

Abstract
Copper and germanium complexes of β-substituted nitrocorroles were reacted with 4-amino-4H-1,2,4-triazole to give the corresponding β-amino-β-nitro derivatives, in moderate to good yields. This is the first successful example of a vicarious nucleophilic substitution performed on corrole derivatives, because the same reaction carried out on silver complexes afforded the corresponding 6-azahemiporphycenes by way of corrole ring expansion. The first step of this work is related to the modification of a synthetic protocol for preparation of the β-substituted nitro corroles. The nitration reaction was carried out on a copper corrole using NaNO2 as the primary source of NO2− coupled with AgNO2 used as oxidant. By variation of the molar ratio of the reagents it was possible to direct the product distribution towards mono- and di-nitro derivatives. The reaction between mono- and di-nitro derivatives of (TtBuCorrCu) with 4-amino-4H-1,2,4-triazole gave good results, leading to the isolation of 2,3-(NH2)(NO2)-TtBuCorrCu and 2,18-(NH2)2-3,17-(NO2)2-TtBuCorrCu in moderate yields. To elucidate factors that influence the reaction, and to highlight the different behavior observed for different metal complex substrates, the electrochemistry of three copper complexes, TtBuPCorrCu, (NO2)TtBuPCorrCu and (NO2)2TtBuPCorrCu, were studied by cyclic voltammetry and thin-layer UV-visible spectroelectrochemistry. The nitro groups on (NO2)xTtBuPCorrCu are highly electron-withdrawing, which leads not only to a substantial positive shift of all redox potentials, but also to a unique redox behavior and UV-vis spectrum of the singly reduced product as compared to the parent compound, TtBuPCorrCu. Finally, the amination reaction was carried out on a Ge(IV) nitrocorrolate, giving in good yield the 2-amino-3-nitroderivative, which was structurally characterized by single crystal X-ray crystallography.
Introduction
The development of new and efficient routes for functionalization of the porphyrinoid skeleton represents a mandatory task for researchers interested in investigating both the macrocycle reactivity and in exploring potential uses of these compounds in a variety of practical applications, from molecular materials and catalysts to sensors and medical uses. Among the known pyrrolic macrocycles, porphyrins have been the most intensively studied and different examples of macrocycle “decoration by introduction of peripheral groups” have been reported in the literature.1 As expected for an aromatic ring, the porphyrin β-pyrrolic positions can undergo attack by both electrophilic and nucleophilic agents, although reactions with electrophiles are more common. It is important to note that the macrocyclic reactivity can be finely tuned by the presence of other substituents and/or by the nature of the inner core coordinated metal ion. From this point of view one of the most useful peripheral substituents is the nitro group and the electron withdrawing character of such a substituent has been used to direct the orientation of further functionalizations2 or to facilitate aromatic nucleophilic substitution. In this latter case the reaction with a variety of nucleophiles3 such as alkoxides, cyanide, hydride or active methylene compounds yields 2-substituted, 2-nitro-3-substituted porphyrins, or stable chlorins as products, depending on the experimental conditions and the nature of the nucleophile. Furthermore, starting from 2-nitroporphyrins, it is possible to introduce useful functionalities on the porphyrin β-pyrrolic positions, namely pyrrole4 and di- or tetraone respectively,5 allowing for the construction of elaborated β-fused oligoporphyrin architectures in which the extended aromatic system could potentially be exploited in molecular electronics.
In the case of corroles, peripheral functionalizations of the macrocycle have been far less explored compared to the dramatic development observed for synthesis of the macrocycle itself. The quite original chemistry shown by the corroles has led to the first seminal exploitations of corroles in different fields6 and for this reason the modulation of corrole properties by synthetic manipulations has become an attractive task for many researchers. However, only a few examples of corrole functionalizations have so far been reported in the literature,7 this fact being due in part to the inherent lower stability of corroles compared with porphyrins.
This problem of decreased compound stability is evident in the case of β-nitrocorroles, where efficient routes for preparation of the free-base macrocycles has been limited by the facile oxidation of the macrocycle which occurs under the utilized reaction conditions. For this reason, the preparation of β-nitrocorroles has been limited to the synthesis of metal derivatives,8,9 using a variety of reaction conditions and nitrating agents.
We have been particularly interested in elucidating new synthetic routes to β-nitrocorroles, with the aim of investigating both the macrocycle reactivity and using these derivatives as a starting point for the preparation of covalently linked corrole units. Since the preparation of di-substituted corroles is the first necessary step toward new chromophores as well as highly conjugated fused oligomeric systems, we have investigated the role of the β-nitro group in activating nucleophilic substitution on the vicinal positions of the corrole ring. The role of the nitro group in the nucleophilic aromatic substitution has been detailed in the literature;10 in each reaction, formation of the nitronate σ-adduct is followed by an elimination step that necessarily involves release of a hydride anion that is not a spontaneous event. The H− departure requires additional factors to favor its removal from the negative adduct after which conversion into the product of nucleophilic substitution can occur. The σH-adduct can be transformed according to different pathways depending on the substrates and the reaction conditions, as described by an exhaustive review on the nucleophilic substitution of hydrogen in heterocyclic chemistry.11 The fate of the σH-complex is strictly related to two distinctive outcomes; one is the susceptibility towards oxidation of the adduct (the so-called ONSH path) and the other the presence on the reacting nucleophile of a leaving group X assisting the base-induced elimination of H− as HX. In the latter case, the reaction is called vicarious nucleophilic substitution of hydrogen (VNS) thanks to the supporting action of the nucleophile.
In present paper, we first describe an improved route for the nitration of copper meso-triarylcorroles, which affords mono- and dinitro derivatives in good yields. The Cu(III) β-dinitrocorrolate was prepared for the first time and the influence of the nitro group on corrole redox behavior is investigated in nonaqueous solvents. The synthesized β-nitrocorrole copper complexes were reacted with 4-amino-4H-1,2,4-triazole to obtain the corresponding difunctionalized β-amino-β-nitrocorrole derivatives. The reaction was also carried out in the case of a Ge(IV) complex, so as to study the influence of the coordinated metal ion and its oxidation state on the nucleophilic aromatic substitution reaction.
Structures of the novel corroles presented in this work are illustrated in Chart 1.
Chart 1.

Molecular Structures of the Synthetized Corroles
Experimental Section
Reagents and solvents (Sigma-Aldrich, Fluka and Carlo Erba Reagenti) for synthesis and purification were of synthetic grade and used as received. AgNO2 reagent (Sigma-Aldrich) should be of high purity grade (99.98%) and freshly used. Chromatography was performed on silica gel 60 (70-230 mesh) and neutral alumina (Brockmann grade III). 1H NMR spectra were recorded on a Bruker AV300 (300 MHz) spectrometer. Chemical shifts are given in ppm relative to tetramethylsilane (TMS). UV-vis spectra were measured on a Cary 50 spectrophotometer. Mass spectra were recorded on a VG Quattro spectrometer in the positive-ion mode, using m-nitrobenzyl alcohol (NBA, Aldrich) as a matrix (FAB), or on a Voyager DE STR Biospectrometry workstation in the positive mode, using α-cyano-4-hydroxycinnamic acid as a matrix (MALDI). Electron spin resonance (ESR) spectra were obtained on an IBM model ESP 300 apparatus. Low temperature measurements were carried out using a liquid-nitrogen finger dewar.
