ) are nuclear receptors (NRs) for the vitamin A metabolite, 9-cis-retinoic acid (9cRA), and are involved in a broad spectrum of biological processes, including cell growth, differentiation, metabolism, morphogenesis, and embryogenic development (Mangelsdorf et al. 1993; Giguere 1994). When bound with 9cRA, RXRs can bind to DNA and activate transcription as homodimers (Mangelsdorf et al. 1992). In addition, RXRs also serve as obligate heterodimer partners for many of the subfamily 1 NRs (Committee 1999), including receptors for retinoic acid (RAR), thyroid hormone (TR), vitamin D (VDR), and peroxisome proliferator activators (PPARs) (Mangelsdorf and Evans 1995). Thus RXRs play an essential role in multiple nuclear-hormone-signaling pathways.
As members of the NR superfamily, RXRs are primarily made up of two modular domains: a central DNA-binding domain (DBD) and a carboxy-terminal ligand-binding domain (LBD). In addition to its role in binding of ligands, the LBD contains dimerization motifs and an activation function 2 (AF-2) domain, which requires an integral helical structure (the AF-2 helix) at the carboxyl terminus. In the absence of an activating ligand (the inactive state), some receptors also repress transcription of target genes through interactions with corepressor proteins such as nuclear corepressor (N-CoR) (Horlein et al. 1995) and silencing mediator for retinoid and thyroid receptors (SMRT) (Chen and Evans 1995). Ligand binding induces a conformational change of the AF-2 helix that allows release of corepressor proteins and formation of a charge clamp that is capable of recruiting coactivator complexes formed by proteins from the p160 family or the DRIP/TRAP family (Freedman 1999). These events initiated by ligand binding eventually lead to activation of target genes and subsequent effects on various physiological processes.
Crystal structures of either apo- or ligand-bound LBDs have been determined for several NRs (Bourguet et al. 1995; Renaud et al. 1995; Wagner et al. 1995; Brzozowski et al. 1997; Nolte et al. 1998; Williams and Sigler 1998; Xu et al. 1999). In summary, these crystal structures reveal that the LBDs fold into a three-layered α-helical sandwich that envelopes a hydrophobic ligand-binding pocket. In the apo-RXR structure, the AF-2 helix extends downward from the body of the LBD and leaves an entry point for the ligand. In contrast, all of the agonist-bound structures have the AF-2 helix packed against the body of the receptor, forming an essential part of the charge clamp for recruitment of coactivator proteins (Darimont et al. 1998; Nolte et al. 1998; Shiau et al. 1998). Based on these observations, a mousetrap model was proposed for ligand binding and activation of NRs (Renaud et al. 1995). In this model, the ligand would enter the pocket through the entry point left by the AF-2 helix. With the ligand bound, the AF-2 helix would move into its active position to cover the entry of the pocket and generate a receptor that is competent to recruit coactivators. Our recent structure of 9cRA bound to RXRα in the PPARγ/RXRα heterodimer shows that the activated receptor has the AF-2 helix folded into the active position, thereby validating the mousetrap hypothesis for RXR (Gampe et al. 2000). However, in the apo-structures of PPARγ (Nolte et al. 1998) and PPARδ (Xu et al. 1999), the AF-2 helix does not rotate away from the LBD body as seen in the apo-RXR. Instead, the AF-2 helix in the apo-PPARs is packed against the body of LBD in a position that approximates an active conformation, suggesting that PPAR receptors provide an alternative entry point for their ligands. Indeed a channel that could serve as a ligand entry point exists between helix 3 and the β-strands. The bound ligand would further stabilize the active receptor conformation through direct hydrogen bonds with the AF-2 helix. Thus it appears that there are different ligand-dependent mechanisms for activating different NRs.
In addition to the above mechanisms of ligand-dependent activation, ligand binding can also induce rearrangement of the oligomeric structures of RXR. In vitro studies have indicated that the RXR LBD self-associates into tetramers with high affinity (Kd = ∼3–5 nm for dimer–dimer association) and suggest that at endogenous levels, RXR could exist predominately in the tetrameric form in the absence of ligand (Kersten et al. 1995a). Because titration with 9cRA induces rapid dissociation of RXR tetramers into dimers and monomers (Kersten et al. 1995b,c), it was proposed that the RXR tetramer is transcriptionally inactive and that ligand-induced dissociation of the tetramer might be the first step toward activating RXR (Kersten et al. 1997). This proposal is consistent with the transcriptional activities of RXR mutants that change the properties of the tetramer formation (Kersten et al. 1998). For example, a mutated mouse RXRα tetramer (R321A) fails to dissociate upon ligand binding and is also defective in ligand-dependent activation (Kersten et al. 1998). In contrast, an alternative mutant RXRα (F318A) that fails to form a stable tetramer has the opposite phenotype and shows high levels of transcriptional activity even in the absence of ligand. Tetramers of RXR also have a functional role in the regulation of the cellular retinol-binding protein II (CRBPII) gene (Mangelsdorf et al. 1991). RXR tetramers effectively regulated transcription through various CRBPII response elements that were poorly recognized by RXR dimers. Moreover, the cooperative formation of RXR tetramers on the CRBPII promoter was shown to be essential for the activation of this gene by 9cRA (Chen and Privalsky 1995). Together, these results establish the existence of an RXR tetramer and support its physiological role in gene regulation.
The previous 2.7 Å structure of the apo-RXRα LBD (Bourguet et al. 1995) did not reveal a tetrameric arrangement that is consistent with the in vitro data, nor does it provide structural insights that are related to understanding autorepression or gene regulation. Here we report crystal structures of the RXR LBD tetramer, either with or without a nonactivating isomer of retinoic acid at resolutions of 2.0 Å and 2.5 Å, respectively. These structures elucidate the molecular interactions in the RXR tetramer that are consistent with biochemical and mutagenesis studies and they suggest an autorepression mechanism of the tetramer in which the AF-2 helix blocks coactivator binding by occupying the coactivator- binding site of the adjacent dimer. Moreover, the tetramer interface is assembled from residues that are conserved across a subfamily of receptors related to RXR and may therefore provide a model for understanding the structures and regulation of this subfamily of NRs.
Results
Structure determination and the tetramer organization
The RXR LBD was crystallized in the P21 space group with four LBD monomers in each asymmetry unit (see Materials and Methods). Molecular replacement solutions were obtained using the previous 2.7 Å apo-RXR structure (Bourguet et al. 1995), but these solutions failed to produce an interpretable electron density map for many regions of the protein, including the AF-2 helix. Independent phase information was subsequently obtained by the combined use of multiple isomorphous replacement (MIR) and multiwavelength anomalous diffraction (MAD) with data from several derivative crystals containing xenon and/or selenomethionines (Table 1). The experimental map showed clear density for the entire chain of all four monomers except for the H2 helix of each LBD. H2 is also absent from the RXR LBD in the PPARγ/RXRα heterodimer structure (Gampe et al. 2000). The data and refinement statistics are summarized in Table 1.
Table 1.
