

Abstract
To study the late β-cell-specific function of the homeodomain protein IPF1/PDX1 we have generated mice in which the Ipf1/Pdx1 gene has been disrupted specifically in β cells. These mice develop diabetes with age, and we show that IPF1/PDX1 is required for maintaining the β cell identity by positively regulating insulin and islet amyloid polypeptide expression and by repressing glucagon expression. We also provide evidence that IPF1/PDX1 regulates the expression of Glut2 in a dosage-dependent manner suggesting that lowered IPF1/PDX1 activity may contribute to the development of type II diabetes by causing impaired expression of both Glut2 and insulin.
Keywords: Ipf1/Pdx1, β-cell-specific mutants, hormone production, glucose-sensing, diabetes
At present, very little is known about the sources and the nature of the inductive molecules that control the development of the pancreas and the generation of individual differentiated cell types. In contrast, genes encoding a number of different transcription factors that exert important functions at different levels during pancreas development have been isolated (Jonsson et al. 1994; Offield et al. 1996; Ahlgren et al. 1997; Sosa-Pineda et al. 1997; St-Onge et al. 1997 and references therein). The endocrine cells, which control multiple homeostatic functions, are organized into islets, and the predominant islet cell type, the β cell, responds to glucose by synthesizing and secreting insulin, thus exerting a feedback control of blood glucose levels. The intrinsic regulatory molecules that establish and maintain hormone production and the glucose-sensing system in β cells are, however, still largely unknown.
The homeodomain factor IPF1/PDX1 is initially expressed in the early mouse pancreatic anlagen (Leonard et al. 1993; Ohlsson et al. 1993; Miller et al. 1994; Guz et al. 1995; Ahlgren et al. 1996), but is later restricted to differentiating β cells (Ohlsson et al. 1993). IPF1/PDX1 has been proposed to regulate the expression of a variety of different pancreatic endocrine genes including insulin, somatostatin, glucokinase, islet amyloid polypepride (IAPP), and glucose transporter type 2 (Glut2) (Leonard et al. 1993; Ohlsson et al. 1993; Serup et al. 1995; Waeber et al. 1996; Watada et al. 1996a,b). Mice lacking IPF1/PDX1 fail to form a pancreas, precluding an analysis of later contribution of IPF1/PDX1 to β-cell development and function (Jonsson et al. 1994; Ahlgren et al. 1996; Offield et al. 1996). To address the role of IPF1/PDX1 in adult differentiated β cells we have generated β-cell-specific Ipf1/Pdx1 mutant mice using the CRE–Lox system (Lakso et al. 1992; Gu et al. 1993; Kilby et al. 1993; Tsien et al. 1997). In this paper we show that expression of IPF1/PDX1 is critical for maintaining the hormone-producing phenotype and the homeostatic regulation of the glucose-sensing system in β cells.
Results and Discussion
Homozygous Ipf1/Pdx1–loxP mice carrying the loxP sites flanking exon 2 (Fig. 1A) are indistinguishable from +/+ mice in all respects. By crossing these mice with mice expressing the Cre recombinase under the control of the Rat insulin1-promoter (Rip1) (Dahl et al. 1996), we generated mice that allow CRE-mediated excision of the loxP-flanked exon 2 in insulin-expressing β cells (Fig. 1A). The Rip1/Ipf1▴ mutant mice exhibit an apparently normal pancreas that contains islets (Figs. 1 and 3, below; data not shown), and initially such mice were healthy. However, at ∼3–5 months of age mutant mice (n = 10) exhibited markedly elevated urine (>55 mm, n = 10) and blood glucose levels (Table 1) and thus were overt diabetic (OD).
Figure 1.
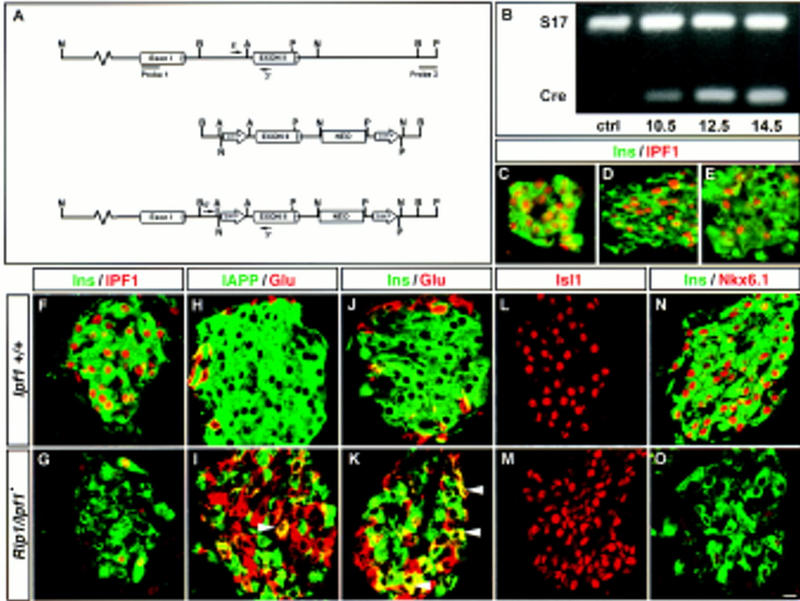
Cre–loxP-mediated targeting of the Ipf1/Pdx1 gene. (A) Schematic representation of targeting construct, genomic DNA, and the expected product of Cre/loxP recombination. The PCR primers used for genotyping and the probes used for confirmation of correct recombination events are shown. (B) RT–PCR analysis using Cre- and rpS17 control primers on total RNA prepared from the alimentary tract of E10.5, E12.5, and E14.5 Rip1/Ipf1▴ and E14.5 Ipf1+/+ (ctrl) embryos. Pancreatic sections from neonatal (C), 3 (D)-, and 5 (E)-week-old prediabetic Rip1/Ipf1▴ mice stained with anti-Ins and anti-IPF1 antibodies. (F–O) β-Cell-specific loss of Ipf1/Pdx1 affects the β-cell phenotype. Confocal images showing Ipf1+/+ (F,H,J,L,N) and diabetic Rip1/Ipf1▴ (G,I,K,M,O) islets from 18-week-old mice. See text for details. (C–O) Bar, 10 μm.
