

Abstract
The spindle assembly checkpoint mechanism delays anaphase initiation until all chromosomes are aligned at the metaphase plate. Activation of the anaphase-promoting complex (APC) by binding of CDC20 and CDH1 is required for exit from mitosis, and APC has been implicated as a target for the checkpoint intervention. We show that the human checkpoint protein hMAD2 prevents activation of APC by forming a hMAD2–CDC20–APC complex. When injected into Xenopus embryos, hMAD2 arrests cells at mitosis with an inactive APC. The recombinant hMAD2 protein exists in two-folded states: a tetramer and a monomer. Both the tetramer and the monomer bind to CDC20, but only the tetramer inhibits activation of APC and blocks cell cycle progression. Thus, hMAD2 binding is not sufficient for inhibition, and a change in hMAD2 structure may play a role in transducing the checkpoint signal. There are at least three different forms of mitotic APC that can be detected in vivo: an inactive hMAD2–CDC20–APC ternary complex present at metaphase, a CDC20–APC binary complex active in degrading specific substrates at anaphase, and a CDH1–APC complex active later in mitosis and in G1. We conclude that the checkpoint-mediated cell cycle arrest involves hMAD2 receiving an upstream signal to inhibit activation of APC.
Keywords: MAD2, CDC20, CDH1, anaphase-promoting complex, spindle assembly checkpoint, anaphase initiation
Cell cycle progression is monitored by surveillance mechanisms that ensure the integrity of the genome and the fidelity of chromosome segregation (Elledge 1996). The spindle assembly checkpoint mechanism is thought to monitor the attachment of the kinetochores of all chromosomes to the mitotic spindle and the tension exerted on the kinetochores by microtubules (Murray 1994, 1995; Rudner and Murray 1996; Nicklas 1997). For example, the presence of a single unattached kinetochore will activate the spindle assembly checkpoint and prevent chromatid separation, anaphase initiation, and loss of Cdc2 kinase activity. The spindle assembly checkpoint can thus prevent aneuploidy from improper chromosome separation. This checkpoint mechanism may also function in every cell cycle to determine the normal timing of anaphase initiation (Taylor and McKeon 1997).
Microtubule destabilizing drugs, such as nocodazole, activate the spindle assembly checkpoint. Using these drugs, genetic studies have identified several components of the checkpoint pathway in budding yeast (Hoyt et al. 1991; Li and Murray 1991). Mutations in any of these genes, MAD1, MAD2, MAD3 and BUB1, BUB2, BUB3, cause aberrant mitosis in the presence of nocodazole and eventually lead to cell death. MPS1, a gene required for spindle pole body duplication, has also been shown to be involved in checkpoint control (Hardwick et al. 1996). A combination of genetic and biochemical studies has shown that Bub1p, Bub3p, and Mps1p act upstream of Mad1p and Mad2p, whereas Bub2p and Mad3p act either downstream of Mad1p and Mad2p, or in a parallel pathway (Elledge 1996; Rudner and Murray 1996).
Human and Xenopus homologs of the yeast Mad2p, hMAD2 and xMAD2, have been cloned and shown to be required for checkpoint control (Chen et al. 1996; Li and Benezra 1996). In addition to a general nuclear distribution, MAD2 protein localizes to only those kinetochores that have not attached to the mitotic spindle. The murine homolog of yeast Bub1p also localizes to unattached kinetochores during mitosis (Taylor and McKeon 1997). Those observations suggest an important role of kinetochore structure in sensing and transducing the checkpoint signal.
Little is known about how the checkpoint signal is transduced to the cell cycle machinery, leading to arrest of cell division. Activation of the anaphasepromoting complex (APC), a ubiquitin-cyclin ligase required for degradation of mitotic cyclins and other cell cycle regulators, has been shown to be required for metaphase → anaphase transition and for exit from mitosis during the normal cell cycle (Holloway et al. 1993; Irniger et al. 1995; King et al. 1995; Sudakin et al. 1995; Tugendreich et al. 1995). Although there could be many points of intervention, it seems logical that APC itself might be a target for the checkpoint control. Recent studies in fission yeast have identified a genetic interaction between MAD2 and APC (He et al. 1997). In addition, hMAD2 has been reported to associate with APC in nocodazole-arrested HeLa cells, and it has been proposed that this association may mediate cell cycle arrest in the checkpoint-activated cells (Li et al. 1997). However, the activity of hMAD2-associated APC has not been measured directly, and therefore, the effect of this association on the APC activity remains to be determined.
