

Abstract
Telomere maintenance has been proposed as an essential prerequisite to human tumor development. The telomerase enzyme is itself a marker for tumor cells, but the genetic alterations that activate the enzyme during neoplastic transformation have remained a mystery. Here, we show that Myc induces telomerase in both normal human mammary epithelial cells (HMECs) and normal human diploid fibroblasts. Myc increases expression of hEST2 (hTRT/TP2), the limiting subunit of telomerase, and both Myc and hEST2 can extend the life span of HMECs. The ability of Myc to activate telomerase may contribute to its ability to promote tumor formation.
Keywords: Myc, telomerase, hEST2, tumorigenesis
Telomerase activity is largely absent from somatic cells in vivo and from normal human cells in culture (Counter et al. 1992). As these cells proliferate, telomeric repeats are progressively lost as a result of incomplete replication of chromosome ends during each division cycle (Watson 1972; Olovnikov 1973; Harley et al. 1990; Hastie et al. 1990). Telomere shortening has been proposed as the mitotic clock that marks the progress of a cell toward the end of its replicative life span. According to this model, erosion of chromosome ends triggers cellular senescence (Harley et al. 1990; for review, see Harley 1991). Bypass of senescence can be accomplished by negation of tumor suppressor pathways (e.g., p53 and Rb/p16). This allows continued proliferation (extended life span) that is accompanied by further telomere loss (Counter et al. 1992). Indefinite proliferation in the absence of a telomere maintenance strategy would eventually result in a complete loss of telomeres and in destabilization of chromosomes (Singer and Gottschling 1994). Because this situation is probably incompatible with survival, cells with an indeterminate life span must adopt strategies for telomere conservation. As predicted, cells that emerge from extended life span as immortal cell lines often activate the telomerase enzyme (Counter et al. 1992; Kim et al. 1994).
Cells that are programmed for continuous proliferation generally maintain telomere length. For example, many stem cell populations possess telomerase activity (Counter et al. 1992; Kim et al. 1994). Telomerase is also induced in mitogen-stimulated lymphocytes and is detected in mitotically active regions of hair follicles and intestinal crypts (for review, see Greider 1998). The association of telomerase with cell proliferation has led to the hypothesis that telomere maintenance is simply a housekeeping function. However, proliferating normal cells in culture generally lack telomerase activity (Counter et al. 1992; Kim et al. 1994).
Stabilization of telomeric repeats may be a prerequisite for tumorigenesis (Counter et al. 1992). Consistent with this notion, telomerase is activated in a high percentage of late-stage human tumors and is present in most tumor-derived cell lines in culture (Counter et al. 1992, 1994; Kim et al. 1994; Shay and Wright 1996). To test the role of telomere maintenance in tumorigenesis, we surveyed known oncogenes for their ability to activate telomerase.
Results and Discussion
Myc activates telomerase
Normal human mammary epithelial cells (HMECs) lack telomerase, whereas immortal HMEC-derivatives and breast tumor cell lines are almost universally telomerase-positive (Shay et al. 1995; Bryan and Reddel 1997; Shay and Bacchetti 1997). Introduction of HPV-16 E6 protein into primary HMECs stimulates telomerase activity, suggesting that, in these cells, a single genetic event can potentiate the enzyme (Shay et al. 1993; Klingelhutz et al. 1996; Fig. 1A,C and 2). Therefore, we asked whether increased expression of other cellular or viral oncogenes could induce telomerase in HMECs. Ectopic expression of mdm-2 failed to induce telomerase, consistent with the observation that activation of telomerase by E6 is separable from the ability of E6 to promote the degradation of p53 (Klingelhutz et al. 1996; Fig. 2). Several other cellular and viral oncoproteins, including E7, activated Ras (V12), cyclin D1, cdc25C, and cdc25A, also failed to induce telomerase (Fig 2). However, introduction of a c-Myc expression cassette stimulated telomerase activity in HMECs (Figs. 1A and 2). Enzyme activity was elevated within one passage after transduction of HMECs with a retrovirus that directs Myc expression (Fig. 1C). The Myc-expressing populations displayed levels of telomerase activity that approximated those seen in breast carcinoma cell lines (Fig. 1A; e.g., T47D).
Figure 1.



Myc activates telomerase. (A) Primary HMECs at passage 12 were infected with empty vector (lanes 1–5), E6 (lanes 6–10), c-Myc (lanes 11–15), cdc25A (lanes 16–20), or hEST2 (lanes 21–25) viruses. Breast cancer cell lines BT549 (lanes 26–30) and T47D (lanes 31–35) were included for comparison. TRAP assays contained lysates from 10,000 (lanes 2,6,7,11,12,17,21,22,26,27,31,32), 1000 (lanes 3,8,13,18,23,28,33), 100 (lanes 4,9,14,19,24,29,34), or 10 (lanes 5,10,15,20,25,30,35) cells. (− and +) Absence or presence of RNase A, respectively. (Mix; lanes 1,16) To exclude the presence of inhibitors in apparently negative lysates, lysate from 10,000 of the indicated cells was mixed with lysate from 10,000 c-Myc-expressing cells. (B) IMR90 cells at passage 14 were infected with empty vector (lanes 1–5), c-Myc (lanes 6–10), E6 (lanes 11–15), or hEST2 (lanes 16–20) viruses. HT1080 cells (lanes 21–25) were included for comparison. TRAP assays were performed with decreasing cell equivalents as in A. (C) HMEC (lanes 1–4), IMR90 (lanes 5–8), or WI38 (lanes 9–12) cells were infected with empty vector (lanes 1,5,9), hEST2 (lanes 2,6,10), c-Myc (lanes 3,7,11), or E6 viruses (lanes 4,8,12). Cells were selected for ∼5 days with puromycin or hygromycin and then lysed for telomerase assay. Each lane corresponds to 10,000 cells.