Statistics of data sets and refinement
| Data sets
|
Tetramer
|
tRA–isomer complex
|
Xe
|
Xe/SeMET
|
SeMET L1
|
SeMET L2
|
SeMET L3
|
|---|---|---|---|---|---|---|---|
| Wavelength (Å) | 1.0 | 1.0 | 1.54 | 1.54 | 0.979338 | 0.979475 | 0.942129 |
| (maximum) | (inflection) | (remote) | |||||
| Resolution (Å) | 2.5 | 2.0 | 2.1 | 2.5 | 2.2 | 2.3 | 2.2 |
| Unique reflections | 32,492 | 64,577 | 52,850 | 29,526 | 96,905 | 85,305 | 92,846 |
| Completeness (%) | 99.8 | 99.8 | 93.8 | 90.2 | 99.5 | 99.8 | 98.1 |
| I/ς | 20.3 | 23.5 | 25.1 | 11.5 | 22.7 | 21.2 | 14.6 |
| Rsyma (%) | 7.2 | 5.0 | 4.8 | 9.9 | 5.4 | 4.9 | 9.6 |
| Risob (%) | — | — | 21.6 | 25.6 | 16.4 | 16.5 | 20.6 |
| <PP>c (acen) iso/ano | — | — | 1.32/0.90 | 1.97/0.91 | 3.02/2.57 | 3.03/2.95 | 2.68/1.56 |
| RCulla (acen) iso/ano | — | — | 0.84/0.80 | 0.80/0.80 | 0.61/0.68 | 0.62/0.67 | 0.73/0.87 |
| Refinement statistics: | |||||||
| R factord (%) (2 ς) | 21.89 23.14e | ||||||
| R free (%) (2 ς) | 27.09 25.41 | ||||||
| r.m.s.d. bond: | |||||||
| lengths (Å) | 0.007 0.008 | ||||||
| angles (degrees) | 1.26 1.38 |
r.m.s.d is the root mean square deviation from ideal geometry.
Rsym = Σ | Iavg − Ii |/Σ Ii.
Riso = Σ | FP − FPH |/Σ FP.
<PP>: Phasing Power = Σ | FPH | / |FPHobs| − FPHcalc|.
Rfactor = Σ | FB − FPcalc|/ΣFP, where Fp and Fpcale are observed and calculated structure factors, Rfree is calculated from a randomly chosen 10% of reflections that were not used for refinement (Brunger et al. 1992b) and Rfactor is calculated for the remaining 90% of reflections.
The underlined numbers are statistics for the tetramer/tRA–isomer complex.
Differing from the previous apo-RXR dimer structure (Bourguet et al. 1995), the four RXR LBD monomers form a compact, disc-shaped tetramer with two symmetric homodimers packed bottom-to-bottom against each other (Fig. 1A). The RXR LBD contains 13 α-helices and two β-strands that are folded into the helical “sandwich” seen in other NRs (Fig. 1B). Each monomer occupies one-fourth of the disc-shaped complex with the dimer interface mainly consisting of H10 of each monomer. The H11 helix of each monomer in the upper dimer (blue and yellow) is packed against the corresponding H11 helix in the lower dimer (white and pink), forming the core of the tetramer interface. The AF-2 helix follows H11, protruding outward from each LBD into the coactivator binding site of the corresponding monomer in the lower dimer. The orientation of the AF-2 helix confirms that each monomer represents an inactive conformation. Thus, the RXR tetramer structure is an autorepressed complex, assembled from two symmetric dimers that cross-repress each other through the extended AF-2 helices.
Figure 1.
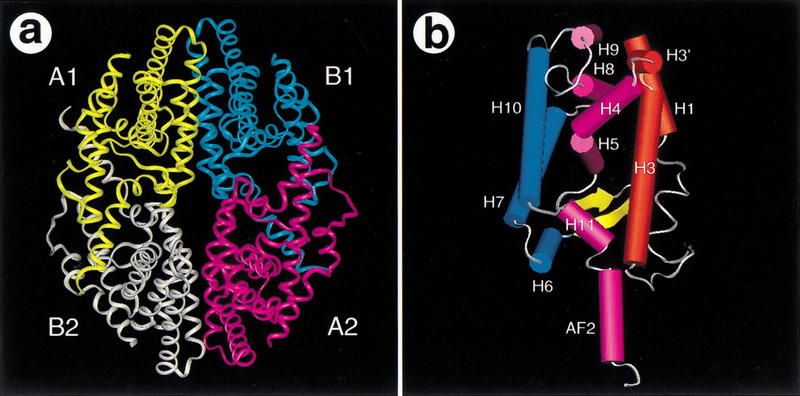
The RXR tetramer. (A) An overview of the RXR tetramer is shown with a uniquely colored ribbon for each protein monomer. The two symmetric dimers are labeled A1 and B1 or A2 and B2. (B) The overall fold of the RXR LBD monomer is presented with solid rendering (cylinders for α-helices and arrows for β-strands). The three layers of α-helices are shown in red, purple, and blue, respectively. Other color codes are yellow for β-strands and white for loops. The secondary structure numbering was adopted from the previous apo-RXR LBD structure, with the addition of H3′ for a short helix between H3 and H4. H3′ also exists in many other LBDs including TR (Wagner et al. 1995), ERα (Brzozowski et al. 1997), PR (Williams and Sigler 1998), and the PPARs (Nolte et al. 1998; Xu et al. 1999).
The tetramer interface
The disc-shaped tetramer is a compact complex where each monomer buries 2630 Å2, or 21% of its total solvent-accessible surface. This accounts for the average buried areas of 1260 Å2 in the homodimer interface and 1370 Å2 in the dimer–dimer interface from each monomer. The vast interface involved in tetramer formation is consistent with the high affinity (Kd ∼3–5 nm) measured for the dimer–dimer dissociation constant (Kersten et al. 1995a). In addition to the homodimer interface, each monomer contributes three distinct areas to the tetramer interfaces: the H3/H3 interface, the H11/H11 interface, and the interface between the AF-2 helix and coactivator binding site (Fig. 2A–C). These intermolecular interfaces are assembled from reciprocal interactions between the two symmetrically opposed dimers that associate to form the tetramer.
Figure 2.
