

Abstract
Late-phase long-term potentiation (L-LTP), a cellular model for long-term memory (LTM), requires de novo protein synthesis. An attractive hypothesis for synapse specificity of long-term memory is “synaptic tagging”: synaptic activity generates a tag, which “captures” the PRPs (plasticity-related proteins) derived outside of synapses. Here we provide evidence that TrkB, the receptor of BDNF (brain-derived neurotrophic factor), may serve as a “synaptic tag.” TrkB is transiently activated by weak theta-burst stimulation (TBS) that induces only early-phase LTP (E-LTP). This TrkB activation is independent of protein synthesis, and confined to stimulated synapses. Induction of L-LTP by strong stimulation in one synaptic pathway converts weak TBS-induced E-LTP to L-LTP in a second, independent pathway. Transient inhibition of TrkB around the time of weak TBS to the second pathway diminished L-LTP in that pathway without affecting the first one. Behaviorally, weak training, which induces short-term memory (STM) but not LTM, can be consolidated into LTM by exposing animals to novel but not familiar environment 1 h before training. Inhibition of TrkB during STM training blocked such consolidation. These results suggest TrkB as a potential tag for synapse-specific expression of L-LTP and LTM.
Introduction
The synaptic tagging hypothesis provides a compelling explanation for synapse specificity of L-LTP (Frey and Morris, 1997; Govindarajan et al., 2006; Morris, 2006). Strong stimulation induces the synthesis of plasticity-related proteins (PRPs) either in soma or dendrites (local synthesis). These PRPs are then transported randomly in dendrites. In the mean time, the stimulation creates “synaptic tags,” active molecules that capture the PRPs, which modify only the stimulated (activated) synapses. A litmus test for the synaptic tagging hypothesis is the two-pathway experiment in which two independent synaptic inputs to the same neuronal population are activated by two stimulating electrodes, while field EPSPs (fEPSPs) are monitored by a single recording electrode in the stratum radiatum of CA1 (see Figs. 5A, 6D). When one afferent pathway (S1) to a specific population of CA1 neurons is activated by strong tetanic stimulation, weak tetanic stimulation applied to a second, independent pathway (S2) usually will induce L-LTP because it creates a tag that captures the PRP induced by the strong stimulation applied to S1 (Frey and Morris, 1997) (see Fig. 6D).
Figure 5.
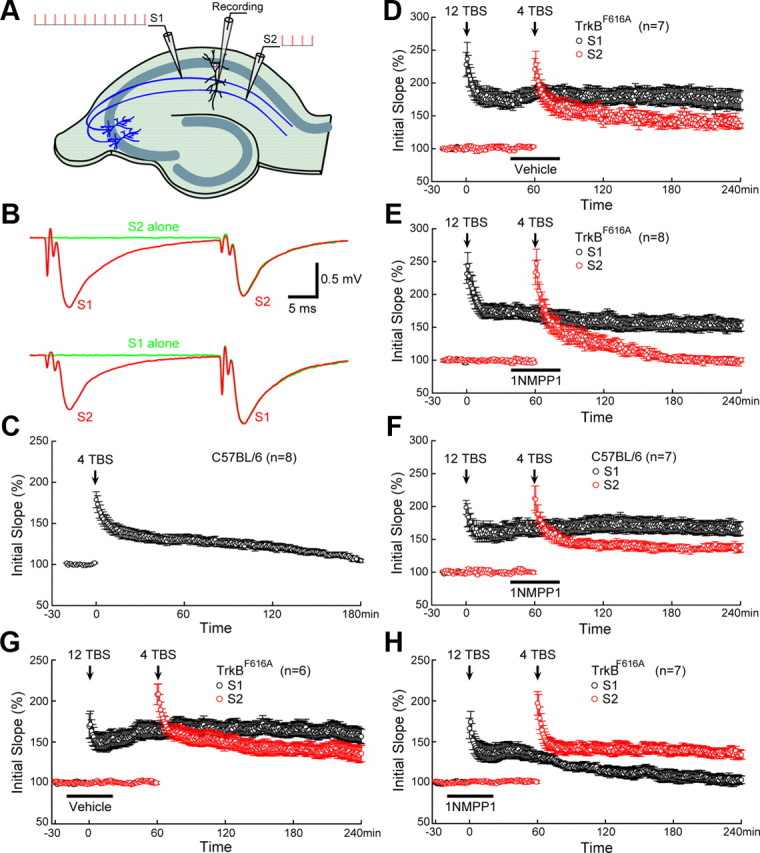
TrkB as synaptic tag for L-LTP. A, Two-pathway recording scheme. Two independent Schafer collateral pathways that project to the same CA1 neurons were stimulated by two stimulating electrodes positioned on either side of the recording electrode. S1 was stimulated by strong TBS (12TBS), whereas S2 was stimulated by weak TBS (4TBS). B, Lack of facilitation between the two Schafer collateral pathways. Top, The superimposed red and green fEPSP traces induced by S2 were same, regardless of whether S1 was stimulated 30 ms earlier. Bottom, S1-induced fEPSPs were the same regardless of S2 stimulation. C, Failure to induce L-LTP by weak TBS (4TBS). Application of 4TBS alone induced only E-LTP (last <2 h), but not L-LTP. D, Weak TBS in S2 induced L-LTP when preceded by strong TBS in S1. In a typical “two-pathway” experiment using slices from TrkBF616A mice, a 12TBS to S1 was followed by a 4TBS to S2. Application of 12TBS resulted in a robust L-LTP in S1. Application of 4TBS to S2, which normally induces only E-LTP, now elicited L-LTP. Treatment with vehicle (indicated by the black bar) around the time of 4TBS had no effect on L-LTP in TrkBF616A slices. E, Inhibition of TrkB activation blocked E-LTP→L-LTP conversion in S2. The “two-pathway” experiment was done exactly the same way as in D, except that 1NMPP1 (5 μm) was applied instead of vehicle (DMSO). Treatment with 1NMPP1 blocked L-LTP in S2, without affecting L-LTP in S1 in TrkBF616A slices. F, Lack of effect of 1NMPP1 on L-LTP in wild-type hippocampal slices. 1NMPP1 was applied to hippocampal slices from C57BL/6 mice around the time of 4TBS (black bar). 12TBS induced a 166 ± 10% potentiation in S1, whereas 4TBS induced a 138 ± 5% potentiation in S2. G, Lack of effect of vehicle on L-LTP in TrkBF616A slices. L-LTP was induced in both S1 (158 ± 9%) and S2 (135 ± 10%) when vehicle was applied at the time of 12TBS. H, 1NMPP1 inhibited L-LTP in S1 (104 ± 5%) but left L-LTP intact in S2 (135 ± 5%) when it was applied at the time of 12TBS. The number of slices tested is indicated in brackets. At least three animals were tested for each experiment.
Figure 6.

Further evidence for TrkB as synaptic tag for L-LTP. The “two-pathway” experiment was done exactly the same way as in Figure 5, except that 4TBS was applied to S2 40 min before (instead of 60 min after) 12TBS applied to S1. Application of 1NMPP1 or vehicle (DMSO) is indicated by the black bar. A, Treatment with 1NMPP1 (5 μm) in TrkBF616A slices blocked L-LTP in S2, without affecting L-LTP in S1. B, L-LTP was induced by 12TBS in S1 and by 4TBS in S2 in TrkBF616A slices treated with vehicle around the time of 4TBS. C, L-LTP was induced by 12TBS in S1 and by 4TBS in S2 in wild-type (C57BL/6) slices treated with 1NMPP1 around the time of 4TBS. D, Synaptic tagging model for two-pathway recording. Short-lived synaptic tag can be generated by strong (12TBS) and weak (4TBS) stimulation, while the PRPs can only be induced by strong stimulation. Overlap of the tag wave and PRP wave indicates the capture of PRPs, leading to L-LTP in that specific pathway.