Figure 2.

Oncogene activation of telomerase. HMECs (lanes 1–9) or IMR90 cells (lanes 10–14) were infected with viruses that direct the expression of the indicated oncogenes (lanes 2–9,11–14) or empty vector (lanes 1,10). Cell extracts were analyzed by TRAP assay.
Introduction of E6 into normal human diploid fibroblasts failed to activate telomerase (Klingelhutz et al. 1996; Shay et al. 1993; Figs. 1B,C and 2). Similar results were observed for E1A (Fig. 2), activated Ras (V12, not shown), or a dominant-negative p53 allele (Fig. 2). However, telomerase was induced by transduction of either IMR-90 (Figs. 1B,C and 2) or WI-38 cells (Fig. 1C) with a retrovirus that directs c-Myc expression. As with HMECs, activity was apparent immediately after drug selection (Fig. 1C). The Myc-expressing cells contained levels of telomerase activity comparable to those seen in a telomerase-positive fibrosarcoma cell line, HT1080 (Fig. 1B).
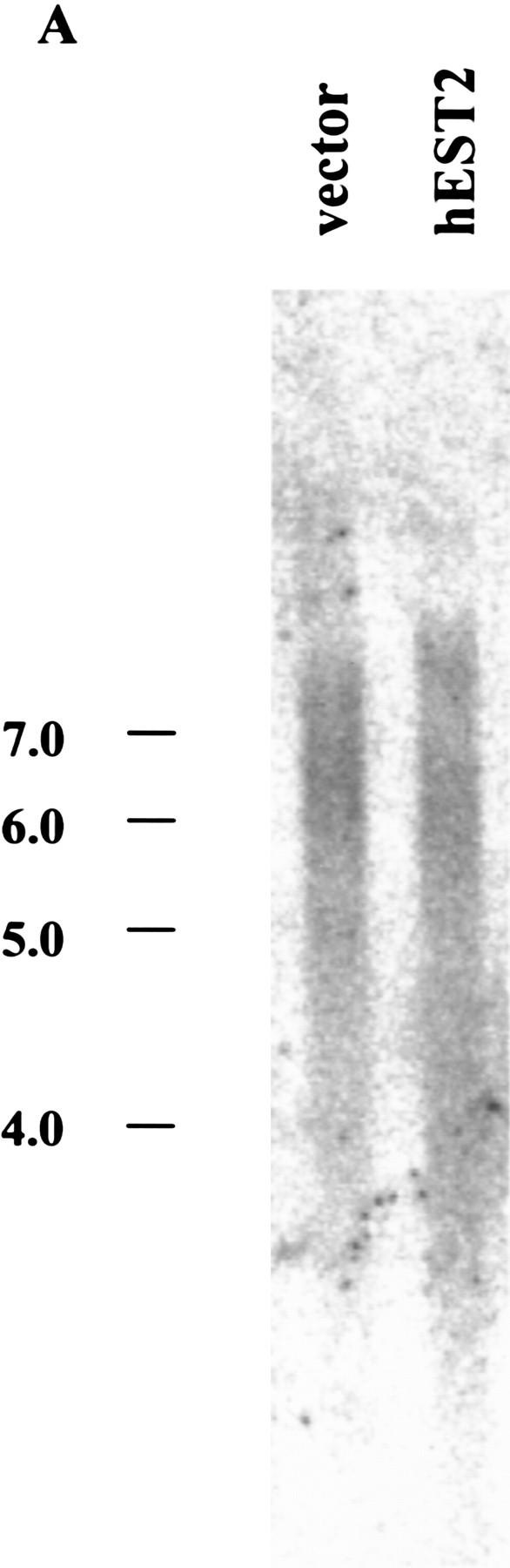
Although c-Myc expression elevates telomerase in both normal epithelial cells and in normal fibroblasts, the HPV-16 E6 protein has been shown to affect telomerase only in epithelial cells (Klingelhutz et al. 1996). Therefore, we questioned the basis of cell-type specific telomerase activation by E6. A recent report suggesting that E6 can activate the Myc promoter (Kinoshita et al. 1997) prompted us to ask whether E6 might regulate telomerase through an effect on Myc expression. In HMECs, expression of E6 induced Myc to levels approaching those achieved upon transduction of HMECs with a Myc retrovirus (Fig. 3A). Surprisingly, E6-induced alterations in Myc protein did not reflect changes in the abundance of myc mRNA (Fig. 3B). Therefore, Myc expression must be controlled post-transcriptionally by E6 in HMECs. In contrast, Myc levels remained unaltered following expression of E6 in IMR-90 cells wherein E6 is incapable of activating telomerase (Fig. 3A). Although E6 may regulate telomerase by other mechanisms, this result is consistent with a model in which E6 regulates telomerase in HMECs by altering the abundance of Myc.
Figure 3.