The activity of APC is cell cycle regulated; it is active from anaphase up to late G1, but not in S-phase or G2 (Amon et al. 1994; King et al. 1995; Brandeis and Hunt 1996; Peters et al. 1996; G. Fang, H. Yu, and M.W. Kirschner, in prep.). Its activity is in part controlled by the regulatory factors CDC20 and CDH1/HCT1 (Schwab et al. 1997; Sigrist and Lehner 1997; Visintin et al. 1997; G. Fang, H. Yu, and M.W. Kirschner, in prep.), and in part by cell cycle-specific phosphorylation (King et al. 1995; Lahav-Baratz et al. 1995; Peters et al. 1996). CDC20 and CDH1 are WD-40 containing proteins, conserved from yeast to human (Weinstein et al. 1994; Dawson et al. 1995; Matsumoto 1997; Yamaguchi et al. 1997). Recently we have shown that the human homologs, hCDC20 and hCDH1, bind directly to and activate APC (G. Fang, H. Yu, and M.W. Kirschner, in prep.). Formation of the hCDC20–APC complex during mitosis correlates roughly with the activation of the ubiquitination machinery during mitosis. However, there is a period in G2 and M where both CDC20 and APC are present, but where cyclin proteolysis does not occur, suggesting the existence of additional regulatory mechanism. Recent genetic studies in yeast show that Cdc20p interacts with Mad2p and is a target for the checkpoint control (Hwang et al. 1998; Kim et al. 1998). The role of hCDH1 in the cell cycle is less clear. Presumably it has different functions from hCDC20 and most likely is responsible for APC activity in G1.
In summary, the discovery of APC, and its regulators CDC20 and CDH1 have complicated our picture of the regulatory events during mitosis. To better understand control of mitosis and G1, it is necessary to examine the progression of different activity states of APC, and to investigate how each is regulated. Because APC might also be the principal effector of the spindle checkpoint control system, it is expected that some of the known checkpoint genes could intervene in specific parts of this APC network. We report here that APC is a target for checkpoint intervention. The checkpoint protein hMAD2 forms a ternary complex with hCDC20 and APC in vivo, and prevents activation of APC when the checkpoint signal is present. Anaphase is initiated by dissociation of hMAD2 from the complex; APC is then activated by bound hCDC20. Later in mitosis, hCDH1 binds to and activates APC, which allows cells to exit from mitosis. The checkpoint-mediated inhibition of APC has been reconstituted in vitro with purified hMAD2, hCDC20, and APC. Interestingly, we found that the recombinant hMAD2 protein exists in two different forms, a tetramer and a monomer. Only the tetramer inhibits activation of APC by hCDC20 in vitro, suggesting a possible role of a hMAD2 structure change in transducing the checkpoint signal.
Results
APC is not active when the spindle assembly checkpoint is activated
To test whether APC is a target for the spindle assembly checkpoint pathway, we analyzed directly the cyclin ubiquitination activity in checkpoint-arrested cells. HeLa cells were synchronized by thymidine, released and then incubated with nocodazole, which arrested >90% of cells at prometaphase (Fig. 1A, lane 3). APC was immunopurified from the arrested cells and assayed for cyclin ubiquitination activity. We found much lower APC activity in nocodazole-treated cells compared with cells going through mitosis (Fig. 1C, cf. lane 3 with lane 6). When cells were released from the nocodazole arrest, a burst of APC activity was detected later in mitosis (Fig. 1C, lanes 5,6). As cells progress through G1, APC activity was reduced (lanes 7,8), but was still higher than that in S phase cells (lane 2). Although the APC activity had not been measured previously in checkpoint-arrested cells, our results are consistent with an earlier report on the stability of the cyclin protein in nocodazole-treated cells (Brandeis and Hunt 1996). Therefore, APC is inhibited in checkpoint-arrested cells, and is a target of the spindle assembly checkpoint.
Figure 1.
