

Abstract
Asymmetric cell division is a widespread mechanism in developing tissues that leads to the generation of cell diversity. In the embryonic central nervous system of Drosophila melanogaster, secondary precursor cells—ganglion mother cells (GMCs)—divide and produce postmitotic neurons that take on different cell fates. In this study, we show that binary fate decision of two pairs of sibling neurons is accomplished through the interplay of Notch (N) signaling and the intrinsic fate determinant Numb. We show that GMCs have apical–basal polarity and Numb localization and the orientation of division are coordinated to segregate Numb to only one sibling cell. The correct positioning of Numb and the proper orientation of division require Inscuteable (Insc). Loss of insc results in the generation of equivalent sibling cells. Our results provide evidence that sibling neuron fate decision is nonstochastic and normally depends on the presence of Numb in one of the two siblings. Moreover, our data suggest that the fate of some sibling neurons may be regulated by signals that do not require lateral interaction between the sibling cells.
Keywords: Asymmetric cell division, neural progenitor, cell fate determinant
The generation of cellular diversity in a variety of developing contexts requires the specification of asymmetric sibling cell fates (Horvitz and Herskowitz 1992; Posakony 1994; Chenn and McConnell 1995). The embryonic central nervous system (CNS) of Drosophila melanogaster is segmented and bilaterally symmetric. Each hemisegment contains ∼300 neurons (Bossing et al. 1996) and 30 glia (Ito et al. 1995) that derive from ∼30 stem cell-like progenitor cells called neuroblast/glioblasts (NB/GB). NBs undergo repeated asymmetric divisions with the mitotic spindle oriented perpendicular to the apical surface of the embryo. Each division yields a NB and a secondary precursor cell—the ganglion mother cell (GMC). GMCs divide once into pairs of postmitotic sibling neurons (and/or glia), which often take on different cell fates. Dividing NBs exhibit an apical–basal polarity that is reflected in the exclusive positioning of the cell fate determinants such as Numb (Uemura et al. 1989) and Prospero (Pros) (Doe et al. 1991; Vaessin et al. 1991; Matsuzaki et al. 1992) at the basal cortex. The asymmetric division of NBs (horizontal cleavage plane) results in the segregation of Numb (Rhyu et al. 1994; Kraut et al. 1996) and Pros (Hirata et al. 1995; Knoblich et al. 1995; Spana and Doe 1995) into GMCs. Recently, a protein—Inscuteable (Insc)—has been identified (Kraut and Campos-Ortega 1996) that acts as an organizer of asymmetry in NBs with respect to spindle orientation as well as protein and RNA localization (Kraut et al. 1996; Li et al. 1997). Consistent with the notion that insc functions in establishing polarity for the NB asymmetric cell division, the Insc protein itself is localized to the apical cell cortex prior to mitosis. Moreover, loss of insc function causes the basal cortical localization of Numb and Pros to become randomized and prevents the mitotic spindle from becoming correctly oriented. In the absense of Insc, several embryonic CNS neurons can be duplicated, suggesting that the mechanisms that govern binary sibling fate determination may be perturbed (Kraut and Campos-Ortega 1996; Knirr et al. 1997).
Studies on asymmetric sibling cell-fate choice in the embryonic CNS have been limited so far to the MP2 lineage. The MP2 precursor delaminates from the neuroectoderm and is morphologically identical to neuroblasts. In contrast to neuroblasts, MP2 divides only once to produce two post–mitotic neurons with distinct identities: The slightly larger dMP2 neuron and the smaller vMP2 neuron. Spana and Doe (1996) have shown that the acquisition of the proper cell fates requires the combined action of extrinsic and intrinsic cues. The MP2 precursor divides asymmetrically with respect to the segregation of Numb protein into the prospective dMP2 cell. Loss of numb function results in the duplication of vMP2 at the expense of dMP2, indicating that the function of numb within the MP2 lineage is to act as an intrinsic fate determinant (Spana et al. 1995). In addition to Numb, several proteins that have been shown previously to act in cell signaling are required for the proper fate choice of the MP2 progeny: Loss of the transmembrane receptor Notch (N) (Artavanis-Tsakonas et al. 1995) or its ligand Delta causes both MP2 progeny to adopt the dMP2 fate, a phenotype that is opposite to that of numb mutations.
The interplay of N-mediated cell signaling and the intrinsic fate determinant Numb has been studied extensively in the process of binary cell fate choice in the external sensory (es) organ (Uemura et al. 1989; Rhyu et al. 1994; for review, see Jan and Jan 1995; Knoblich et al. 1995; Campos-Ortega 1996; Guo et al. 1996). Each es organ is derived from a single precursor cell (SOP) that undergoes two rounds of asymmetric cell divisions to generate four cells with distinct identities. The first SOP division gives rise to the intermediate precursor cells pIIa and pIIb. The acquisition of the correct cell fates is dependent on the asymmetric segregation of Numb into pIIb: Loss of Numb function results in the duplication of pIIa at the expense of pIIb. Conversely, mutations in components of the N signaling pathway (N, Delta, and Suppressor of Hairless) cause the two daughter cells to adopt the fate of pIIb. Accordingly, proper cell fate specification of the daughter cells of pIIa and pIIb is regulated through the interaction of Numb and N signaling.
The effects of N and numb single and double mutations on the SOP and MP2 lineages has led to the following model for binary cell fate determination: Productive N signaling is required in one of the sibling cells to prevent it from adopting the primary (default) fate. In the other sibling, N signaling is aborted by the interaction of the receptor with Numb, thereby enabling the cell to adopt the primary fate (Jan and Jan 1995; Spana and Doe 1996). This model is corroborated by the observation that the Numb protein can interact directly with N (Guo et al. 1996). This model implies that the prospective fate of sibling cells is predetermined by the asymmetric localization of Numb during mitosis of the respective progenitor cell. It has been postulated that in the absence of numb, the realization of asymmetric sibling fates can still be accomplished solely through N signaling (e.g., Jan and Jan 1995). In this context, N signaling itself would be the source of asymmetry: Reciprocal signaling of initially equivalent strength could result in irreversible asymmetry when small, randomly occuring differences are amplified by intercellular feed-back loops. Because each cell has a 50:50 probability of adopting either fate, the outcome of this process would be stochastic and unbiased (N signaling is reviewed by Artavanis-Tsakonas et al. 1995).
With the exception of the progeny of MP2, the issue of sibling neuron fate determination in the embryonic CNS has not been addressed experimentally. In contrast to the MP2 neurons that derive directly from a neuroblast-like cell, the vast majority of neurons derive from intermediate precursor cells—the GMCs. N and Numb are widely expressed at the time when cell fate decisions take place and may interact to specify the fate of the GMC progeny in a process analogous to that of fate specification within the SOP and MP2 lineages. However, the results of laser ablation experiments in the closely related grasshopper suggest that lateral signaling is required to resolve fate specification of at least one pair of sibling neurons in a stochastic and unbiased manner (Kuwada and Goodman 1985). Moreover, mutant alleles of numb that are associated with extensive fate transformations within the SOP and MP2 lineages do not affect the fate of a group of identified sibling neurons (Spana et al. 1995).
In this study we have analyzed the effect of insc-mediated asymmetry on sibling neuron fate determination. We show that the immediate precursors of postmitotic neurons—the GMCs—possess the same apical–basal polarity that is exhibited by NBs. GMCs divide in an asymmetric manner to generate pairs of sibling neurons that are intrinsically different. The organization of asymmetry in dividing GMCs requires the function of Insc protein. The asymmetric division of GMCs leads to the exclusive segregation of Numb protein into one of the sibling neurons. Loss of Insc randomizes this distribution and results in the generation of equivalent sibling neurons. We have analyzed the role of Numb as a potential cell fate determinant using two newly identified alleles. Our results indicate that strong alleles of numb cause a failure to resolve sibling neuron fates. In addition, we found that loss of function of N as well as newly isolated (weak) alleles of mastermind (mam) (Smoller et al. 1990)—a putative downstream effector of N signaling (Fortini and Artavanis-Tsakonas 1994)—cause cell fate transformations that are opposite to those of numb loss-of-function mutants. We conclude that binary cell fate specification of sibling neurons is nonstochastic and is accomplished through the interplay of N-mediated signaling and Insc-dependent asymmetric segregation of Numb; moreover, under conditions in which numb is limiting and/or in the absence of asymmetric segregation of Numb, extrinsic cues mediated by N are insufficient to resolve distinct cell fates for the GMC derived neuronal sibling pairs. Finally, our data suggest that the extrinsic signals regulating the fate of sibling neurons do not require lateral interaction between the sibling cells.
Results
An inherent size difference in sibling neurons RP2 and RP2sib
To analyze the roles of insc, numb, and components of the N-signaling pathway in fate specification, we have focused on the only two pairs of GMC-derived neurons for which sibling relationships have been established: the RP2/RP2sib and the aCC/pCC neurons (Fig. 1A). These neurons are derived from two GMCs that can be identified unambigously by their specific expression of the nuclear protein Even-skipped (Eve) (Frasch et al. 1987; Doe 1992; Patel et al. 1994). GMC1-1a divides into the aCC/pCC neurons that have approximately equal size and continue to express Eve. However, at later stages of development, aCC is distinguished from pCC by the expression of Zfh-1 (a putative transcription factor; Lai et al. 1991; see Figs. 1A and 3A, below) and 22C10 (a membrane associated antigen; Fujita et al. 1988). aCC is a motoneuron and forms an ipsilateral projection that pioneers the intersegmental nerve (Fig. 3B, below). GMC4-2a divides to form the sibling neurons RP2/RP2sib that are morphologically distinguishable: In 88% of the hemisegments, the newborn siblings show a significant difference in the size of their nuclei and cell bodies (Fig. 2A; Table 1). This asymmetry appears to be initiated during cell division: In GA1019 mutant embryos in which GMC4-2a fails to complete cytokinesis (M. Buescher, unpubl.; see Materials and Methods), cells are formed that contain one large and one small nucleus (Fig. 2C; Table 1). This strongly suggests that the difference in size is generated early, prior to the completion of cytokinesis. (For the possible significance of the size difference, see Discussion, second to last paragraph). The larger cell always adopts the RP2 fate that is characterized by the expression of Eve, Zfh-1 (Figs. 1A and 3A), and 22C10. RP2 forms an antero-ipsilateral projection (Fig.3B). The smaller sibling always adopts the RP2sib fate, which is characterized by a further decrease in cell and nuclear size and the loss of Eve immunreactivity. Zfh-1 and 22C10 expression have not been shown in RP2sib. These observations suggest that the cell and nuclear size difference may serve as an early physical marker that will allow us to differentiate between the two progeny of GMC4-2a irrespective of the molecular markers they express later.