Figure 3.
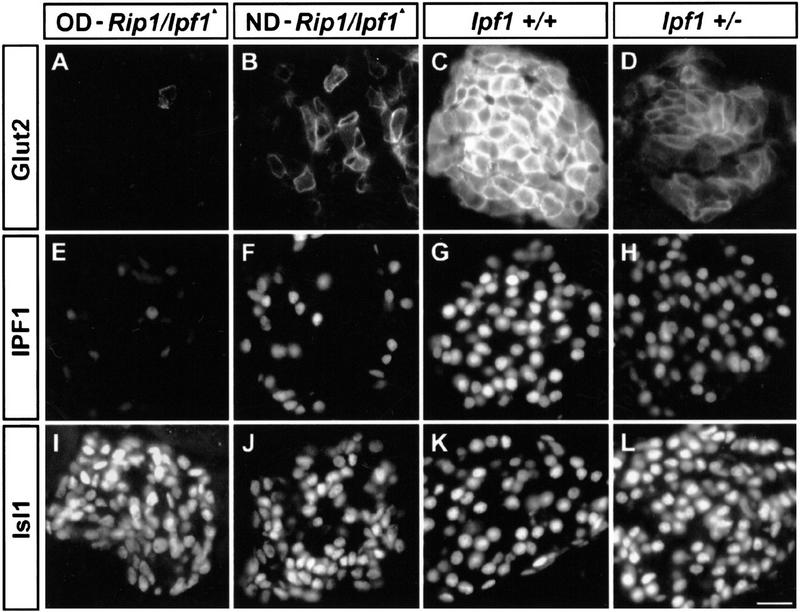
Glut2 expression is dependent on high levels of IPF1/PDX1 expression. Analysis of Glut2 expression in correlation to IPF1 expression by immunohistochemistry on pancreas from 18-week diabetic Rip1/Ipf1▴ (A,E,I), 11-week ND Rip1/Ipf1▴ (B,F,J), 18-week Ipf1+/+ (C,G,K), and 18-week Ipf1+/− mice (D,H,L) (Jonsson et al. 1994). Images are representative of at least 10 different islets. Bar, 20 μm.
Table 1.
Pancreatic insulin content and blood glucose levels
|
|
Blood glucose levelsa | Pancreatic insulin contentb
|
||||
|---|---|---|---|---|---|---|
| nonfasted
|
fasted
|
|||||
| Ipf1+/+ | 5.9 ± 0.5 | (n = 8) | 3.9 ± 0.1 | (n = 8) | 10.0 ± 0.6 | (n = 8) |
| Ipf1+/− | 7.2 ± 0.7 | (n = 8) | 5.0 ± 0.5 | (n = 8) | 9.9 ± 0.6 | (n = 8) |
| ND Rip1/Ipf1▴ | 8.1 ± 1.2 | (n = 3) | 3.9 ± 0.6 | (n = 3) | 3.1 ± 0.5 | (n = 4) |
| OD Rip1/Ipf1▴ | >27.8 | (n = 4) |
11.4 ± 1.7 | (n = 8) | 1.1 ± 0.2 | (n = 4) |
Results are mean ± s.e.m. of (n) animals in each group. ND Rip1/Ipf1▴ mice were 8–12 weeks of age, Ipf1+/+, Ipf1+/− and OD Rip1/Ipf1▴ were 17–19 weeks of age.
Blood glucose levels are expressed as mmoles/liter from nonfasted and over night-fasted animals, respectively.
Insulin is expressed as μg/mg pancreas protein from nonfasted animals.