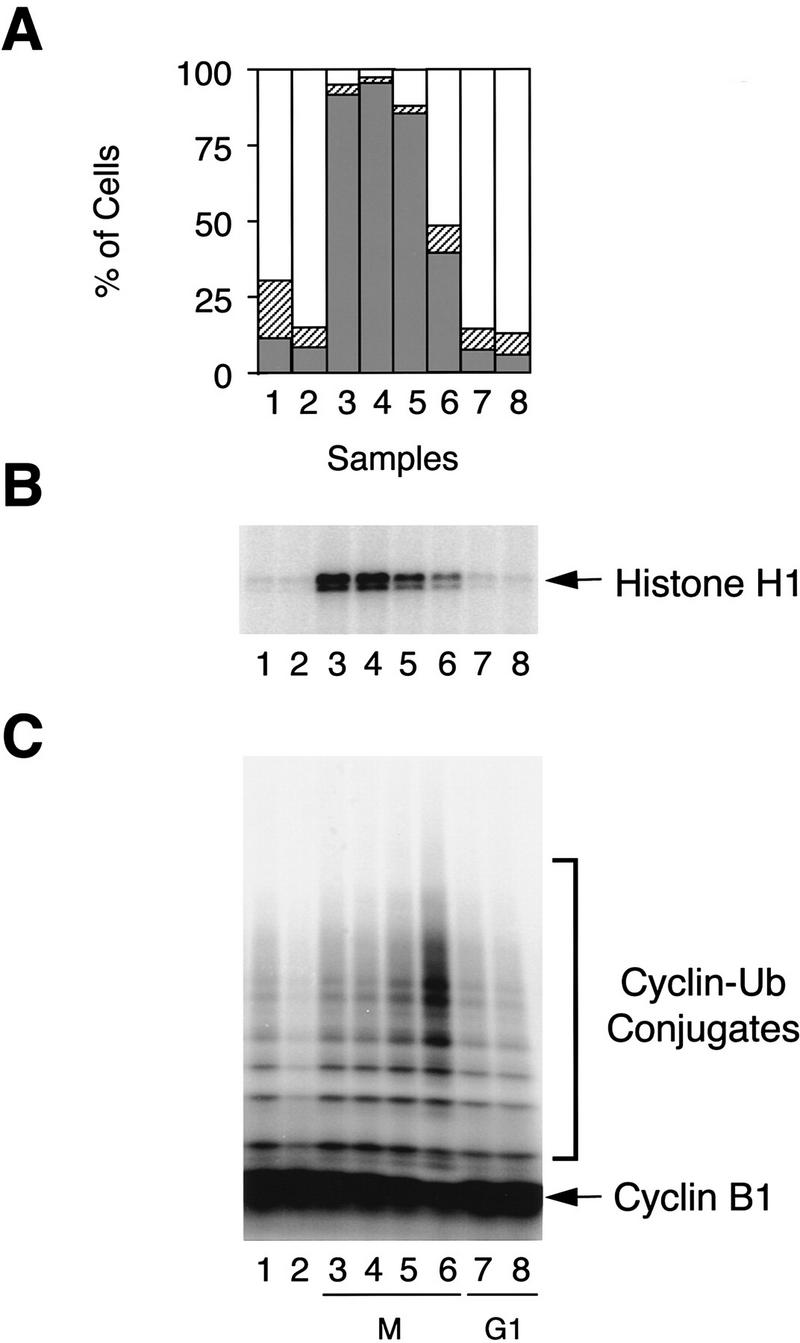
APC activity at different stages of cell cycle. HeLa cells were synchronized at prometaphase by a thymidine–nocodazole block. Cells were collected immediately after nocodazole treatment (lane 3), or released into fresh medium for 0.5, 1, 1.5, 4, and 10 hr (lanes 4–8). Asynchronous cells (lane 1) and cells arrested at G1/S boundary by a double thymidine block (lane 2) were included as controls. (A) Cell cycle stage was determined by FACS analysis. (Open bars) G1; (hatched bars) S; (solid bars) G2/M. (B) Cdc2 kinase was immunopurified from cell lysates and assayed for histone H1 kinase activity. (C) APC was purified from the cell lysates with anti-CDC27/protein A beads and analyzed for its ability to ubiquitinate a 125I-labeled amino-terminal fragment of Xenopus cyclin B1.