Diffraction data were collected at T = 90K on a Bruker Kappa Apex-II diffractometer equipped with graphite-monochromated CuKα radiation (λ=1.54178 Å) and an Oxford Cryostream low temperature device.
Cyclic voltammetry was carried out at 298 K by using an EG&G Princeton Applied Research (PAR) 173 potentiostat/galvanostat. A homemade three-electrode cell was used for cyclic voltammetric measurements and consisted of a glassy carbon working electrode, a platinum counter electrode and a homemade saturated calomel reference electrode (SCE). The SCE was separated from the bulk of the solution by a fritted glass bridge of low porosity that contained the solvent/supporting electrolyte mixture.
Thin-layer UV-visible spectroelectrochemical experiments were performed with a home-built thin-layer cell, which has a light transparent platinum net working electrode. Potentials were applied and monitored with an EG&G PAR Model 173 potentiostat. Time-resolved UV-visible spectra were recorded with a Hewlett-Packard Model 8453 diode array spectrophotometer. High purity N2 from Trigas was used to deoxygenate the solution and kept over the solution during each electrochemical and spectroelectrochemical experiment.
Synthesis
TtBuPCorrH3 and 3NO2-TPCorrGe(OCH3) were prepared as previously described.8a,8b
3,17-(NO2)2-TtBuPCorrCu
Method A: CorrCu/AgNO21:50
TtBuPCorrH3 (100 mg, 0.14 mmol) was dissolved in pyridine (15 mL) and Cu(OAc)2 (29 mg, 0.14 mmol) was added. The mixture was stirred at reflux and the progress of the reaction was monitored by UV-vis spectroscopy. When the metal insertion was complete AgNO2 (1.1 g, 7.1 mmol) was added. After 20 min the starting material was all consumed, as indicated by this UV-vis spectrum and TLC analysis, and a more polar green band was observed as the major product. The reaction mixture was precipitated by adding distilled water (20 mL), then filtered and washed extensively with water. The crude residue was taken up in CHCl3 and filtered again through Na2SO4. The solvent was evaporated under vacuum, the residue was dissolved in CH2Cl2 and purified by chromatography on a silica gel column using CH2Cl2 as eluent. Traces of 3-(NO2)-TtBuPCorrCu were isolated as a first brownish band, and successively traces of 2,17-(NO2)2-TtBuPCorrCu were also collected as orange rust fractions. The third dark green fraction was eluted with CHCl3 and corresponded to 3,17-(NO2)2-TtBuPCorrCu. Crystallization from CH2Cl2/MeOH, gave 62 mg (52% yield) as a dark green powder. Anal. Calcd for C49H45CuN6O4: C, 69.6; H, 5.4; N, 9.9%. Found C, 69.9; H, 5.2; N, 9.8%. λmax(CHCl3)(log ε) nm 370 (4.58), 449 (4.89), 594 (4.46), 699 (4.18). 1HNMR δH(CDCl3, J [Hz]): 8.29 (s, 2H, β-pyrrole), 7.67 (m, 6H, β-pyrrole+phenyls), 7.48 (m, 10H, phenyls), 1.45 (s, 9H, p-tBu), 1.44 (s, 18H, p-tBu). MS (MALDI): m/z 845 (M+).
2,17-(NO2)2TtBuPCorrCu
Anal. Calcd for C49H45CuN6O4: C, 69.6; H, 5.4; N, 9.9%. Found C, 69.7; H, 5.2; N, 9.7%. λmax(CHCl3)(log ε) nm 466 (4.37), 652 (4.37). 1HNMR δH(CDCl3, J [Hz]): 8.36 (s, C18 1H, β-pyrrole), 7.83 (s, C3 1H, β-pyrrole) 7.68 (m, 8H, β-pyrrole+phenyl), 7.55 (d, 2H, J=8.2 Hz, phenyl), 7.44 (m, 6H, phenyl), 1.47 (s, 9H, p-tBu), 1.45 (s, 9H, p-tBu), 1.42 (s, 9H, p-tBu). MS (MALDI): m/z 845 (M+).
Method B: CorrCu/AgNO2/NaNO2 1:2:8
TtBuPCorrH3 (100 mg, 0.14 mmol) was dissolved in DMF or pyridine (15 mL) and Cu(OAc)2 (29 mg, 0.14 mmol) was added. The mixture was stirred at reflux and the progress of the reaction was monitored by UV-vis spectroscopy. When the metal insertion was complete AgNO2 (45 mg, 0.29 mmol) and NaNO2 (83 mg, 1.2 mmol) were added. After 20 min the starting material was all consumed as indicated by UV-vis spectroscopy and TLC analysis that showed a more polar green band as the major product. The product was precipitated by adding distilled water (30 mL), then filtered and washed extensively with water. The crude residue was taken up in CHCl3 and filtered again through Na2SO4. The solvent was evaporated under vacuum, the residue dissolved in CH2Cl2 and purified by chromatography on a silica gel column using CH2Cl2 as eluent. Traces of the 3-mononitro derivative and the 2,17-dinitrocorrole regioisomer were also detected as the first and the second fraction, respectively. The third dark green fraction corresponding to 3,17-(NO2)2-TtBuPCorrCu was eluted with CHCl3, isolated and crystallized from CH2Cl2/MeOH, to give 60 mg (51% yield) as a dark green powder.
Note: It is important to underline that high purity grade AgNO2 should be used; the color of the reagent should be pale yellow; in the case of lower grade reagent, or one having a clearly gray solid color, the molar proportions of the TtBuCorrCu, AgNO2 and NaNO2 reagents reported in Method B must be changed to 1:5:5, respectively, to achieve comparable results.
3-(NO2)TtBuPCorrCu
TtBuPCorrH3 (100 mg, 0.14 mmol) was dissolved in DMF or pyridine (15 mL) and Cu(OAc)2 (29 mg, 0.14 mmol) was added. The mixture was stirred at reflux and the progress of the reaction was monitored by UV-vis spectroscopy. When the metal insertion was complete, AgNO2 (22 mg, 0.14 mmol) and NaNO2 (89 mg, 1.3 mmol) were added and the reaction mixture, following the above-mentioned experimental procedure, afforded in 20 min the copper mono- and dinitro compounds. In particular, purification on a silica column eluting with a CH2Cl2/hexane (7:3) solvent mixture afforded the first brownish fraction corresponding to 3-(NO2)TtBuPCorrCu (86 mg) and second green fraction of 3,17-(NO2)2-TtBuPCorrCu (18 mg) in 75 and 15% yields, respectively. Spectroscopic data for the copper 3-nitrocorrolate were in full agreement with those previously reported in the literature8a
Note: The scale-up of the reaction, typically starting from 300 mg of TtBuPCorrH3, afforded as a first fraction a small amount of the 2-(NO2)TtBuPCorrCu regioisomer, just enough for complete spectroscopic characterization:
2-(NO2)TtBuPCorrCu: Anal. Calcd for C49H46CuN5O2: C, 73.5; H, 5.8; N, 8.7. % Found: C, 73.6; H, 6.0; N, 8.7. UV-vis: λmax(CHCl3)(log ε) nm: 370 (4.40), 442 (4.82), 636 (3.96). 1HNMR δH(CDCl3, J [Hz]): 7.91 (s, 1H, β-pyrrole), 7.80 (d, 1H, J=4.1 Hz, β-pyrrole), 7.73 (m, 6H, β-pyrrole+phenyl), 7.64 (d, 1H, J=4.3 Hz, β-pyrrole), 7.58 (m, 7H, β-pyrrole+ phenyl), 7.43 (d, 1H, J=4.6 Hz, β-pyrrole), 7.38 (d, 2H, J=8.2 Hz, phenyl), 1.49 (br s, 18H, p-tBu), 1.41 (br s, 9H, p-tBu). MS (MALDI): m/z 801 (M+).