Figure 1.
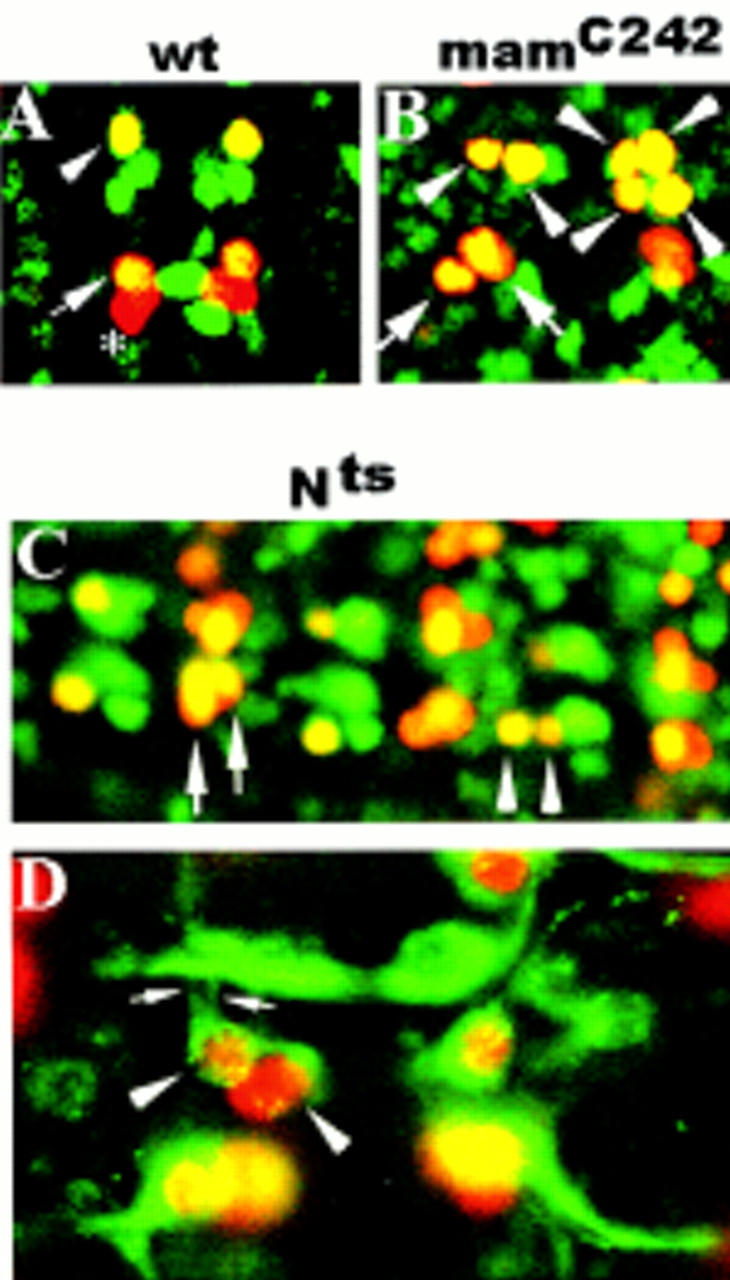
Mutations in mam and N are associated with RP2sib to RP2 and pCC to aCC fate transformations. (A,B,C) Ventral views of stage 15 embryos double stained with rabbit anti-Eve (red) and mouse anti-Zfh-1 (green). (A,B) Anterior is up; (C) anterior is left. (A) wt RP2 (arrowhead) and aCC (large arrow), but not pCC (asterisk), coexpress Eve and Zfh-1 (yellow). (B) mamC242. (C) Nts1. Note the different sizes of the RP2 nuclei in B and C (arrowheads). The right hemisegment in B depicts four RP2 neurons that presumably derive from two parental NB4-2. (D) Ventral view of a stage 15 whole-mount Nts1 embryo stained with rabbit anti-Eve (red) and mouse-mAb 22C10 (green). Anterior is up. Note the ipsilateral axon projections of both RP2s (small arrows).
Figure 3.
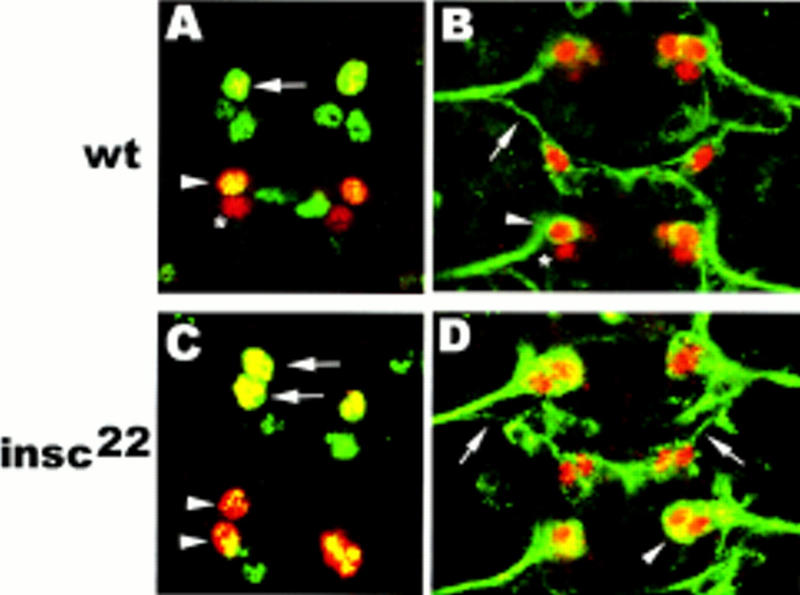
Loss of insc causes RP2sib to RP2 and pCC to aCC fate transformations. Ventral view of whole-mount stage 15 embryos. Anterior is up. (A,C) Double staining with rabbit anti-Eve (red) and mouse anti-Zfh-1 (green). (B,D) Double staining with rabbit anti-Eve (red) and mouse mAb 22C10 (green). (A,B) wt: RP2 (arrow) and aCC (arrowheads) coexpress Eve and Zfh-1 (yellow in A) and form a 22C10-positive, ipsilateral axon projection (green in B); pCC (asterisk) neither expresses Zfh-1 (A) nor 22C10 (B). (C,D) insc22. Note that both GMC1-1a and GMC4-2a progeny coexpress Eve and Zfh-1 (yellow in C) and form an ipsilateral axon projection (D).
Figure 2.
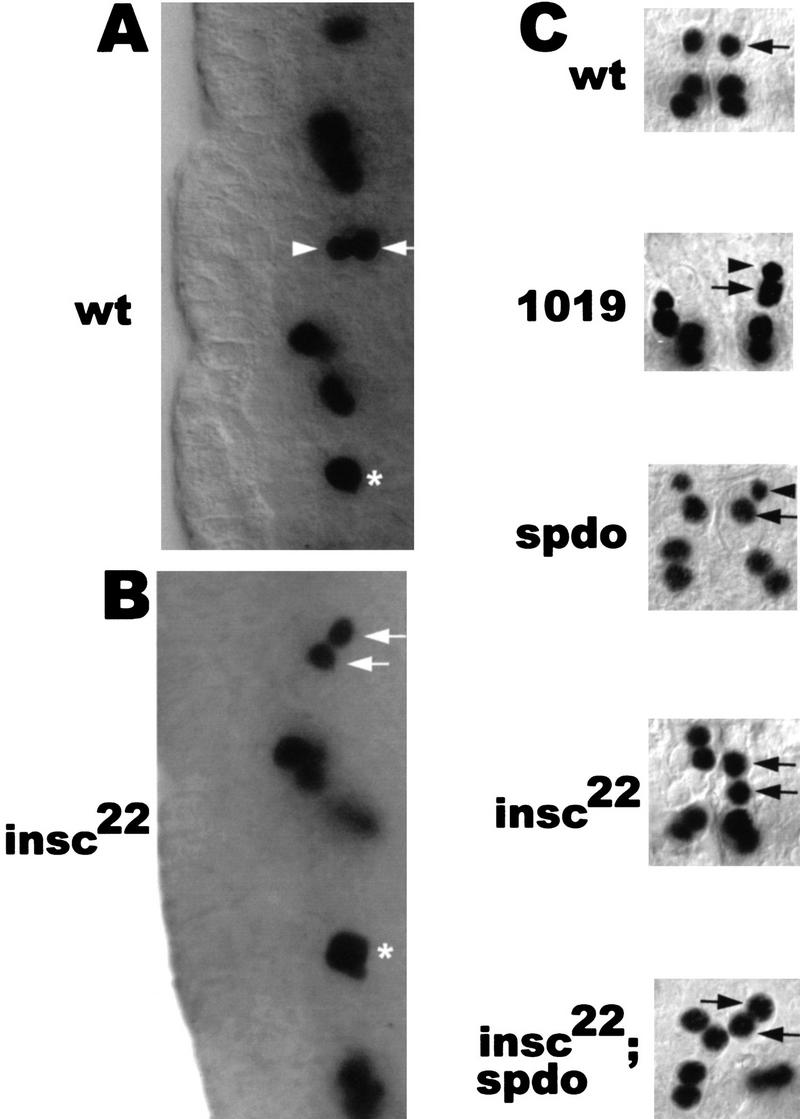
Mutations in spdo result in RP2sib to RP2 fate transformation with respect to marker gene expression but not with respect to cell/nuclear size. (A,B) Lateral views of stage 11 embryos stained with anti-Eve. Ventral (apical) is to the left. (A) In wt, GMC4-2a divides into a smaller and a larger cell. The newborn siblings are oriented perpendicular to the apical surface with the larger cell (future RP2) in the more dorsal position. (B) In insc22, GMC4-2a divides into sibling cells of equal size that are rarely oriented perpendicular to the apical surface. (Arrows) RP2 neuron; (arrowheads) RP2sib; (asterisks) undivided GMC4-2a. (C) Dorsal views of dissected wild-type and various mutant stage 15 embryos stained with anti-Eve. Anterior is up. In GA1019 (1019) embryos, GMC4-2a does not undergo cytokinesis and binucleated cells with two Eve-expressing nuclei are formed. Note the difference in the sizes of the two nuclei (arrowhead vs. arrow) derived from GMC4-2a in spdo and GA1019 embryos.