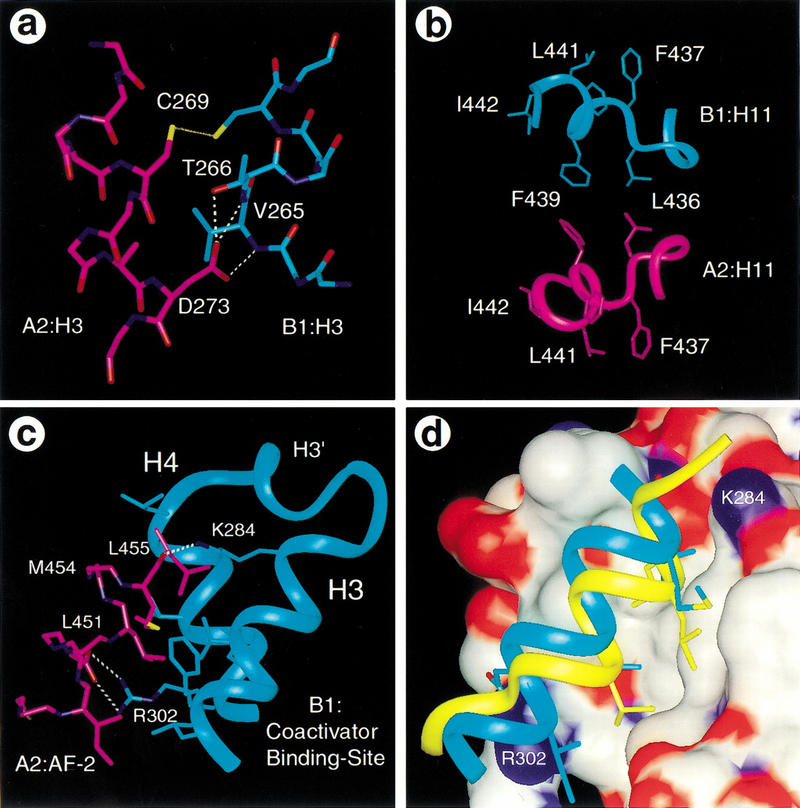
The tetramer interface. (A–C). The RXR tetramer interface. Each LBD contributes three different areas to the tetramer interface: the H3/H3 interface (A), the H11/H11 interface (B), and the AF-2/coactivator binding site interface (C). Hydrogen bonds are indicated with dotted lines. (D). Superpositon of the coactivator binding site between the current structure and the PPARγ/SRC-1 complex (Nolte et al 1998) indicates that the RXR AF-2 (blue) overlaps the binding site of the SRC-1 LXXLL motif (yellow). The RXR coactivator binding site is shown with a surface presentation that is colored by atom types: (white) carbons, (red) oxygens, (blue) nitrogens.
The H3/H3 interface is composed of the amino-terminal end of H3 from one monomer in the upper dimer (e.g., residues 264–269 of monomer B1) and the amino-terminal end of H3 of the adjacent monomer (residues 269–273 of monomer A2) from the lower dimer (Figs. 1A,2A). The core of this interface is assembled from the Van der Waals contacts between the side chain of B1:V265 and residues A272 and D273 of monomer A2. These central hydrophobic interactions are surrounded by polar interactions between each C269 in both monomers and the capping interactions of the backbone amide protons of residues 265 and 266 from B1:H3 by the side chain of A2:D273 (Fig. 2A).
The H11/H11 interface is composed of residues L436 and F439 of H11 from each monomer within the tetramer. H11 (residues 436–442) is predominately composed of large hydrophobic residues and sits between H3 and H10 as part of the hydrophobic core in the bottom half of the LBD (Figs. 1B and 2B). The orientation of H11 is directed toward H3 by a kink at residue H435 of H10. Four of six hydrophobic residues (F437, F438, L441, and I442) (Fig. 2B) in H11 are facing into the hydrophobic core of the protein and the other two (L436 and F439) are facing outward from the protein, forming part of the tetramer interface (Fig. 2B).
The AF-2/coactivator binding site interface is made up of the extended AF-2 helix from each monomer that binds the coactivator site in the adjacent monomer within the symmetric dimer (Figs. 1A and 2C,D). The AF-2 residues (451-LMEML-455, underlined residues) resemble the sequence of the LXXLL motif of coactivators and make similar interactions. As is the case for coactivator binding, the hydrophobic side of the AF-2 helix (L451, M454, and L455) fits directly into the hydrophobic groove formed by helices 3 and 4. The carbonyl group of L455 is capped by a hydrogen bond with the side chain of K284 from H3. The amino terminus of the AF-2 helix extends one additional turn to residue D444 and partially seals off the pocket of this adjacent monomer. A pair of hydrogen bonds between the conserved D448 and R302 may further stabilize the AF-2/coactivator binding site interface.
The homodimer interface
The RXR homodimer is butterfly shaped, with two symmetric monomers rotated 180° relative to each other around the twofold axis (Fig. 3A). The overall dimeric arrangement is identical between the two dimers within the tetramer. The monomer–monomer interface within each dimer is made up of an extensive network of hydrophobic and polar interactions mediated by H7, H9, H10, and the loop between H8 and H9 of both LBDs. The major portion of dimer interface is assembled from the amino-terminal half of H10 (residues 415–434), which forms a parallel coiled–coil structure (Fig. 3A). Most residues in the interface from H7, H9, and the loop between H8 and H9 are charged and are primarily involved in formation of complementary hydrogen bonds (Fig. 3B). Many of the residues involved in the homodimer interface also contribute to the heterodimer interface in the PPARγ/RXRα crystal structure (Gampe et al. 2000). However, the dimer interface is asymmetric in the PPARγ/RXRα crystal structure. Several pairs of polar interactions in the heterodimer are absent in the RXR homodimer interface, and this may account for preferential formation of heterodimers.
Figure 3.

The RXR dimer interface. (A). The butterfly shape of the RXR LBD homodimer is presented with a ribbon representation of the secondary structures. See Fig. 1B for key to color scheme. The insertion of E352 in the middle of H7 (red E) and the approximate C2 symmetry axis are indicated. (B). Interactions in the dimer interface between each monomer. The double arrows represent two-way symmetric interactions between the two monomers and single arrows indicate one-way asymmetric interactions. The residues are clustered based on the secondary structure of monomer A1 and are colored with the residue types: (white) hydrophobic residues, (red) hydrogen-bond acceptors, (blue) hydrogen-bond donors. (C). The experimental evidence for the E-insert in H7 is presented with a superposition of helix 7 between the current structure (blue) and the previous apo-structure (green). The displayed electron density (purple) is the difference between the selenomethionine and the native data sets, and is contoured at 5.0 ς. The density superimposes with the selenium atom positions of the three selenomethionines in the current structures. However, the K356 side chain in the previous RXR structure superimposes onto the density peak for M357, indicating a different registration for the residues in this region. (D). Sequence alignment for H7, H11, and the AF-2 helix. All residues are colored according to their side-chain properties: (white) hydrophobic, (blue) positively charged, (red) negatively charged, (green) polar. The dots represent residues that were omitted in the alignment. The conserved residues in H11 and AF-2 in the members of subfamily 2 are boxed.
Although the dimeric arrangement within the tetramer resembles that of the previous apo-RXR structure, a distinct difference exists around E352 of H7. Each monomer in the tetramer contains a helical geometry approaching the hypothetical π-type helix with 4.3 residues per turn, rather than 3.6 residues per turn, which is associated with a normal α-helix (Schulz and Schirmer 1979). The structure can alternatively be described as a single-residue insertion of E352 into the α-helix (blue helix in Fig. 3C). An identical “E-insert” conformation was observed in all four monomers and is confirmed by the electron density map across the region. In contrast, H7 was modeled as a more nearly ideal α-helix in the previous apo-RXR structure (green helix in Fig. 3C), leaving the amino acid sequence one residue out of register relative to the current structure between E352 and D359. This unusual π-helical geometry in the tetramer structure forces E352 to bulge outward from the H7 axis toward the partner monomer, thereby improving the surface fit in the monomer–monomer interface. The improved fit can be quantitated with the buried surface area in the dimer interface. With the Connolly molecular surface representation, the buried surface increases from 430 Å2 in the original structure (Bourguet et al. 1995) to 580 Å2 in this structure. Alternatively, using the solvent-accessible surface representation, the buried surface increases from 1180 Å2 to 1265 Å2.