We examined the onset of Ipf1/Pdx1 inactivation by performing immunohistochemistry using anti-insulin and anti-IPF1 antibodies on pancreases derived from mice at different developmental stages. Although RT–PCR analysis of Rip1/Cre expression revealed that the Cre transgene is activated during the fetal stage (Fig. 1B), a very limited extent of Ipf1/Pdx1 inactivation was observed in pancreases derived from postnatal day 1 (P1) Rip1/Ipf1▴ mice (Fig. 1C). However, by 3 (Fig. 1D) and 5 weeks (Fig. 1E) of age, inactivation of Ipf1/Pdx1 becomes more prominent. To determine the cause of diabetes and the role of IPF1/PDX1 in β cell function, we examined the formation of islets and the state of differentiation of β cells in OD Rip1/Ipf1▴ mice. No significant reduction in the number of islets was observed in the mutant mice (data not shown). In normal mice, 82% of pancreatic islet cells are insulin-expressing (Ins+) β cells of which >99% coexpress IPF1/PDX1 (Figs. 1F and 2A; data not shown). In the OD Rip1/Ipf1▴ mutants the number of Ins+ cells was reduced by ∼40%, and of the remaining Ins+ cells only 17% (n = 96/559) coexpressed IPF1/PDX1 (Figs. 1G and 2A), indicating that Cre/loxP-mediated recombination of the Ipf1/Pdx1 gene had occurred in at least 80% of β cells.
Figure 2.
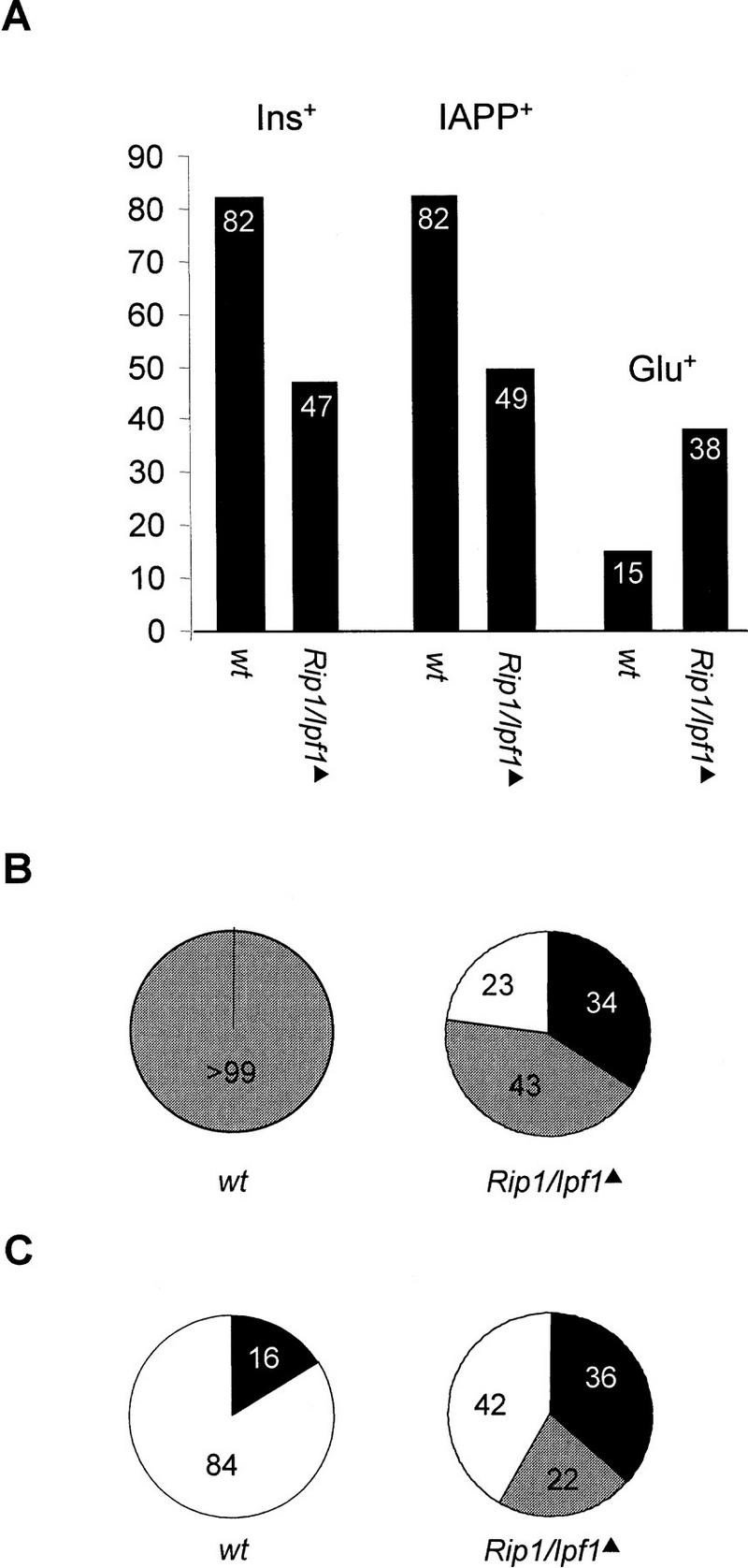
Rip1/Ipf1▴ mice show abnormal islet composition. (A) The distribution of insulin (Ins+)-, IAPP (IAPP+)-, or glucagon (Glu+)-expressing cells per total number of islet cells in wild-type and Rip1/Ipf1 mice. (B) Ratio of Ins+ to IAPP+ cells in islets from wild-type and Rip1/Ipf1 mice. (Open area) Ins+ only; (solid area) IAPP+ only; (shaded area) Ins+ and IAPP+ coexpressed. (C) Ratio of Ins+ to Glu+ cells in islets from wild-type and Rip1/Ipf1 mice. (Open area) Ins+ only; (solid area) Glu+ only; (shaded area) Ins+ and Glu+ coexpressed. (A–C) n = 600–1050 cells.