E6 increases c-Myc protein in HMECs. (A) Cell lysates from E6 (lane 1)- and vector (lane 2)-infected IMR90 cells and lysates from c-Myc (lane 3)-, E6 (lane 4)-, and vector (lane 5)-infected HMECs were analyzed by Western blot with a polyclonal Myc antibody. Tumor cell lines, HT1080 (lane 6), HBL100 (lane 7), BT549 (lane 8), and T47D (lane 9), were included for comparison. The expression of TFIIB was used to normalize loading. (B) Northern analysis of Myc RNA levels in total RNA. GAPDH was probed as a loading control.
Myc induces hEST2
The presence of the mRNA encoding hEST2, the catalytic subunit of telomerase, strictly correlates with telomerase activity. The hEST2 message is undetectable in normal tissue and in normal cell lines but is present in immortal and tumor-derived cell lines (Harrington et al. 1997; Meyerson et al. 1997; Nakamura et al. 1997). Moreover, hEST2 expression and telomerase are suppressed concomitantly when cells are induced to differentiate (Meyerson et al. 1997). These results suggest that availability of hEST2 may limit telomerase activity, as was demonstrated recently in a number of normal cell lines (Weinrich et al. 1997; Bodnar et al. 1998). Expression of hEST2 could also induce telomerase in HMECs and WI38 and IMR-90 cells (Fig. 1A–C). Activity was apparent immediately following selection of hEST2-expressing cells (Fig. 1C), and the level of telomerase activity observed in hEST2-expressing populations consistently exceeded that observed in cell populations containing c-Myc (Fig. 1A–C).
Because increased expression of hEST2 was sufficient to activate telomerase in both HMECs and in IMR-90 cells (Fig. 1), we asked whether Myc activates telomerase through an effect on hEST2. As expected, hEST2 mRNA was not detectable in normal HMECs, but was induced at least 50-fold following transduction with a Myc retrovirus (Fig. 4A). Thus, Myc regulates telomerase by controlling the expression of a limiting telomerase subunit. Because Myc enhances the expression of responsive genes, its action on hEST2 could be either direct or indirect.
Figure 4.

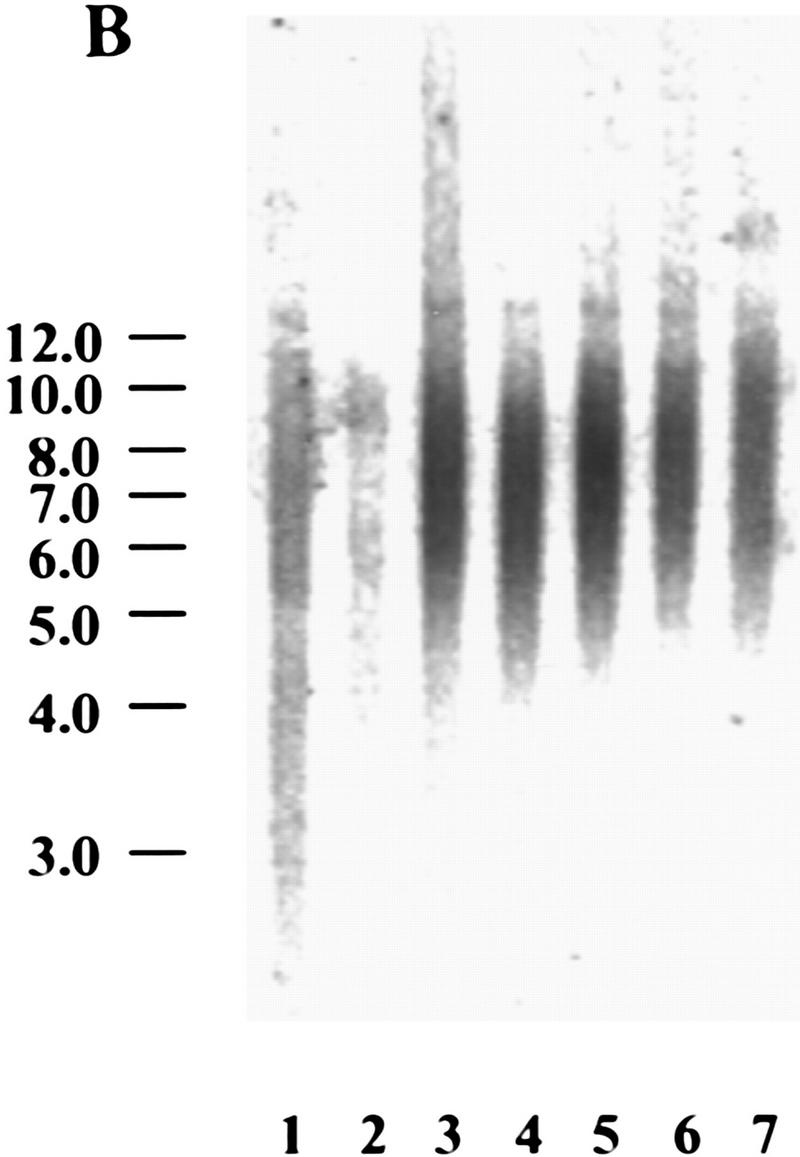

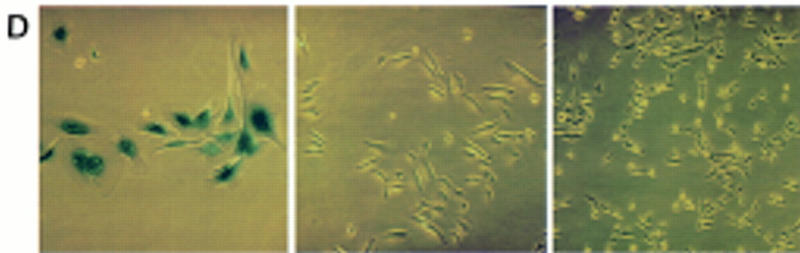
Myc regulates hEST2 and extends cellular life span in HMECs. (A) hEST2 Northern analysis of poly(A)+ RNA from normal HMECs and from HMECs that had been infected with a Myc retrovirus. A Northern blot with GAPDH was performed as a loading control. (B) Genomic DNA (3 μg) from early-passage HMECs (passage 12, lane 1), late-passage HMECs (passage 26, lane 2), and hEST2-expressing HMECs [infected at passage 12 and cultured for 3 (lane 3), 6 (lane 4), 8 (lane 5), 10 (lane 6), or 14 (lane 7) additional passages] was digested with RsaI and HinfI. Fragments were separated on a 0.8% agarose gel, and telomeric restriction fragments were visualized with a 32P-labeled human telomeric sequence (TTAGGG)3 probe. (C) Genomic DNA (3 μg) from early-passage HMECs (passage 12, lane 1), vector-infected HMEC (infected at passage 12 and cultured for six additional passages or ∼12–14 PD, lane 2), hEST2-expressing HMECs (infected at passage 