2-NH2-3NO2-TtBuPCorrCu
3NO2-TtBuPCorrCu (253 mg, 0.32 mmol) was dissolved in 132 mL of toluene/ethanol (120:12) solvent mixture and then NaOH (63 mg, 1.58 mmol) and 4-amino-4H-1,2,4-triazole (319 mg, 3.8 mmol) were added. The reaction mixture was stirred at room temperature and monitored by TLC. The reaction was complete after 45 min. The solvent was then removed under vacuum, the residue dissolved in CHCl3, washed twice with the aqueous Na2S2O3 and dried over Na2SO4. The crude mixture was purified by chromatography on silica gel eluting with CHCl3. The first red-brown fraction was collected and crystallized from CH2Cl2/MeOH, affording the title compound as a brown-reddish powder (47 mg, 18% yield). Anal. Calcd for C49H47CuN6O2: C, 72.2; H, 5.8; N, 10.3 %. Found C, 72.4; H, 3.9; N, 11.9 %. UV-vis: λmax(CHCl3) (log ε) nm 440 (4.52), 579 (3.99)
1HNMR δH(CDCl3, J [Hz]): 7.56 (d, 2H, J=7.6 Hz, phenyls), 7.42 (m, 12H, β-pyrrole+phenyls), 7.03 (m, 3H, β-pyrrole), 6.96 (d, 1H, J=4.3 Hz, β-pyrrole), 6.04 (s, 2H, −NH2), 1.44 (s, 9H, p-tBu), 1.41 (s, 9H, p-tBu),1.40 (s, 9H, p-tBu). MS (MALDI): m/z 815 (M+).
2,18-(NH2)2-3,17-(NO2)2-TtBuPCorrCu
3,17-(NO2)2-TtBuPCorrCu (160 mg, 0.19 mmol) was dissolved in 33 mL of a toluene/ethanol (30:3) solvent mixture and then NaOH (38 mg, 0.95 mmol) and 4-amino-4H-1,2,4-triazole (191 mg, 2.27 mmol) were added. The reaction mixture was stirred at room temperature and monitored by TLC. All of the starting material was consumed in 20 min, as indicated by TLC analysis, which showed a more polar olive green band together with decomposition products. The solvent was then removed under vacuum, the residue dissolved in CHCl3, washed twice with aqueous Na2S2O3 and dried over Na2SO4. The crude mixture was purified by chromatography on silica gel. Traces of the starting material were eluted with CH2Cl2. Then the use of CHCl3 as eluent afforded a dark green fraction which was collected and crystallized from CH2Cl2/MeOH, giving the title compound as a greenish powder (50 mg, 30% yield). Anal. Calcd for C49H47CuN8O4: C, 67.2; H, 5.3; N, 12.8 %. Found C, 67.4; H, 5.4; N, 12.7 %. UV-vis: λmax(CHCl3) (log ε) nm 366 (4.67), 465 (4.84), 604 (4.23).
1HNMR δH(CDCl3, J [Hz]): 7.41 (d, 2H, J=8.0 Hz, phenyl), 7.29 (m, 6H, phenyl), 7.14 (d, 4H, J=7.6 Hz, phenyl), 6.86 (d, 2H, J=4.4 Hz, β-pyrrole), 6.72 (d, 2H, J=4.2 Hz, β-pyrrole), 5.66 (s, 4H, -NH2), 1.45 (s, 9H, p-tBu), 1.37 (s, 18H, p-tBu). MS (MALDI): m/z 876(M+).
2-NH2-3-NO2-TPCorrGe(OCH3)
3-NO2-TPCorrGe(OCH3) (40 mg, 0.06 mmol) and 4-amino-4H-1,2,4-triazole (50 mg, 0.6 mmol) were dissolved in toluene/ethanol (20:1) (32 mL) and the solution was stirred at room temperature until complete dissolution of all reagents. The temperature was then raised to 80 °C and NaOH (24 mg, 0,60 mmol) was added. After 1 h the reaction mixture was cooled, the solvent removed and the residue was taken up in CH2Cl2 and washed with H2O. The organic phase was dried over anhydrous Na2SO4, concentrated, and then purified using an alumina Brockmann Grade IV column eluted with CHCl3. The major green fraction, after adding few drops of MeOH, was precipitated from CH2Cl2/hexane, affording the title compound as a dark green powder (20 mg, yield 50%). Anal. Calcd for C38H26GeN6O3: C, 66.4; H, 3.8; N, 12.2%. Found C, 66.5; H, 3.8; N, 12.0 %. UV-vis: λmax(CHCl3) (log ε) nm 384 (4.64), 448 (5.01), 624 (4.36). HNMR δH(CDCl3, J [Hz]): 9.21 (d, 1H, J=3.8, β-pyrrole), 9.10 (m, 2H, β-pyrrole), 9.02 (d, 1H, J= 4.8, β-pyrrole), 8.83 (d, 2H, J= 3.8, β-pyrrole), 7.5-8.5 (br, m, 15H, phenyl), 7.22 (br. s, 2H, −NH2), −0.77 (s, 3H, −OCH3). MS (FAB): m/z 687 (M+). Crystal data: C38H26GeN6O3, monoclinic, space group P21/c, a = 10.6310(5), b = 18.9383(10), c = 15.0218(9) Å, β = 92.646(5)°, V = 3021.2(3) Å3, Z = 4, Dcalc = 1.511 g cm−3, = 1.797 mm−1, T = 90 K, 23268 reflections collected with θmax<68.9°, 5398 independent reflections (Rint = 0.033) which were used in all the refinement, using SHELXL12. An electron-density peak 1.1 Å from the Ge position, slightly out of the corrole plane opposite Ge, was interpreted as an alternate position for Ge, and was included in the refinement with 2.0% occupancy. All H atoms were visible in difference maps. Coordinates for NH2 hydrogen atoms were refined, while all other H atoms were in idealized positions, with a torsional parameter refined for the Me group. Final residuals (for 445 parameters) were R1 [I >2σ(I)] = 0.031, wR2 (all data) = 0.083, max. resid. density 0.40 e Å−3.
Results and Discussion
Synthesis of (NO2)xTtBuPCorrCu
We recently reported the preparation of [3-(NO2)triarylcorrolato]Ag(III) by reaction of the corresponding free-base corrole with AgNO2 as nitrating agent.8a Although this approach leads to formation of the silver corrole complex, the low stability of these compounds under both acidic and basic conditions allowed us to define efficient demetalation procedures,13 which opened the way to preparation of otherwise unavailable novel 3-(NO2)metallocorrolates using other nitration methods, the exception being the gallium9 and germanium8b corrole complexes. In this case, reductive demetalation in basic conditions (DBU/THF) was particularly useful to obtain the corresponding free-base macrocycle, while acidic conditions led to the formation of isocorrole species, in which oxidation regioselectively occurred at the 5-position of the ring. In light of such a finding, we were able to identify as a by-product of the nitration reaction,8a a [3-(NO2)5-(OH)] isocorrole species, which was previously supposed to be an open chain compound. The formation of such a compound, both during the reaction with AgNO2 and during chromatographic purification on silica gel, represents the main drawback of the nitration reaction, causing a substantial decrease in the yield of the silver 3-nitrocorrolate. This drawback could be addressed by subsequent reduction of the isocorrole to the corresponding corrole14 or by the direct application of the demetalation protocol to the crude mixture of the nitration reaction, which reduced the isocorrole formation. We therefore decided to follow an alternative route, which can use the same reaction protocol on corrole metal complexes, with the aim to improve the reaction yields by completely avoiding the isocorrole formation. Copper was the metal ion of choice, since it has the double advantage of being readily inserted and removed from the macrocyclic core,15 and its corrole complexes are diamagnetic at room temperature, hence easy to characterize.