Several lines of evidence suggest that brain-derived neurotrophic factor (BDNF) is a candidate PRP. First, enhanced BDNF expression has been demonstrated in CA1 pyramidal neurons upon L-LTP-inducing stimulation (Patterson et al., 1992), probably mediated by enhanced BDNF transcription through activity-dependent activation of promoter IV (Lu, 2003; Greer and Greenberg, 2008). Second, strong theta-burst stimulation (12 bursts, each of 4 pulses at 100 Hz, or 12TBS, which should create a tag) failed to induce L-LTP in mice with reduced BDNF expression, but application of mature BDNF (mBDNF) rescues the L-LTP deficit (Patterson et al., 2001; Pang et al., 2004). Third, BDNF can transform E-LTP into L-LTP by weak TBS, which can create a “synaptic tag” but not PRP (BDNF) expression (Pang et al., 2004). In VP16-CREB mice in which BDNF levels are elevated, weak stimulation was able to induce L-LTP, which could be reversed by the BDNF scavenger TrkB-IgG (Barco et al., 2005). In mice in which the BDNF gene is deleted in the hippocampus (or more specifically in postsynaptic CA1 neurons which could produce PRPs), weak stimulation can no longer capture the PRP to sustain L-LTP in S2 in the “two-pathway” experiment (Barco et al., 2005). Finally, when L-LTP is blocked by a protein synthesis inhibitor, it can be completely rescued by application of exogenous BDNF (Pang et al., 2004). It is known that de novo BDNF expression is required for L-LTP within 70 min after stimulation, but not later (Kang et al., 1997).
After its secretion, BDNF binds and activates its receptor TrkB, which is localized to the dendritic spines (Drake et al., 1999; Ji et al., 2005). If BDNF is a PRP, TrkB is an obvious candidate for a synaptic tag. In theory, a synaptic tag must satisfy several criteria (Martin and Kosik, 2002; Kelleher et al., 2004; Lu et al., 2008): (1) a tag can be activated by weak stimulation that induces only E-LTP; (2) the lifetime of a tag is ∼1–2 h; (3) the activation of a tag does not require protein synthesis; (4) a tag is induced in an input-specific manner and is relatively immobile; and (5) a tag must interact with (and therefore capture) PRP for L-LTP.
In the present study, we tested the hypothesis that TrkB serves as synaptic tag for L-LTP in vitro and behavioral tag for LTM in vivo. We demonstrate that TrkB can be activated by both weak and strong TBS in a transient and protein synthesis-independent manner. The activation is local and immobile according to our imaging results. Moreover, using the two-pathway protocol and the TrkBF616A mutant mice, in which TrkB can be reversibly inhibited through genetic modification, we show that TrkB is essential for the capture of L-LTP. Finally, using a recently developed behavioral tagging paradigm, we provide evidence that TrkB may serve as a behavioral tag for long-term memory consolidation in vivo. Our findings suggest that TrkB is a potential tag for synapse-specific expression of L-LTP and LTM.
Materials and Methods
TrkBF616A knock-in mice.
All procedures involving animals were approved by the NIH Animal Care and Use Committee. TrkBF616A knock-in mice were generated by replacing the ATP binding pocket of endogenous TrkB with the one in which the phenylalanine (F616) is mutated to alanine (A). The tyrosine kinase activity of TrkBF616A can be selectively inhibited by a membrane-permeable small molecule 1NMPP1 (Calbiochem) (Chen et al., 2005).
Preparation of BDNF-conjugated beads.
Generation of BDNF-beads is based on a noncovalent but very tight binding interaction between streptavidin and biotin. To biotinylate BDNF in mild physiological condition, FluoReporter Biotin-XX protein labeling kit was used (Invitrogen). Briefly, 50 μg of BDNF was diluted with 200 μl of distilled water and 20 μl of 1 m sodium bicarbonate (0.84 g of NaHCO3 in 10 ml of distilled water; pH 8.4) was added. Two microliters of reactive form of Biotin-XX and sulfosuccinimidyl ester sodium salt were added into a reaction tube that contained BDNF in bicarbonate solution and incubated for 2 h at room temperature. Biotinylated BDNF was collected and purified by a spin column. Approximately 80–90% of biotinylated BDNF was recovered after column purification. The biological activity of BDNF-biotin was accessed by calcium imaging using Fura-2 orange. To quantify biotinylated BDNF, we measured the protein concentration of biotin-labeled BDNF by analyzing the absorbance at 280 nm using a NanoDrop spectrophotometer (Nanodrop Technology), because biotin does not absorb at 280 nm. Then, the number of biotin labels on a protein was determined by using the biotin quantification assay kit (Invitrogen). To make BDNF-beads, 100 μl of streptavidin beads (1.25% w/v; Bangs Laboratories) with binding capacity of 7.5 μmol of biotinylated conjugates per 1 μl of bead were rinsed three times with PBS, centrifuged, and resuspended in 100 μl of 0.1 m PBS, pH 7.4. To this, 30 μg of freshly made BDNF-biotin was added and allowed to mix on a rotary shaker overnight (12–14 h) at 4°C. BDNF-beads were centrifuged and rinsed three times to remove any unbound BDNF-biotin. The pelleted BDNF-beads were resuspended in 100 μl of 0.1 m PBS and stored at 4°C until use. The diameter of a single bead is ∼40 nm.
pTrkB staining and quantification.
Cultured hippocampal neurons of 14 d in vitro (Ji et al., 2005) were treated with 50 ng/ml BDNF, which was conjugated on beads, for 30 min with or without k252a applied 1 h before the BDNF bead incubation. The cells were carefully fixed without any disturbance using 4% PFA for 20 min at room temperature and permeabilized for 10 min with 0.1% Triton X-100 at room temperature. Samples were washed three times with PBS, pH 7.4, and incubated with corresponding primary antibodies for TrkB [TrkB (794): sc-12; Santa Cruz Biotechnology] and pTrkB (Arévalo et al., 2006; Spencer-Segal et al., 2011) using 1:2000 dilutions at 4°C overnight. Samples were washed and probed with rhodamine-conjugated secondary antibody and incubated for 1 h at room temperature. Finally the cells were washed again for three times with PBS and mounted using Mowiol premixed with antifade reagents. Slides were imaged using a 63× objective in an inverted Zeiss confocal laser scanning microscope (LSM-510-Meta series). Images were processed and analyzed using LSM Meta analyzer and ImageJ (NIH).