Thus, although the recombination event commences at the neonatal stage the OD state develops first when ∼80% of the Ins+ cells have lost IPF1/PDX1. The time required to reach the state of 80% inactivation of both alleles is dependent on the amount of Cre recombinase (Lakso et al. 1992; Tsien et al. 1997). Transgene expression is known to vary between transgenic lines and also between cells in a given transgenic line (Martin and Whitelaw 1996; Kioussis and Festenstein 1997). In addition, islet neogenesis, that is, emergence of new β cells, is known to continue for as long as 3 weeks postnatally (Githens 1993; Sander and German 1997), and this possibly also prolongs the time required for enough β cells to have lost IPF1/ PDX1. Finally, excision of Ipf1/Pdx1, that is, an insulin gene transactivator, most likely negatively affects the level of Rip1/Cre transcription, and this is also likely to influence the time required for both Ipf1/Pdx1 alleles to become inactivated.
β cells also express IAPP (Figs. 1H and 2A) but in the Rip1/Ipf1▴ mutants the number of IAPP+ cells was decreased by 40% (Figs. 1I and 2A), and only 43% of all Ins+ and/or IAPP+ cells coexpressed the two proteins (Fig. 2B). In addition, cells coexpressing IAPP and glucagon were present (Fig. 1I, arrowhead). In normal mice the ratio of Ins+ to glucagon-expressing (Glu+) cells is ∼5:1 and no islet cells coexpress the hormones (Figs. 1J and 2A,C). In the Rip1/Ipf1▴ mice, however, there was a significant increase in the number of Glu+ cells, from 15% in Ipf1+/+ mice to 38% in the mutants, resulting in an ∼1:1 ratio of Ins+/Glu+ cells (Figs. 1J,K and 2A,C). In addition, ∼22% of all Ins+ and/or Glu+ cells coexpressed the two hormones (Figs. 1K, arrowheads, and 2C). The increase in the number of glucagon cells in the mutant mice was also associated with a more homogenous distribution of Glu+ cells within the islets as compared to the normal, peripheral location of Glu+ cells in wild-type islets (Fig. 1H–K). No significant change in the number of somatostatin-expressing (Som+) cells was observed in Rip1/Ipf1▴ mutants, and no Som+ cells coexpressed either insulin or glucagon (data not shown). The expression of the LIM–homeodomain protein ISL1 (Karlsson et al. 1990; Thor et al. 1991) (Fig. 1L,M), which is required for the generation of all islet cell types (Ahlgren et al. 1997), was also unaffected, showing that islets do form and cells within islets still retain endocrine properties in mutant mice.
These results show that IPF1/PDX1 is required for maintaining the hormone-producing phenotype of the β cell by positively regulating insulin and IAPP expression and by repressing Glu+ in β cells. The homeodomain protein Nkx6.1 is expressed in β cells (Fig. 1N; Jensen et al. 1996), and virtually no Nkx6.1+ cells were detected in Rip1/Ipf1▴ mutants (Fig 1O). Interestingly, Nkx6.1 is homologous to the homeodomain protein Gtx which can act as a transcriptional repressor (Komuro et al. 1993). The loss of Nkx6.1 in the mutants may therefore suggest that IPF1/PDX1 positively regulates the expression of Nkx6.1, which in turn might repress glucagon expression in β cells.
The pancreas of the OD Rip1/Ipf1▴ mutant mice contained ∼60% of the normal number of β cells (Fig. 2A) and ∼10% of the normal total pancreatic insulin content (Table 1). This suggests that a progressive decrease in insulin expression may be the causative agent underlying the development of diabetes in the Rip1/Ipf1▴ mice. The diabetic mutants responded to insulin administration by transiently reverting to normoglycemia (data not shown), excluding insulin resistance as a cause of diabetes. IPF1/PDX1 has, however, been suggested to regulate the expression of the Glut2 and glucokinase genes (Waeber et al. 1996; Watada et al. 1996a), two key components of the β cell glucose-sensing system. Glut2-deficient mice show an impaired glucose-induced insulin secretion and develop diabetes (Guillam et al. 1997), and mutation and/or inactivation of the glucokinase gene results in impaired insulin secretion and development of diabetes (Frougel et al. 1992; Bali et al. 1995; Grupe et al. 1995) both in mouse and man. Thus, an impaired expression of these genes could also possibly contribute to the diabetic phenotype of Rip1/Ipf1▴ mutant mice.