Normally, the π-helical geometry is energetically unfavorable, however the RXR tetramer appears to stabilize this conformation through a series of intramolecular hydrogen bonds that exist between the insertion residue, E352, and residues R348, R421, and S421. These residues, together with T351, S355, K356, and D359, generate an intricate network of hydrogen bonds that extend to the H8–H9 loop region of the other monomer in the same dimer (Fig. 3B). Similar interactions were observed in the PPARγ/RXRα heterodimer structure (Gampe et al. 2000). The unusual π-helical geometry near E352 is essential for this hydrogen bond network because it rotates the amino acid side chains by ∼90° about the helical axis, relative to the positions that would occur in a regular α-helix. Thus the E-insert appears to facilitate homodimerization as well as heterodimerization in RXR. Sequence comparison of several representative NRs shows that this unusual α-helical insertion improves the alignment of amino acids in H7 (Fig. 3D). The alignment further suggests that the π-helical E-insert conformation will also occur in hepatocyte nuclear factor 4 (HNF4) and the chicken ovalbumin upstream promoter (COUP) transcription factors, which belong to the RXR subfamily of NRs and also function as homodimers (Cooney et al 1992; Jiang et al. 1995).
The tetramer ligand-binding pocket
Each of the RXR LBDs in the tetramer contains a ligand-binding pocket (Fig. 1B) that is surrounded by the following secondary structure elements: H5 on the top, H11 on the bottom, H3 on the right, H7 and H10 on the left, and the β-hairpin on the back. This pocket is open to solvent beyond the left sides of H11 and H3 in Fig. 4A orientation. The gross fold of each monomer within the tetramer is essentially identical to that of the other monomers. Comparisons of the monomer structure from the tetramer with the previous RXR structure (Fig. 4A) reveal significant differences in the positions of the AF-2 helix, H11, and the amino terminus of H3, which are major components of the tetramer interface, and the E-insert in H7, which is part of the homodimer interface. Although the scaffold of the pocket of the tetramer structure is similar to that of the previous apo-RXR dimer structure, the binding pocket in this structure is significantly different and substantially larger (boxed area in Fig. 4B,C). The previous apo-RXR structure shows very little space for binding a ligand because of the side chain conformation of residues that surround the pocket. In contrast, each LBD in the tetramer possesses a similar I-shaped pocket (boxed area in Fig. 4C). This I-shaped pocket is distinctly different from the L-shaped pocket in the activated RXR structure (Gampe et al. 2000), which reveals that receptor activation involves a substantial change in the shape of the ligand-binding pocket. This suggests the possibility of designing ligands with specificity for the activated or inactivated forms of the receptor.
Figure 4.

The apo-RXR pocket. (A) Overlap of a monomer structure from the tetramer (yellow) with that of the previous apo-RXR structure (blue). (B,C). Comparison of the overall fold and the ligand-binding pocket between the current RXR structure (yellow) and the previous apo-RXR structure (blue). The protein backbone is shown with a ribbon and the ligand-binding site is marked with a white box. The ligand-binding pocket of RXR is shown with water molecules (magenta spheres) that are docked into the available space within the protein.
Structure of the tetramer/tRA-isomer complex
Because 9cRA is an endogenous activator of RXR, we attempted to crystallize RXR in the presence of 9cRA. One approach, using RXR, 9cRA, PPARγ, rosiglitazone, and SRC-1 coactivator peptides, was successful and led to the structures of the activated PPARγ/RXRα heterodimer (Gampe et al. 2000). However, the approach described here, using only RXR and 9cRA, unexpectedly led to the inactivated tetrameric form, which is essentially identical to the apo-tetramer described above (Fig. 5A). A number of large hydrophobic interfaces are buried within the tetramer and it is possible that the protein concentrations and the salt conditions used in the crystallization shift the equilibrium between various oligomer states of the RXRα LBD toward the tetramer even in the presence of an agonist. The electron density map at a 2.0 Å resolution shows strong density for the protein, but only weak density for a ligand in one monomer within each dimer. The electron density in the pocket does not fit 9cRA (Fig. 5B), but instead it resembles an alternative trans-isomer of retinoic acid (tRA-isomer). Because retinoids are known to isomerize in the presence of light and reducing agents (Curley et al. 1988; Shil et al. 1997), the tRA-isomer most likely arose and bound to the RXR during the course of the protein concentration and crystallization (4–6 weeks). The electron density map is clear for the acid group, for the two methyl groups (C19 and C20), and for the tetraene linker between the β-ionone ring and the acid group (Fig. 5B). The tRA-isomer fits straight into the I-shaped pocket but does not adopt the low-energy all-trans retinoic acid (atRA) conformation seen in the structure of the atRA/RARγ complex (Renaud et al. 1995), and this may explain the low occupancy of the tRA-isomer in the tetramer. The ligand is constrained with a trans-like configuration at the C9–C10 double bond. The steric hindrance from H11, which lies on the edge of the ligand-binding pocket, would prevent binding of 9cRA.
Figure 5.

Structure of the tetramer/tRA-isomer complex. (A) An overview of the RXR tetramer/tRA complex. See Fig. 1A for key to the color scheme. The two tRA-isomers are shown in space-filling representation: (green) carbons, (red) oxygens. (B) Electron density map for the tRA-isomer. The map was calculated in the absence of the compound with 2Fo–Fc coefficients and contoured at 1.0 ς. (C). Molecular interactions between the RXR and the tRA-isomer. Hydrogen bonds are indicated with solid arrows and hydrophobic interactions are indicated with dashed lines. The solvent channel between the β-ionone ring and the AF-2 helix from the neighbor monomer (B2) is purple.
The tRA-isomer ligand makes both hydrophobic and polar interactions with the protein (Fig. 5C). The extended tetraene linker and the β-ionone ring make hydrophobic interactions with the surrounding residues, including I268, A271, A272, N275, L276 from H3, W305, N306, L309, and F313 from H5, and F438 and L441 from H11. The carboxylate makes several hydrogen bonds with the side chain of R316 and the main chain NH of A327. The same hydrogen bond network was observed in the low-energy 9cRA isomer when bound to RXRα in the PPARγ/RXRα heterodimer structure (Gampe et al. 2000). Although binding of the tRA-isomer was unexpected, these observations still identified the RXR residues that directly interact with the carboxylate and the tetraene linker of a retinoic acid before we had determined the PPARγ/RXRα heterodimer structure. It is difficult to attribute any particular significance or function to the bound tRA-isomer, except to state that it can bind to the tetramer as a nonactivating ligand. The presence of a tRA-isomer in the I-shaped apo-pocket may further suggest the possibility of designing small molecules to stabilize the tetramer conformation, thereby antagonizing receptor activation by endogenous ligands such as 9cRA.