12 and cultured for six additional passages or ∼12–14 PD, lane 3), and Myc-expressing cells (infected at passage 12 and cultured for six additional passages or ∼18 PD, lane 4) were digested with RsaI and HinfI. Fragments were probed with a telomeric probe as described in B. TRF intensity was quantitated on a Fuji BAS2000 PhosphorImager. Normalizing vector-containing HMECs (lane 2) to 100 units of intensity, both early passage HMECs (lane 1) and Myc-expressing HMECs (lane 4) gave ∼150 units of intensity and hEST2-expressing HMECs (lane 3) gave ∼200 units of intensity. (D) HMECs transduced with empty vector (left), hEST2 (middle), or c-Myc viruses (right) were grown to a PDL of ∼56–60. At this PDL, vector cells adopted a senescent morphology and ceased growth. Cells expressing c-Myc and hEST2 continued to proliferate. To assess the percentage of senescent cells in the population, each culture was stained for senescence-associated β-galactosidase. Greater than 95% of the vector-containing cells were β-galactosidase positive whereas <10% of cells expressing hEST2 or Myc were stained.
hEST2 increases replicative life span in HMECs but not in IMR-90 cells
Preservation of telomeric repeats requires either the telomerase enzyme or the activation of an alternative pathway for telomere maintenance (Kim et al. 1994; Broccoli et al. 1995; Strahl and Blackburn 1996; Wright et al. 1996; Bryan and Reddel 1997). In addition, telomere length can be controlled by telomere-binding proteins (van Steensel and de Lange 1997). To determine whether activation of telomerase in HMECs is sufficient to stabilize telomere length, we followed telomeric restriction fragment (TRF) size as HMECs proliferated either in the presence or absence of telomerase activity. In normal HMECs, telomere length and the abundance of telomeric sequences diminished slightly as cells underwent multiple rounds of division (Fig. 4B). Activation of telomerase by expression of hEST2 not only prevented telomere shrinkage but also increased both the overall abundance of telomeric sequences and the average length of telomeres (Fig. 4B). In Myc-expressing cells, however, the abundance of telomeric sequences was intermediate between that observed in cells expressing hEST2 and that observed in control cells (Fig. 4C). Telomere lengths followed a similar pattern (Fig. 4C).
Generally, in tumors and in immortal cell lines, telomeres are short but stable (Hastie et al. 1990). Comparison of TRF levels in Myc-expressing HMECs to those in early-passage HMECs suggested that Myc probably stabilized telomeres rather than promoted an increase in TRFs as occurs in hEST2-expressing HMECs (Fig. 4C). Thus, alterations in telomere dynamics after Myc transduction mimic the situation in tumors. Telomerase was ∼10-fold more active in hEST2-expressing cells than in Myc-expressing cells. Thus, differences in telomerase activity likely reflect differences in hEST2 expression as the abundance of viral hEST2 mRNA greatly exceeded native hEST2 mRNA levels present in either Myc-expressing HMECs or in any of the tumor cell lines tested to date.