In OD Rip1/Ipf1▴ mice Glut2 expression was virtually extinct (Fig. 3A), whereas the expression of glucokinase appears unaffected (data not shown). In nondiabetic (ND) Rip1/Ipf1▴ 11-week mice the pancreatic insulin content is ∼30% of the normal level (Table 1), the number of Glut2+ cells is reduced, and the level of Glut2 expression is highly variable (Fig. 3B). Thus, excision of the Ipf1/Pdx1 gene in β cells leads to an early loss of Glut2 expression (Fig. 3A–C,E–G), which via impaired glucose-stimulated insulin release probably contributes to the development of hyperglycemia. The combined, progressive loss of both Glut2 and insulin expression as a result of Ipf1/Pdx1 inactivation would then lead to the subsequent development of OD in the Rip1/Ipf1▴ mice. Interestingly, Glut2-deficient mice also show an increased number of α versus β cells (Guillam et al. 1997) but, unlike in Rip1/Ipf1▴ mice, the α cells still remain located at the periphery of the islets (B. Thorens, pers. comm.). This suggests that the change in the ratio of Glu+ versus Ins+ cells in Rip1/Ipf1▴ mice primarily is mediated by the loss of IPF1/PDX1 in β cells, resulting in a change in the pattern of hormone production.
A strong family history of early onset type II diabetes is associated with heterozygosity for a point mutation in the human Ipf1 gene (Stoffers et al. 1997a,b). Our results raise the possibility that a dosage-dependent regulation of Glut2 and insulin expression by IPF1/PDX1 could be causative. Mice heterozygous for the original Ipf1/Pdx1 null mutation are ND and show blood glucose levels in the upper range of normoglycemia but were found to be glucose intolerant. In Ipf1+/+ mice intraperitoneal (IP) injections of glucose (1 gram/kg body weight) resulted in an increase in blood glucose levels to 12–13 mm 15–20 min postinjection (Fig. 4). In Ipf1/Pdx1+/− mice, the blood glucose levels increased for 40–50 min, reaching 22–23 mm, and did not return to basal levels 180 min after challenge (Fig. 4). The glucose intolerance could reflect either impaired insulin, Glut2 expression, or both. The Ipf1/Pdx1-null (+/−) mice, however, showed a normal total pancreatic insulin content and islet architecture (Fig. 3D,H,L; Table 1; data not shown), and the pattern and levels of expression of ISL1 (Fig. 3I–L), insulin, glucagon, IAPP, and Nkx6.1 appeared normal (data not shown). In contrast, Glut2 was expressed at homogeneous but markedly reduced levels and IPF1/PDX1 at decreased levels (Fig. 3C,D,G,H) providing evidence that IPF1/PDX1 exerts a dosage-dependent regulation of Glut2 expression.
Figure 4.

Heterozygosity for the Ipf1/Pdx1 null mutation results in impaired glucose tolerance. Glucose challenge of 18-week-old Ipf1+/+ (▪) and Ipf1+/− (♦) (Jonsson et al. 1994) mice. Blood glucose levels are shown at indicated time points after IP administration of glucose. Results are mean ± s.e.m. of eight animals in each group.
Hence, these results offer a plausible explanation for the development of maturity onset diabetes in Ipf1/Pdx1 haploinsufficient individuals and identify Ipf1/Pdx1-null (+/−) mice as a relevant animal model for studying this disease. These results also strengthen the idea that an early loss of Glut2 expression is involved in the development of hyperglycemia in Ipf1/Pdx1 β-cell-specific mutants and that the combined loss of Glut2 and gradual decrease of insulin expression together leads to the manifestation of diabetes. Thus, lowered IPF1/PDX1 expression, or activity, resulting in impaired expression of both Glut2 and insulin could be a more general cause of development of hyperglycemia that in turn may progress to type II diabetes. In conclusion, the results presented here provide direct evidence that IPF1/PDX1 exhibits dual functions during pancreas development. Loss of the early function results in an inability to form a pancreas (Jonsson et al. 1994; Ahlgren et al. 1996; Offield et al. 1996; Stoffers et al. 1997a), whereas the later β-cell expression is required to maintain the β-cell pattern of hormone production, Glut2 expression, and normoglycemia.