For pTrkB staining in hippocampal slices, CA1 areas were stimulated by stimulating Schaffer collaterals with 12TBS (each with 4 pulses at 100 Hz and an interburst interval of 200 ms) after slices recovery. Slices were incubated in ACSF for another 30 min before 4% PFA fixation. Slices were dehydrated in 30% sucrose overnight after 2 h fixation in 4% PFA. Slices were resection to 40 μm in optimal cutting temperature compound (OCT). Slices were incubated in blocking buffer (3% donkey serum + 0.3% Triton X-100 in PBS) at room temperature for 1 h, and incubated with rabbit anti-pTrkB816 antibody (1:500) (Arévalo et al., 2006) at 4°C overnight. The secondary antibodies used were Alexa Flour 488 donkey anti-rabbit IgG (1:500, Invitrogen). Slices were incubated with DAPI (Sigma) for 3–5 min before mounted. Images were taken by Nikon A1R Confocal microscope (4× or 63×; Nikon Instruments) and analyzed by Image-Pro Plus 6.3 (Media Cybernetics).
Preparation of acute hippocampal slices.
Coronal hippocampal slices (400 μm) were prepared from 8- to 10-week-old male mice, in accordance with NIH guidelines. Briefly, hippocampal slices were cut using a vibrating microtome (NVSLM1, WPI) in ice-cold slicing buffer (in mm: 127 NaCl, 26 NaHCO3, 1.2 KH2PO4, 1.9 KCl, 1.1 CaCl2, 2 MgSO4, 10 d-glucose) bubbled with 95% O2 and 5% CO2. Slices were then transferred to a holding chamber containing oxygenated artificial CSF (ACSF; in mm: 127 NaCl, 26 NaHCO3, 1.2 KH2PO4, 1.9 KCl, 2.2 CaCl2, 1 MgSO4, 10 d-glucose) for 30 min at 34°C and for another 30 min at 22°C for recovery, and then transferred to a submersion recording chamber continually perfused with 32°C oxygenated ACSF (rate: 2 ml/min). Slices were equilibrated in the recording chamber for at least 15 min before recording. Stable long recordings were obtained in slices with such preparation (see Fig. 4E).
Figure 4.

Critical role of TrkB activation within the first 40 min of L-LTP, but not in STP. A, 1NMPP1 blocked 12TBS-induced TrkB activation in TrkBF616A mice. The CA1 regions of hippocampal slices from TrkBF616A mice were microdissected 1 h after 12TBS. For each experiment, three mice were used, and for each condition, four CA1 slices were used. The same experiment was repeated five times (n = 5), each with new batch of animals. 1NMPP1 (5 μm), but not vehicle control (DMSO), blocked the 12TBS-induced increase of pTrkB in TrkBF616A mice (*p < 0.003; one-way ANOVA). B, Blockade of 12TBS induced L-LTP by inhibiting TrkB activation with 1NMPP1 in adult hippocampal slices derived from TrkBF616A mice. 1NMPP1 (5 μm), applied to slices throughout the experiment, completely blocked L-LTP (blue diamonds), while vehicle control had no effect (black circles). 1NMPP1 did not affect L-LTP in the control mice (C57BL/6) (pink diamonds). C, 1NMPP1 applied immediately after 12TBS also blocked L-LTP in TrkBF616A slices. D, Inhibition of L-LTP by 1NMPP1 applied between 0 and 40 min after 12TBS (red) but not 45 min after 12TBS in TrkBF616A slices (black). E, Stable baseline recording were obtained for >4 h without any treatment. F, Indistinguishable STPs in 1NMPP1- and vehicle-treated TrkBF616A slices. STPs were induced by 2TBS. In both cases, fEPSPs returned to baseline at 60 min after 2TBS.
Electrophysiology.
ACSF-filled glass electrodes (resistance <1 MΩ) were positioned in the stratum radiatum of area CA1 for extracellular recording. Synaptic responses were evoked by stimulating Schaffer collaterals with 0.2 ms pulses once every 15 s. For two-pathway recordings, the stimulating electrodes were placed on either side of the recording electrode in stratum radiatum. The independence of the two pathways was tested with a paired-pulse facilitation protocol (see Fig. 5B). The stimulation intensity was systematically increased to determine the maximal fEPSP slope and then adjusted to yield 40–60% of the maximal (fEPSP) slope. Experiments with maximal fEPSPs of <0.5 mV, with large fiber volleys, or with substantial changes in the fiber volley during recording were rejected.
Field EPSPs were recorded (AxoClamp 2B amplifier, Molecular Devices), filtered at 1 kHz, digitized at 10 kHz (Axon Digidata 1322A), and stored for off-line analysis (Clampfit 9). Initial slopes of fEPSPs were expressed as percentages of baseline averages. In summary graphs, each point represents the average of 4 consecutive responses. The time-matched, normalized data were averaged across experiments and expressed as means ± SEM, and the number of tested slices is indicated in each figure.
Western blotting for total TrkB and phosphorylated TrkB.
Slices were taken out of the recording chamber at different time points after stimulation. CA1 area was quickly dissected on ice and stored on dry ice. Four pieces of CA1 for each condition were lysed in an ice-cold RIPA lysis buffer. The lysates were mixed with sample buffer for SDS-PAGE. Samples were boiled for 5 min, subjected to SDS-PAGE, and immunoblotted using rabbit anti-Phospho-TrkA (Tyr490) (1:1000, Neuromics) and rabbit anti-TrkB [1:1000; TrkB (794): sc-12; Santa Cruz Biotechnology]. Densitometric analysis was conducted using Molecular Analysis software of Bio-Rad. The same experiments (with multiple time points) were repeated at least six times (n = 6).
Behavioral tasks.
We used a mouse behavioral tagging paradigm according to the one previously established in rat by Moncada and Viola (2007). Male mice (8–12 weeks) were tested blindly for intraperitoneal administration of vehicle (DMSO) or 1NMPP1 (1 mm, 400 μl each mouse). Mice were habituated in the test room for at least 30 min before experiment. Inhibitory avoidance (IA) tasks were performed in a computer-controlled Gemini Avoidance System (San Diego Instruments) that contains two equal chambers divided by a guillotine door. During tasks, the right chamber remained illuminated while the left chamber remained dark. Training began by placing mouse into illuminated chamber facing away from the shut guillotine door. Mouse was allowed to explore the illuminated chamber for 2 min. The door was then opened to let the mouse explore both illuminated and dark chamber for 5 min. At the end of this exploration period, door was shut after the mouse returned into the illuminated chamber. Two minutes later, the door was opened. Latency to step into dark chamber was recorded by computer. Upon entering the dark chamber, the door was shut and 2 weak footshocks (0.1 mA, 100 ms, 1 s interval) were delivered. Ten seconds later, the mouse was removed form the dark chamber and put back to the home cage. At 60 min or 24 h after training, memory was assessed by placing mouse in the illuminated chamber facing away from the shut guillotine door. After 2 min, door was opened and latency to step into dark chamber was recorded as the measurement of memory.
Behavioral tagging experiments were performed by exposing mice to open field arena (OF) before IA training. The OF is a plastic box with the dimension of 35 cm (width) × 35 cm (length) × 35 cm (height). Four objects were randomly placed in the arena for a mouse to explore. After 15 min exploration, mouse was removed from the box and placed back to the home cage for 1 h before subjected to IA training.
Hot plate tests were performed with Hotplate Analgesia Meter (Columbus Instrument). Temperature was set at 55°C. The latencies to lick forepaw and hindpaw were hand recorded as measurement of foot pain sensation.
Elevated zero maze tests were performed using the ring-shaped platform consisting of two walled (white Plexiglas) sections separated by open sections of equal length. Each mouse was placed such that it was in an open section but directly facing a walled section. Activity was recorded with a video camera for 5 min and the time spent in open sections was analyzed using Clever Systems TopScan (Clever Sys).