Discussion
Structural correlation with biochemical data
A number of biochemical studies have established the existence of an RXR tetramer and support the tetramer structures observed here. For example, in vitro studies at low salt concentrations have indicated that full-length RXR self-associates into tetramers and that the LBD alone is sufficient to mediate tetramer formation with 3–5 nm affinity between the dimers (Kersten et al. 1995a,c). The high affinity for the tetramer formation is consistent with the compact organization and the extensive interface displayed in the tetramer structure. Furthermore, the tetramer structures explain the results of mutagenesis studies, which show that two phenylalanine residues (F438 and 439) in H11 are critical for tetramer formation of RXR (Kersten et al. 1997). Mutating these two phenylalanines to alanines abolishes the ability of RXR to form a tetramer. This result is consistent with the structure in which H11 from each monomer donates an extensive hydrophobic surface to the tetramer interface (Fig. 2B). These mutations most likely disrupt the H11/H11 interface by directly affecting either the intermolecular (F439A) or the intramolecular (F438A) packing of H11. It is interesting that RXR mutated in both phenylalanines is also transcriptionally inactive (Kersten et al 1997). These two phenylalanines are located at the edge of the AF-2 helix and the coactivator binding site as seen in the RXR/9cRA/SRC-1 half of the heterodimer structure (Gampe et al. 2000). This observation is consistent with the in vitro binding studies, which show that the double mutated protein fails to bind coactivator proteins (Kersten et al. 1998).
It has also been proposed that the RXR tetramer is transcriptionally silent based on the correlation between the transcription activity of RXR mutants and their ability to form tetramers (Kersten et al. 1998). The data show that mutated mouse RXRα (R321A), which forms tetramers that do not dissociate upon ligand binding, is also defective in ligand-dependent activation. In contrast, another mutant, RXRα (F318A), which fails to form a stable tetramer, shows a high basal transcription activity even in the absence of ligand (Kersten et al. 1998). These mutagenesis data are consistent with the observed tetramer structures. The position of the AF-2 helix overlaps the coactivator binding site (Fig. 2D) in the tetramer and would physically exclude the binding of coactivators to the receptor. Thus, the structures reported here provide a molecular basis for autorepression of the RXR tetramer.
It is interesting that RXR can interact with N-CoRs only when the AF-2 is deleted, suggesting that the AF-2 helix masks the corepressor binding site in apo-RXR (Zhang et al. 1999). The AF-2 helix does mask the coactivator binding site in the tetramer structure, suggesting that the corepressor and coactivator binding sites are nearly the same or at least that they overlap. This is consistent with recent mutagenesis data, which show that coactivator and corepressor binding require many of the same residues in NR helices 3, 3′, and 4 (Hu and Lazar 1999; Perissi et al. 1999). Whereas coactivators interact with NRs through the LxxLL motifs, corepressor proteins interact with NRs through two L/IxxI/VI motifs (Hu and Lazar 1999; Perissi et al. 1999). Further work suggests that the α-helix in corepressors differs from that in coactivators by extending at least three residues farther in the amino-terminal direction, allowing the corepressor to fill some of the volume otherwise occupied by the AF-2 helix (Perissi et al. 1999). This leads to the extended LxxI/HIxxxI/L motif and explains why corepressors bind in the absence of ligand, whereas coactivators bind in the presence of ligand (Perissi et al. 1999). The AF-2 helix in the RXR tetramer is also extended one more helical turn at the amino terminus and may thus provide a model of the bound conformation of corepressors as well.
The tetramer arrangement presented here is different from the dimer configuration of the previous apo-RXR structure (Bourguet et al. 1995). This may arise from the different crystallization conditions. In addition to not being a tetramer, the two phenylalanines (F438 and 439) in H11 in the previous structure do not participate in protein–protein interactions as seen in the tetramer. It is clear that a structure consisting only of dimers does not correlate with the biochemical and mutagenesis data that support the existence of tetramers as discussed above. In addition, the tetramer structure is more compact than the previous dimer structure and naturally avoids excessive exposure of the extensive hydrophobic surfaces to the solvent. It is evident that a dimer-only RXR structure would show a number of large solvent-exposed hydrophobic surfaces, including H11, AF-2, and the coactivator binding site. A protein with such large solvent-exposed hydrophobic surfaces is inherently unstable and tends to be associated with heat shock proteins or subjected to rapid proteolysis in vivo. There is no evidence for either of the above scenarios for RXR and shielding the hydrophobic surface through the tetramer formation as described in our structure may be an alternative solution to this problem.
The role of RXR tetramer in gene regulation
The existence and functional role of RXR tetramers in vivo have also been shown in the context of the CRBPII gene, which is regulated by RXR (Mangelsdorf et al. 1991). The CRBPII promoter contains two nonoptimal DR1 sites that are poorly recognized by an RXR dimer, and the RXR tetramer is required for DNA binding to this promoter (Chen and Privalsky 1995). Either RXRα or RXRγ can form tetramers cooperatively on the CRBPII promoter and regulate this gene efficiently. In explanation, the ability of RXRα and RXRγ to form tetramers increases their DNA-binding affinity to the CRBPII promoter and leads to the synergistic activation of this gene by 9cRA. In contrast, RXRβ, which fails to form tetramers, neither binds to the CRBPII promoter cooperatively nor activates the gene. Deletion of the amino-terminal portion of RXRβ allows the truncated protein to form tetramers and to bind and activate the CRBPII gene in the presence of 9cRA. Furthermore, the transcription activities of a series of synthetic promoters are correlated with their abilities to be recognized by the RXR tetramer (Lin et al. 1997). These data clearly indicate that RXR tetramers play a direct role in gene regulation in vivo through cooperative DNA-binding to the CRBPII promoter. Although the exact mechanism and origin of the cooperative activation remain unclear, Chen and Privalsky (1995) concluded that the DBDs may account for the specificity of DNA recognition but that the interacting surfaces leading to the tetramer formation would lie in the carboxy-terminal domains. Indeed, a recent crystal structure of the RXR DBD/DNA complex reveals that RXR binds to two tandem repeats of an optimal DR1 element as two head-to-tail dimers (Zhao et al. 2000). However, there are too few protein–protein interactions to support this as the sole basis for cooperative recruitment and regulation. It is possible that the extensive dimer–dimer interactions observed in the RXR LBD tetramer represent the carboxy-terminal domain interactions proposed by Chen and Privalsky. The vast surface area shared between the LBDs of the tetramer would provide a physical explanation for the reported synergy of RXR tetramers binding to the CRBPII promoter.
In addition to their functions as homodimers and tetramers, RXRs also serve as obligate heterodimer partners with a number of nuclear receptors, including RAR, TR, VDR and PPARs. However, the RXR tetramer has been shown to be a stable complex that does not spontaneously dissociate (Kersten et al 1995b). In fact, in the absence of an activating ligand, RXR tetramers fail to dissociate and form a heterodimer with either RAR or VDR, two well characterized heterodimer partners. Only after titration with 9cRA does the tetramer population rapidly dissociate and form a heterodimer (Dong and Noy 1998). This result appears to be in paradox with the ability of RXR to form heterodimers even in the absence of an obvious activating ligand. However, given that RXR homo and heterodimers have specific biological profiles, it seems more probable to assume that the intracellular state of RXR exists in a dynamic equilibrium between heterodimers and homodimers or tetramers whose populations are dependent on dietary flux and homeostasis. Thus, the RXR tetramer may provide a mechanism for the cell to store excess receptor until needed or activated. Increased cellular levels of either vitamin A, 9cRA or other various lower affinity fatty acids (Kitareewan et al. 1996; Bourguet et al. 2000) could stimulate dissociation of the tetramer and lead to the formation and activation of RXR homo- and heterodimers. Furthermore, a rapid induction of RXR heterodimers from a pool of RXR tetramers may in part account for the synergistic response that is observed for some RXR heterodimers (Schulman et al. 1998).