Table 1.
Size difference of sibling progeny derived from GMC4-2a cell division
|
|
Percent unequal cell/ nucleus size
|
|---|---|
| Wild type | 88 (n = 79) |
| 1019 (binucleated cells) | 85 (n = 22) |
| spdozz27 | 83 (n = 106) |
| insc22 | 17 (n = 88) |
| insc22; spdozz27 | 11 (n = 39) |
(n) Number of hemisegments.
Mutations in components of the Notch signaling pathway are associated with sibling cell-fate transformations
From a collection of second chromosome lethal mutations (Seeger et al. 1993), we isolated novel, hypomorphic alleles of mam (mamC242, mamGA424, mamGA1037; see Materials and Methods). In addition to a general, mild hypertrophy in the CNS, these alleles were found to be associated with specific sibling cell-fate transformations. For a detailed analysis, we chose the weakest of these alleles, mamC242. Frequently, we observed two neurons in the aCC/pCC position that both display a combination of marker gene expression characteristic for the aCC but not the pCC fate (Fig. 1B). Moreover, in mamC242 mutant embryos, both progeny of GMC4-2a can adopt the RP2 fate with respect to Eve, Zfh-1 (Fig. 1B), and 22C10 (data not shown) expression. However, despite this apparent change of the RP2sib to the RP2 cell fate, the unequal size of the GMC4-2a daughter cells remains unaffected (Fig. 1B). It is interesting to note that in mamC242 mutant embryos, the number of Eve-expressing CQ neurons is strongly reduced (see below and Discussion).
These results show that mam is required for the correct fate specification of RP2sib and pCC but not for that of RP2 and aCC. The requirement for mam suggests that N signaling may be involved in the resolution of distinct sibling neuron cell fate. We analyzed the CNS phenotype of a temperature-sensitive allele of N (Nts1; Shellenbarger and Mohler 1975). When Nts1 embryos were allowed to develop at the permissive temperature (18°C), aCC/pCC and RP2/RP2sib cell fates were found to be correctly resolved (data not shown). In contrast, embryos that were shifted to the nonpermissive temperature (29°C) 2–4 hr after egg lay displayed sibling cell-fate transformations in addition to a general hypertrophy (Fig. 1C,D). Specifically, we observed pCC to aCC and RP2sib to RP2 cell fate transformations as judged by marker gene expression (Fig. 1C) and axon trajectory (Fig. 1D). Moreover, as in mam mutant embryos, the size difference of the two GMC4-2a progeny was found to be unaltered. These data show that mutations in mam and N result in similar defects and support the notion that N signaling is required for the resolution of sibling neuron fate. Similar conclusions have been reached independently for the CNS (Skeath and Doe 1998) and PNS (Dye et al. 1998).
Mutations in mam, sanpodo, and Notch equalize aspects of sibling cell fate but retain the difference in cell and nuclear size of sibling neurons
Recently, a novel gene has been identified that is required for the correct cell-fate choice of RP2 and RP2sib. sanpodo (spdo) encodes a Drosophila tropomodulin, which is an actin-tropomyosin binding protein (Salzberg et al. 1994; Dye et al. 1998). Loss of spdo has been shown to equalize the fate of PNS sibling cells. Many pairs of cells take on a neuronal identity instead of a neuronal and ectodermal/glia identity in the PNS (Dye et al. 1998). Moreover, in the embryonic CNS, loss of spdo is associated with several sibling neuron fate transformations (Skeath and Doe 1998). In the absence of spdo (spdozz27) function, the fate of RP2sib is transformed to that of RP2 with respect to marker gene expression; however, the difference in nuclear and cell size is retained (Fig. 2C; Table 1). It is important to note that in this mutant, the GMC4-2a identity does not appear to be altered—it is born at the right time, expresses normal levels of Eve and Pdm-1 (Yeo et al. 1995), and, in addition, divides to produce neurons (RP2/RP2sib or RP2/RP2) that are normally produced by the wild-type GMC4-2a. These results suggest that mam, spdo, and N play a role in fate determination at the level of the postmitotic sibling neurons but affect neither GMC4-2a identity nor the asymmetry of the GMC4-2a division (see below).
Loss of insc function equalizes sibling neuron fate as well as cell and nuclear size
For this study, we used the insc22 allele (Knirr et al. 1997), which represents an EMS-induced premature translational termination mutation near the amino terminus of the insc coding region (M. Zavortink, unpubl.). insc22 homozygous embryos do not produce detectable amounts of Insc protein in the embryonic nervous system as judged by immunofluorescence (data not shown). With respect to NB cell division, insc22 mutant embryos exhibit defects in Pros localization as well as mitotic spindle orientation (M. Zavortink, unpubl.) similar to those described previously for insc alleles P49 and P72, which are multigenic deletions that remove other genes besides insc (Kraut and Campos-Ortega 1996; Knirr et al. 1997).
In insc22 mutant embryos, GMC1-1a and GMC4-2a are correctly formed and express normal levels of Eve (and in the case of GMC4-2a, also Pdm-1) (data not shown). However, GMC1-1a divides to form two sibling neurons that both adopt the aCC fate (94%) with respect to marker gene expression (Fig. 3C). Similarly, GMC4-2a division results in two sibling cells, both of which adopt the RP2 fate (96%) with respect to expression of Eve, Zfh-1 (Fig. 3C), and 22C10 as well as axon morphology (Fig. 3D). This strongly suggests that in wild-type embryos, the divisions of GMC1-1a and GMC4-2a are asymmetric in an insc-dependent manner and produce sibling cells that are intrinsically different (see below); loss of insc function leads to the generation of sibling neurons with equivalent cellular identities.
Moreover, in contrast to mam, spdo, and N mutant embryos, the duplicated RP2s seen in insc22 are equal with respect to their cell and nuclear size (Fig. 2B,C; Table 1). These observations are consistent with the idea that the size difference seen in wild-type embryos is generated by an insc-dependent process during the GMC cell division and occurs prior to the events mediated by mam, spdo, and N that presumably act at the level of the postmitotic sibling cells. If this interpretation is correct, no size asymmetry between the sibling neurons should be generated in an insc background regardless of whether the other functions (e.g., spdo) are present or not. To test this, we constructed an insc22; spdozz27 double mutant. In the double mutant embryos stained with anti-Eve, we observed that 89% of GMC4-2a progeny have equal size, indicating that with respect to this morphological trait, the insc22 phenotype (equal sibling size) predominates over those of spdo (unequal sibling size) (Fig. 2C, Table 1). These findings are consistent with the idea that Insc acts to impose size asymmetry on GMC4-2a cell division, whereas the other gene products like Spdo, N, and Mam act later in sibling cell-fate choice. To date, insc is the only gene known to effect both asymmetric fate choice and unequal cell/nuclear size of RP2/RP2sib.
GMCs divide asymmetrically with Insc localized as apical and Numb as basal crescents
Staining of wild-type embryos with anti-Insc antibody revealed that Insc is expressed in many, and possibly all, GMCs. During interphase, Insc protein is found cortically (n = 11; data not shown). In dividing GMCs, anti-Insc immunoreactivity is always seen as an apical/near apical crescent with respect to the surface of the embryo (n = 78). Figure 4A depicts the apical localization of Insc in metaphase GMCs. Figure 4B,C shows the apical localization of Insc protein specifically in GMC4-2a and GMC1-1a. These observations suggest that GMCs possess apical–basal polarity and that their division may be asymmetric in an insc-dependent manner.
Figure 4.
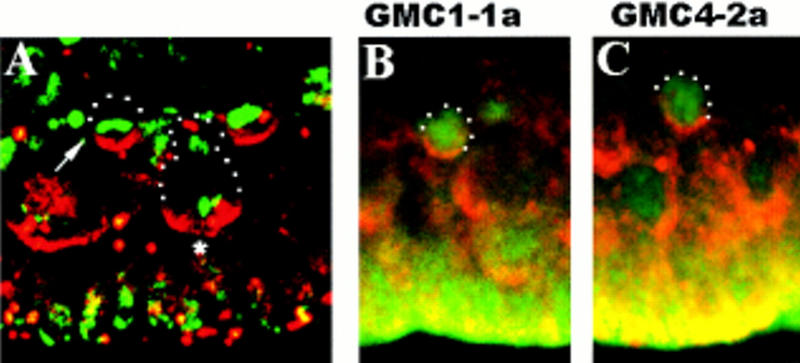
Insc protein forms apical crescents in dividing GMCs. Lateral views of wt stage 11 embryos; basal (dorsal) is up. (A) Double staining with rabbit anti-Insc (red) and DNA stain (green). (Arrow) GMC in metaphase. Note that the metaphase plate is oriented horizontally with respect to the apical surface. (Asterisk) Dividing neuroblast. (B,C) GMC1-1a and GMC4-2a double stained with anti-Insc (red) and mouse anti-Eve (green).
If the asymmetry of GMC division leads to the generation of pairs of sibling cells that are nonequivalent, then which are the cell fate determinants that are segregated differentially? One such candidate is Numb: It is widely expressed in the developing CNS and has been shown to act as an intrinsic fate determinant in the SOP and MP2 lineages (Rhyu et al. 1994; Spana and Doe 1996). Moreover, asymmetric Numb localization has been shown to be Insc-dependent in NBs (Kraut et al. 1996). To analyze if Numb is asymmetrically localized in GMC1-1a and GMC4-2a in an Insc-dependent manner, we stained wild-type and insc22 embryos with anti-Numb, anti-Eve, and DNA stain (Fig. 5A–H). In wild-type embryos, Numb always forms a crescent at or near the basal cortex of dividing GMCs (n = 80), whereas in insc22 mutant embryos, crescents are either not formed (72%) or appear in basal (20%) and occasionally in lateral or apical positions (8%) (n = 54).
Figure 5.