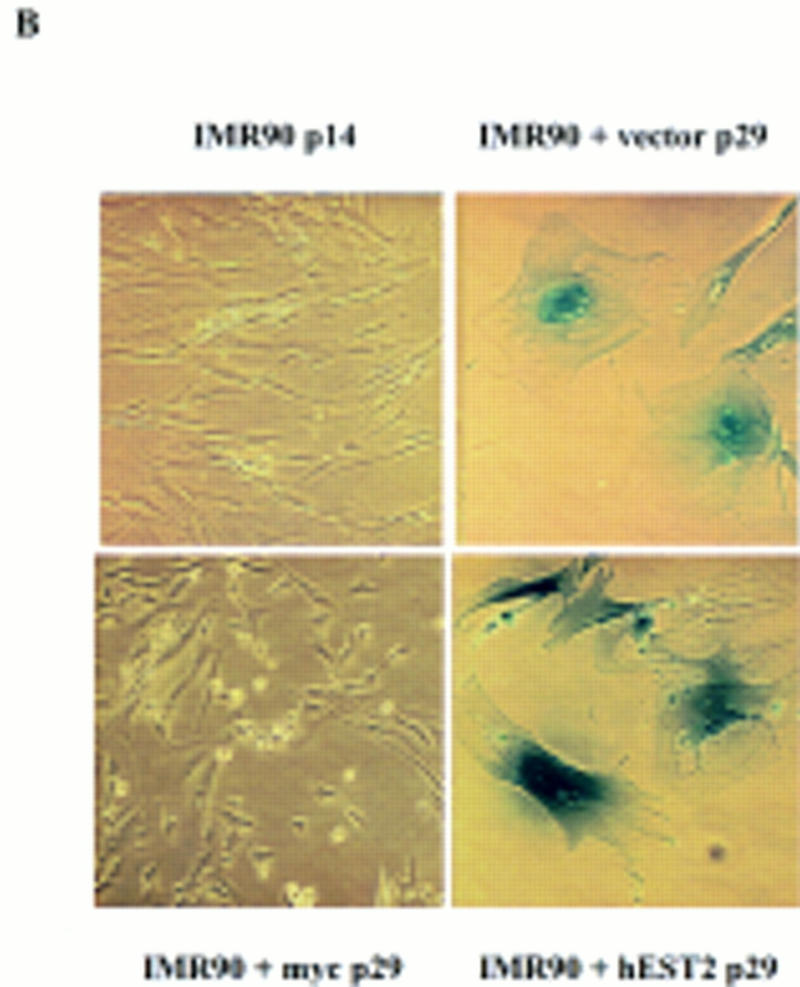
Telomere length has been proposed as the counting mechanism that determines the replicative life span of a cell (Harley 1990; Harley et al. 1991). At a population doubling level (PDL) of ∼55–60, vector-containing HMECs ceased proliferation, adopted a senescent morphology (for review, see Stein and Dulic 1995), and stained positive for senescence-associated β-galactosidase (Dimri et al. 1995; Fig. 4D). These cells also showed increased expression of PAI, another senescence marker (Goldstein et al. 1994; data not shown). In contrast, normal HMECs that had received either hEST2 or c-Myc at early passage displayed an extended life span. Cells expressing either c-Myc or hEST2 continued to proliferate beyond the normal senescence point and did not show evident β-galactosidase staining or increased PAI expression (Fig. 4D; data not shown). In both c-Myc and hEST2-expressing cell populations, <10% of cells showed any senescence-associated phenotype at the point at which vector-infected cells senesced. Furthermore, neither population has shown any accumulation of senescent cells during subsequent growth. At present, hEST2- and Myc-expressing populations are a minimum of 40 population doublings (PD) beyond the normal senescence point.
In IMR-90 cells, the consequences of telomerase activation differed from those observed in HMECs. Although hEST2 induced telomerase activity to high levels in early-passage (p14) IMR-90 cells (Fig. 1B,C), this activity was not accompanied by an increase in either the abundance or the length of telomeric sequences (Fig. 5A). To examine the consequences of telomerase activation in normal fibroblasts, vector-containing IMR-90 cells and telomerase-positive hEST2-expressing IMR-90 cells were cultured continuously until the replicative life span of the normal IMR-90 cells was exhausted. Consistent with the idea that telomeres but not telomerase activity per se regulate replicative senescence, hEST2 expression failed to alter the life span of IMR-90 cells. These cells entered replicative senescence within two passages of the point at which normal IMR-90 cells senesced (Fig. 5B). Even after >1 month of maintenance, not a single cell from a population of >106 cells escaped the senescence block. Senescence was not attributable to a loss of telomerase as the arrested hEST2-expressing IMR-90 population maintained activity (Fig. 5C).
Figure 5.
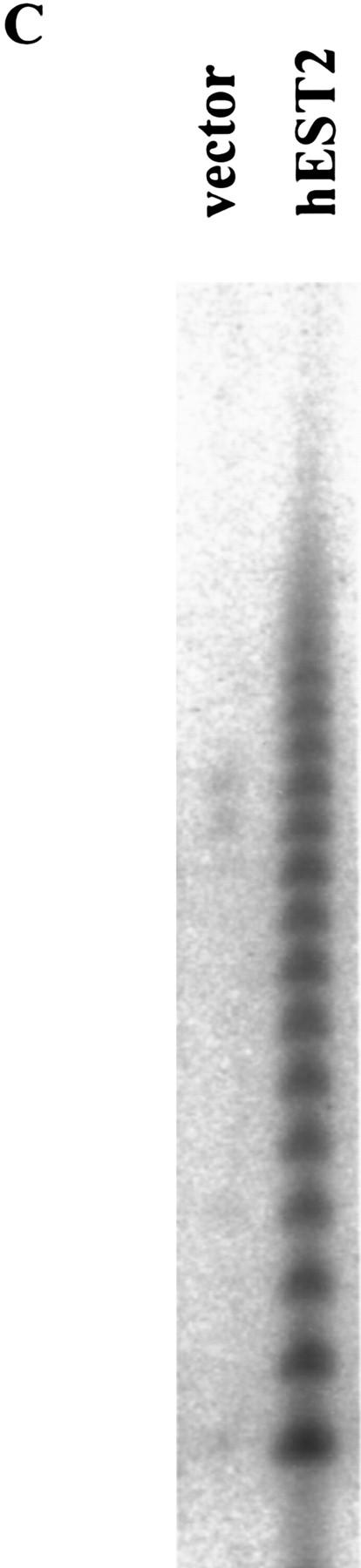
Telomerase activation does not affect life span in IMR-90 cells. (A) TRF length of senescent vector-containing IMR-90 and hEST2-expressing IMR-90 cells was analyzed as in Fig. 4. (B) Early-passage IMR-90 cells (passage 14) were infected with empty vector, a hEST2 retrovirus, or a Myc retrovirus as indicated. Cells were passaged until the vector-infected cells reached senescence (∼15 additional passages). At this time, hEST2 cells also senesced, but Myc-expressing IMR-90 cells continued to proliferate. Shown are senescence-associated β-galactosidase stains of early-passage IMR-90 cells, senescent vector-containing IMR-90 cells, senescent hEST2-expressing IMR-90 cells and Myc-expressing IMR-90 cells that have bypassed the senescence point and entered extended life span. (C) Telomerase assays of lysates derived from senescent vector-containing IMR-90 and hEST2-expressing IMR-90 populations. Each lane corresponds to 10,000 cells.