We have earlier reported the preparation of copper 3-nitrocorrolates by reacting Cu triarylcorroles with a large excess of NaNO2 in refluxing DMF or CH3CN.8a Evidence for the π-radical cation nature of copper corrolates was obtained, although the synthetic relevance of this route was lessened by the low yields obtained. In order to reduce the reaction steps in preparation of the corrole nitro derivatives, we decided to study the feasibility of a one-pot reaction, starting from the free-base corrole TtBuCorrH3 (Chart 2).
Chart 2.

Synthesis of (NO2)xTtBuPCorrCu
The corrole was first reacted with Cu(OAc)2 in refluxing pyridine to obtain the metal complexes. When the metalation was complete a 50-fold excess of AgNO2 was added and the reaction progress was monitored by UV-vis spectroscopy. After 20 minutes, the color of the solution changed from brownish to dark green, and the UV-vis spectrum showed a novel compound having a Soret band which was red-shifted by about 25 nm compared with the starting copper complex, along with two new bands at ca. 591 and 690 nm. Chromatographic purification afforded traces of the Cu 3-mononitrocorrole, followed by an orange rust fraction having a peculiar UV-vis spectrum, characterized by an intense Soret band at 465 nm and broadened absorptions in the Q band region. Although only traces of such a product were isolated, we were able to identify it by 1H NMR spectroscopic analysis. The proton spectrum revealed the presence of two proton singlets at 8.36 and 7.83 ppm and three different tert-butyl proton signals of equal intensity at 1.47, 1.45 and 1.42 ppm, respectively, indicating an asymmetric product (Figure S1). Furthermore, the integral calculations are consistent with a disubstituted compound and the MALDI mass spectrum affording a molecular peak at m/z 845 led us to identify the compound as 2,17-(NO2)2TtBuCorr Cu. The main reaction product was isolated in 52% yield and fully characterized as 3,17-(NO2)2TtBuCorrCu. The 3,17-substitution pattern observed is in fact identical with that recently reported by us and others for the selective nitration of a Ge triphenylcorrole8b and a gallium(III) 5,10,15-tris(pentafluorophenyl)corrole, respectively.9 Introduction of the second nitro group at the C17 position restored the molecular symmetry, as reflected by the proton resonance distribution in the 1H NMR spectrum: the number of signals decreases and the tert-butyl protons return to a 2:1 ratio, as found for the starting Cu complex. (Figure S2).
Although the above-mentioned conditions are convenient for preparation of 3,17-(NO2)2-5,10,15-triarylcorrolato Cu complexes in satisfactory yield starting from the corresponding corrole free-base macrocycles, it should be noted that a large excess of the quite expensive AgNO2 reagent was necessary. For this reason we decided to decrease the amount of silver nitrite, further surmising that formation of the mono-nitroderivative would be favored; when we employed from 1.2- up to a 5-fold molar excess of AgNO2 relative to the copper complex, we generally obtained a mixture of three products; these were the starting Cu complex, and the mono- and di-nitro derivatives, with relative yields of each depending on the molar excess of AgNO2 used.
These results indicate that an excess of nitrite ion is necessary for completion of the reaction, and for this reason we turned our attention toward other nitrating systems that would combine the oxidation properties of AgNO2, allowing also the required nitrite excess.
The first system investigated utilized NaNO2 as the nitrite source and I2 as oxidant, but the results were not satisfying since only the complete decomposition of the macrocycle was observed. Considering this failure we decided to use AgNO2 as oxidant, mixing it in appropriate amounts with NaNO2, to ensure the nitrite ion excess. The first attempt was carried out in refluxing DMF or pyridine and made use of a TtBuCorrCu/AgNO2/NaNO2 molar ratio of 1:2:8. After formation of the copper complex, silver and sodium nitrite were added and in 20 minutes formation of the copper 3,17-dinitroderivative was observed by TLC analysis and UV-visible spectroscopy. Chromatographic purification afforded the same fractions as when using the previous method, namely traces of the 3-mononitro derivative and the 2,17-dinitro regioisomer along with a main green fraction corresponding to the Cu 3,17-dinitrocorrole in 52% yield. This latter value is similar to what was obtained when only AgNO2 was used. However, it is worth mentioning that the yields of the nitration reaction are remarkably affected by the purity grade of the silver nitrite reagent utilized. We found that when the color of the salt is quite gray, indicative of a less pure grade of AgNO2, the TtBuCorrCu/AgNO2/NaNO2 molar ratio needed to be scaled up to obtain similar results as described in the Experimental Section.
The encouraging results led us to explore the possible formation of a mononitro product by modification of the AgNO2/NaNO2 ratio. It was found that a Cu complex/silver nitrite/sodium nitrite molar ratio of 1:1:9 was the best choice to obtain 3-(NO2)TtBuCorrCu as the main product, together with the corresponding 3,17-dinitroderivative, in 75 and 15% yields, respectively.
It is worth mentioning that the scale-up of the reaction under these conditions led us to detect, during purification on chromatography columns, traces of a less polar compound having similar UV-vis spectral features to that of the 3-nitro derivative, (see Experimental Section); the identity of this compound was established by mass spectrometry and 1H NMR spectroscopy. The MALDI mass spectrum showed the new compound to have a molecular peak at m/z 801, as found for 3-(NO2)TtBuCorrCu, and the proton NMR spectrum (Figure S3) was consistent with a mono-substituted product. The presence of a singlet at 7.91 ppm allowed us to identify this new compound as the isomer, 2-(NO2)TtBuCorrCu, in which nitro substitution had occurred at position 2 of the macrocycle.
Amination reaction on (NO2)xTtBuPCorrCu
The establishment of these synthetic procedures for preparation of nitrocorrole copper derivatives allowed us to explore their possible further functionalization. Among possible peripheral functionalizations we were most interested in the amination reaction, with the aim to use the resulting di-functionalized corroles as building blocks for the preparation of oligomeric species, following an approach reported by Crossley, who utilized 2-amino-3-nitroporphyrin derivatives as excellent starting compounds for the construction of elaborated laterally-bridged oligoporphyrin arrays.5
We had previously studied the amination of 3-(NO2)TtBuCorrAg using 4-amino-4H-1,2,4-triazole, an aminating reagent introduced by Katritzky16 for introduction of an NH2 group onto electron-deficient arenes, and recently used for the synthesis of 2-amino-3-nitroporphyrins.17 In this case the reaction failed and we observed the unexpected formation of 6-azahemiporphycene, a novel porphyrin analog, arising from macrocyclic ring expansion.18 In our studies we demonstrated that formation of the 6-azahemiporphycene species is not dependent on the presence of the nitro group because the same corrole ring-expansion was observed when the reaction was carried out on unsubstituted corroles. We discovered that the macrocycle oxidation is a key step for the reaction, considering that 5- and a 10-hydroxyisocorrole could be used as starting materials.19 In this case, silver corroles are not useful as starting materials, because they are readily demetalated under the basic conditions necessary for the reaction. For this reason copper corroles seem to be more promising for the reaction, providing that their stability would allow the vicarious substitution to occur instead of the azahemipophycene ring expansion.