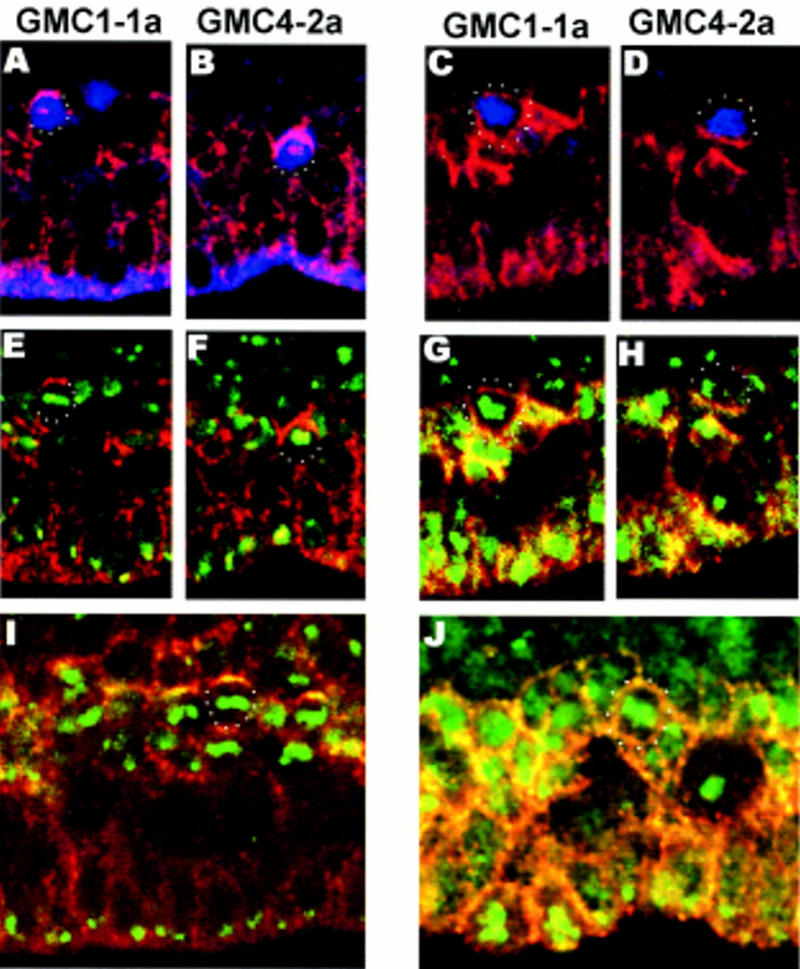
insc is required for the coordination of Numb localization and the orientation of the metaphase plate in GMCs. Lateral views of dividing GMCs in stage 11 embryos triple stained with anti-Numb (with rhodamine conjugated secondary antibody, red), mouse anti-Eve (with FITC conjugated secondary antibody, blue) and DNA stain (green). Basal (dorsal) is up. (A,B,E,I, left) Wild type; (C,D,G,H,I, right) insc22. Top panels (A–D) depict the anti-Numb and anti-Eve staining; middle panels (E–H) show the anti-Numb and DNA staining of the same samples; bottom panels (I,J) show the anti-Numb and DNA staining in randomly chosen GMCs. (A,B,E,F,I) wt. Note the basal Numb crescents in all samples and the horizontal orientation of the metaphase plates in E and I. (C,D,G,H,J) insc22. Note the cortical Numb staining with no distinct crescents in C,G,J and the apical crescent in D and H. The metaphase plates are not oriented horizontally with respect to the apical surface (G,J). Relevant cells are circled with dots.
Although it is difficult to directly visualize the orientation of the mitotic spindle in GMCs, the positioning of the metaphase plate in wild-type strongly suggests a perpendicular orientation with respect to the apical surface of the embryo (n = 12 for GMC1-1a and GMC4-2a) (Fig. 5E). Consistent with a horizontally placed cleavage plane, we always observe the newborn RP2/RP2sib cells oriented perpendicular or nearly perpendicular to the apical surface with the larger cell located in the more dorsal (basal) position (n = 114) (Fig. 2A). We conclude that during GMC4-2a division, Numb will be segregated predominantly or exclusively to the future RP2 neuron (we cannot predict which of the GMC1-1a progeny will inherit Numb, because the size difference between aCC and pCC is too small to serve as an unambigous marker). In addition, we frequently observed that the orientation of metaphase plate in dividing GMCs other than GMC4-2a is also horizontal with respect to the apical surface (n = 34, in random GMCs) (Figs. 4A and 5I), indicating that the division of many, possibly all, GMCs is stereotyped in the same manner as NB divisions. Consistent with its function in NBs, loss of insc alters the orientation of GMC division (Fig. 5G,H,J): In 74% of the samples (n = 38) the metaphase plate is oriented perpendicular or close to perpendicular to the apical surface. Accordingly, the newborn RP2/RP2sib are frequently oriented horizontally with respect to the apical surface (see Fig. 2B). Taken together, our results indicate that the apical–basal polarity, which is found in NBs is maintained in their daughter cells, the GMCs, and that Numb localization as well as spindle orientation are coordinated to ensure asymmetric segregation of Numb into one sibling only. So far, insc appears to be the only gene that is required for the apical–basal polarity of GMCs: The analysis of mam, spdo, and N mutant embryos revealed that none of these mutations alter the plane of GMC division (data not shown). This observation is in agreement with the notion that these genes act later than insc in sibling neuron fate determination (see above).
insc acts through numb to effect asymmetric sibling cell fate
The sibling cell-fate changes that are found in insc22 mutant embryos coincide with the mislocalization of numb in the corresponding GMCs. To address the question of whether incorrect localization/segregation of Numb may be the cause for the observed cell fate changes, we analyzed the phenotype of two previously described alleles of numb, nb1 and nb3, as well as two novel alleles, nb796 and nb2758, isolated from a collection of second chromosome lethal mutations (see Materials and Methods; Seeger et al. 1993) and a deficiency [Df(2L)30A-C] that removes numb as well as other genes. nb1 and nb3 have been shown to cause extensive cell-fate changes in the developing PNS (Uemera et al. 1989; Rhyu et al. 1994). In agreement with Spana et al. (1995), we did not observe any cell-fate changes of RP2/RP2sib or the aCC/pCC sibling neuron pairs in nb1 and nb3 embryos. However, in the EMS-induced allele nb796, 27% of mutant hemisegments fail to exhibit an Eve-positive RP2 neuron (nb2758:19%) (Fig. 6; Table 2). Detailed analysis revealed that Eve-expressing GMC4-2a(s) were properly formed and their division produced two sibling cells of unequal size (not shown). However, Eve immunoreactivity was gradually lost in both progeny (Fig. 6G) and Zfh-1 expression was not initiated in either cell (data not shown) suggesting that both siblings adopt the fate of RP2sib. These results indicate that asymmetrically segregated Numb is an important component of RP2/RP2sib fate specification and loss or reduction of numb function causes a phenotype that is opposite to that of mam, spdo, and N mutations. Consistent with this idea is the observation that the number of Eve-positive CQ neurons is increased in nb796 embryos (mamC242 is associated with a loss of Eve-positive CQ neurons; see above). The aCC/pCC fate could not be determined because these neurons cannot be identified because of the increase in CQ neurons.
Figure 6.
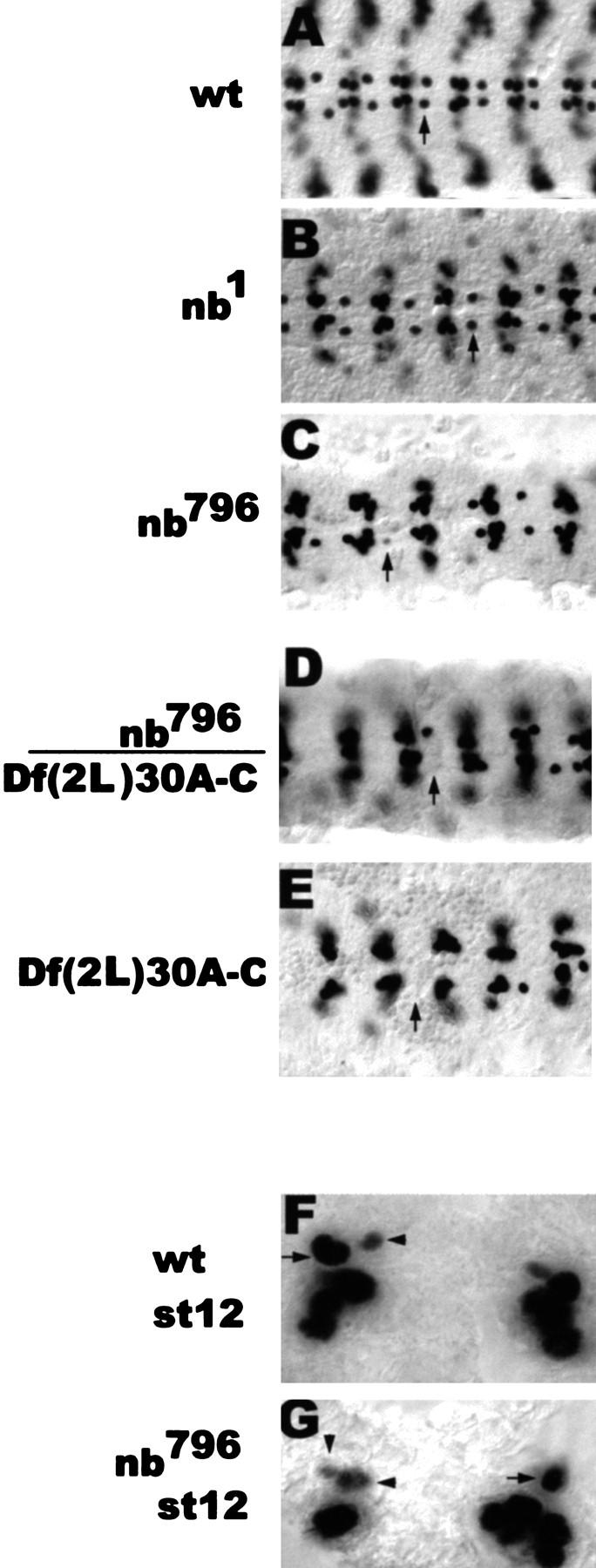
Mutations in numb cause RP2 to RP2sib fate transformation. (A–E) Dorsal views of dissected stage 15 embryos stained with rabbit anti-Eve. Anterior is to the left. (Arrows) RP2 neuron (A) or the RP2 position (C–E). (F,G) Ventral views of stage 12 whole-mount embryos stained with rabbit anti-Eve. Anterior is up. (Arrowheads) RP2sib. Compare the fading Eve staining in the smaller and the larger cell in nb796 (G) with the fading Eve staining in the smaller cell only in wt (F).