Recombinant hMAD2 exists in two folded states
Genetic studies in Saccharomyces cerevisiae have identified Mad2p as the component of the checkpoint pathway most distal from the sensory machinery (Elledge 1996; Rudner and Murray 1996). Therefore, we tested whether hMAD2 regulates APC. Recombinant hMAD2 tagged with His6 was expressed in Escherichia coli and purified to apparent homogeneity. The hMAD2 protein fractionated on a gel filtration column as two separate peaks, 30 and 120 kD, corresponding to the monomer and tetramer of hMAD2 (Fig. 2). The existence of the tetramer and monomer is further supported by velocity gradient sedimentation and chemical cross-linking experiments. By gradient sedimentation, the hMAD2 tetramer and monomer sediment as 4.4S and 2.2S particles, respectively (data not shown). Assuming an average partial specific volume for proteins (0.74 gram/ml3), they correspond to molecular masses of 72 and 19 kD. In chemical cross-linking experiments, the tetramer was cross-linked between subunits to form multiple 50- to 55-kD products by both EDC [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride] and DST (disuccinimidyl tartrate), but the monomer was not (data not shown). We have named the monomeric form hMAD2m, and the tetrameric form hMAD2t; as described below (Fig. 3 and 4), they have different activities.
Figure 2.

Identification of different oligomerization states of the recombinant hMAD2 protein. Oligomerization states of the recombinant hMAD2 proteins were determined by gel filtration chromatography with a S100 column. Column fractions were analyzed by SDS-PAGE. The elution peaks of molecular mass standards are labeled at the top of the UV trace. (*) Peak corresponding to void volume. (WT) Wild-type hMAD2; (ΔN) hMAD2ΔN; (ΔC) hMAD2ΔC.
Figure 3.
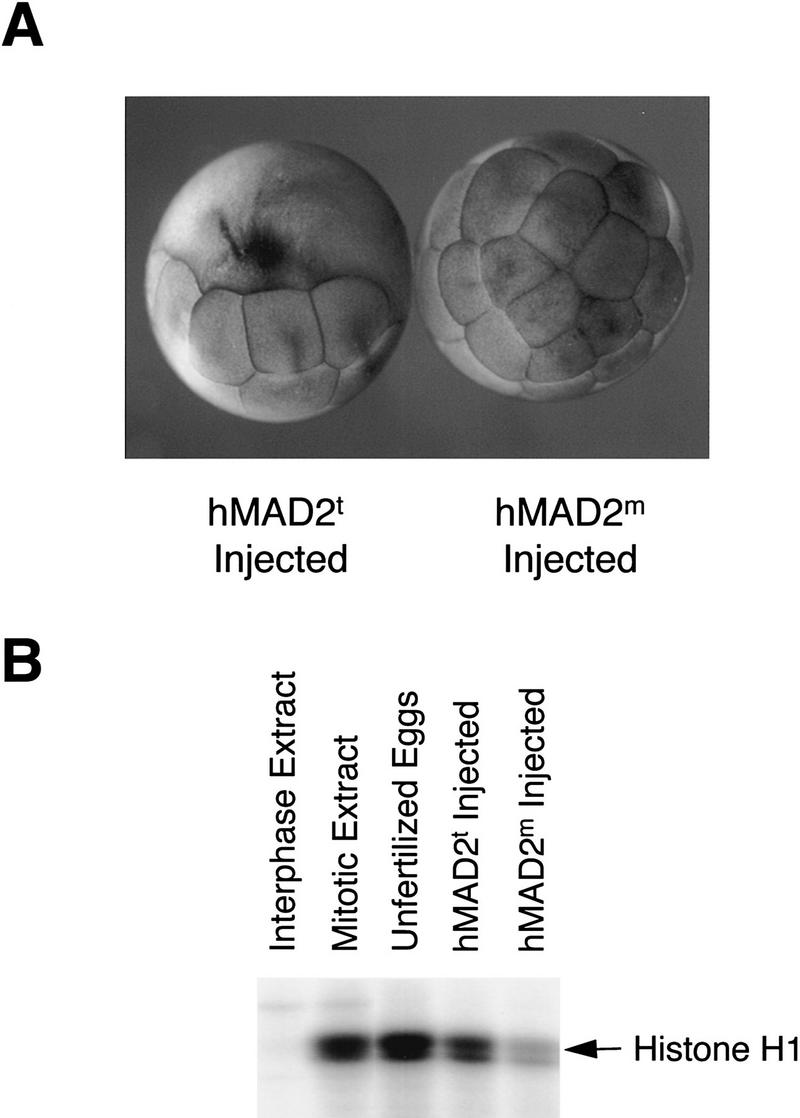
Block of cell division in Xenopus embryos by the tetrameric hMAD2. (A) Twenty-five nanoliters of 10 mg/ml recombinant hMAD2t and hMAD2m proteins were microinjected into one of the blastomeres in two-cell stage Xenopus embryos. The embryos were collected and analyzed 2 hr after injection. Cell cycle arrest by hMAD2t is stable for at least 8 hr before cells degenerate. (B) To assay histone H1 kinase assays, both blastomeres in two-cell stage embryos were injected with hMAD2, and extracts from injected embryos were analyzed for histone H1 kinase activity. Unfertilized eggs and equivalent aliquots of interphase and mitotic extracts were also assayed as controls.