The amination on 3-(NO2)TtBuCorrCu was performed using 4-amino-4H-1,2,4-triazole and NaOH as reagents, in the molar ratio of 1:12:5 following the same protocol employed for the preparation of 2-amino-3-nitroporphyrins (Chart 3). When we carried out the reaction at reflux temperatures, rapid decomposition of the starting complex was observed, as shown by both TLC analysis and UV-vis spectroscopy. When all of the reagents were stirred at room temperature, TLC analysis showed after a few minutes the formation of a second band having a lower Rf value with respect to the starting complex. After 45 minutes the copper 3-nitrocorrolate was completely consumed and the crude reaction mixture was subjected to purification on a chromatography column. A single brownish fraction was isolated and analyzed by UV-vis and 1H NMR spectroscopies. The UV-vis spectral features are quite similar to those of the starting material, although a slight red-shift of the Soret band is clearly visible.
Chart 3.

Amination reaction of (NO2)xTtBuPCorrCu
The 1H NMR spectrum (Figure S4) showed a disappearance of the proton singlet of the β-pyrrolic carbon 2 and an upfield shifting of some pyrrolic resonances below ca. 7 ppm. Furthermore, the presence of a broad singlet at 6.04 ppm can be ascribed to the amino group, the corresponding proton signal of which is strongly deshielded by the presence of the adjacent nitro group, as recently reported in the case of 2-amino-3-nitroporphyrins. The MALDI mass spectrum shows a molecular peak at 815 m/z, enabling unambiguous identification of the product as the desired 2-NH2-3-NO2-TtBuCorrCu.
The reaction was performed on 3,17-(NO2)2TtBuCorrCu under the same experimental conditions, affording, after 20 minutes the tetra-substituted 2,18-diamino-3,17-dinitrocorrole copper complex, whose aromatic region of the 1H NMR spectrum is presented in Figure 1. Introduction of the two NH2 groups at the 2 and 18 β-pyrrole positions of the macrocycle results in a deshielding of both the aromatic and the pyrrolic proton signals. The effect is particularly pronounced for the β-hydrogens of the B and C pyrroles that resonate at 6.86 and 6.72 ppm, uncommonly low values for such resonances.
Figure 1.

1H NMR spectrum of 2,18-(NH2)2-3,17-(NO2)2-TtBuPCorrCu
It is worth mentioning that when 4-amino-4H-1,2,4-triazole was reacted with an unsubstituted copper corrolate TtBuCorrCu, the copper 6-azahemiporphycene was obtained as the main product, and was identified by comparison with the same compound prepared by metalation of [6-Aza-5,11,16-tris(4-tert-butylphenyl)hemiporphycene] with Cu(OAc).19
To investigate in greater detail the different reactivity shown by the unsubstituted copper complex with respect to the corresponding 3-nitro- and 3,17-dinitro- derivatives, we decided to study the electrochemistry of the species. This is described on the following pages.
Electrochemistry
Since the seminal paper by Vogel, Kadish and collaborators on electrochemical and spectroscopic studies of (OECorr)Cu,20 different triphenylcorroles (TPCorr) containing electron-donating or electron-withdrawing substituents on the meso and/or β-pyrrole positions of the macrocycle have been investigated in detail.20-29
While the early characterization of copper corrole complexes proposed an equilibrium between the two forms reported in Equation 1, subsequent investigations now favor the formulation of the complex as a copper(II) corrole π cation radical rather than as a metal(III) corrole with an unoxidized π ring system. Such an assignment has been discussed in the literature in light of the non-innocent nature of the corrole macrocycle.20, 27-32
| (1) |
Previous electrochemical characterization of Cu corroles with a variety of structures have shown up to six different redox reactions to occur for these complexes, with the number of processes and half-wave potentials being dependent upon the solvent and specific electron-donating or electron-withdrawing properties of the substituents. For example, TPCorrCu undergoes three ring-centered oxidations in CH2Cl2, with only one reduction process,29 while corroles with electron-withdrawing substituents on the meso and/or β-pyrrole positions undergo a maximum of two ring centered oxidations within the potential solvent limit (~+1.80 V in CH2Cl2 or PhCN) and one additional ring centered reduction process under the same solution condition.
In the absence of nitro substituents, TtBuPCorrCu undergoes a reversible reduction process located at E1/2 = −0.17 V in CH2Cl2 and −0.18 V in PhCN, both containing 0.1 M TBAP. A similar redox behavior has been reported for copper triphenylcorrole, TPCorrCu and 17 related corroles29 containing electron-donating or electron-withdrawing substituents on the meso and/or β-pyrrole positions of the macrocycle. The parent triphenylcorrole is reduced at −0.16 V in CH2Cl2 while the same process for the other corroles varied from E1/2 = −0.22 to +0.64 V, depending upon the type and position of the electron-withdrawing or electron-donating substituents.
As described below, this behavior differs from what is seen for the currently investigated nitrocorroles in non-aqueous media. Cyclic voltammograms of the three (NO2)xTtBuPCorrCu complexes are illustrated in Figure 2 (CH2Cl2) and Figure S12 (PhCN) and a summary of half-wave and peak potentials are given in Table 1. The nitro groups on (NO2)xTtBuPCorrCu are highly electron-withdrawing and this leads to a substantial positive shift of all redox potentials as compared to the parent compound, TtBuPCorrCu. Two one-electron reductions, not observed for TtBuPCorrCu, are also seen for the mono- and dinitrocorrole and these are located at E1/2 = −1.11 and −1.44 V vs SCE for the case of (NO2)xTtBuPCorrCu in CH2Cl2.
Figure 2.

Cyclic voltammograms of (NO2)xTtBuPCorrCu in CH2Cl2, 0.1 M TBAP.
Table 1.
Half-Wave Potentials (V vs SCE) of (NO2)xTtBuPCorrCu in CH2Cl2 or PhCN containing 0.1 M TBAP.
| Solvent | # of NO2 groups, x |
Oxidation |
Reduction |
||||
|---|---|---|---|---|---|---|---|
| 3rd | 2nd | 1st | 1st | 2nd | 3rd | ||
| CH2Cl2 | 0 | 1.75a | 1.31 | 0.71 | −0.17 | ||
| 1 | 1.50 | 0.90 | 0.00 | −1.34 | −1.74a | ||
| 2 | 1.61 | 1.11 | 0.20 | −1.11 | −1.44 | ||
| PhCN | 0 | 1.69a | 1.39 | 0.74 | −0.18 | ||
| 1 | 1.86a | 1.58 | 0.92 | 0.04 | −1.40 | ||
| 2 | 1.65 | 1.12 | 0.24 | −1.12 | −1.47 | ||
Irreversible peak potential at a scan rate of 0.10 V/s.