Results
Transient, local, and protein synthesis-independent activation of TrkB
As the receptor, TrkB naturally fits the fifth criterion for a synaptic tag as highlighted above. To investigate whether TrkB fits other criteria, we examined TrkB activation (phosphorylation) levels after strong or weak stimulation. According to previous findings, BDNF is essential for L-LTP induced by TBS (Patterson et al., 2001; Pang et al., 2004). The Schaffer collateral region of CA1 was stimulated with 12TBS or 4TBS (12 or 4 bursts, each of 4 pulses at 100 Hz). At different time points after stimulation, CA1 regions were quickly dissected out on ice and frozen in dry ice. Collected tissues were subjected to Western blotting for detection of phosphorylated TrkB (pTrkB), and TrkB phosphorylation was expressed as the ratio of pTrkB to total TrkB levels. For each experiment, pTrkB/TrkB ratios were normalized against the control condition in which slices were not stimulated. TrkB phosphorylation was significantly increased following strong TBS (12TBS) as well as weak TBS (4TBS) (Fig. 1A). The pTrkB level quickly increased, peaked around 30 min, and gradually declined to baseline levels ∼2 h after TBS stimulation (Fig. 1B). This transient activation of TrkB was not affected by protein synthesis inhibition (40 μm anisomycin) (Fonseca et al., 2006), regardless of whether weak or strong TBS was delivered (Fig. 1B,C). In addition, the extent of TrkB phosphorylation is not dependent on the strength of stimulation (12TBS vs 4TBS) (Fig. 1A). Thus, neurons may not use TrkB phosphorylation as the way to discern the strength of stimulation at synapses, but whether the synapse has been stimulated. Instead, the strength of stimulation is more likely to be discerned with the amount of Ca2+ influx (Malenka and Siegelbaum, 2001; Dan and Poo, 2006). Thus, TrkB appears to satisfy the first three criteria for a synaptic tag as previously described in the introduction.
Figure 1.

Transient and protein synthesis independent activation of TrkB by TBS. CA1 areas were stimulated by stimulating Schaffer collaterals with 12TBS or 4TBS, dissected out quickly at different time points after TBS, and processed for Western blotting. For each individual time point, the ratio of pTrkB signal to total TrkB signal was normalized against the control condition from the same experiment. Three mice were used for each experiment with multiple time points, and each time point contains CA1 from four hippocampal slices. The same experiment was repeated at least six times (n = 6) in A and B, each with new batch of animals. A, Examples of TrkB activation in hippocampal slices induced by 12TBS and 4TBS. No obvious difference was found in the pattern of TrkB activation. B, Quantification of time courses of TrkB activation induced by 4TBS with and without the protein synthesis inhibitor anisomycin (40 μm). The pTrkB time course induced by 12TBS is included for comparison. C, Protein synthesis independence of TrkB activation measured 1 h after 12TBS. Both example (top) and quantification (bottom, n = 4) are shown. Inhibition of protein synthesis by anisomycin (40 μm) does not block the TBS-induced increase in pTrkB (*p < 0.05, one-way ANOVA; no difference was found between “12TBS” and “12TBS+anisomycin” groups).
To examine the fourth criterion, whether TrkB phosphorylation is restricted to the site of BDNF release and physically immobile, we first used hippocampal cultures. To stimulate individual spots of the dendrite rather than homogeneously the whole dendrite with exogenously applied BDNF, we conjugate BDNF with small beads (40 nm). BDNF-conjugated fluorescent beads (green) were applied to cultured hippocampal neurons, which express TrkB throughout their dendrites (red, Fig. 2E). Thirty minutes after BDNF-bead application, cells were fixed for pTrkB staining (Bath et al., 2008). As shown in Figure 2A, some BDNF-beads were bound to neuronal dendrites, while others were not. BDNF-beads specifically induced pTrkB staining without affecting the total TrkB signals (Fig. 2G–I). Remarkably, TrkB phosphorylation (red spots) appeared to be confined to the vicinity of BDNF-beads in touch with dendritic membranes, and pTrkB staining was the strongest in dendritic spots juxtaposed with BDNF beads (Fig. 2A). In dendrites not in contact with the BDNF-beads, there was very low level of pTrkB signal (Fig. 2A). Since cases existed where multiple beads were clustered, we only quantified instances in which beads were not clustered. Quantitative analyses of fluorescence spots showed that pTrkB signals decreased dramatically in dendritic regions 5–10 μm away from the point of contact with BDNF-beads, and 20 μm beyond BDNF-bead contact pTrkB staining diminished to near baseline levels (Fig. 2F). The pTrkB signals were extremely low in cases where BDNF-beads were in close proximity, but not actually touching the dendrites (“BDNF w/o touch” in Fig. 2F). TrkB phosphorylation was almost not detectable on dendritic branches that were free of BDNF-beads, when a separate branch of the same neuron was touched by BDNF-beads (Fig. 2F).
Figure 2.

Local activation of TrkB by BDNF. Hippocampal neurons were treated with BDNF-beads (green) for 30 min, and stained with an antibody against phosphorylated TrkB (pTrkB, red). Local pTrkB immunoreactivity is dramatically increased in dendrites touched by BDNF-beads (arrow). A, Preferential activation of TrkB on dendrites in contact with BDNF-beads. The area highlighted by the blue box is enlarged and shown in the lower right. Note the strong pTrkB signals (red) associated with the BDNF-beads (green), indicated by arrows. B, Blockade of BDNF-beads induced TrkB activation by k252a. Neurons treated with k252a and BDNF-beads (green) show little pTrkB immunoreactivity even when dendrites were touched by BDNF-beads (arrow). C, Touching by beads alone does not activate TrkB. Neurons were treated with uncoupled beads (green) without BDNF. Little pTrkB immunoreactivity was detected along the dendrites even when dendrites were touched by beads (arrow). D, Low levels of basal TrkB activation in cultured hippocampal neurons. Little pTrkB immunoreactivity was detected in dendrites in absence of BDNF beads (asterisk). E, Abundant TrkB immunoreactivity (red) along the dendrites of cultured hippocampal neurons. Arrows show the place where BDNF-bead touched dendrite. F, Quantification of pTrkB immunoreactivity under different conditions. The intensity of an individual pTrkB immunofluorescent spot along the dendrite was quantified by ImageJ. The intensities of all spots from the point touched by BDNF beads (0–5 μm, 5–10 μm, 20–50 μm) were summed and presented as arbitrary units (AU). pTrkB signal gradually decreases along the dendrites from the point of BDNF-bead contact. Note that there is little pTrkB signal on the dendritic branch not contacted by BDNF-bead, despite the fact that other branches are contacted by BDNF-beads. The measured regions of interest are indicated as “n” for each condition. ***p < 0.001; Newman–Keuls analysis following one-way ANOVA. G–I, Confocal images of total TrkB immunoreactivity (red) along the dendrites of cultured hippocampal neurons treated with (H) or without (G) BDNF-conjugated beads, and the quantifications (I). Note that total TrkB level with BDNF-bead treatment was comparable to control without BDNF-conjugated beads.