In contrast, IMR-90 cells engineered to express c-Myc display an extended life span (Fig. 5B), even though these cells do not show an obvious stabilization of telomeres. At present Myc-expressing IMR-90 populations have grown for >17 passages (∼68 PD) beyond the normal senescence point. Therefore, Myc can extend the life span of a cell even when telomerase activation fails to do so.
These results indicate that the ability of telomerase to extend life span is not universal. Telomerase-positive cells may senesce and still maintain telomerase activity. The mechanisms that regulate the ability of telomerase to extend telomeres and life span may provide an additional level of control over indefinite proliferation and thus tumorigenesis. Furthermore, telomerase-negative cells may adopt alternative strategies for telomere maintenance [alternative lengthening of telomeres (ALT); Bryan and Reddel 1997] and, therefore, achieve immortality without activation of telomerase. The long-term strategy for telomere maintenance adopted by an individual cell will likely depend on the constellation of genetic alterations that such a cell acquires along the pathway to immortality and possibly neoplastic transformation.
The myc oncogene is activated by overexpression, gene amplification, translocation, and possibly mutation in a wide variety of different tumor types (Alitalo et al. 1987). Because Myc can elevate telomerase in normal epithelial and fibroblast cells to a level approximating that observed in tumor cell lines, increased Myc activity could account for the presence of telomerase in many late-stage tumors. In this regard, a study of 100 neuroblastomas revealed that ∼20% (16/100) had exceptionally high telomerase activity. Of these, 11 showed amplification of the N-Myc locus (Hiyama et al. 1995). Thus, in this case, telomerase levels correlated well with Myc activation. Although the myc oncogene may induce telomerase in a significant proportion of tumors, telomerase may also be regulated by other pathways that contribute to transformation (Holt et al. 1997).
Although telomerase activation has been suggested to be a housekeeping component of a variety proliferative programs (Greider 1998), oncogenic transformation is often achieved through constitutive activation of elements of normal growth control. In this regard, Myc expression accompanies the proliferation of diverse cell types in vivo, and there is significant overlap between contexts in which Myc is expressed and contexts in which telomerase is detected in normal cells. For example, mitogenic stimulation of normal lymphocytes increases Myc levels (Lacy et al. 1986; Kelly and Siebenlist 1988), and stimulated lymphocytes express telomerase (for review, see Greider 1998). Telomerase activity and Myc are also found in human endometrial tissues during the menstrual cycle. Coincidentally, both Myc and telomerase are high during the proliferative phase but are low during the secretory phase (Odom et al. 1989; Kyo et al. 1997). Conversely, Myc is lost as proliferating cells differentiate and exit the cell cycle (e.g., HL-60; Mitchell et al. 1992). Differentiation of these same cells results in loss of both hEST2 expression and telomerase (Meyerson et al. 1997).
The results presented here, considered together with the overlap between Myc activation and telomerase expression in normal tissues, suggest a model in which telomerase may respond to Myc both during the execution of normal proliferation programs and in tumors. Promotion of cell proliferation and oncogenic transformation by Myc probably requires induction of a number of different target genes (for review, see Grandori and Eisenman 1997). In fact, we show that Myc can bypass replicative senescence under circumstances in which telomerase activation alone is ineffective. Thus, telomerase activity in tumors may simply reflect activation of oncogenes such as Myc. However, it is likely that telomere maintenance contributes to the long-term proliferative potential of tumor cells, and therefore telomerase activation may be one component of the ability of Myc to facilitate tumor formation.
Materials and methods
Retroviral plasmids
The following viral plasmids were used: pBabe-puro (Morgenstern and Land 1990), MarXII-hygro, mouse c-myc/MarXII-hygro, mdm-2/MarXII-hygro (from Dr. P. Sun, CSHL), E6/pBabe-puro, cdc25A/MarXII-hygro, cyclin D1/pBabe-puro, rasV12/pBabe-puro, E1A/pWzl-hygro, p53175H/pWzl-hygro, cdc25C/pBabe-puro, and E7/pBabe-puro. The full-length hEST2 cDNA (from Dr. R. Weinberg, MIT, Cambridge, MA) was cloned into pBabe-puro vector at the EcoRI and SalI sites.