Plots of E1/2 for first reduction and first oxidation vs the number of NO2 groups on the molecule are linear (ΔE1/2/ΔNO2 ≈ 200 mV) indicating a similar redox mechanism for all three compounds. A linear correlation also exists between the number of NO2 groups and λmax for the Soret band of the neutral compound (Figure 3) or between E1/2 and the Soret band λmax when plotted in wavenumbers (Figure 4).
Figure 3.

Correlation between a) E1/2 for the first reduction of (NO2)xTtBuPCorrCu in PhCN and b) the Soret band maximum of the neutral compounds.
Figure 4.

a) Cyclic voltammograms of (NO2)xTtBuPCorrCu in PhCN, 0.1 M TBAP and b) plots of half wave potential for the first oxidation and first reduction of the corrole vs the Soret band maximum of (NO2)xTtBuPCorrCu in wavenumbers. Values of λmax are given in Table 2.
The above linear correlations suggest the same initial electron transfer site for reduction of all three compounds but, as earlier indicated, the redox properties of the singly reduced product are quite different in the case of (NO2)TtBuPCorrCu and (NO2)2TtBuPCorrCu as compared to all previously investigated triphenylcorroles with electron-withdrawing Br, F or CF3 substituents. For example, TFPCorrCu and Br8TPCorrCu are reduced at 0.21 V and 0.19 V vs SCE in CH2Cl2,29, which are potentials almost identical to those of (NO2)2TtBuPCorrCu, where the first reduction occurs at 0.20 V (see Table 1). The oxidation potentials of these three compounds are also similar, being 1.12 V for TFPCorrCu, 1.16 V for Br8TPCorrCu and 1.11 V for (NO2)2TtBuPCorrCu in CH2Cl2. In contrast, the singly reduced products of TFPCorrCu and Br8TPCorrCu exhibit only one additional reduction at E1/2 = −1.70 and −1.48 V, respectively, in CH2Cl2 as compared to two reversible one-electron reductions for [(NO2)2TtBuPCorrCu]− which are located at E1/2 = −1.11 and −1.44 V under the same solution conditions. Two well-defined reductions are also seen for singly reduced (NO2)TtBuPCorrCu in CH2Cl2 (Fig. 3) or PhCN (Fig. S12) and this occurrence is unprecedented in the literature of copper corroles. It is also markedly different from what is seen in the case of TtBuPCorrCu, TFPCorrCu and all previously investigated corroles with electron-withdrawing substituents, which exhibit UV-visible spectra for the electroreduced product that are characteristic of a Cu(II) corrole with an unreduced π ring system.29 UV-visible spectra obtained in a thin-layer cell during controlled potential reduction of the currently investigated compounds in PhCN are shown in Figure 5; UV-visible spectra of the singly oxidized species were also obtained in PhCN and examples of thin-layer spectra are given in Figure S13. A summary of the spectral data is given in Table 2.
Figure 5.

Spectra changes during first reduction of (NO2)xTtBuPCorrCu in PhCN, 0.1 M TBAP.
Table 2.
Absorption Maximum (λmax, nm) and Molar Absorptivities (logε in parentheses) of [(Corr)Cu]n Where Corr = (NO2)xTtBuPCorr and n = 0, −1 and +1.
| Solvent | x | (Corr)Cu |
[(Corr)Cu]−1 |
[(Corr)Cu]+1 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CH2Cl2 | 0 | 418 (5.08) | 547 (4.02) | 636 (3.74) | 428 (5.08) | 613 (4.22) | 418 (4.97) | |||||
| 1 | 371 (4.73) | 436 (5.05) | 578 (4.33) | 682 (4.03) | 378 (4.78) | 408 (4.81) | 477 (4.74) | 616 (4.58) | 682 (4.39) | 446 (4.82) | ||
| 2 | 372 (4.59) | 450 (4.88) | 594 (4.45) | 700 (4.34) | 397 (4.62) | 450 (4.78) | 594 (4.40) | 700 (4.41) | 450 (4.82) | |||
| PhCN | 0 | 422 (4.96) | 548 (3.84) | 640 (3.52) | 435 (5.03) | 614 (4.26) | 422 (4.65) | |||||
| 1 | 377 (4.33) | 438 (4.69) | 575 (4.00) | 686 (3.77) | 377 (4.40) | 408 (4.45) | 480 (4.42) | 618 (4.27) | 686 (4.03) | 438 (4.46) | ||
| 2 | 377 (4.17) | 453 (4.49) | 596 (4.08) | 703 (3.82) | 327 (4.23) | 356 (4.23) | 399 (4.24) | 508 (4.17) | 636 (4.12) | 703 (4.15) | 453 (4.29) | |
In summary, electrochemistry and thin-layer spectroelectrochemistry of the investigated (NO2)xTtBuPCorrCu derivatives are self-consistent in indicating the same initial site of electron transfer for all three compounds, although they show very different UV-visible changes. The peripheral nitro groups are responsible for this different behavior, but the mechanism of their influence should be clarified. While the initial product of the one electron addition is assigned as (Corr)CuII for all three compounds, some contrasting results make difficult to elucidate the nature of the final electroreduced products in the case of (NO2)TtBuPCorrCu and (NO2)2TtBuPCorrCu (see Fig. 5).
For example it is interesting to note that the same UV-visible changes were obtained by addition of DBU to the (NO2)xTtBuPCorrCu solutions; this DBU influence is similar to what observed in the case of silver corrole complexes, which are reductively demetallated by addition of DBU.13 These UV-vis spectra are also shown in Figure 6 and closely resemble spectra reported for structurally similar Ge(IV) corroles after a one-electron reduction,8b where only a corrole π-anion radical is possible. However low temperature ESR characterization of singly reduced (NO2)xTtBuPCorrCu shows clear signatures of a Cu(II) corrole (Figure 6) and does not support this hypothesis; the above conflicting results in the case of singly reduced (NO2)xTtBuPCorrCu may be related to the effect of temperature, but we were unable to obtain room temperature ESR measurements to resolve this point. Furthermore, the peculiar spectra for the singly reduced corroles with nitro groups might be ascribed to a mesomeric rather than an inductive electron-withdrawing effect of the β-pyrrole NO2 substituents, which would stabilize the anion by delocalization of the negative charge via conjugation with the aromatic corrole ring, thus resulting in the significant UV-visible changes.33
Figure 6.

(a) UV-vis spectra of (NO2)xTtBuPCorrCu before (----) and after ( ) reduction with 1000 eqs DBU in PhCN, 0.1 M TBAP and (b) ESR spectra of singly reduced (NO2)xTtBuPCorrCu in the same solution at 77 K.
) reduction with 1000 eqs DBU in PhCN, 0.1 M TBAP and (b) ESR spectra of singly reduced (NO2)xTtBuPCorrCu in the same solution at 77 K.
In any event, the observed behavior is very specific to the currently investigated nitrocorroles and does not seem to occur for other related corroles having Br, F or CF3 substituents on the macrocycle. These highly electron-withdrawing groups will lead in some cases to more positive redox potentials than in the case of (NO2)TtBuPCorrCu or (NO2)2TtBuPCorrCu, but despite the more facile reduction, the currently investigated nitrocorroles appear to be unique in their redox behavior and in the UV-vis spectrum of the singly reduced product.