Mechanism of RXR activation by 9cRA
The mousetrap model for ligand activation of NRs is based on comparisons between the previous apo-RXR and the ligand-bound RARγ structures (Renaud et al. 1995). Specifically, our recent PPARγ/RXRα heterodimer structures show that the RXR AF-2 helix closes the pocket when bound with 9cRA and contributes to the charge clamp required for coactivator recruitment, which means that the activation of RXR by 9cRA is consistent with the mousetrap model. However, the crystal structure described here, combined with the previous biochemical studies, suggests that the activation of RXR by 9cRA requires an additional step that would involve dissociation of the tetramer into activated homodimers (Fig. 6B). In this scenario, the apo-RXR would form a tetramer complex that is transcriptionally inactive because the binding of coactivators is precluded by the AF-2 helix. Binding of 9cRA generates an activated receptor by inducing three major conformation changes in the RXR LBD that are best shown by the superposition of the current apo-RXR structure with the 9cRA-bound RXR from the RXR/PPARγ heterodimer structure (Fig. 6A) (Gampe et al. 2000). First, the β-ionone ring of the 9cRA protrudes into the ligand-binding site that was once occupied by H11 in the apo-state, allowing H11 to rejoin with H10 as a single uninterrupted H10/H11 helix. This leaves some empty space because the β-ionone ring is smaller in volume than H11 (residues 436–442). In the second step, the amino-terminal end of H3 collapses into the void left by H11 and H10. This motion bends H3 gently around H4/H5, still leaving a substantial binding site for 9cRA in the space between H3, H4/H5, and H10/H11. Third, the AF-2 helix rotates back against the main body of the LBD, fitting into the hydrophobic site formed by H3, H4/H5, and H10/H11. The tetramer interface, which consists largely of H3, H11, and the AF-2 helix, would be disrupted by these conformational changes and lead to the cooperative dissociation of the tetramer as proposed by Kersten et al. (1995b). The activated AF-2 helix, together with residues from H3, H3′, and H4, would contribute to the charge clamp that is competent for coactivator recruitment and complete the activation of the receptor. This scenario is consistent with the biochemical characterization of the solution-state RXR tetramers that rapidly dissociate in response to ligand (Kersten et al. 1995b). The tetramer structure and the 9cRA-bound RXR structure thus provide a more complete picture of how 9cRA binds and activates RXR (Fig. 6B).
Figure 6.
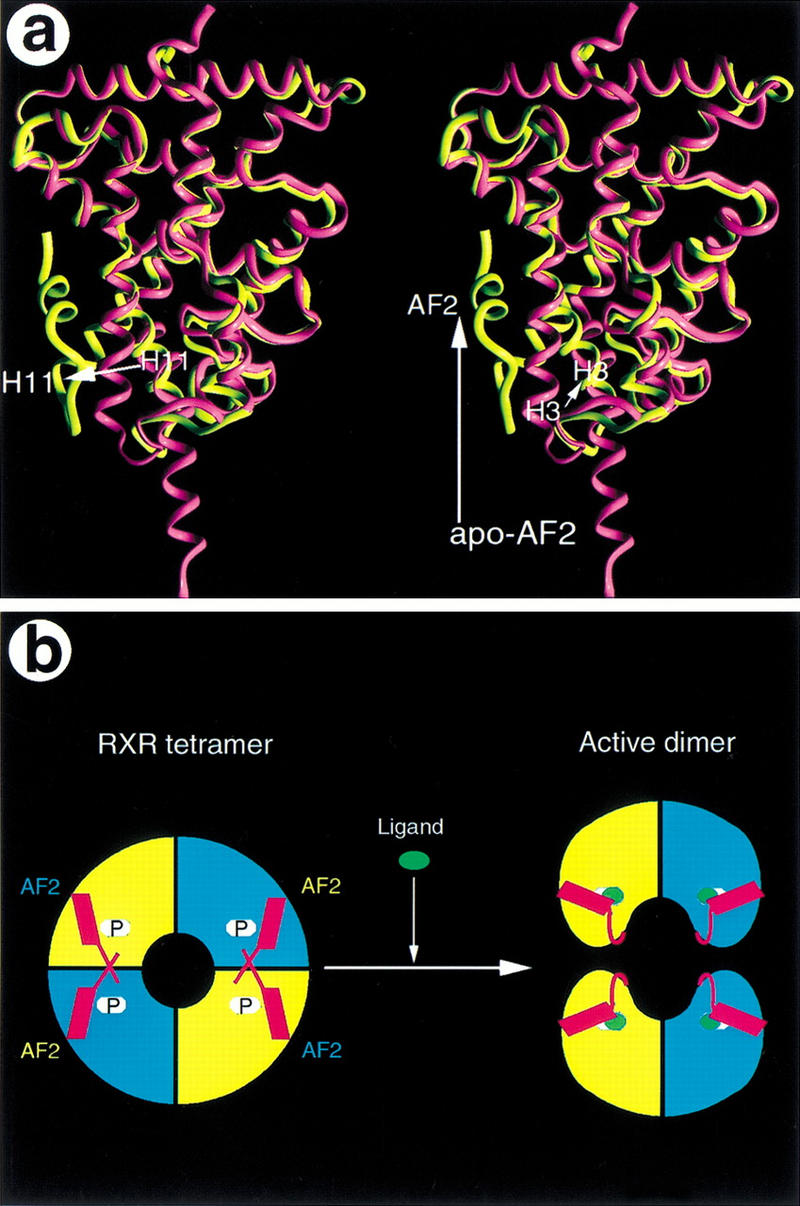
Mechanism of RXR activation by 9cRA. (A) Stereo view of the overlap between the structures of the apo-RXR (purple) from the tetramer and the 9cRA-bound RXR (yellow) from the PPARγ/RXRα heterodimer. Three major structural changes between the apo-structure and the 9cRA-bound structure are indicated with arrows along with their secondary elements (H3, H11, and the AF-2 helix). (B) A model that depicts the activation process of RXR by 9cRA. The apo-RXR forms an autorepressed tetramer (left, P, pockets), in which the AF-2 helix (red) from one monomer crosses over to occupy the coactivator binding site of the adjacent monomer from the symmetric dimer. Binding of 9cRA (green ovals) destabilizes the tetramer and induces the formation of the active RXR dimer in which the AF-2 helix is rearranged to form a charge clamp for binding of coactivators.