Materials and methods
Preparation of the Ipf1/Pdx1-loxP gene targeting construct
The two loxP sites (Kilby et al. 1993) (5′ and 3′ of exon 2 in Ipf1/Pdx1; see Fig. 1A) were constructed using single-stranded oligonucleotides. The upstream loxP recognition and core sequence contains ApaI sites at its 5′ and 3′ ends and an internal NcoI site. Similarly, the downstream loxP recognition and core sequence has NcoI sites at its ends and an internal PstI site. The internal sites were used to verify homologous recombination. A BamHI–XhoI fragment carrying the gene for neomycin resistance (neoR) (Jonsson et al. 1994) was cloned into the BglII and XhoI sites of the downstream loxP site (Fig. 1A). The downstream neoR–loxP construct was inserted into the NcoI site 3′ of exon 2A of the genomic mouse Ipf1/Pdx1 DNA (Fig. 1A; Jonsson et al. 1994). The upstream loxP site was subsequently inserted in the ApaI site 5′ of exon 2 in the Ipf1/Pdx1 gene. The functionality of the targeting construct was verified by incubating the final 8.7-kb Ipf1/Pdx1–neoRloxP fragment with Cre enzyme (NEN–DuPont, Inc.). Bands corresponding to 8.7 and 3.7 kb and the supercoiled circular loxP product were identified on an 8% polyacrylamide gel.
Generation of Ipf1/Pdx1–loxP mice
E14-1 ES cells (Jonsson et al. 1994) were cultured and electroporated using standard procedures as described previously (Hogan et al. 1994). ES cells carrying the Ipf1/Pdx1–loxP mutation were injected into blastocysts of C57BL/6 mice (Hogan et al. 1994), and the resulting male chimeras were subsequently backcrossed to C57BL/6 mice to generate heterozygous mutants and wild-type mice.
RIP1/CRE transgenic mouse production
The Cre expression vector pRip1/Cre was constructed by inserting the 0.9-kb XbaI–MluI CRE fragment, derived from phCMV/Cre (Sauer and Henderson 1990; Baubonis and Sauer 1993), and blunted at the MluI site, into XbaI–HindIII-digested pRip1ΔEcad (Dahl et al. 1996), blunted at the HindIII site. A 2.6-kb linearized DNA AatII–SalI fragment containing the Rip1/Cre construct was used to generate transgenic founders by pronuclear injection of the linearized DNA into F2 hybrid oocytes from B6/CBA parents as described (Hogan et al. 1994). The Cre founders were backcrossed into the B6 background for production of transgenic offspring and then subsequently also crossed with the Ipf1/Pdx1–loxP mice to generate homozygous Ipf1/Pdx1–loxP mice carrying the Rip1/Cre transgene.
Genotyping and RT–PCR
The genotypes of all offspring were performed on genomic DNA isolated from ES cells or the tail tip of 2- to 4-week-old mice by Southern blot or PCR (Hogan et al. 1994). The 5′ and 3′ primers for the Rip1/Cre transgene (∼700 bp amplified) were 5′-GGTGCTTTGGACTATAAAGC-3′ and 5′-GTCAGTACGTGAGATATCTTTA-3′ under the following conditions: 35 cycles of 94°C for 30 sec, 50°C for 1 min, 72°C for 1 min, followed by 1 cycle of 72°C for 7 min. The 5′ and 3′ primers for the Ipf1/Pdx1 gene were 5′-TCAACAGCTGCGATCAGTA-3′ and 5′-AACATCACTGCCAGCTCCACC-3′ under the following conditions: 1 cycle of 96°C for 5 min, 55°C for 2 min, 72°C for 3 min, followed by 29 cycles of 94°C for 1 min, 55°C for 1 min, 72°C for 3 min, and, finally, 1 cycle of 72°C for 7 min, which results in a ∼300-bp amplified fragment for the wild-type Ipf1/Pdx1 allele and a ∼240-bp amplified fragment for the Ipf1/Pdx1–loxP allele. For Ipf1/Pdx1 Southern blot analysis, DNA was digested with NcoI or PstI and hybridized with probes 1 and 2, respectively. RT–PCR analysis were performed on total RNA prepared from embryonic tissues essentially as described (Tanabe et al. 1995). The primers used for RT–PCR were Cre-primers, 5′-GGACATGTTCAGGGATCGCCAGGCG-3′ and 5′-GCATAACCAGTGAAACAGCATTGCTG-3′ (Lakso et al. 1992); mouse ribosomal protein S17, 5′-TCGCACCAAGACTGTGAAGAAGGC-3′ and 5′-TGGCATAACAGATTAAACAGCTCCACG-3′. The conditions used were 35 cycles of 94°C for 30 sec, 60°C for 1 min, 72°C for 1 min, followed by 1 cycle of 72°C for 7 min. Sizes of the resulting PCR products are 269 bp for Cre and 398 bp for S17.
Glucose challenge and insulin measurements
Overnight-fasted wild-type and Ipf1/Pdx1-null (+/−) mice were injected IP with 1 gram of glucose per kg of body weight. Blood samples were obtained from the tail vein, and glucose levels were measured immediately before and 15, 30, 45, 60, 90, 120, and 180 min after injection using a Glucometer Elite (Bayer, Inc.). Pancreatic insulin was measured using a commercially available radioimmuno assay for rat insulin (Linco), and total pancreatic protein concentration was determined using the Bio-Rad protein assay.