Amination reaction on 3-NO2-TPCorrGeOCH3
Finally, we have also investigated the role of the corrole central metal ion on the reaction with 4-amino-4H-1,2,4-triazole, with the intention to increase the moderate yields obtained in the case of copper derivatives (18 and 30% for the mono- and di-nitro compounds, respectively). According to the similar redox behavior, we decided to carry out the same reaction on germanium(IV) 3-(NO2)triphenylcorrolate, which was recently prepared and characterized in our laboratories.8b In this complex the Ge ion is in a high oxidation state (+4), so it can inductively cooperate with the electron-withdrawing character of the nitro groups for the amination reaction, without the strong electronic interaction with the π-system of the corrole ring.
3-NO2-TPCorrGe(OCH3) was reacted with 4-amino-4H-1,2,4-triazole following a slightly modified amination protocol (see Experimental Section), and a single fraction was isolated in good yield (50%) (Chart 4).
Chart 4.

Amination reaction of 3-NO2-TPCorrGe(OCH3)
The isolated compound displayed a UV-vis spectrum similar in shape to that of the starting complex, although the absorbances were red-shifted. The 1H NMR spectrum are consistent with a disubstituted germanium complex, showing in particular the disappearance of the singlet at 9.71 ppm corresponding to the proton on C2 of the starting compound and a broad singlet at 7.22 ppm ascribable to the amino group (Figure S5). The FAB mass spectrum indicated a molecular peak at m/z 687, confirming the identity of this compound as the difunctionalized β-aminõβ-nitro-corrole derivative. In this case we were able to grow single crystals by slow diffusion of hexane into a diluted dichloromethane solution of the product. The X-ray crystal structure determination unambiguously confirmed it to be 2-NH2-3-NO2-TPCorrGe(OCH3). (Figure 7).
Figure 7.

Molecular Structure of 2-NH2-3-NO2-TPCorrGe(OCH3)
The Ge atom has square-pyramidal coordination geometry, lying 0.4323(2) Å out of the N4 plane. The apical Ge-O distance is 1.7879(14) Å, and the Ge-N distances range from 1.8923(16) to 1.9463(16) Å, averaging 1.913 Å, with Ge-N2 the shortest and Ge-N1 the longest. The O-Me bond is nearly eclipsed with Ge-N4, forming a N-Ge-O-C torsion angle 9.89(13)°. The NO2 group is twisted by 28° out of the plane of the pyrrole to which it is bonded and accepts an intramolecular hydrogen bond (N…O 2.770(2) Å, H…O 2.22(3) Å, N-H…O 120(2)°) from the NH2 group. The NH2 group also donates an intermolecular hydrogen bond (2.953(2) Å) to methoxy O.
Conclusions
Modification of the reaction protocol established for the preparation of 3-nitro- and 3,17-dinitro-corrole copper complexes allows for modulation of the number of the peripheral substituents while also leading to an increase in the reaction yields and a reduction in the quantity of the expensive AgNO2 reagent utilized. These complexes were reacted with 4-amino-4H-1,2,4-triazole to obtain the corresponding amino-nitrocorrole derivatives. This is the first successful preparation of such derivatives, because corrole ring expansion was observed to give the corresponding 6-azahemiporphycene in the case of the corresponding silver complexes. To the best of our knowledge, this is also the first example for nucleophilic aromatic substitution carried out on a corrole ring. To investigate in detail the influence of the coordinated metal on the reaction pathway, an electrochemical characterization of the copper corrole nitro derivatives was carried out. The highly electron-withdrawing character of the nitro groups leads to a substantial positive shift of all redox potentials of (NO2)xTtBuPCorrCu as compared to the parent compound, TtBuPCorrCu, inducing also unique UV-vis spectra of the singly reduced products. The scope of the reaction was tested on the Ge(IV) corrole complex, which afforded higher yields of the amino-derivative than with the copper analog; the obtained Ge(IV) product was characterized by X-ray crystallography. A synthetic route to these bis-functionalized corroles, which are promising building blocks for the development of more complex molecular architectures based on corroles, has been defined.
Supplementary Material
Scheme 1.
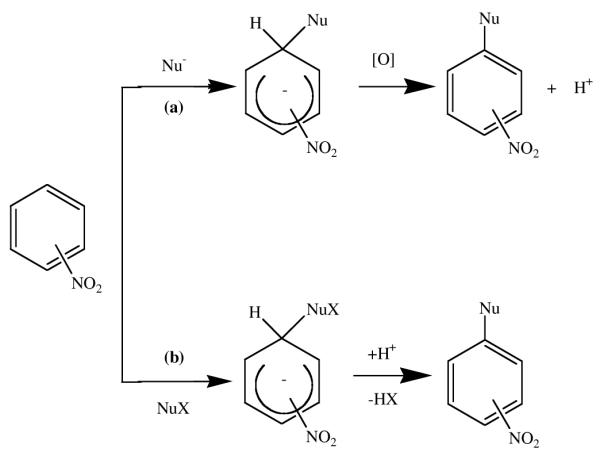
Conversion of σ-adducts: (a) the oxidative nucleophilic substitution (ONSH) and (b) the vicarious nucleophilic substitution (VNS) pathways.
ACKNOWLEDGMENT
We gratefully acknowledge the support of MIUR, Italy (PRIN project 2007C8RW53), the Robert A. Welch Foundation (KMK, Grant E-680), and the US National Institutes of Health (KMS, Grant CA 132861).
Footnotes
SUPPORTING INFORMATION. 1H NMR spectra, UV-vis, mass spectra, cyclic voltammograms, and CIF of 2-NH2-3-NO2-TPCorrGe(OCH3). This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- (1).Senge MO. Acc. Chem. Res. 2005;38:733–743. doi: 10.1021/ar0500012. [DOI] [PubMed] [Google Scholar]
- (2).Shea KM, Jaquinod L, Khoury RG, Smith KM. Chem. Commun. 1998:759–760. [Google Scholar]
- (3).Jaquinod L. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 1. Academic Press; San Diego: 2000. pp. 201–237. [Google Scholar]
- (4).Gros CP, Jaquinod L, Khoury RG, Olmstead MM, Smith KM. J. Porphyrins Phthalocyanines. 1997;1:201–212. [Google Scholar]
- (5).(a) Crossley MJ, Govenlock LJ, Prashar JK. Chem. Commun. 1995:2379–2380. [Google Scholar]; (b) Crossley MJ, Burn PL. Chem. Commun. 1991:1569–1571. [Google Scholar]