Tetramer formation as a regulating mechanism for the RXR subfamily
RXR belongs to subfamily 2 of the NR superfamily, which includes HNF4, Drosophila Seven-up, and COUP transcription factors (Committee 1999). Many members of subfamily 2 can function as homodimers (Cooney et al. 1992; Jiang et al. 1995; Mangelsdorf and Evans 1995). Sequence alignments indicate that the E-insert in H7 is a common feature for these receptors (Fig. 3D), and that it may perform an evolutionarily conserved function in the receptor dimerization as well. Furthermore, many residues that are involved in tetramerization are also conserved among members of subfamily 2, particularly, the hydrophobic nature of H11 and the AF-2 helix (Fig. 3D), which makes up the core interface of the tetramer. These observations suggest that the receptors from subfamily 2 may also form tetramers as part of their regulation and that the RXR tetramer structure may represent a prototype fold for this subfamily of receptors.
Conclusion
We have provided a detailed analysis of the RXR LBD tetramer structures, which reveal a novel tetramer interface that is consistent with the available biochemical and mutagenesis data. The tetramer arrangement suggests a mechanism of RXR autorepression that is mediated by the AF-2 helix. This conformation of the AF-2 helix has important implications for the binding of either coactivators or corepressors to RXR. The extended AF-2 helix into the coactivator site could also provide a structural model for binding of corepressors. The E352 insert in H7 creates an unusual helical geometry that improves the fit between the two RXR monomers and elucidates an important structural feature of the RXR dimer interface. The members of NR subfamily 2 share the highly conserved E-insert as well as many of the residues that form the tetramer interface. This suggests that the RXR tetramer structure may represent a prototype for this subfamily in which a self-repressed tetramer is part of receptor regulation.
Materials and methods
Protein preparation
The RXRα LBD (amino acids 231–462) tagged with MKKGHHHHHHG was expressed from the T7 promoter of plasmid vector pET14. The native and selenomethionine-labeled RXRα proteins were purified through a Ni-agarose column and an S-Sepharose column with procedures similar to those for the PPARδ LBD (Xu et al. 1999). Aliquots of RXRα were diluted to a protein concentration of ∼0.05 mg/ml or ∼2 μm with a buffer containing 0.5 m ammonium acetate, 2 mm EDTA, 5.0 mm dithiothreitol (DTT), 2.5% propanediol, 0.1% β-octyl-glucoside, and 10% glycerol. The diluted protein solutions were mixed with a 5 m equivalent excess of authenticated 9cRA or LG100069, known activators of RXRα, and were progressively concentrated to 18 mg/ml, then aliquoted and stored at −80°C. The 9-cis isomer content of 9cRA was confirmed to be greater than 95% by Raman spectroscopy.
Crystallization and data collection
The crystals for this study were grown at room temperature in hanging drops containing 1 μl of the above protein–ligand solutions, and 1 μl of well buffer (20 mm HEPES at pH 7.5, 1.5 m Li2SO4, and 10 mm DTT). Crystals also grew with 2% PEG4000, 350 mm Li2SO4 and 10% glycerol. The Xenon derivatives were prepared with a Cryo-X-citer (Molecular Structure Corporation). Before data collection, crystals were transiently mixed with the well buffer that contained an additional 20% glycerol and then flash frozen in liquid nitrogen.
The crystals for the tetramer/tRA-isomer complex formed in the P21 space group, with a = 51.1 Å, b = 99.7 Å, c = 96.3 Å, and β = 96.8°. The apo-tetramer crystals also formed in the P21 space group, with a = 51.1 Å, b = 99.3 Å, c = 95.0 Å, and β = 97.4°. Each asymmetry unit contains a single RXR tetramer with 39% of solvent content. Data were collected with a Rigaku R-Axis II detector in house, or with a MAR CCD detector in the IMCA 17ID beam line at the Argonne National Laboratories, and the observed reflections were reduced, merged, and scaled with DENZO and SCALEPACK in the HKL2000 package (Otwinowski et al. 1993).
Structure determination and refinement
We initially attempted to solve the structure by simple molecular replacement using the previous apo-RXR dimer structure as the searching model. Two molecular replacement solutions that differ ∼180° rotation were obtained with the Collaborative Computational Project Number 4 (CCP4) AMoRe program (Navaza et al. 1992), and were in agreement with two dimers in each asymmetry unit. However, the electron density map calculated from the phases from the molecular replacement solution was ambiguous for many regions of the protein, including the AF-2 helix. Thus we decided to obtain additional phase information by preparing the xenon derivatives and crystals of the proteins labeled with selenomethionines.
The experimental phases were determined with data sets from three heavy-atom derivatives and three wavelength data sets from a crystal containing the selenomethionine protein (Table 1). The derivative and native data sets were merged and scaled together with the CCP4 truncate and cad programs (Collaborative Computational Project Number 4 1994). The initial positions for four xenon atoms and 10 selenium atoms in each RXR tetramer were determined by the difference fourier method with phases from the molecular replacement solution. Heavy-atom parameters were refined with the CCP4 MLPHARE program (Collaborative Computational Project Number 4 1994) and the SHARP program (La Fortelle and Bricogne 1997). The initial MIR and MAD phases had a mean figure of merit of 0.54 with the data to 2.2 Å, and this experimental map was further improved with solvent flattening, histogram matching, and NCS averaging as implemented in the CCP4 DM program (Cowtan 1994). The modified map showed clear density for the majority of the protein. In addition, weak density was observed for a small molecule ligand in two of the four LBDs. The density for residues 242–263 was not clear, and these residues were built as polyglycine chains. Model building was performed with Quanta (Molecular Simulation Inc.) and refinement was done with XPLOR (Brunger 1992a) and CNS (Brunger et al. 1998). The structure of the tetramer/tRA-isomer complex contains four LBDs, two tRA-isomers, and 497 water molecules with good stereochemistry (Table 1). The structure for the tetramer without the tRA-isomer was determined by molecular replacement with the tetramer/tRA-isomer structure, and includes four RXR LBDs and 309 water molecules. The refined statistics are shown in Table 1.
Computational analysis
Surface areas calculated with the Connolly MS program (Connolly 1983) and the MVP program (Lambert 1997). The C2-symmetry axis, sequence alignments and binding site accessible waters were calculated with MVP.
Acknowledgments
We thank W. Burkart, M. Moyer, and T. Consler for protein analysis; R. Nolte and R. Xu for help with data collection and the SHARP program; D. Minick for validation of the 9cRA purity; T. Willson, S. Kliewer, and A. Miller for discussion. We also thank J. Chrzas and the IMCA beamline staff at Argonne National Laboratories for assistance with data collection. Correspondence and request for materials should be addressed to H. Eric Xu.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL ex11957@glaxowellcome.com; FAX (919) 483-0368.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.802300.