Figure 4.

Inhibition of APC by hMAD2 oligomers in Xenopus mitotic extracts. (A) Effect of hMAD2 on cyclin degradation in Xenopus extracts. hMAD2t, hMAD2m, hMAD2ΔNd, hMAD2ΔNm, and hMAD2ΔC were incubated with Xenopus mitotic extracts for 20 min at a final concentration of 0.5 mg/ml before addition of the radioactive amino-terminal fragment of the Xenopus cyclin B1. Aliquots of the extracts were sampled at different times and the stability of the radioactive cyclin B1 was analyzed by SDS-PAGE. (B–D) The state of extracts and of APC in extracts treated with hMAD2 tetramer and monomer. hMAD2t and hMAD2m were incubated with Xenopus mitotic extracts for 20 min at a final concentration of 0.5 mg/ml. Aliquots of extracts were then assayed for histone H1 kinase activity and for the phosphorylation state of CDC27 by Western blot analysis (B). APC was immunopurified from the hMAD2-treated extracts with anti-CDC27 antibody beads and analyzed for the cyclin ubiquitination activity in the presence of recombinant E1 and E2 (C) and for subunit composition by silver staining (D). The mitotic APC subunits are labeled as APC1–APC8 in (D). Because of phosphorylation, the electrophoretic mobility of APC1(BIME), APC3(CDC27), APC8(CDC23) is retarded in mitotic APC; the corresponding interphase subunits are labeled by stars. (I) Interphase extracts; (M) mitotic extracts; (t) hMAD2 tetramer; (m) hMAD2 monomer. (E) Reactivation of hMAD2t-inhibited APC. hMAD2t was incubated with Xenopus mitotic extracts for 20 min at a final concentration of 0.5 mg/ml. APC was immunopurified from extracts with anti-CDC27 antibody beads and incubated with fresh mitotic extracts (lane 3) or interphase extracts (lane 5) in the absence of hMAD2. As a negative control, the APC beads was also incubated with mitotic extracts that have been preincubated with hMAD tetramer (lane 2). As a positive control, APC was immunopurified directly from mitotic extracts in the absence of hMAD2, incubated with mitotic extracts again, and then assayed for cyclin ubiquitination activity (lane 1). The APC beads were then washed stringently and assayed for cyclin ubiquitination activity. Activity detected in lane 3 is not attributable to an exchange of hMAD2t-treated APC with mitotic APC in extracts, as hMAD2t-treated APC is also reactivated in APC-depleted mitotic extracts (lane 4). (F) The dominant effect of monomeric hMAD2 on the inhibition by hMAD2 tetramer. hMAD2t was mixed with either hMAD2m or hMAD2ΔC and then added to Xenopus mitotic extracts. After addition of the radioactive amino-terminal fragment of cyclin B, aliquots of extracts were sampled at different times, and the stability of radioactive cyclin B was analyzed by SDS-PAGE.