A series of control experiments were performed to show that the pTrkB staining is specific and BDNF dependent: (1) the baseline pTrkB signals in the absence of BDNF were very low (Fig. 2D); (2) the BDNF-bead-induced TrkB phosphorylation was completely blocked by the trk kinase inhibitor k252a (Fig. 2B) (200 nm k252a alone had no effect on pTrkB, data not shown); and (3) beads not conjugated with BDNF had no effect on TrkB phosphorylation (Fig. 2C).
We next examined whether TBS stimulation to hippocampal slices, which presumably results in local BDNF secretion, could induce TrkB phosphorylation locally. A train of high-frequency stimulation (12TBS) was applied to Shaffer collaterals in the distal part of the CA1 pyramidal neurons of adult hippocampal slices. Thirty minutes later, the slices were fixed, sectioned, and subject to pTrkB immunohistochemistry staining. Careful confocal microscopy was performed in the areas proximal (yellow box) or distal (red box) to the CA1 neurons (Fig. 3A). We found that 12TBS elicited significant increase in pTrkB immunoreactivity in distal region (Fig. 3D,E) but not proximal region (Fig. 3B,C) of CA1 area. Quantitative analysis of the pTrkB-positive fluorescent spots by Image Pro Plus revealed a 2.3-fold increase in the number of positive puncta only in the area close to the TBS stimulated (distal) neurites (Fig. 3F,G). It is conceivable, therefore, that TBS induces local BDNF secretion, which in turn activates TrkB located within 5–10 μm of dendrites, creating a tag at or near the stimulated site (Lu et al., 2008).
Figure 3.
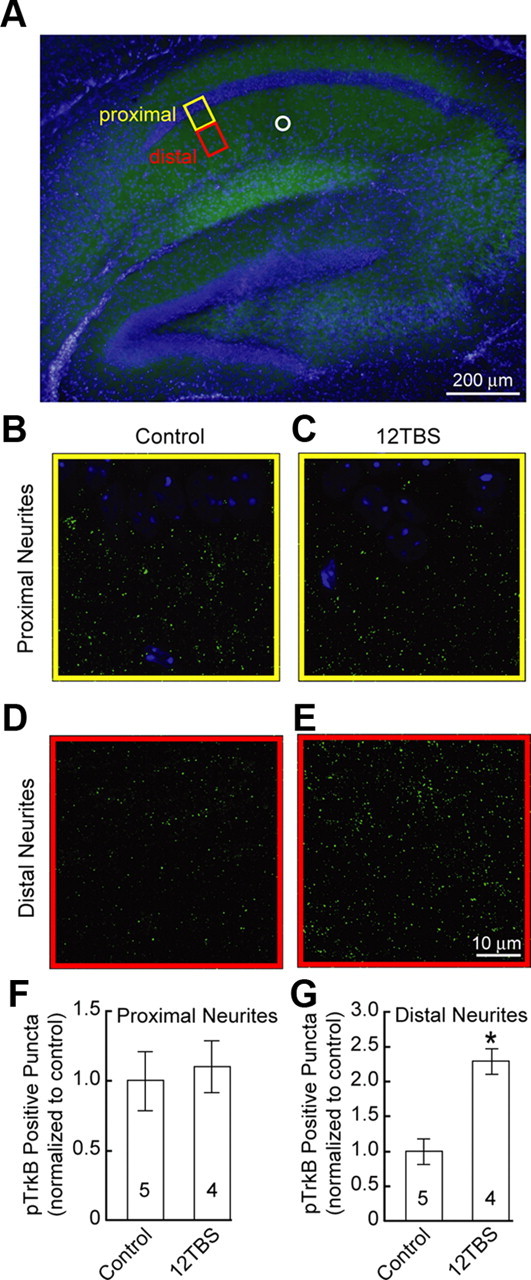
TrkB activation induced by 12TBS in CA1 dendritic area of hippocampal slice. A, Double staining of pTrkBY816 (green) and DAPI (blue) of hippocampus slice. The proximal and distal areas of hippocampal CA1 neurons are highlighted by the yellow and red boxes, respectively. The position of the stimulating electrode is indicated by the white circle. B, C, Double staining of pTrkBY816 (green) and DAPI (blue) of proximal region from cell body with or without 12TBS. Note the CA1 neuronal cell body stained by DAPI located in the upper part of the images. D, E, Double staining of pTrkBY816 and DAPI of distal region from cell body with or without 12TBS. Note the marked increase in pTrkB staining after 12TBS. F, G, Quantification of the number of pTrkB-positive puncta in the proximal and distal regions from cell body of CA1 neurons, respectively (*p < 0.001, t test).
TrkB as synaptic tag for L-LTP
To determine the time window in which TrkB phosphorylation is required in L-LTP, we took advantages of the TrkBF616A mice, in which a single phenylalanine (F616) located in the ATP binding pocket of TrkB subdomain V was mutated to alanine (A). This mutation provided a system in which TrkB phosphorylation can be specifically and reversibly inhibited by the compound 1NMPP1 (Fig. 4A) (Chen et al., 2005). The TrkBF616A slices exhibited a very stable baseline over >4 h (Fig. 4E). As expected, L-LTP at CA1 synapses was completely blocked in TrkBF616A slices pretreated with 1NMPP1 (5 μm) (Fig. 4B) (vehicle, 153 ± 11%; 1NMPP1, 98 ± 6%, p < 0.0002, t test). Application of 1NMPP1 to wild-type (C57BL/6) slices had no effect on L-LTP (Fig. 4B) (158 ± 8%), suggesting that 1NMPP1 acts via TrkBF616A, rather than nonspecifically, to inhibit L-LTP. Since a tag is thought to be created transiently by the stimulation to capture PRPs (Martin and Kosik, 2002; Kelleher et al., 2004; Barco et al., 2008; Lu et al., 2008), inhibition of the tagging within a short time window after stimulation should block L-LTP. Indeed, application of 1NMPP1 right after 12TBS also blocked L-LTP (Fig. 4C) (101 ± 6%). Further, application of 1NMPP1 within 40 min following 12TBS was able to block L-LTP (Fig. 4D) (110 ± 5%), while L-LTP was preserved when 1NMPP1 was applied after 45 min (152 ± 5%). Thus, TrkB phosphorylation is essential for L-LTP within a short time window (∼40 min) after TBS stimulation.
Previous studies have shown a “housekeeping plasticity” role of tyrosine kinases in the induction of E-LTP (O'Dell et al., 1991). TrkB signaling has also shown to be required for the induction of E-LTP (Minichiello et al., 1999). To determine whether TrkB also has a “housekeeping plasticity role,” we tested short-term plasticity (STP) in TrkBF616A slices using stimulation protocol (2TBS) that is unable to induce E-LTP. We found that 2TBS induced a rapid increase in the synaptic efficacy which returned to baseline within ∼30 min. Inhibition of TrkB with 1NMPP1 did not affect at all the magnitude or during of STP (Fig. 4F). Thus, TrkB does not seem to play a “housekeeping plasticity role” at the Shaffer collateral–CA1 synapses.