References
- Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXRα. Nature. 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- Bourguet W, Vivat V, Wurtz J-M, Chambon P, Gronemeyer H, Moras D. Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol Cell. 2000;5:289–298. doi: 10.1016/s1097-2765(00)80424-4. [DOI] [PubMed] [Google Scholar]
- Brunger AT. XPLOR, v. 3.0. New Haven, CT: Yale University Press; 1992a. [Google Scholar]
- ————— The free R-value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992b;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- Chen H, Privalsky ML. Cooperative formation of high-order oligomers by retinoid X receptors: An unexpected mode of DNA recognition. Proc Natl Acad Sci. 1995;92:422–426. doi: 10.1073/pnas.92.2.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–776. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Committee NRN. A unified nomenclature system for the nuclear receptor superfamily [letter] Cell. 1999;97:161–163. doi: 10.1016/s0092-8674(00)80726-6. [DOI] [PubMed] [Google Scholar]
- Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221:709–713. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- Cooney AJ, Tsai SY, O' Malley BW, Tsai MJ. Chicken ovalbumin upstream promoter transcription factor (COUP-TF) dimers bind to different GGTCA response elements, allowing COUP-TF to repress hormonal induction of the vitamin D3, thyroid hormone, and retinoic acid receptors. Mol Cell Biol. 1992;12:4153–4163. doi: 10.1128/mcb.12.9.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. ‘DM’: An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM. Newsletter on Protein Crystallogr. 1994;31:34–38. [Google Scholar]
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes & Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong D, Noy N. Heterodimer formation by retinoid X receptor: Regulation by ligands and by the receptor's self-association properties. Biochemistry. 1998;37:10691–10700. doi: 10.1021/bi980561r. [DOI] [PubMed] [Google Scholar]
- Freedman LP. Increasing the complexity of coactivation in nuclear receptor signaling. Cell. 1999;97:5–8. doi: 10.1016/s0092-8674(00)80708-4. [DOI] [PubMed] [Google Scholar]
- Gampe RT, Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, Kliewer SA, Willson TM, Xu HE. Asymmetry in the PPARγ/RXRα crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol Cell. 2000;5:545–555. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- Giguere V. Retinoic acid receptors and cellular retinoid binding proteins: Complex interplay in retinoid signaling. Endocr Rev. 1994;15:61–79. doi: 10.1210/edrv-15-1-61. [DOI] [PubMed] [Google Scholar]
- Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- Hu X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–96. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- Jiang G, Nepomuceno L, Hopkins K, Sladek FM. Exclusive homodimerization of the orphan receptor hepatocyte nuclear factor 4 defines a new subclass of nuclear receptors. Mol Cell Biol. 1995;15:5131–5143. doi: 10.1128/mcb.15.9.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Kelleher D, Chambon P, Gronemeyer H, Noy N. Retinoid X receptor α forms tetramers in solution. Proc Natl Acad Sci. 1995a;92:8645–8469. doi: 10.1073/pnas.92.19.8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Pan L, Noy N. On the role of ligand in retinoid signaling: positive cooperativity in the interactions of 9-cis-retinoic acid with tetramers of the retinoid X receptor. Biochemistry. 1995b;34:14263–14269. doi: 10.1021/bi00043a034. [DOI] [PubMed] [Google Scholar]
- Kersten S, Pan L, Chambon P, Gronemeyer H, Noy N. Role of ligand in retinoid signaling. 9-cis-retinoic acid modulates the oligomeric state of the retinoid X receptor. Biochemistry. 1995c;34:13717–13721. doi: 10.1021/bi00042a001. [DOI] [PubMed] [Google Scholar]
- Kersten S, Reczek PR, Noy N. The tetramerization region of the retinoid X receptor is important for transcriptional activation by the receptor. J Biol Chem. 1997;272:29759–29768. doi: 10.1074/jbc.272.47.29759. [DOI] [PubMed] [Google Scholar]
- Kersten S, Dong D, Lee W, Reczek PR, Noy N. Auto-silencing by the retinoid X receptor. J Mol Biol. 1998;284:21–32. doi: 10.1006/jmbi.1998.2168. [DOI] [PubMed] [Google Scholar]
- Kitareewan S, Burka LT, Tomer KB, Parker CE, Deterding LJ, Stevens RD, Forman BM, Mais DE, Heyman RA, McMorris T, et al. Phytol metabolites are circulating dietary factors that activate the nuclear receptor RXR. Mol Biol Cell. 1996;7:1153–1166. doi: 10.1091/mbc.7.8.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Fortelle ED, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology. New York, NY: Academic Press; 1997. pp. 472–494. [DOI] [PubMed] [Google Scholar]
- Lambert MH. Docking conformationally flexible molecules into protein binding sites. In: Charifson PS, editor. Practical Application of Computer-Aided Drug Design. New York, NY: Marcel-Dekker; 1997. pp. 243–303. [Google Scholar]
- Lin BC, Wong CW, Chen HW, Privalsky ML. Plasticity of tetramer formation by retinoid X receptors. An alternative paradigm for DNA recognition. J Biol Chem. 1997;272:9860–9867. doi: 10.1074/jbc.272.15.9860. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Umesono K, Kliewer SA, Borgmeyer U, Ong ES, Evans RM. A direct repeat in the cellular retinol-binding protein type II gene confers differential regulation by RXR and RAR. Cell. 1991;66:555–561. doi: 10.1016/0092-8674(81)90018-0. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, Kakizuka A, Evans RM. Characterization of three RXR genes that mediate the action of 9-cis-retinoic acid. Genes & Dev. 1992;6:329–344. doi: 10.1101/gad.6.3.329. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Kliewer SA, Kakizuka A, Umesono K, Evans RM. Retinoid receptors. Recent Prog Horm Res. 1993;48:99–121. doi: 10.1016/b978-0-12-571148-7.50008-7. [DOI] [PubMed] [Google Scholar]
- Navaza J, Gover S, Wolf W. AMoRe: A new package for molecular replacement. In: Dodson EJ, editor. Molecular Replacement: Proceedings of the CCP4 Study Weekend. Daresbury, UK: SERC; 1992. pp. 87–90. [Google Scholar]
- Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator- activated receptor-γ. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Isaacs N, Burley S. Oscillation data reduction program. In: Sawyer L, editor. Proceedings of the CCP4 Study Weekend. Daresbury, UK: SERC, Daresbury Laboratory; 1993. pp. 56–62. [Google Scholar]
- Perissi V, Staszewski LM, McInerney EM, Kurokawa R, Krones A, Rose DW, Lambert MH, Milburn MV, Glass CK, Rosenfeld MG. Molecular determinants of nuclear receptor-corepressor interaction. Genes & Dev. 1999;13:3198–3208. doi: 10.1101/gad.13.24.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud JP, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Crystal structure of the RARγ ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- Schulman I G, Shao G, Heyman RA. Transactivation by retinoid X receptor-peroxisome proliferator-activated receptor γ (PPARγ) heterodimers: Intermolecular synergy requires only the PPARγ hormone-dependent activation function. Mol Cell Biol. 1998;18:3483–3494. doi: 10.1128/mcb.18.6.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- Schulz GE, Schirmer RH. Principles of Protein Structure. Springer-Verlag; 1979. pp. 69–71. [Google Scholar]
- Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;378:690–697. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- Williams SP, Sigler PB. Atomic structure of progesterone complexed with its receptor. Nature. 1998;393:392–396. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]
- Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Chasse SA, Devarakonda S, Sierk ML, Ahvazi B, Rastinejad F. Structural basis of RXR–DNA interactions. J Mol Biol. 2000;296:509–520. doi: 10.1006/jmbi.1999.3457. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hu X, Lazar MA. A novel role for helix 12 of retinoid X receptor in regulating repression. Mol Cell Biol. 1999;19:6448–6457. doi: 10.1128/mcb.19.9.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]