The difference between monomer and tetramer cannot be attributed to a post-translational modification, as they have exactly the same molecular mass by mass spectrometric analysis (25,540.8 ± 1.3 Da for each tetramer subunit and 25,542.5 ± 1.8 Da for the monomer). The tetramer can convert to a dimer, but not to the monomer. When the purified tetramer was diluted to 0.4 mg/ml and loaded onto a Superdex 200 column, we detected a peak of 60 kD, corresponding to a dimer of hMAD2. No absorbance was detected in the monomeric region (data not shown). When fractions from this dimer peak were reloaded onto the column, hMAD2 protein still eluted as a dimer, not as a monomer (data not shown). On the other hand, the monomer does not seem to convert to the tetramer even at high protein concentrations under nondenaturing conditions. When the purified monomer was concentrated to 1.6 mg/ml and loaded onto the Superdex 200 column, it still elutes as a monomer of 30 kD (data not shown). However, when the monomer was denatured with 6 m guanidine hydrochloride and then slowly refolded at 0.15 mg/ml, >60% of the monomer is converted to the dimer (data not shown). The hMAD2 dimer has the same biochemical activity as the tetramer when assayed for inhibition of cyclin ubiquitination and degradation in Xenopus extracts (see below). Thus, it seems possible that the hMAD2 monomer and oligomers (dimer and tetramer) are kinetically stable conformers of the same protein. Neither the tetramer nor the monomer seems to be an unfolded form of the protein. When the protein conformation was analyzed by limited proteolysis under native conditions with trypsin, chymotrypsin, or endoproteinase C, hMAD2t, and hMAD2m gave rise to the same sets of digestion products, although they differed somewhat in proteolytic sensitivity (G. Fang, X. Luo, and M.W. Kirschner, unpubl.). Finally, hMAD2t, hMAD2m, and hMAD2ΔC (see below) have similar α-helical contents by circular dichroism, and thus, have a similar global structure (X. Luo, H. Yu, G. Fang, and M.W. Kirschner, unpubl.). Therefore, the ability for hMAD2 to oligomerize must be attributable to subtle folding differences. To begin to understand the structural basis of the monomer and tetramer conformation, we constructed two mutants, hMAD2ΔN and hMAD2ΔC, with 10 amino acids truncated from either the amino or the carboxyl terminus of hMAD2, respectively. By gel filtration chromatography, hMAD2ΔN exists as a dimer (hMAD2ΔNd) of 53 kD and a monomer (hMAD2ΔNm) of 28 kD, and hMAD2ΔC only exists as a monomer of 28 kD (see Fig. 2).
Tetrameric hMAD2 causes mitotic arrest when injected into Xenopus embryos
To test the biological function of hMAD2, we microinjected the recombinant hMAD2 tetramer or monomer into one of the blastomeres in two-cell stage Xenopus embryos. In all 40 injected embryos, hMAD2t caused an immediate and stable arrest of cell cycle progression on injected blastomeres, whereas the uninjected blastomeres continued to divide normally (Fig. 3A). Injection of hMAD2m had no effect on cell division (Fig. 3A). When the injected embryos were analyzed for histone H1 kinase activity, hMAD2t-injected blastomeres had a high level of the kinase activity, similar to that detected in extracts arrested at mitosis, whereas hMAD2m-injected blastomeres had a much lower histone H1 kinase activity (Fig. 3B). Thus, ectopic expression of hMAD2t arrests cells at mitosis, consistent with observations that overexpression of yeast Mad2p causes a cell cycle arrest in Schizosaccharomyces pombe (He et al. 1997), and that addition of the recombinant MAD2 protein to Xenopus cycling extracts arrests cell cycle progression (Li et al. 1997).
Oligomeric hMAD2 inhibits APC in Xenopus mitotic extracts
We then tested whether hMAD2t arrests the cell cycle by inhibiting APC. To assay this inhibition, hMAD2 protein was incubated with Xenopus egg extracts, which were arrested in mitosis by addition of a nondegradable form of cyclin B (mitotic extracts) (King et al. 1995). Radioactive amino-terminal fragment of Xenopus cyclin B was then added, and its stability analyzed. Without exogenously added hMAD2, >90% of cyclin was degraded within 20 min (Fig. 4A, panel 1). If the extracts were preincubated with hMAD2t or hMAD2ΔNd, no cyclin degradation was observed up to 60 min (panels 2, 4). Like hMAD2t, the wild-type hMAD2 dimer, generated either from a dilution of hMAD2t or from refolding of hMAD2m (see above), is active in inhibiting cyclin degradation (data not shown). Consistent with the micro-injection experiments, monomeric forms of hMAD2, hMAD2m, hMAD2ΔNm, and hMAD2ΔC, have no effect on cyclin degradation (panels 3, 5, 6). Addition of hMAD2t to extracts does not change the mitotic state of the extracts, as histone H1 kinase activity remains high and the CDC27 subunit of APC remains hyperphosphorylated (Fig. 4B, lane 3). These observations are consistent with an earlier report on the effect of MAD2 protein on Xenopus cycling extracts (Li et al. 1997).