Table 2.
Frequency of RP2 to RP2sib fate transformations associated with different alleles of numb
| Allele
|
Percent frequency
|
|---|---|
| nb1 | 4 (n = 64) |
| nb3 | 2 (n = 56) |
| nb796 | 27 (n = 89) |
| nb2758 | 19 (n = 101) |
| nb1/nb796 | 11 (n = 56) |
| nb1/Df(2L)30A–C | 23 (n = 79) |
| nb796/Df(2L)30A–C | 62 (n = 74) |
| nb2758/Df(2L)30A–C | 31 (n = 112) |
| Df(2L)30A–C | 83 (n = 47) |
| HSNintra | 34 (n = 82) |
| insc22 nb796 | 97 (n = 112) |
| GA339 nb796 | 32 (n = 153) |
(n) Number of hemisegments.
The rather low penetrance of the numb phenotype has prompted us to determine the relative strength of the various alleles by analyzing their phenotype in hemizygosity with the corresponding deficiency. On the basis of our results (see Table 2) we conclude first, the phenotypes of nb2758 and nb796 are most probably the result of lesions in the numb gene alone and not the result of interactions with potential second site mutations (see also Table 2: nb796/nb1). Second, as expected from their homozygous phenotypes, the different numb alleles can be classified according to their strength: nb1, nb3, nb2758, and nb796. Third, even the strongest allele—nb796—does not represent a total loss-of-function mutation because the phenotype of the nb796 homozygote is weaker than that of the nb796/deficiency (Table 2). To date, none of the numb alleles described appear to be amorphic with respect to their CNS cell fate phenotype. However, the phenotype of homozygous deficiency—Df(2L)30A-C—embryos is quite informative. Despite some pleiotrophic morphological defects, most Eve-expressing neurons can be identified unambigously. We observed a loss of RP2 from 83% of the hemisegments. In those hemisegments in which RP2/RP2sib fate is correctly resolved (17% of all hemisegments), we found that the larger cell always adopts the RP2 fate, whereas the smaller cell differentiates into RP2sib. This suggests that even in the absence of all zygotic numb, RP2/RP2sib cell fates are not resolved via a stochastic mechanism because we never see the cell with the smaller nucleus adopting the RP2 fate.
These results prompted us to further analyze the role of numb in cell fate specification in a situation in which equivalent sibling cells are generated. This situation appears to exist in insc mutant embryos and, therefore, we made the double mutant insc22 nb1. Staining with anti-Eve revealed a near complete loss of Eve-positive cells in the RP2 position (98% loss; this is a conservative estimate because in the 2% of the hemisegments in which an Eve+ cell can be seen at or near the RP2 position, we cannot rule out that this cell might be a misplaced CQ neuron) (Fig. 7D,E). This is a dramatic effect because nb796 homozygotes show only a moderate loss of RP2 (27% loss) and insc22 homozygotes show RP2 duplication at close to complete expressivity (96% duplication) (Fig. 7B,C). Yet, the double mutant exhibits a phenotype that is stronger than that seen for the strongest numb allele, for example, embryos homozygous for the numb deficiency. Our data show that in a situation in which equivalent sibling cells are generated, cell fate specification is largely dependent on numb function. A possible rationalization of the double-mutant phenotype is that nb796 produces sufficient function to resolve RP2/RP2sib fate most of the time; however, in the double mutant, numb function is diluted in the cell destined to become RP2 because in the absence of insc, the residual Numb is distributed to both sibling neurons rather than just one cell. As a consequence, RP2 cell fate is almost never realized. These results are consistent with and support the notion that numb acts downstream of insc.
Figure 7.
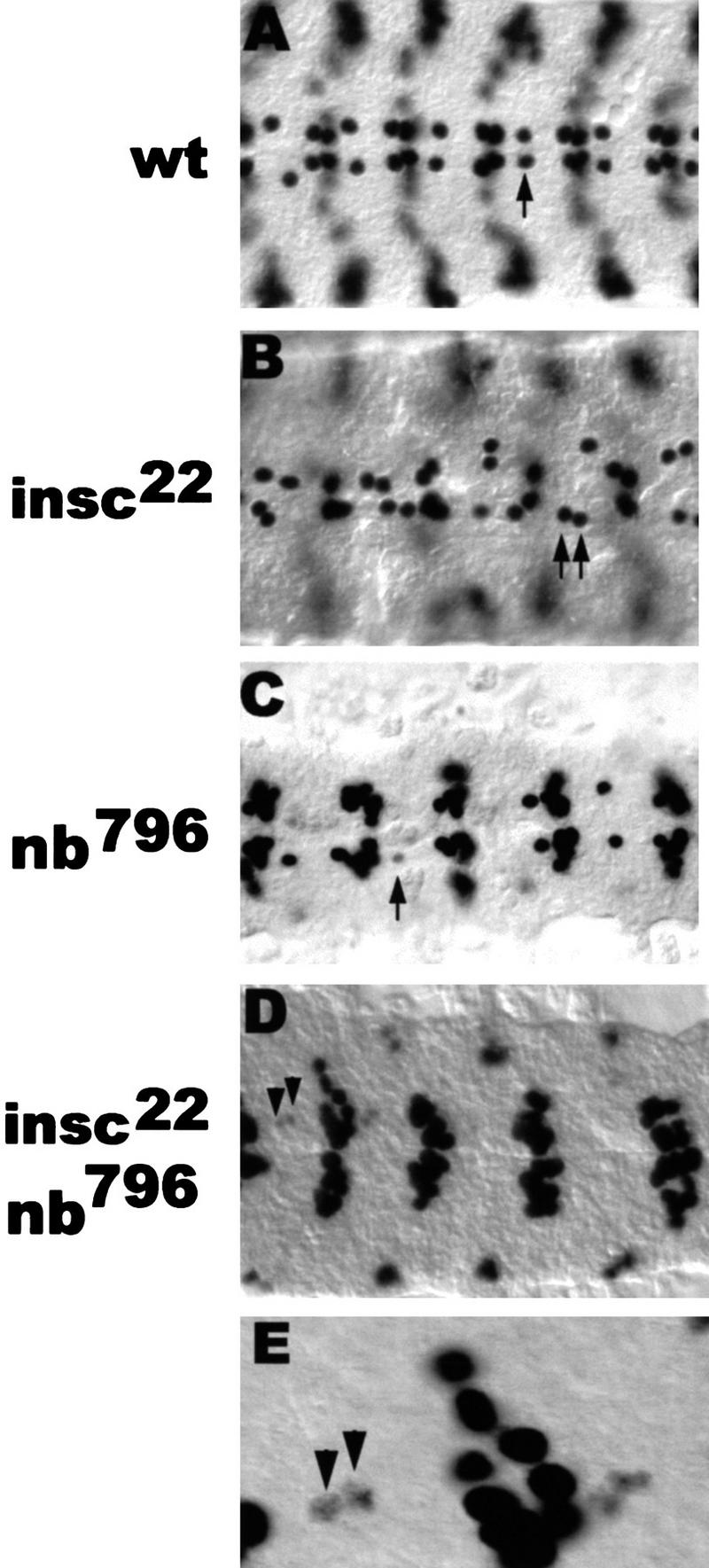
In equalized sibling neurons (insc background), RP2/RP2sib fate specification is only dependent on Numb. Dorsal view of dissected stage 15 embryos stained with rabbit anti-Eve. Anterior is to the left. (Arrows) RP2 neuron; (arrowheads) RP2sib. (E) Enlarged detail of D. Note the fading Eve staining in two RPsibs of equal size in the insc22 nb796 double-mutant embryo (E). Also note the increase in the number of Eve-expressing cells at the position of the CQ neurons, see Discussion.
In the absence of cell division, GMC4-2a differentiation is dependent on Numb
Our results suggest that asymmetric cell fate determination is resolved through the interplay of cell signaling mediated by N and at least one intrinsic fate determinant, Numb. In the absence of cell signaling, both progeny adopt the RP2 fate, indicating that the realization of the RP2sib fate requires cell signaling. This raises the question as to which cells provide the signal source. The signal source could reside strictly within the lineage and thus would require both GMC4-2a progeny to realize RP2sib fate (lateral signaling). Alternatively, the signal source could lie outside the lineage, and RP2sib fate realization can occur even in the absence of its sibling (inductive signaling). To distinguish between these possibilities, we analyzed the fate of GMC4-2a in the cell division mutant line GA339. In GA339 mutant embryos, each hemisegment contains an Eve-positive cell at the RP2 position (Fig. 8A). Moreover, these cells coexpress Zfh-1 (Fig. 8B) and 22C10 and, in addition, can exhibit an axon with an ipsilateral projection like the wild-type RP2 (Fig. 8C). The NB4-2 cell division producing GMC4-2a appears not to be affected. However, detailed analysis of stage 11/12 embryos revealed that only 47% of GMC4-2a undergo cell division, whereas 53% of the GMC4-2a remain undivided (n = 153). We conclude that in the absence of cell divison, GMC4-2a adopts RP2 fate. The observation that GMC4-2a can adopt an RP2-like identity is not a peculiarity of GA339 because we have observed a similar GMC4-2a to RP2-like transformation in alleles of cyclin A (data not shown). These observations are in agreement with our conclusion that the sibling cell that inherits Numb will differentiate into RP2 (see above).
Figure 8.