To examine whether TrkB truly serves as a synaptic tag during L-LTP, we performed the two-pathway experiment (Frey and Morris, 1997) using the TrkBF616A slices. Field EPSPs were monitored by a single recording electrode in the stratum radiatum of CA1. S1 was stimulated by strong TBS (12TBS), while S2 was stimulated by weak TBS (4TBS) (Fig. 5A). Stimulation of one pathway (S2) consistently elicited fEPSPs of the same size (Fig. 5B, top; the red and green fEPSP traces were superimposed), regardless of whether the other pathway (S1) was stimulated 30 ms earlier. The converse was true too: S1-induced fEPSPs were the same regardless of S2 stimulation (Fig. 5B, bottom). The lack of facilitation between the two pathways suggests the independence of the two inputs (S1 and S2). Weak stimulation (4TBS) typically does not induce L-LTP (Fig. 5C) (106 ± 2%). However, when one afferent pathway (e.g., S1) was activated by strong stimulation (12TBS), weak stimulation (4TBS) applied to a second independent pathway (e.g., S2) usually induced L-LTP(Frey and Morris, 1998a). This was also true in either TrkBF616A slices treated with vehicle (Fig. 5D) (S1: 174 ± 14%; S2: 139 ± 6%) or wild-type slices treated with 1NMPP1 (Fig. 5F) (S1: 166 ± 10%; S2: 138 ± 5%). However, application of 1NMPP1 to slices from TrkBF616 mice 40–80 min after 12TBS in S1 blocked the capture of L-LTP in S2 (Fig. 5E) (S2: 98 ± 4%). Synaptic potentiation decayed to baseline in <2 h after 4TBS (Fig. 5E). On the other hand, 1NMPP1 had no effect on L-LTP of S1 in the same TrkBF616 slice (Fig. 5E) (S1: 152 ± 8%). L-LTP of S1 was indistinguishable among all three groups (WT treated with 1NMPP1, TrkBF616A treated with 1NMPP1, and TrkBF616A treated with vehicle), while potentiation of S2 was significantly decreased in TrkBF616A slices treated with 1NMPP1 compared to the other two groups (p < 0.001, one-way ANOVA followed by Newman–Keuls test). These results strongly support the notion that a weak TBS creates a tag in S2, which can capture the PRP induced by the strong TBS in S1. Blockade of TrkB around the time of 4TBS by 1NMPP1 appears to prevent the synaptic capture of the PRP. However, strong stimulation (e.g., 12TBS) can induce not only tag but also PRP. To examine the possibility that TrkB is essential for tagging, PRP synthesis, or both, TrkB activation was blocked with 1NMPP1 at the time of 12TBS (S1) but not 4TBS (S2). Interestingly, L-LTP was abolished in S1 but intact in S2 (Fig. 5G,H). This strongly suggests that inhibition of TrkB in S1 prevents the formation of the tag and therefore impaired L-LTP in this pathway, but had no effect on PRP production. Thus, application of 4TBS at S2 should still be able to generate a tag at S2 to capture the newly synthesized PRP, leading to L-LTP.
When a strong stimulation precedes weak stimulation (S1 strong–S2 weak), it is possible that the strong stimulation can induce the synthesis of some factors, and that their diffusion may lower the threshold for L-LTP induction (Frey and Morris, 1998a,b; Barco et al., 2008). To exclude this possibility, a second set of two-pathway experiments was performed in which the weak TBS preceded strong TBS (S2 weak–S1 strong). In either TrkBF616A slices treated with vehicle (Fig. 6B) or wild-type slices treated with 1NMPP1 (Fig. 6C), 12TBS to S1 at 40 min after 4TBS converted E-LTP to L-LTP at S2 synapses (4TBS to S2: 133 ± 5% in Fig. 6B and 132 ± 5% in Fig. 6C). In contrast, application of 1NMPP1 to slices from TrkBF616 mice between 20 min before and after 4TBS to S2 blocked the capture of L-LTP in S2 (Fig. 6A) (105 ± 4%). Similarly, L-LTP of S1 was indistinguishable among all three groups (WT treated with 1NMPP1, TrkBF616A treated with 1NMPP1, and TrkBF616A treated with vehicle), while potentiation of S2 was significantly decreased in TrkBF616A slices treated with 1NMPP1 compared to the other two groups (p < 0.01, Newman–Keuls analysis after one-way ANOVA). These weak-before-strong experiments strongly support our hypothesis that TrkB works as synaptic tag in the BDNF-dependent L-LTP.
TrkB as behavioral tag for LTM
To determine whether TrkB also serves as a synaptic tag to mediate formation of long-term memory (LTM) in vivo, we performed experiments for behavioral tagging, a newly developed behavior paradigm thought to be the analog of synaptic tagging (Moncada and Viola, 2007). Briefly, a short-term memory (STM) induced by weak IA training could be consolidated into LTM by exploration of novel environment. We used a single-trial IA task. The apparatus contains two chambers, one illuminated and one dark, separated by a guillotine door. During training session, mouse was initially placed in the illuminated chamber. Once mouse stepped into the dark chamber, two weak footshocks (0.1 mA, 100 ms) were delivered at the interval of 1 s. In the test session 60 min or 24 h later, memory was assessed by measuring the latency to step into dark chamber. We adjusted the footshocks so that the weak training task induced STM but not LTM in both wild-type (C57BL/6) and TrkBF616A (Fig. 7A,C) mice. Such STM was intact when TrkB activation was blocked by administration of 1NMPP1 in TrkBF616A mice (Fig. 8A).
Figure 7.

TrkB as behavioral tag for LTM. Mice were trained in an IA task with two weak footshocks (0.1 mA, 100 ms) at 1 s interval. Latency to step through the door from illuminated chamber to dark chamber was recorded as measurement of memory. Data were plotted as mean ± SEM, and Newman–Keuls analysis was performed after one-way ANOVA. The number of tested mice is indicated in each column. A, C, STM (60 min) and LTM (24 h) were assessed with different groups of wild-type mice or TrkBF616A mice, respectively. The weak IA training only induced STM but not LTM. B, Exploration in novel environment (open filed with four objects, OF) consolidated weak IA induced STM into LTM. 1NMPP1 did not block the consolidation in wild-type mice. D, Novelty-promoted LTM in TrkBF616A mice was blocked by intraperitoneal injection of 1NMPP1.
Figure 8.

Normal short-term memory, pain sensation, emotional state, and locomotion after TrkB inhibition by 1NMPP1. A, Normal short-term memory after TrkB inhibition by 1NMPP1. At 10 min after intraperitoneal injection with 1NMPP1 or vehicle, mice were trained in an IA task with two weak footshocks (0.1 mA, 100 ms) at 1 s interval. Latency to step through the door from illuminated chamber to dark chamber was recorded as measurement of memory and plotted as mean ± SEM. B, Normal pain sensation after TrkB inhibition by 1NMPP1. Hot plate test was performed at 55°C. Latency to lick forepaw and hindpaw was plotted as mean ± SEM. C–E, Normal emotional state and locomotion after TrkB inhibition by 1NMPP1. Elevated zero maze tests were performed using the ring-shaped platform consisting of two walled (white Plexiglas) sections separated by open sections of equal length. Activity of each mouse was recorded for 5 min. Mice injected with 1NMPP1 spent similar amount of time in open arms as compared with those injected with vehicle (C). In addition, 1NMPP1 did not affect the number of times that mice entered the open arms (D) and the total distance that mice traveled in the maze (E).