- (6).(a) Aviv I, Gross Z. Chem. Eur. J. 2009;15:8382–8394. doi: 10.1002/chem.200900920. [DOI] [PubMed] [Google Scholar]; (b) Kupershmidt L, Okun Z, Amit T, Mandel S, Saltsman I, Mahammed A, Bar-Am O, Gross Z, Youdim MBH. Journal of Neurochemistry. 2010;113:363–373. doi: 10.1111/j.1471-4159.2010.06619.x. [DOI] [PubMed] [Google Scholar]; (c) Kanamori A, Catrinescu MM, Mahammed A, Gross Z, Levin LA. Journal of Neurochemistry. 2010;114:488–498. doi: 10.1111/j.1471-4159.2010.06781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Paolesse R. Synlett. 2008;15:2215–30. and references therein. [Google Scholar]; (b) Mahammed A, Goldberg I, Gross Z. Org. Lett. 2001;3:3443–3446. doi: 10.1021/ol0163878. [DOI] [PubMed] [Google Scholar]; (c) Barata JFB, Neves MGPMS, Tomè AC, Cavaleiro JAS. J. Porphyrins Phthalocyanines. 2009;13:415–418. [Google Scholar]; (d) Du RB, Liu C, Shen DM, Chen QY. Synlett. 2009;16:2701–2705. [Google Scholar]
- (8).(a) Stefanelli M, Mastroianni M, Nardis S, Licoccia S, Fronczek FR, Smith KM, Zhu W, Ou Z, Kadish KM, Paolesse R. Inorg. Chem. 2007;46:10791–10799. doi: 10.1021/ic7014572. [DOI] [PubMed] [Google Scholar]; (b) Mastroianni M, Zhu W, Stefanelli M, Nardis S, Fronczek FR, Smith KM, Ou Z, Kadish KM, Paolesse R. Inorg. Chem. 2008;47:11680–11687. doi: 10.1021/ic801421a. [DOI] [PubMed] [Google Scholar]; (c) Stefanelli M, Nardis S, Tortora L, Fronczeck FR, Smith KM, Licoccia S, Paolesse R. Chem. Commun. 2011;47:4255–4257. doi: 10.1039/c0cc05491g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Saltsman I, Mahammed A, Goldberg I, Tkachenko E, Botoshansky M, Gross Z. J. Am. Chem. Soc. 2002;124:7411–7420. doi: 10.1021/ja025851g. [DOI] [PubMed] [Google Scholar]
- (10).(a) Chupakhin ON, Chupakhin VN, Van der Plas HC. Nucleophilic Aromatic Substitution of Hydrogen. Academic Press; London: 1994. [Google Scholar]; (b) Terrier F. In: Nucleophilic Aromatic Displacement. Feuer H, editor. VCH; New York: 1991. p. 257. Chapter 5. [Google Scholar]; (c) Ma̧kosza M, Winiarski J. Acc. Chem. Res. 1987;20:282–289. [Google Scholar]; (d) Ma̧kosza M. Pol. J. Chem. 1992;66:3–23. [Google Scholar]
- (11).Ma̧kosza M, Wojciechowski K. Chem. Rev. 2004;104:2631–2666. doi: 10.1021/cr020086+. [DOI] [PubMed] [Google Scholar]
- (12).Sheldrick GM. Acta Cryst. 2008;A64:112–122. [Google Scholar]
- (13).Stefanelli M, Shen J, Zhu W, Mastroianni M, Mandoj F, Nardis S, Ou Z, Kadish KM, Fronczek FR, Smith KM, Paolesse R. Inorg. Chem. 2009;48:6879–6887. doi: 10.1021/ic900859a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tortora L, Nardis S, Fronczeck FR, Smith KM, Paolesse R. Chem. Commun. 2011;47:4243–4245. doi: 10.1039/c0cc05837h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Mandoj F, Nardis S, Pomarico G, Paolesse R. J. Porphyrins Phthalocyanines. 2008;12:19–26. doi: 10.1142/S1088424610002513. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ngo TH, Van Rossom W, Dehaen W, Maes W. Org. Biomol.Chem. 2009;7:439–443. doi: 10.1039/b819185a. [DOI] [PubMed] [Google Scholar]; (c) Capar C, Thomas KE, Ghosh A. J. Porphyrins Phthalocyanines. 2008;12:964–967. [Google Scholar]
- (16).(a) Katritzcky AR, Laurenzo KS. J. Org. Chem. 1986;51:5039–5040. [Google Scholar]; (b) Katritzcky AR, Laurenzo KS. J. Org. Chem. 1988;53:3978–3982. [Google Scholar]
- (17).Richeter S, Hadj-Aïssa A, Taffin C, van der Lee A, Leclercq D. Chem. Commun. 2007:2148–2150. doi: 10.1039/b704681b. [DOI] [PubMed] [Google Scholar]
- (18).Mandoj F, Stefanelli M, Nardis S, Mastroianni M, Fronczek FR, Smith KM, Paolesse R. Chem. Commun. 2009:1580–1582. doi: 10.1039/b821390a. [DOI] [PubMed] [Google Scholar]
- (19).Mandoj F, Nardis S, Pomarico G, Stefanelli M, Schiaffino L, Ercolani G, Prodi L, Genovese D, Zaccheroni N, Fronczek FR, Smith KM, Xiao X, Shen J, Kadish KM, Paolesse R. Inorg. Chem. 2009;48:10346–10357. doi: 10.1021/ic9014866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kadish KM, Adamian VA, Van-Caemelbecke E, Gueletii E, Will S, Erben C, Vogel E. J. Am. Chem. Soc. 1998;120:11986–11993. [Google Scholar]
- (21).Sridevi B, Narayanan SJ, Chandrashekar TK, Englich U, Ruhlandt-Senge K. Chem. Eur. J. 2000;6:2554–2563. doi: 10.1002/1521-3765(20000717)6:14<2554::aid-chem2554>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- (22).Wasbotten IH, Wondimagegn T, Ghosh A. J. Am. Chem. Soc. 2002;124:8104–8116. doi: 10.1021/ja0113697. [DOI] [PubMed] [Google Scholar]
- (23).Guilard R, Gros CP, Barbe JM, Espinosa E, Jerome F, Tabard A, Latour JM, Shao J, Ou Z, Kadish KM. Inorg. Chem. 2004;43:7441–7455. doi: 10.1021/ic049651c. [DOI] [PubMed] [Google Scholar]
- (24).Kadish KM, Royal G, Caemelbecke EV, Gueletti L. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 9. Academic Press; San Diego: 2000. pp. 1–220. [Google Scholar]
- (25).Will S, Lex J, Vogel E, Schmickler H, Gisselbrecht JP, Haubtmann C, Bernard M, Gross M. Angew. Chem. Int. Ed. 1997;36:357–361. [Google Scholar]
- (26).Hush NS, Dyke JM. J. Inorg. Nucl. Chem. 1973;35:4341–4347. [Google Scholar]
- (27).(a) Ghosh A, Wondimagegn T, Parusel AB. J. Am. Chem. Soc. 2000;122:5100–5104. [Google Scholar]; (b) Steene E, Dey A, Ghosh A. J. Am. Chem. Soc. 2003;125:16300–16309. doi: 10.1021/ja021158h. [DOI] [PubMed] [Google Scholar]
- (28).(a) Bruckner C, Brinas RP, Bauer JA. Inorg. Chem. 2003;42:4495–4497. doi: 10.1021/ic034080u. [DOI] [PubMed] [Google Scholar]; (b) Luobeznova I, Simkhovich L, Goldberg I, Gross Z. Eur. J. Inorg. Chem. 2004:1724–1732. [Google Scholar]
- (29).Ou Z, Shao J, Zhao H, Ohkubo K, Wasbotten IH, Fukuzumi S, Ghosh A, Kadish KM. J. Porphyrins Phthalocyanines. 2004;8:1236–1247. [Google Scholar]
- (30).Oort B. v., Tangen E, Ghosh A. Eur. J. Inorg. Chem. 2004:2442–2445. [Google Scholar]
- (31).Alemayehu AB, Gonzalez E, Hansen LK, Ghosh A. Inorganic Chemistry. 2009;48(16):7794–7799. doi: 10.1021/ic900744v. [DOI] [PubMed] [Google Scholar]
- (32).Bröring M, Brégier F, Tejero EC, Hell C, Holthausen MC. Angew. Chem. Int. Ed. 2007;46:445–448. doi: 10.1002/anie.200603676. [DOI] [PubMed] [Google Scholar]
- (33).We thank a referee for the helpful suggestions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.