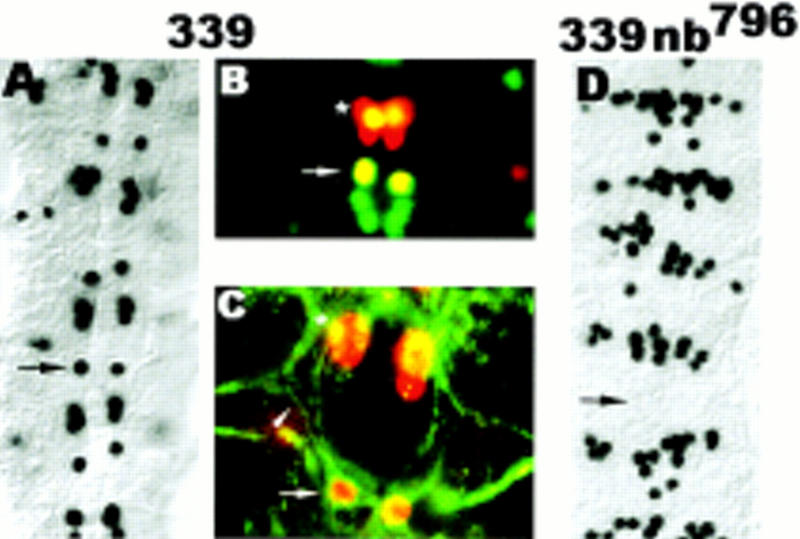
In the absence of cell division, GMC4-2a differentiation is dependent on Numb. Dorsal view of dissected stage 15 embryos; anterior is up. (A) GA339 stained with rabbit anti-Eve, note that each hemisegment has an Eve-positive cell at the RP2 position; note the decrease in the number of Eve-expressing cells in the position of the CQ neurons, see Discussion. (B) GA339 double stained with rabbit anti-Eve (red) and mouse anti-Zfh-1 (green). Note that the undivided GMC4-2a expresses Zfh-1, a marker gene, which in wild-type embryos is not expressed in GMC4-2a but only the mature RP2 neuron. (C) GA339 double stained with rabbit anti-Eve (red) and mouse mAb 22C10 (green). Note that the undivided GMC4-2a forms an ipsilateral axon projection characteristic for the RP2 neuron. (D) nb796 GA339 double mutant, note the loss of Eve-positive cells at the RP2 position and the increase in the number of Eve-expressing cells in the position of the CQ neurons. (Arrows) Position of the RP2 neuron; (arrowhead in C) the RP2 axon; [asterisk (*) in C] aCC neuron.
To analyze if the reduction of Numb function can alter the differentiation program of GMC4-2a, we examined the phenotype of the double-mutant nb796 GA339. We observed that a loss of Eve-positive cells from the RP2 position occurs with similar frequency in nb796 and nb796 GA339 mutant embryos (27% and 32%, respectively) (Fig. 8D). These data suggest that RP2sib fate realization occurs regardless of whether GMC4-2a divides or not. Our results do not exclude that lateral signaling can occur in wild-type embryos. However, in a situation in which no partner cell for lateral signaling is generated, signaling from outside the sublineage is sufficient for RP2sib fate realization.
Discussion
In this study we have analyzed how insc affects asymmetric fate specification of the sibling neuron pairs RP2/RP2sib and aCC/pCC. We show that cell-fate specification is accomplished through the interplay of the N signaling pathway and the intrinsic determinant Numb; the immediate precursor cells of neurons—the GMCs—exhibit the same apical–basal polarity that is seen in NBs and Numb localization as well as spindle orientation are coordinated in a stereotyped manner that ensures that Numb is segregated into the more dorsally located sibling. The correct positioning of Numb and the proper orientation of the cleavage plane require the function of insc. Therefore, Insc acts on asymmetric fate specification by making sibling cells intrinsically different. In addition, we demonstrate that sibling neuron fate specification is nonstochastic both in wild-type and under conditions in which numb function is limiting.
Binary fate decision of the GMC4-2a progeny is a deterministic process
GMC4-2a divides into a larger and a smaller daughter cell that differentiate into distinct neurons: The larger cells always adopt the RP2 fate, whereas the smaller cells will become RP2sib. The specification of RP2 appears not to require N-signaling because mutations in N and mam do not interfere with RP2 formation. Thus, RP2 can be considered the primary (default) fate. The realization of the RP2 fate does require the presence of numb because a reduction of numb activity or removal of the numb gene results in an apparent RP2 to RP2sib fate transformation. This conclusion is consistent with the observation that during GMC4-2a division, Numb is segregated into the more dorsal, larger sibling cell. Moreover, in GA339 (and alleles of cyclin A) mutant embryos in which GMC4-2a fails to undergo division and therefore retains Numb protein, the undivided GMC4-2a differentiates into a RP2-like cell as assessed by marker gene expression and axon morphology. In light of these results, the phenotype of insc mutants can be rationalized as follows: In insc mutant embryos, Numb localization and spindle orientation are not coordinated and, therefore, GMC4-2a division will result in two Numb-containing progeny, both of which will differentiate into RP2.
It has been suggested that asymmetric cell-fate specification in the SOP lineage is accomplished through lateral cell signaling and asymmetrically segregated Numb serves to impose a bias on the outcome of binary fate decision (Jan and Jan 1995). These models propose that in the absence of Numb, asymmetric cell fates can still be realized; albeit the outcome will be stochastic and unbiased. With respect to the GMC4-2a/RP2/RP2sib lineage, our data suggest that RP2-fate specification is fully deterministic and cannot be resolved solely by lateral signaling. First, if fate specification could be resolved by lateral signaling alone when numb is limiting [nb796 and nb796/Df(2L)30A-C], both the smaller and the larger sibling cell should have the capacity to adopt the RP2 fate. We always observed that the larger cell adopts the RP2 fate (we never see the smaller cell adopting the RP2 fate), indicating that the fate decision is nonstochastic. Second, when equivalent GMC4-2a daughter cells are generated (in insc mutant background), fate specification is solely dependent on the amount of Numb activity (insc22: 96% RP2sib to RP2 transformation; insc22 nb796:>98% RP2 to RP2sib transformation). Because no significant numbers of RP2 are generated in insc22 nb796 mutant embryos, RP2 fate realization appears not to occur via a stochastic process. However, the requirement for Numb as an intrinsic determinant for RP2 fate does not exclude that additional factors might aid the realization of the RP2 fate. In the total absence of zygotic Numb, RP2 is formed in 17% of the hemisegments. We conclude that at least some of the GMC4-2a progeny remain intrinsically different [Df(2L)30A-C mutant embryos]. At present, we do not know if the additional intrinsic determinant(s) is maternally provided Numb and/or factors (protein or mRNA) that are unrelated to Numb. We do not think that N itself or any downstream effectors of N signaling are asymmetrically segregated: A reduction of numb function is sufficient to cause RP2 to RP2sib fate transformation (on the basis of marker expression) indicating that both GMC4-2a progeny have inherited the potential to respond to cell signaling. Consistent with this conclusion is our observation that Mam is present in both siblings of the respective neuron pairs (data not shown).
Our experiments with the cell division mutant GA339 suggest that the signal source can originate outside the RP2/RP2sib lineage. This is in agreement with the observation that the correct fate specification of the vMP2 and dMP2 neurons in culture requires the presence of high cell density (Spana and Doe 1996). Moreover, in GA339 nb796 double-mutant embryos and nb796 mutant embryos, RP2 to RP2sib fate transformations occur with similar frequencies, indicating that the competence to respond to N signaling does not require cytokinesis but is already present in the GMC. However, a recent study on sensory organ development based on clonal analysis indicates that signaling between sibling cells is necessary for the resolution of alternative cell fates (Zeng et al. 1998). Although there are likely to be differences in the mechanisms of cell fate determination between the CNS and PNS, our results based on cell division defects are indirect and should be interpreted with caution.
Taken together, our data show that asymmetric fate determination in the GMC4-2a lineage involves the same components as fate specification in the MP2 and SOP lineages and may be accomplished through productive (effective) N signaling in one sibling (RP2sib) and inhibited (ineffective) N signaling in the Numb-containing sibling (RP2). Consistent with this notion, we observed that the ectopic expression of N-intra (a constitutively active form of N; Struhl et al. 1993) causes the same RP2 to RP2sib fate transformation as numb loss of function (Table 2), implying that N-intra ectopic expression can override the effects of Numb.
Besides RP2/RP2sib, sibling relationship has only been established for the aCC/pCC neuron pair. aCC/pCC fate determination has been studied in grasshopper embryos by laser ablation techniques (Kuwada and Goodman 1985). It was observed that random ablation of one newborn sibling always caused the remaining sibling to adopt the aCC fate. This result can be rationalized if both siblings are equivalent at the time of birth and asymmetric fate choice depends on lateral signaling with a stochastic outcome. In the absence of its partner cell, the remaining sibling will adopt the default fate. Is aCC/pCC fate resolved through lateral signaling in Drosophila embryos? A prerequisite for aCC/pCC fate determination through a stochastic process would require the equivalence of both siblings at the time of birth. Accordingly, loss of insc function, which results in random distribution of intrinsic fate determinants, should not affect aCC/pCC formation. However, in insc mutant embryos, nearly all GMC1-1a progeny differentiate into aCC, indicating that in wild-type both siblings are intrinsically different in an insc-dependent manner. This is consistent with the observation that Numb is asymmetrically localized in dividing GMC1-1a. Furthermore, aCC appears to be the primary fate because Numb segregation into both siblings—as it occurs in insc mutant embryos—results in pCC to aCC fate transformation. This is corroborated by the observation that nondividing GMC1-1a (as seen in GA339) always differentiate into aCC (data not shown). Although we cannot analyze the fate of aCC and pCC in numb mutant embryos, we conclude that asymmetrically segregated Numb is an important component of aCC/pCC fate specification.
The discrepancy between the results in grasshopper and Drosophila embryos may indicate that sibling fate specification in related species can rely on different mechanisms. Experiments on the ventral uterine and the gonadal anchor cell in different nematode species have demonstrated that fate specification of the same sibling pair shows phylogenetic variations that range from deterministic to stochastic mechanisms (Felix and Sternberg 1996).
In addition to the aCC/pCC and RP2/RP2sib fate, the fate of the CQ neurons appears to be affected by mutations in mam, insc, and numb. Specifically, mutations in mam and insc lead to a decrease of Eve-expressing CQ neurons. Conversely, numb mutations result in an increase of Eve-positive CQ neurons. In agreement with lineage tracing experiments that have identified NB7-1 as the progenitor of all CQ neurons (Bossing et al. 1996), we speculate that sibling relationships exist among the CQ neurons and that fate specification is resolved through the same mechanisms as RP2/RP2sib and aCC/pCC fate.