Immunohistochemistry and confocal image analysis
Immunohistochemistry and confocal image analysis was performed essentially as described previously (Ahlgren et al. 1996). Primary antibodies used were rabbit anti-IPF1/PDX1 (Ohlsson et al. 1993), rabbit anti-ISL1 (Thor et al. 1991), guinea pig anti-insulin C-peptide (Linco), guinea pig anti-glucagon (Linco), rabbit anti-glucagon (Linco), rabbit anti-somatostatin (DAKO), rabbit anti-IAPP/amylin (Euro Diagnostics), sheep anti-glucokinase (H.J. Seitz, University of Hamburg, Germany), rabbit anti-Glut2, kindly provided by Dr. B. Thorens (Institute of Phamacology and Toxicology, Lausanne, Switzerland), and rabbit anti-Nkx6.1 (Jensen et al. 1996), kind gift from Dr. P. Serup. Secondary antibodies used were Cy3-conjugated goat anti-rabbit IgG (Jackson) and Fluorescein (DTAF)-conjugated goat anti-GP IgG (Jackson). For multiple labelling a blocking step using swine anti-Rabbit IgG (DAKO) was included.
Acknowledgments
We thank K. Falk, U.B. Backman, and I. Berglund for skillful technical assistance; Drs. T. Edlund, T.M. Jessell for critical reading and comments, U. Dahl, J. Petterson, S. Norlin, and members from our laboratory for helpful discussions. We are indebted to Dr. B. Thorens for the Glut2 antibodies, Dr. P. Serup for the Nkx6.1 antibodies, and Dr. M. Cogné for phCMV/CRE. This work was supported by grants from the Swedish Medical Research Council and the Juvenile Diabetes Foundation, New York (to H.E).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL helena.edlund@micro.umu.se; FAX 46-90-772630.
References
- Ahlgren U, Jonsson J, Edlund H. Arrested development of the pancreas in IPF1/PDX1 deficient mice reveals that the pancreatic mesenchyme develops independently of the pancreatic epithelium. Development. 1996;122:1409–1416. doi: 10.1242/dev.122.5.1409. [DOI] [PubMed] [Google Scholar]
- Ahlgren U, Pfaff SL, Jessell TM, Edlund T, Edlund H. Independent requirement for ISL1 in the formation of pancreatic mesenchyme and islet cells. Nature. 1997;385:257–260. doi: 10.1038/385257a0. [DOI] [PubMed] [Google Scholar]
- Bali D, Svetlanov A, Lee HW, Fusco-DeMane D, Leiser M, Li B, Barzilai N, Surana M, Hou H, Fleischer N, DePinho R, Rossetti L, Efrat S. Animal model for maturity-onset diabetes of the young generated by disruption of the mouse glucokinase gene. J Biol Chem. 1995;270:21464–21467. doi: 10.1074/jbc.270.37.21464. [DOI] [PubMed] [Google Scholar]
- Baubonis W, Sauer B. Genomic targeting with purified Cre recombinase. Nucleic Acids Res. 1993;21:2025–2029. doi: 10.1093/nar/21.9.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl U, Sjödin A, Semb H. Cadherins regulate aggregation of pancreatic β-cells in vivo. Development. 1996;122:2895–2902. doi: 10.1242/dev.122.9.2895. [DOI] [PubMed] [Google Scholar]
- Froguel Ph, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, Lesage S, Vionnet N, Clement K, Fougerousse F, Tanizawa Y, Weissenbach J, Beckmann JS, Lathrop GM, Passa Ph, Permutt MA, Cohen D. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356:162–164. doi: 10.1038/356162a0. [DOI] [PubMed] [Google Scholar]
- Githens S. Differentiation and development of the pancreas in animals. In: Go VLW, et al., editors. The pancreas: Biology, pathobiology, and disease. New York, NY: Raven Press; 1993. pp. 21–55. [Google Scholar]
- Grupe A, Hultgren B, Ryan A, Ma YH, Bauer M, Stewart TA. Transgenic knockouts reveal a critical requirement for pancreatic β cell glucokinase in maintaining glucose homeostasis. Cell. 1995;83:69–78. doi: 10.1016/0092-8674(95)90235-x. [DOI] [PubMed] [Google Scholar]
- Gu H, Zou Y-R, Rajewski K. Independent control of immunoglobulin switch recombination at individual swotch regions evidenced through Cre-loxP-mediated targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- Guillam M-T, Hümmler E, Schaerer E, Wu J-Y, Birnbaum MJ, Beermann F, Schmidt A, Dériaz N, Thorens B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nature Genet. 1997;17:327–330. doi: 10.1038/ng1197-327. [DOI] [PubMed] [Google Scholar]
- Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CV, Teitelman G. Expression of murine STF-1, a putative insulin gene transcription factor, in β-cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development. 1995;121:11–18. doi: 10.1242/dev.121.1.11. [DOI] [PubMed] [Google Scholar]
- Hogan B, Constantini F, Lacey E. Manipulating the mouse embryo: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Jensen J, Serup P, Karlsen C, Funder T, Madsen OD. mRNA profiling of rat islet tumors reveals Nkx6.1 as a β-cell-specific homeodomain transcription factor. J Biol Chem. 1996;271:18749–18758. doi: 10.1074/jbc.271.31.18749. [DOI] [PubMed] [Google Scholar]