To determine whether this inhibitory effect is directly on APC, APC was immunopurified from hMAD2t-treated extracts. As shown in Figure 4C (lane 3), APC isolated from extracts that had been incubated with hMAD2t is much less active in cyclin ubiquitination than interphase APC. Preincubation with hMAD2m, as expected, had no effect on mitotic APC activity, and by 60 min, 100% of the cyclin B1 was converted to conjugates (lane 4). This inhibition of APC by hMAD2t is not attributable to dissociation of APC into subcomplexes, as all eight subunits still form a tight complex in hMAD2t-treated extracts (Fig. 4D).
The checkpoint-mediated inhibition is expected to be reversible once the checkpoint signal disappears. To test the reversibility of inhibition by hMAD2, hMAD2t-inhibited APC was immunopurified and then added back to interphase extracts in the absence of exogenous hMAD2 protein. A short incubation reactivates the APC activity (Fig. 4E, lane 5), suggesting that there is an activity in interphase extracts that reverts the hMAD2t-mediated inhibition of APC. However, a longer incubation of hMAD2t-treated APC with interphase extracts inactivates this transiently active APC (data not shown). In control experiments, we found that incubation of mitotic APC with interphase extracts results in a continuous reduction in APC activity over time and leads to eventual inactivation of mitotic APC (data not shown). Thus, the hMAD2t-treated APC is different from both interphase and mitotic APC, as it can be activated transiently by an interphase activity. Like mitotic APC, this transiently active APC is eventually inactivated in interphase extracts. In addition, mitotic extracts can also reactivate hMAD2t-inhibited APC (Fig. 4E, lanes 3,4), and therefore, if there is a single reactivating activity, this activity is not cell cycle dependent.
The failure of hMAD2m to inhibit APC does not seem to be attributable to hMAD2m misfolding in E. coli. In addition to the biophysical data described above, we show in Figure 4F that the monomeric form of hMAD2 can act dominantly to abolish the inhibitory effect by hMAD2t. When hMAD2t was incubated with either hMAD2m or hMAD2ΔC and then added to Xenopus mitotic extracts, cyclin B was degraded with the same kinetics as when no exogenous hMAD2t protein was added to mitotic extracts.
Tetrameric hMAD2 inhibits activation of APC through association with hCDC20
CDC20, an activator of APC, has been shown to be required for the metaphase → anaphase transition (Dawson et al. 1995; Visintin et al. 1997; G. Fang, H. Yu, and M.W. Kirschner, in prep.). Our data on temporal activation of APC during mitosis also suggests a critical role of hCDC20 for anaphase initiation (see Fig. 6, below). One simple model for the inhibition of APC by the checkpoint pathway is that hMAD2 may prevent activation of APC by inhibiting hCDC20. To test this possibility directly, in vitro-translated hCDC20 was first incubated with hMAD2 tetramer or hMAD2ΔC monomer and then added to interphase APC beads. After incubation, the APC beads were washed and assayed for the cyclin ubiquitination activity. hCDC20 by itself activated APC very efficiently (Fig. 5A, lane 4). Preincubation of hCDC20 with hMAD2t inhibited completely the ability of hCDC20 to activate APC (cf. lane 5 with lane 4), whereas preincubation with hMAD2ΔC or hMAD2m had no effect on activation (lane 6; data not shown). Neither hMAD2 tetramer nor hMAD2ΔC monomer alone affected the basal activity level of interphase APC (lanes 1–3). This inhibition of APC by hMAD2 is not attributable to inhibition of binding of hCDC20 to APC, as we detected the same amount of APC-associated hCDC20 in the presence of either hMAD2 tetramer or monomer (Fig. 5A, lanes 5,6; data not shown).
Figure 6.