When mice were introduced into a novel environment (open field, or OF, with 4 objects) for 15 min at 1 h before IA training, STM induced by the weak training task was consolidated into LTM, as evidenced by increased latency at a test session 24 h later (Fig. 7B,D). On the other hand, latencies during training were indistinguishable across all wild-type groups in Figure 7, A and B (p = 0.36, one-way ANOVA). We next tested whether inhibition of the TrkB tag induced by weak training could prevent the novelty-facilitated conversion of STM to LTM. 1NMPP1 was administered to TrkBF616A or wild-type mice 10 min before IA training session (Fig. 7B, top). Remarkably, a single intraperitoneal injection of 1NMPP1 at a concentration known to block TrkB activation (Johnson et al., 2008) dramatically impaired LTM in TrkBF616A mice (Fig. 7D) but not wild-type mice (Fig. 7B). Thus, inhibition of TrkB prevents the novelty-induced conversion of STM to LTM in a standard behavioral tagging paradigm, further supporting the notion that TrkB may serve as a synaptic tag in vivo.
To exclude the possibility that inhibition of TrkB by 1NMPP1 reduces the pain threshold in TrkBF616A mice, mice injected with 1NMPP1 were subjected to hot plate test. Both latencies to lick forepaw and hindpaw were recorded and were indistinguishable between 1NMPP1 group and vehicle group (Fig. 8B). Administration of 1NMPP1 in TrkBF616A mice may have altered their mood status, leading to impairment in memory. To address this issue, elevated zero maze was carried out. Mice injected with 1NMPP1, compared with those injected with vehicle, spent similar amount of time in the open arms (Fig. 8C) and entered open arms for comparable number of times (Fig. 8D). In addition, the total traveling distance in the maze was indistinguishable between mice injected with 1NMPP1 and vehicle (Fig. 8E). Together, our results suggest TrkB is a synaptic tag as well as a behavioral tag for certain forms of LTM.
Discussion
Since its inception, the “synaptic tagging hypothesis” has inspired many to search for synaptic tags. However, very few molecules have been proposed as candidates (Frey and Frey, 2008). The nature and identity of PRPs and synaptic tags are under intensive investigations (Frey and Frey, 2008). Two pathway experiments showed that blockade of protein kinase A (PKA) or its interaction with A kinase-anchoring proteins (AKAPs) prevents synaptic capture, suggesting that PKA or its anchoring at active synapses may serve as a synaptic tag for L-LTP (Huang et al., 2006; Young et al., 2006). Calcium/calmodulin-dependent protein kinase II (CaMKII) is also implicated as an L-LTP-specific tag (Sajikumar et al., 2007). Studies thus far have been based on the use of pharmacological inhibitors in two-pathway experiments. However, it is necessary to show that these tags are transiently and locally activated in a protein synthesis-independent manner by weak stimulation. In a recent study, NMDA-dependent, input-specific “spine entry” of Homer-1a has been suggested as an L-LTP tag (Okada et al., 2009). However, it remains to be established whether Homer-1a is required in L-LTP and LTM. Using a combined genetic, pharmacological and biochemical approach, we now show that TrkB fits four of the five criteria insightfully proposed by Kelleher et al. (2004), for an L-LTP tag. The fifth criterion is a given, since TrkB is a natural receptor that binds BDNF. It should be pointed out that although strongly implicated as PRP in the “two-pathway” experiments (Barco et al., 2005), BDNF may play a more complex role in the tagging process (see below). Most importantly, we show that TrkB is involved in behavioral tagging in vivo.
Although multiple molecules may be involved in the synaptic tagging processes, TrkB as synaptic tagging has several interesting features. First, compared with previously proposed tags, which are mostly cytosolic molecules, TrkB is a relatively immobile membrane protein, and thus more likely to be activated in a local and synapse-specific manner. Indeed, our imaging experiments using hippocampal neuronal cultures and acute slices show that TrkB activation is confined to dendritic spots near the site of local BDNF secretion induced by high-frequency synaptic activity. BDNF is a positively charged molecule that tends to be associated with cell surface membrane after its secretion, with very limited diffusion (Nagappan et al., 2009). It is therefore conceivable that the presynaptic secretion of BDNF into synaptic cleft after stimulation could activate the TrkB tag within the vicinity of the stimulated synapses. Second, activity regulation of TrkB trafficking and signaling may provide links between TrkB and other postulated tags and PRPs in L-LTP. For example, increases in synaptic activity, Ca2+ influx through Ca2+ channels and NMDA receptors, and activation of CaMKII have been shown to facilitate membrane insertion of TrkB in hippocampal neurons (Du et al., 2000). Synaptic activity, through local secretion of BDNF and the rise of intracellular cAMP, also enhances the recruitment of TrkB into PSD95-containing dendritic spines (Suzuki et al., 2004; Ji et al., 2005; Yoshii and Constantine-Paton, 2007). Moreover, TrkB phosphorylation is markedly attenuated when the cAMP–PKA pathway is blocked, suggesting that cAMP–PKA gates TrkB signaling (Ji et al., 2005). These results suggest that CaMKII and cAMP–PKA may act upstream to facilitate the TrkB tag. In contrast, BDNF–TrkB signaling has been shown to enhance Homer-1a expression in the hippocampus, placing it downstream of TrkB (Sato et al., 2001; Rosenblum et al., 2002; Schratt et al., 2004). In addition, different forms of synaptic plasticity (e.g., LTP and LTD) at different neuronal compartments may recruit different molecules for synaptic tagging (Sajikumar et al., 2007). Since our study has been focused on the role of BDNF–TrkB during synaptic tagging of TBS-induced LTP in apical dendrites of hippocampal CA1 region, it will be interesting to determine whether they are also involved in the tagging process of other regions or other plasticity forms.
Together, the present study supports a model for BDNF/TrkB's involvement in the synaptic tagging process at Schaffer collateral–CA1 synapses: synaptic stimulation, which presumably triggers the secretion of BDNF from the existing BDNF-containing vesicles either presynaptically or postsynaptically, activates postsynaptic TrkB. In this sense, both pre-existing BDNF and TrkB should be considered as part of the initial tag. Moreover, the local synaptic activity/cAMP–PKA/BDNF recruits more TrkB to the dendritic spines (Ji et al., 2005), as well as facilitates TrkB signaling (Du et al., 2000, 2003; Suzuki et al., 2004). Thus, this initial synaptic stimulation–TrkB activation can lead to further accumulation of TrkB available transiently (<2 h) at the stimulated synapses (Nagappan and Lu, 2005). In parallel, the strong stimulation initiates a later wave of de novo synthesis of PRP (BDNF). This postsynaptically derived BDNF is transported to the apical dendrites, secreted, and “captured” by TrkB accumulated at the stimulated synapses. Thus, it might be helpful to imagine two waves of BDNF induced by synaptic stimulation. The first is activity-dependent secretion of BDNF, which contributes to the creation (accumulation) of the tag (TrkB) locally. The synaptic stimulation also triggers BDNF synthesis in the soma or dendrites. The newly synthesized BDNF binds and acts on the accumulated TrkB at the synapses previously stimulated, thus ensuring synapse specificity. This second wave of BDNF does not fit to the conventional view of PRP because the BDNF–TrkB signaling is necessary for only a transient period of ∼45 min after afferent stimulation (Fig. 6D). The activation of signaling events and effectors downstream of TrkB may be responsible for the maintenance of L-LTP and LTM. The TrkB tag can also be created by weak stimulation at a different synapse to capture newly synthesized BDNF, leading to transformation of E-LTP into L-LTP, if the weak stimulation is within a short period of time before or after the strong stimulation. Our behavioral study, revealing TrkB as a potential behavioral tag, implicates a similar scenario for the consolidation of STM to LTM. Identification of TrkB as potential synaptic tag and behavioral tag provides an opportunity for further investigations into the maintenance phases of LTP and LTM.