Our data demonstrate that asymmetric fate specification of the few identified sibling neurons is accomplished through the same deterministic mechanism. Is asymmetric sibling fate specification in the Drosophila embryonic CNS always accomplished through the same mechanism? A definite answer would require more information about sibling relationships of identified neurons (and glia) than is available to date. Our observations that the asymmetric localization of Numb protein at the basal cortex is stereotypically coordinated with the orientation of division in many GMCs (unidentified) suggest that many sibling neurons are born intrinsically different and may differentiate according to the mechanism described above. However, we do not know if asymmetrically segregated Numb always results in a deterministic outcome of binary fate decision or, in some cases, may only serve to impose a bias (probabalistic outcome). Moreover, we cannot exclude that some binary fate decisions may not require numb.
GMCs have apical–basal polarity
In this study we have shown that GMCs exhibit the same apical–basal polarity that is seen in NBs. Specifically, we observed that Insc protein is located at the apical and Numb protein at the basal cortex of dividing GMCs. From the orientation of the metaphase plate and the position of the newborn siblings, we deduce that most, if not all, GMCs divide with the cleavage plane placed horizontally to the apical surface (perpendicular spindle orientation). It is tempting to speculate about the events in the genesis of the GMCs that might provide the cues for their polarization. A possible candidate for the initial asymmetric cue is the position of the previous cell division site that will become the apical part of the GMC, which coincides with the location of the Insc protein crescent. This would imply that GMCs memorize their previous cell-division site in the form of specialized domains in the cortical cytoskeleton. These structures have been shown to serve as asymmetric cues in Saccharomyces cerevisiae budding site selection (for review, see Goenczy and Hyman 1996). Alternatively, GMCs may be born without an inherent polarity and environmental (extrinsic) cues are required to impose asymmetry on GMCs. At present, we cannot distinguish between these possibilities.
GMC4-2a divides asymmetrically not only with respect to Numb distribution, but also with respect to the morphology of its progeny: In wild-type embryos in 88% of the hemisegments, the newborn sibling neurons are unequal in cell and nuclear size. The smaller cell always adopts the RP2sib fate that is characterized by a further decrease in cell and nuclear size. We speculate that GMC4-2a cytokinesis and cell-fate realization of its progeny do not occur strictly sequentially but are temporally overlapping. Thus, the unequal size of the sibling cells at the time of birth may reflect a partial commitment to their future fates. In this study we describe several mutations that cause RP2sib to RP2 fate transformations with respect to marker gene expression but not with respect to size (mam, spdo, N). We postulate that in wild-type embryos, the products of these genes are required in the postmitotic neurons for their full commitment and subsequent differentiation. This is consistent with the observation that in loss-of-function mutations the initial physical asymmetry remains unaltered, but a further decrease in cell and nuclear size does not occur. In contrast, insc appears to be the only mutation that equalizes cell/nuclear size and acts prior to mam, spdo, and N. This is in agreement with insc functioning as an organizer of asymmetry in GMCs.
The loss of size asymmetry in insc mutant embryos is not restricted to the RP2/RP2sib lineage but is also observed in muscle progenitor cell divisions. For example, when the muscle progenitor P15 (Carmena et al. 1995) divides, the daughter cells are not equal in size; it is the larger of the two daughter cells that preferentially inherits the Numb that is asymmetrically localized in the dividing muscle progenitor. Similar to the RP2/RP2sib situation, removing insc function appears to equalize the size of the daughter cells derived from the P15 cell division (see Fig. 5 in Carmena et al. 1998). In contrast, this equalizing effect does not occur in NBs. NB division is highly asymmetric: Each division generates a new NB and a GMC that is several times smaller than the NB. In insc mutants, NBs often bud off GMCs in lateral (rather than basal) positions, but the size asymmetry is retained. At present, we do not understand how size asymmetry is generated during progenitor cell division and if, and how, it might be linked to spindle orientation.
Materials and methods
Fly strains
The deficiency Df(2L)30A-C was obtained from the Bloomington Stock Center. insc22 was provided by R. Renkawitz-Pohl (Burchard et al. 1995; Knirr at al. 1997); nb1 and nb3 by L.Y. Jan and Y.N. Jan (Uemura et al. 1989); spdozz27 was provided by J. Skeath and C.Q. Doe; Nts1 (Shellenbarger and Mohler 1975) was provided by V. Rodrigues. The mutant stocks mamC242, mamGA1037, mamGA424 nb796, nb2758, GA339, and GA1019 were isolated from a collection of EMS-induced lethal second chromosome mutations that had been prescreened for axonal defects (Seeger et al. 1993). The identities of mamC242, nb796, and nb2758 were confirmed by the lack of complementation with the respective deficiencies and with previously identified alleles. The genes affected in the mutant lines GA339 and GA1019 are unknown. GA1019 is associated with an ana-/telophase defect.
Immunocytochemistry
Embryos were collected in plastic vials, dechorionated by 2 min incubation in 50% Clorox and subsequently washed several times with PBS/0.1% Triton X-100 (PBT). After the removal of all PBT, 700 μl of n-heptane and 700 μl of 37% formaldehyde were added to the embryos and the mixture was shaken vigorously for 2–3 min. Subsequently, the formaldehyde phase was removed and 700 μl of methanol were added. After 1 min of vigorous shaking, the embryos were transferred into fresh vials, washed twice with methanol, and subsequently stained as described previously (Yang et al. 1993). For immunofluorescence, FITC-conjugated anti-mouse antibody (Boehringer Mannheim) and biotinylated anti-rabbit antibody in combination with avidin-Texas Red (Vector Laboratories) were used. DNA was visualized by use of 1 mg/ml sonicated paraphenylene-diamine (Sigma) in 90% glycerol (Lundell and Hirsh 1994). Mutant alleles were balanced over a blue balancer chromosome carrying a ftz–lacZ or Ubx–lacZ insertion to facilitate the identification of homozygous mutant embryos.
The following antibodies were used: anti-Eve (rabbit polyclonal; M. Frasch, The Mount Sinai Medical Center, New York, NY); anti-Eve (mouse monoclonal; K. Zinn, Caltech); anti-β-galactosidase (rabbit polyclonal; Cappel); anti-Zfh1 (mouse polyclonal; Z. Lai, University of Pennsylvania); rabbit anti-Numb (L.Y. Jan and Y.N. Jan, University of California at San Francisco); anti-Mam (rabbit polyclonal; B. Yedvobnick, Emory University, Atlanta, GA); anti-Insc (rabbit polyclonal).
Preparation of Nts1 and HS Nintra embryos
Nts1 flies were propagated at the permissive temperature of 18°C. Nts1 embryos were collected for 2 hr at 18°C and aged for an additional 2 hr at 18°C. Subsequently, the embryos were shifted for 6 hr to the nonpermissive temperature (29°C) and finally allowed to develop for an additional 6 hr at 18°C. Embryos were collected and fixed as described above.
HS Nintra embryos were collected for 2 hr and allowed to develop for an additional 6 hr at 25°C. Subsequently, the embryos were collected and placed into a moist chamber. Heat shock was administered for 20 min at 37°C. Finally, the embryos were allowed to develop for 10 hr at 16°C and then fixed as described above.
Microscopy and staging of embryos
Embryos were staged according to Campos-Ortega and Hartenstein (1985). Embryos were observed by DIC optics with a Zeiss Axiophot microscope. Confocal microscopy was performed with a BioRad MRC600 scan head equipped with a krypton/argon laser and a Zeiss Axiophot microscope. Adobe Photoshop was used for image processing.
Acknowledgments
We thank Hugo Bellen, Chris Doe, and Jim Skeath for sharing information, reagents, and flies prior to publication; Manfred Frasch, Yuh-Nung Jan, Kai Zinn, Zhi-Chun Lai, and B. Yedvobnick for antibodies and/or flies; Kathy Matthews and the Bloomington Stock Center for providing stocks; Veronica Rodrigues for comments on the manuscript; the Institute of Molecular and Cell Biology and the Human Science Frontier Programme for financial support. We thank the referees for some very helpful comments.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mcbwchia@imcb.nus.edu.sg; FAX (65) 779 1117.