Cell culture and retroviral-mediated gene transfer
HMEC 184 spiral K cells were from Dr. M. Stampter (Lawrence Berkeley Laboratory, Berkeley, CA); IMR90 and WI38 and human breast cancer cell lines BT549, T47D, and HBL100 were from ATCC; and HT1080 cells were from G. Stark (Cleveland Clinic Foundation, OH). The amphotropic packaging line, LinX-A, was produced in our laboratory (L.Y. Xie, D. Beach, and G. Hannon, unpubl.). HMEC were cultured in complete mammary epithelium growth medium (MEGM) (Clonetics). Fibroblasts and LinX-A cells were maintained in DMEM (GIBCO-BRL) plus 10% FBS (Sigma). BT549, HBL100, and T47D were maintained as directed by the supplier. LinX-A cells were transfected by calcium-phosphate precipitation with a mixture containing 15 μg of retroviral plasmid and 15 μg of sonicated salmon sperm DNA. Transfected cells were incubated at 37°C for 24 hr and then shifted to 30°C for virus production. After 48 hr, virus was collected, and the virus-containing medium was filtered to remove packaging cells (0.45-μm filter; Millipore). Target cells were infected with virus supernatants supplemented with 4 μg/ml polybrene (Sigma) by centrifugation for 1 hr at 1000g and incubation at 30°C overnight. Infected cells were selected 48 hr after infection with the appropriate drugs (hygromycin, G418, or puromycin).
Telomerase assays and expression analyses
The TRAP assay was performed essentially as described (Kim et al. 1994) with some modification. Briefly, extracts were prepared in lysis buffer (10 mm Tris at pH 7.5, 1 mm MgCl2, 1 mm EGTA, 10% glycerol) and cleared by centrifugation for 30 min at 50,000g. Lysate corresponding to from 10 to 104 cells was used. Telomeric repeats were synthesized onto an oligonucleotide, TS (5′-AATCCGTCGAGCAGAGTT-3′), in an extension reaction that proceeded at 30°C for 1 hr. Extension products were amplified by PCR in the presence of [32P]dATP with TS and a downstream anchor primer (5′-GCGCGGCTAACCCTAACCCTAACC-3′). Five microliters of each reaction was analyzed on a 6% acrylamide/8 m urea gel.
TRF length was measured as described by Strahl and Blackburn (1996) and senescence-associated β-galactosidase activity was determined as described by Serrano et al. (1997).
For Northern blotting, total RNA was isolated from subconfluent cultures by use of Trizol reagent (GIBCO-BRL). Total RNA (10 μg) or poly(A)+ RNA (5 μg) was resolved by electrophoresis and transferred to Hybond-N+ membranes according to the manufacturer’s instructions. hEST2 was visualized after hybridization with a labeled StuI fragment of hEST2 (Meyerson et al. 1997). Myc and GAPDH were visualized with probes derived from cDNAs.
Western blotting was performed essentially as described by Harlow and Lane (1988). Cells were washed with cold PBS and lysed in Laemmli loading buffer. Lysates were heated at 95°C for 10 min. Samples were separated on 8% SDS–polyacrylamide gels and transferred to nitrocellulose membranes (Schleicher & Schuell). Blots were incubated with either a c-Myc rabbit polyclonal antibody (N-262; Santa Crutz) or a TFIIB rabbit polyclonal antibody (from Dr. B. Tansey, CSHL). Immune complexes were visualized by secondary incubation with 125I-labeled protein A (ICN).
Acknowledgments
We thank R. Weinberg for providing the full-length hEST2 cDNA. HMECs were a kind gift of M. Stampfer. We thank B. Tansey for helpful discussions and for providing the TFIIB antibody. This work was supported by a grant from the U.S. Army Breast Cancer research program (DAMD17-96-1-6053) and in part by funds from the National Institutes of Health. D. Beach is supported by the Hugh and Catherine Stevenson Fund. G. Hannon is a Pew Scholar in the Biomedical Sciences.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL hannon@cshl.org; FAX (516) 367-8874.