Footnotes
This work was supported by the Intramural Research Programs of NIMH and NICHD and by GSK R&D China. We thank Dr. David D. Ginty for providing the TrkBF616A knock-in mice. We also thank Drs. Kimberly M. Christian and KuanHong Wang for critical comments and constructive suggestions for this manuscript, and Shiyi Jin for assistance with the confocal microscopy.
References
- Arévalo JC, Waite J, Rajagopal R, Beyna M, Chen ZY, Lee FS, Chao MV. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron. 2006;50:549–559. doi: 10.1016/j.neuron.2006.03.044. [DOI] [PubMed] [Google Scholar]
- Barco A, Patterson S, Alarcon JM, Gromova P, Mata-Roig M, Morozov A, Kandel ER. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron. 2005;48:123–137. doi: 10.1016/j.neuron.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Barco A, Lopez de Armentia M, Alarcon JM. Synapse-specific stabilization of plasticity processes: the synaptic tagging and capture hypothesis revisited 10 years later. Neurosci Biobehav Rev. 2008;32:831–851. doi: 10.1016/j.neubiorev.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Bath KG, Mandairon N, Jing D, Rajagopal R, Kapoor R, Chen ZY, Khan T, Proenca CC, Kraemer R, Cleland TA, Hempstead BL, Chao MV, Lee FS. Variant brain-derived neurotrophic factor (Val66Met) alters adult olfactory bulb neurogenesis and spontaneous olfactory discrimination. J Neurosci. 2008;28:2383–2393. doi: 10.1523/JNEUROSCI.4387-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- Drake CT, Milner TA, Patterson SL. Ultrastructural localization of full-length trkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci. 1999;19:8009–8026. doi: 10.1523/JNEUROSCI.19-18-08009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, Lu B. Activity- and Ca(2+)-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol. 2000;150:1423–1434. doi: 10.1083/jcb.150.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Feng L, Zaitsev E, Je HS, Liu XW, Lu B. Regulation of TrkB receptor tyrosine kinase and its internalization by neuronal activity and Ca2+ influx. J Cell Biol. 2003;163:385–395. doi: 10.1083/jcb.200305134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nägerl UV. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron. 2006;52:239–245. doi: 10.1016/j.neuron.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Frey S, Frey JU. ‘Synaptic tagging’ and ‘cross-tagging’ and related associative reinforcement processes of functional plasticity as the cellular basis for memory formation. Prog Brain Res. 2008;169:117–143. doi: 10.1016/S0079-6123(07)00007-6. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 1998a;21:181–188. doi: 10.1016/s0166-2236(97)01189-2. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Weak before strong: dissociating synaptic tagging and plasticity-factor accounts of late-LTP. Neuropharmacology. 1998b;37:545–552. doi: 10.1016/s0028-3908(98)00040-9. [DOI] [PubMed] [Google Scholar]
- Govindarajan A, Kelleher RJ, Tonegawa S. A clustered plasticity model of long-term memory engrams. Nat Rev Neurosci. 2006;7:575–583. doi: 10.1038/nrn1937. [DOI] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Huang T, McDonough CB, Abel T. Compartmentalized PKA signaling events are required for synaptic tagging and capture during hippocampal late-phase long-term potentiation. Eur J Cell Biol. 2006;85:635–642. doi: 10.1016/j.ejcb.2006.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Pang PT, Feng L, Lu B. Cyclic AMP controls BDNF-induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat Neurosci. 2005;8:164–172. doi: 10.1038/nn1381. [DOI] [PubMed] [Google Scholar]
- Johnson AW, Chen X, Crombag HS, Zhang C, Smith DR, Shokat KM, Gallagher M, Holland PC, Ginty DD. The brain-derived neurotrophic factor receptor TrkB is critical for the acquisition but not expression of conditioned incentive value. Eur J Neurosci. 2008;28:997–1002. doi: 10.1111/j.1460-9568.2008.06383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2008;89:312–323. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka R, Siegelbaum S. Synaptic plasticity. In: Cowan W, Sudhof T, Stevens C, editors. Synapses. Baltimore: Johns Hopkins UP; 2001. [Google Scholar]
- Martin KC, Kosik KS. Synaptic tagging—who's it? Nat Rev Neurosci. 2002;3:813–820. doi: 10.1038/nrn942. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Moncada D, Viola H. Induction of long-term memory by exposure to novelty requires protein synthesis: evidence for a behavioral tagging. J Neurosci. 2007;27:7476–7481. doi: 10.1523/JNEUROSCI.1083-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG. Elements of a neurobiological theory of hippocampal function: the role of synaptic plasticity, synaptic tagging and schemas. Eur J Neurosci. 2006;23:2829–2846. doi: 10.1111/j.1460-9568.2006.04888.x. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Lu B. Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci. 2005;28:464–471. doi: 10.1016/j.tins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Zaitsev E, Senatorov VV, Jr, Yang J, Hempstead BL, Lu B. Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc Natl Acad Sci U S A. 2009;106:1267–1272. doi: 10.1073/pnas.0807322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Kandel ER, Grant SG. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature. 1991;353:558–560. doi: 10.1038/353558a0. [DOI] [PubMed] [Google Scholar]
- Okada D, Ozawa F, Inokuchi K. Input-specific spine entry of soma-derived Vesl-1S protein conforms to synaptic tagging. Science. 2009;324:904–909. doi: 10.1126/science.1171498. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- Rosenblum K, Futter M, Voss K, Erent M, Skehel PA, French P, Obosi L, Jones MW, Bliss TV. The role of extracellular regulated kinases I/II in late-phase long-term potentiation. J Neurosci. 2002;22:5432–5441. doi: 10.1523/JNEUROSCI.22-13-05432.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajikumar S, Navakkode S, Frey JU. Identification of compartment- and process-specific molecules required for “synaptic tagging” during long-term potentiation and long-term depression in hippocampal CA1. J Neurosci. 2007;27:5068–5080. doi: 10.1523/JNEUROSCI.4940-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suzuki K, Nakanishi S. NMDA receptor stimulation and brain-derived neurotrophic factor upregulate homer 1a mRNA via the mitogen-activated protein kinase cascade in cultured cerebellar granule cells. J Neurosci. 2001;21:3797–3805. doi: 10.1523/JNEUROSCI.21-11-03797.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J Neurosci. 2004;24:7366–7377. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Segal JL, Waters EM, Bath KG, Chao MV, McEwen BS, Milner TA. Distribution of phosphorylated TrkB receptor in the mouse hippocampal formation depends on sex and estrous cycle stage. J Neurosci. 2011;31:6780–6790. doi: 10.1523/JNEUROSCI.0910-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Numakawa T, Shimazu K, Koshimizu H, Hara T, Hatanaka H, Mei L, Lu B, Kojima M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J Cell Biol. 2004;167:1205–1215. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M. BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat Neurosci. 2007;10:702–711. doi: 10.1038/nn1903. [DOI] [PubMed] [Google Scholar]
- Young JZ, Isiegas C, Abel T, Nguyen PV. Metaplasticity of the late-phase of long-term potentiation: a critical role for protein kinase A in synaptic tagging. Eur J Neurosci. 2006;23:1784–1794. doi: 10.1111/j.1460-9568.2006.04707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]