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- Karlsson O, Thor S, Norberg T, Ohlsson H, Edlund T. Insulin gene enhancer binding protein Isl-1 is a member of a novel class of proteins containing both a homeo- and a Cys-His domain. Nature. 1990;344:879–882. doi: 10.1038/344879a0. [DOI] [PubMed] [Google Scholar]
- Kilby NJ, Snaith MR, Murray JAH. Site-specific recombinases: Tools for genome egineering. Trends Genet. 1993;9:413–421. doi: 10.1016/0168-9525(93)90104-p. [DOI] [PubMed] [Google Scholar]
- Kioussis D, Festenstein R. Locus control regions: Overcoming heterochromatin-induced gene inactivation in mammals. Curr Opin Genet Dev. 1997;5:614–619. doi: 10.1016/s0959-437x(97)80008-1. [DOI] [PubMed] [Google Scholar]
- Komuro I, Schalling M, Jahn L, Bodmer R, Jenkins NA, Copeland NG, Izumo S. Gtx: A novel murine homeobox-containing gene, expressed specifically in glial cells of the brain and germ cells of testis, has a transcriptional repressor activity in vitro for a serum-inducible promoter. EMBO J. 1993;12:1387–1401. doi: 10.1002/j.1460-2075.1993.tb05783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Sauer B, Mosinger JR, Lee EJ, Manning RW, Yu S-H, Mulder KL, Westphal H. Targeted oncogene activation by site-specific recombination in transgenic mice. Proc Natl Acad Sci. 1992;89:6232–6236. doi: 10.1073/pnas.89.14.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J, Peers B, Johnson T, Ferreri K, Lee S, Montminy MR. Characterization of somatostatin transactivating factor-1, a novel homeobox fcator that stimulates somatostatin expression in pancreatic islets. Mol Endocrinol. 1993;7:1275–1283. doi: 10.1210/mend.7.10.7505393. [DOI] [PubMed] [Google Scholar]
- Martin DI, Whitelaw E. The vagaries of variegating transgenes. BioEssays. 1996;11:919–923. doi: 10.1002/bies.950181111. [DOI] [PubMed] [Google Scholar]
- Miller CP, McGehee RE, Jr, Habener JF. IDX-1: A new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO J. 1994;13:1145–1158. doi: 10.1002/j.1460-2075.1994.tb06363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. 1993;12:4251–4259. doi: 10.1002/j.1460-2075.1993.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, German MS. The β-cell transcription factors and development of the pancreas. J Mol Med. 1997;75:327–340. doi: 10.1007/s001090050118. [DOI] [PubMed] [Google Scholar]
- Sauer B, Henderson N. Targeted insertion of exogenous DNA into the eukaryotic genome by the Cre recombinase. New Biol. 1990;2:441–449. [PubMed] [Google Scholar]
- Serup P, Petersen HV, Pedersen EE, Edlund H, Leonard J, Petersen JS, Larsson LI, Madsen OD. The homeodomain protein IPF-1/STF-1 is expressed in a subset of islet cells and promotes rat insulin 1 gene expression dependent on an intact E1 helix-loop-helix factor binding site. Biochem J. 1995;310:997–1003. doi: 10.1042/bj3100997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature. 1997;386:399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997;387:406–409. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nature Genet. 1997a;15:106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nature Genet. 1997b;17:138–139. doi: 10.1038/ng1097-138. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Roelink H, Jessell TM. Induction of motor neurons by sonic hedgehog is independent of floor plate differentiation. Curr Biol. 1995;5:651–658. doi: 10.1016/s0960-9822(95)00130-8. [DOI] [PubMed] [Google Scholar]
- Thor S, Ericson J, Brännström T, Edlund T. The homeodomain LIM protein Isl-1 is expressed in subsets of neurons and endocrine cells in the adult rat. Neuron. 1991;7:881–889. doi: 10.1016/0896-6273(91)90334-v. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1997;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Waeber G, Thompson N, Nicod P, Bonny C. Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol Endocrinol. 1996;10:1327–1334. doi: 10.1210/mend.10.11.8923459. [DOI] [PubMed] [Google Scholar]
- Watada H, Kajimoto Y, Umayahara Y, Matsuoka T, Kaneto H, Fujitani Y, Kamada T, Kawamori R, Yamasaki Y. The human glucokinase gene β-cell-type promoter: An essential role of insulin promoter factor 1/PDX-1 in its activation in Hit-T15 cells. Diabetes. 1996a;45:1478–1488. doi: 10.2337/diab.45.11.1478. [DOI] [PubMed] [Google Scholar]
- Watada H, Kajimoto Y, Kaneto H, Matsuoka T, Fujitani Y, Miyazaki J, Yamasaki Y. Involvement of the homeodomain-containing transcription factor PDX-1 in islet amyloid polypeptide gene transcription. Biochem Biophys Res Commun. 1996b;229:746–751. doi: 10.1006/bbrc.1996.1875. [DOI] [PubMed] [Google Scholar]