References
- Alitalo K, Koskinen P, Makela TP, Saksela K, Sistonen L, Winqvist R. myc oncogenes: Activation and amplification. Biochim Biophys Acta. 1987;907:1–32. doi: 10.1016/0304-419x(87)90016-3. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Broccoli D, Young JW, de Lange T. Telomerase activity in normal and malignant hematopoietic cells. Proc Natl Acad Sci. 1995;92:9082–9086. doi: 10.1073/pnas.92.20.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan TM, Reddel RR. Telomere dynamics and telomerase activity in in vitro immortalised human cells. Eur J Cancer. 1997;33:767–773. doi: 10.1016/S0959-8049(97)00065-8. [DOI] [PubMed] [Google Scholar]
- Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counter CM, Hirte HW, Bacchetti S, Harley CB. Telomerase activity in human ovarian carcinoma. Proc Natl Acad Sci. 1994;91:2900–2904. doi: 10.1073/pnas.91.8.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S, Moerman EJ, Fujii S, Sobel BE. Overexpression of plasminogen activator inhibitor type-1 in senescent fibroblasts from normal subjects and those with Werner syndrome. J Cell Physiol. 1994;161:571–579. doi: 10.1002/jcp.1041610321. [DOI] [PubMed] [Google Scholar]
- Grandori C, Eisenman RN. Myc target genes. Trends Biochem Sci. 1997;22:177–181. doi: 10.1016/s0968-0004(97)01025-6. [DOI] [PubMed] [Google Scholar]
- Greider CW. Telomerase activity, cell proliferation, and cancer. Proc Natl Acad Sci. 1998;95:90–92. doi: 10.1073/pnas.95.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CB. Telomere loss: Mitotic clock or genetic time bomb? Mutat Res. 1991;256:271–282. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during aging of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Harlow EL, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Harrington L, Zhou W, McPhail T, Oulton R, Yeung DS, Mar V, Bass MB, Robinson MO. Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes & Dev. 1997;11:3109–3115. doi: 10.1101/gad.11.23.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC. Telomere reduction in human colorectal carcinoma and with aging. Nature. 1990;346:866–868. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- Hiyama E, Hiyama K, Yokoyama T, Matsuura Y, Piatyszek MA, Shay JW. Correlating telomerase activity levels with human neuroblastoma outcomes. Nature Med. 1995;1:249–255. doi: 10.1038/nm0395-249. [DOI] [PubMed] [Google Scholar]
- Holt SE, Wright WE, Shay JW. Multiple pathways for the regulation of telomerase activity. Eur J Cancer. 1997;33:761–766. doi: 10.1016/S0959-8049(97)00066-X. [DOI] [PubMed] [Google Scholar]
- Kelly K, Siebenlist U. Mitogenic activation of normal T cells leads to increased initiation of transcription in the c-myc locus. J Biol Chem. 1988;263:4828–4831. [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- Kinoshita T, Shirasawa H, Shino Y, Moriya H, Desbarats L, Eilers M, Simizu B. Transactivation of prothymosin alpha and c-myc promoters by human papillomavirus type 16 E6 protein. Virology. 1997;232:53–61. doi: 10.1006/viro.1997.8536. [DOI] [PubMed] [Google Scholar]
- Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380:79–82. doi: 10.1038/380079a0. [DOI] [PubMed] [Google Scholar]
- Kyo S, Takakura M, Kohama T, Inoue M. Telomerase activity in human endometrium. Cancer Res. 1997;57:610–614. [PubMed] [Google Scholar]
- Lacy J, Sarkar SN, Summers WC. Induction of c-myc expression in human B lymphocytes by B-cell growth factor and anti-immunoglobulin. Proc Natl Acad Sci. 1986;83:1458–1462. doi: 10.1073/pnas.83.5.1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, Bacchetti S, Haber DA, Weinberg RA. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- Mitchell LS, Neill RA, Birnie GD. Temporal relationships between induced changes in c-myc mRNA abundance, proliferation, and differentiation in HL60 cells. Differentiation. 1992;49:119–125. doi: 10.1111/j.1432-0436.1992.tb00776.x. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- Odom LD, Barrett JM, Pantazis CG, Stoddard LD, McDonough PG. Immunocytochemical study of ras and myc proto-oncogene polypeptide expression in the human menstrual cycle. Am J Obstet Gynecol. 1989;161:1663–1668. doi: 10.1016/0002-9378(89)90946-0. [DOI] [PubMed] [Google Scholar]
- Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973;41:181–190. doi: 10.1016/0022-5193(73)90198-7. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE. Telomerase activity in human cancer. Curr Opin Oncol. 1996;8:66–71. doi: 10.1097/00001622-199601000-00012. [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE, Brasiskyte D, Van der Haegen BA. E6 of human papillomavirus type 16 can overcome the M1 stage of immortalization in human mammary epithelial cells but not in human fibroblasts. Oncogene. 1993;8:1407–1413. [PubMed] [Google Scholar]
- Shay JW, Tomlinson G, Piatyszek MA, Gollahon LS. Spontaneous in vitro immortalization of breast epithelial cells from a patient with Li-Fraumeni syndrome. Mol Cell Biol. 1995;15:425–432. doi: 10.1128/mcb.15.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer MS, Gottschling DE. TLC1: Template RNA component of Saccharomyces cerevisiae telomerase. Science. 1994;266:404–409. doi: 10.1126/science.7545955. [DOI] [PubMed] [Google Scholar]
- Stein GH, Dulic V. Origins of G1 arrest in senescent human fibroblasts. BioEssays. 1995;17:537–543. doi: 10.1002/bies.950170610. [DOI] [PubMed] [Google Scholar]
- Strahl C, Blackburn EH. Effects of reverse transcriptase inhibitors on telomere length and telomerase activity in two immortalized human cell lines. Mol Cell Biol. 1996;16:53–65. doi: 10.1128/mcb.16.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997;385:740–743. doi: 10.1038/385740a0. [DOI] [PubMed] [Google Scholar]
- Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972;239:197–201. doi: 10.1038/newbio239197a0. [DOI] [PubMed] [Google Scholar]
- Weinrich SL, Pruzan R, Ma L, Ouellette M, Tesmer VM, Holt SE, Bodnar AG, Lichtsteiner S, Kim NW, Trager JB, Taylor RD, Carlos R, Andrews WH, Wright WE, Shay JW, Harley CB, Morin GB. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nature Genet. 1997;17:498–502. doi: 10.1038/ng1297-498. [DOI] [PubMed] [Google Scholar]
- Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]