References
- Artavanis-Tsakonas A, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- Bossing T, Udolph G, Doe CQ, Technau G. The embryonic CNS lineages of Drosophila melanogaster: I. Neuroblast lineages derived from the ventral half of the neuroectoderm. Dev Biol. 1996;179:41–64. doi: 10.1006/dbio.1996.0240. [DOI] [PubMed] [Google Scholar]
- Burchard S, Paululat A, Hinz U, Renkawitz-Pohl R. The mutant not enough muscles (nem) reveals reduction of the Drosophila embryonic muscle pattern. J Cell Sci. 1995;108:1443–1454. doi: 10.1242/jcs.108.4.1443. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega JA. Numb diverts Notch pathway off the Tramtrack. Neuron. 1996;17:1–4. doi: 10.1016/s0896-6273(00)80274-3. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega JA, Hartenstein V. The embryonic development of Drosophila melanogaster. Berlin, Germany: Springer-Verlag; 1985. [Google Scholar]
- Carmena A, Bate M, Jimenez F. lethal of scute, a proneural gene, participates in the specification of muscle progenitors during Drosophila embryogenesis. Genes & Dev. 1995;9:2373–2383. doi: 10.1101/gad.9.19.2373. [DOI] [PubMed] [Google Scholar]
- Carmena A, Murugasu-Oei B, Menon D, Jimenez F, Chia W. inscuteable and numb mediate asymmetric muscle progenitor cell divisions during Drosophila myogenesis. Genes & Dev. 1998;12:304–315. doi: 10.1101/gad.12.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, McConnell SK. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell. 1995;82:631–641. doi: 10.1016/0092-8674(95)90035-7. [DOI] [PubMed] [Google Scholar]
- Doe CQ. Molecular markers for identified neuroblasts and ganglion mother cell in the Drosophila embryonic central nervous system. Development. 1992;116:855–863. doi: 10.1242/dev.116.4.855. [DOI] [PubMed] [Google Scholar]
- Doe CQ, Chu-LaGraff Q, Wright DM, Scott MP. The prospero gene specifies cell fate in the Drosophila central nervous system. Cell. 1991;65:451–465. doi: 10.1016/0092-8674(91)90463-9. [DOI] [PubMed] [Google Scholar]
- Dye C, Lee J, Atkinson R, Brewster R, Han P, Bellen HJ. The Drosophila sanpodo gene controls sibling cell fate and encodes a Tropomodulin homolog, an actin/tropomyosin associated protein. Development. 1998;125:1845–1856. doi: 10.1242/dev.125.10.1845. [DOI] [PubMed] [Google Scholar]
- Felix MA, Sternberg PW. Symmetry breakage in the development of one-armed gonads in Nematodes. Development. 1996;122:2129–2142. doi: 10.1242/dev.122.7.2129. [DOI] [PubMed] [Google Scholar]
- Fortini ME, Artavanis-Tsakonis S. The suppressor of Hairless protein participates in Notch receptor signaling. Cell. 1994;79:273–282. doi: 10.1016/0092-8674(94)90196-1. [DOI] [PubMed] [Google Scholar]
- Frasch M, Hoey T, Rushlow C, Doyle H, Levine M. Characterisation and localisation of the even-skipped protein of Drosophila. EMBO J. 1987;6:749–759. doi: 10.1002/j.1460-2075.1987.tb04817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita SC, Zipursky SL, Benzer S, Ferrus A, Shotwell SL. Monoclonal antibodies against the Drosophila nervous system. Proc Natl Acad Sci. 1982;79:7929–7933. doi: 10.1073/pnas.79.24.7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goenczy P, Hyman A. Cortical domains and the mechanisms of asymmetric cell division. Trends Cell Biol. 1996;6:382–387. doi: 10.1016/0962-8924(96)10035-0. [DOI] [PubMed] [Google Scholar]
- Guo M, Jan LY, Jan YN. Control of daughter cell fates during asymmetric division: Interaction of Numb and Notch. Neuron. 1996;17:27–41. doi: 10.1016/s0896-6273(00)80278-0. [DOI] [PubMed] [Google Scholar]
- Hirata J, Nakagoshi H, Nabeshima Y, Matsuzaki F. Asymmetric segregation of a homeoprotein, Prospero, during cell division in neural and endodermal development. Nature. 1995;377:627–630. doi: 10.1038/377627a0. [DOI] [PubMed] [Google Scholar]
- Horvitz HR, Herskowitz I. Mechanisms of asymmetric cell divisions: Two Bs or not two Bs, that is the question. Cell. 1992;68:237–255. doi: 10.1016/0092-8674(92)90468-r. [DOI] [PubMed] [Google Scholar]
- Ito K, Urban J, Technau GM. Distribution, classification and development of Drosophila glial cells in the late embryonic and early larval ventral nerve cord. Roux’s Arch Dev Biol. 1995;204:284–307. doi: 10.1007/BF02179499. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Maggot’s hair and bug’s eye: Role of cell interactions and intrinsic factors in cell fate specification. Neuron. 1995;14:1–5. doi: 10.1016/0896-6273(95)90235-x. [DOI] [PubMed] [Google Scholar]
- Knirr S, Breuer S, Paululat A, Renkawitz-Pohl R. Somatic mesoderm differentiation and the development of a subset of pericardial cells depend on the not enough muscles (nem) locus, which contains the inscuteable gene and the intron located gene, skittles. Mech Dev. 1997;67:69–81. doi: 10.1016/s0925-4773(97)00109-3. [DOI] [PubMed] [Google Scholar]
- Knoblich JA, Jan LY, Jan YN. Asymmetric segregation of Numb and Prospero during cell division. Nature. 1995;377:624–627. doi: 10.1038/377624a0. [DOI] [PubMed] [Google Scholar]
- Kraut R, Campos-Ortega JA. inscuteable, a neural precursor gene of Drosophila, encodes a candidate for a cytoskeleton adaptor protein. Dev Biol. 1996;174:65–81. doi: 10.1006/dbio.1996.0052. [DOI] [PubMed] [Google Scholar]
- Kraut R, Chia W, Jan LY, Jan YN, Knoblich JA. Role of inscuteable in orienting asymmetric cell divisions in Drosophila. Nature. 1996;383:50–55. doi: 10.1038/383050a0. [DOI] [PubMed] [Google Scholar]
- Kuwada JW, Goodman CS. Neuronal determination during embryonic development of the grasshopper nervous system. Dev Biol. 1985;110:114–126. doi: 10.1016/0012-1606(85)90069-7. [DOI] [PubMed] [Google Scholar]
- Lai Z, Fortini ME, Rubin GM. The embryonic expression pattern of zfh1 and zfh2, two Drosophila genes encoding novel zinc-finger homeodomain proteins. Mech Dev. 1991;34:123–134. doi: 10.1016/0925-4773(91)90049-c. [DOI] [PubMed] [Google Scholar]
- Li P, Yang X, Wasser M, Cai Y, Chia W. Inscuteable and Staufen mediate asymmetric localisation and segregation of prospero RNA during Drosophila neuroblast cell divisions. Cell. 1997;90:437–447. doi: 10.1016/s0092-8674(00)80504-8. [DOI] [PubMed] [Google Scholar]
- Lundell MJ, Hirsh J. A new visible light DNA fluorochrome for confocal microscopy. Biotechniques. 1994;16:434–440. [PubMed] [Google Scholar]
- Matsuzaki F, Koizumi K, Hama C, Yoshioka T, Nabeshima Y. Cloning of the Drosophila prospero gene and its expression in ganglion mother cells. Biochem Biophys Res Comm. 1992;182:1326–1332. doi: 10.1016/0006-291x(92)91878-t. [DOI] [PubMed] [Google Scholar]
- Patel NH, Condron BG, Zinn K. Pair-rule expression patterns of even-skipped are found in both long and short band beetles. Nature. 1994;367:429–434. doi: 10.1038/367429a0. [DOI] [PubMed] [Google Scholar]
- Posakony JW. Nature versus nurture: Asymmetric cell divisions in Drosophila bristle development. Cell. 1994;76:415–418. doi: 10.1016/0092-8674(94)90105-8. [DOI] [PubMed] [Google Scholar]
- Rhyu MS, Jan LY, Jan YN. Asymmetric distribution of Numb protein during division of the sensory organ precursor cell confers distinct fates to daughter cells. Cell. 1994;76:477–491. doi: 10.1016/0092-8674(94)90112-0. [DOI] [PubMed] [Google Scholar]
- Salzberg A, D’Evelyn D, Schulze KL, Lee JK, Strumpf D, Tsai L, Bellen HJ. Mutations affecting the pattern of the PNS in Drosophila reveal novel aspects of neuronal development. Neuron. 1994;13:269–287. doi: 10.1016/0896-6273(94)90346-8. [DOI] [PubMed] [Google Scholar]
- Seeger M, Tear G, Goodman CS. Mutations affecting the growth cone guidance in Drosophila: Genes necessary for guidance toward and away from the midline. Neuron. 1993;10:409–426. doi: 10.1016/0896-6273(93)90330-t. [DOI] [PubMed] [Google Scholar]
- Shellenbarger DL, Mohler JD. Temperature-sensitive mutations of the Notch locus in Drosophila melanogaster. Genetics. 1975;81:143–162. doi: 10.1093/genetics/81.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeath JB, Doe CQ. Sanpodo and Notch act in opposition to Numb to distinguish sibling neuron fate in the Drosophila CNS. Development. 1998;125:1857–1865. doi: 10.1242/dev.125.10.1857. [DOI] [PubMed] [Google Scholar]
- Smoller DA, Friedel C, Schmid A, Bettler D, Lam L, Yedvobnick B. The Drosophila neurogenic locus mastermind encodes a nuclear protein unusually rich in amino acid homopolymers. Genes & Dev. 1990;4:1688–1700. doi: 10.1101/gad.4.10.1688. [DOI] [PubMed] [Google Scholar]
- Spana E, Doe CQ. The prospero transcription factor is asymmetrically localised to the cortex during neuroblast mitosis in Drosophila. Development. 1995;121:3187–3195. doi: 10.1242/dev.121.10.3187. [DOI] [PubMed] [Google Scholar]
- ————— Numb antagonises Notch signaling to specify sibling neuron cell fate. Neuron. 1996;17:21–26. doi: 10.1016/s0896-6273(00)80277-9. [DOI] [PubMed] [Google Scholar]
- Spana E, Kopczynski C, Goodman CS, Doe CQ. Asymmetric localisation of Numb autonomously determines sibling neuron identity in the Drosophila CNS. Development. 1995;121:3489–3494. doi: 10.1242/dev.121.11.3489. [DOI] [PubMed] [Google Scholar]
- Struhl G, Fitzgerald K, Greenwald I. Intrinsic activity of the Lin-12 and Notch intracellular domains in vivo. Cell. 1993;74:331–345. doi: 10.1016/0092-8674(93)90424-o. [DOI] [PubMed] [Google Scholar]
- Uemura T, Shepherd S, Ackerman L, Jan LY, Jan YN. numb, a gene required in determination of cell fate during sensory organ formation in Drosophila embryos. Cell. 1989;5:349–360. doi: 10.1016/0092-8674(89)90849-0. [DOI] [PubMed] [Google Scholar]
- Vaessin H, Grell E, Wolff E, Bier E, Jan LY, Jan YN. prospero is expressed in neuronal precursors and encodes a nuclear protein that is involved in the control of axonal outgrowth in Drosophila. Cell. 1991;67:941–953. doi: 10.1016/0092-8674(91)90367-8. [DOI] [PubMed] [Google Scholar]
- Yang X, Yeo S, Dick T, Chia W. The role of a Drosophila POU homeodomain gene in the specification of neural precursor cell identity in the developing embryonic central nervous system. Genes & Dev. 1993;7:504–516. doi: 10.1101/gad.7.3.504. [DOI] [PubMed] [Google Scholar]
- Yeo S, Lloyd A, Kozak K, Dinh A, Dick T, Yang X, Sakonju S, Chia W. On the functional overlap between two Drosophila POU homeodomain genes and the cell fate specification of a CNS neural precursor. Genes & Dev. 1995;9:1223–1236. doi: 10.1101/gad.9.10.1223. [DOI] [PubMed] [Google Scholar]
- Zeng C, Younger-Shepherd S, Jan LY, Jan YN. Delta and Serrate are redundant Notch ligands required for asymmetric cell divisions within the Drosophila sensory organ lineage. Genes & Dev. 1998;812:1086–1091. doi: 10.1101/gad.12.8.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]