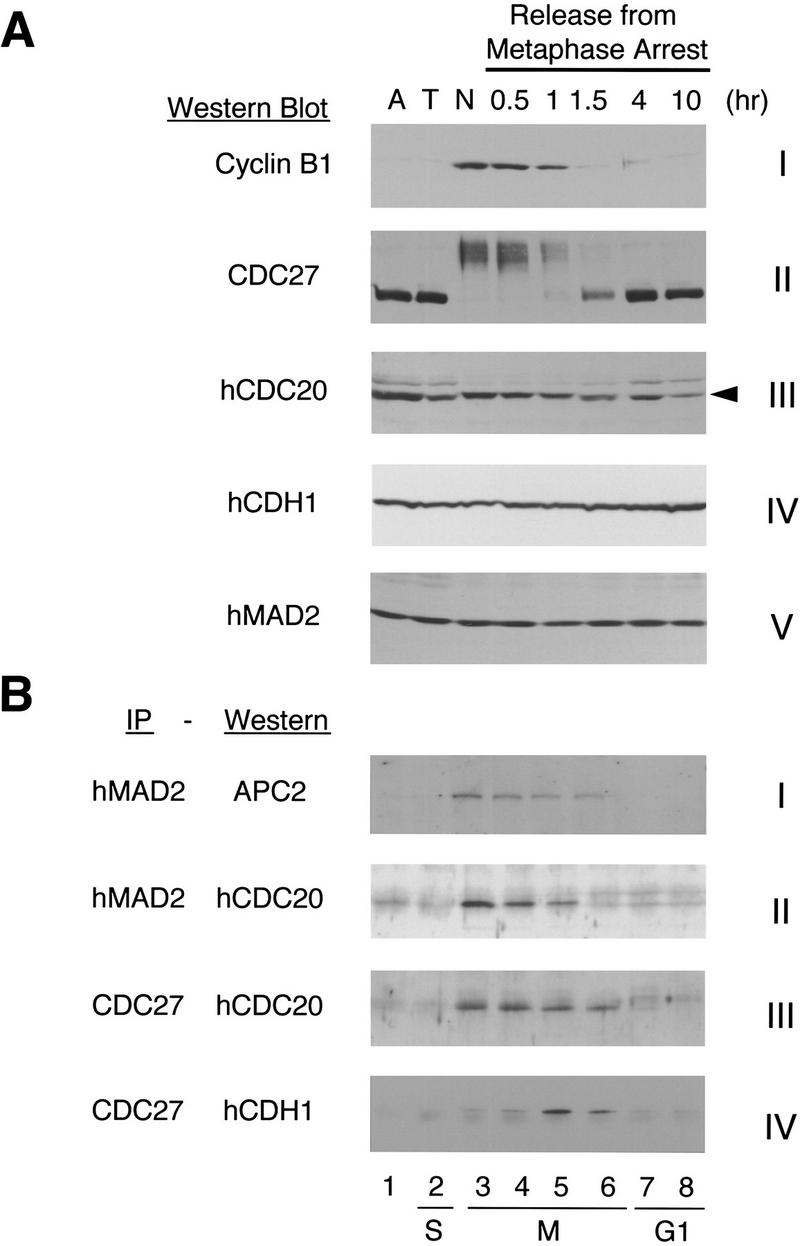
Cell cycle-regulated association of APC with hCDC20, hCDH1, and hMAD2. HeLa cells were synchronized at the prometaphase by a thymidine–nocodazole block. Cells were collected immediately after nocodazole treatment (lane 3, N), or released into fresh medium for 0.5, 1, 1.5, 4, and 10 hr (lanes 4–8). Asynchronous cells (lane 1, A) and cells arrested at G1/S boundary by a double thymidine block (lane 2, T) were included as controls. (A) HeLa cells were lysed with the SDS sample buffer and the levels of cyclin B1, CDC27, hCDC20, hCDH1, and hMAD2 proteins were determined by Western blot analysis. The arrowhead in panel III points to hCDC20. The band above is a cross-reacting protein. (B) Extracts from HeLa cells were immunoprecipitated with either anti-hMAD2 antibody beads or with anti-CDC27 antibody beads, and the immunoprecipitates were analyzed by Western blotting with anti-APC2, anti-hCDC20, or anti-hCDH1 antibodies.
Figure 5.


Inhibition of hCDC20-mediated activation of APC by hMAD2t. (A) 35S-labeled hCDC20 (lanes 4–6) was incubated for 30 min with hMAD2t (lane 5), hMAD2ΔC (lane 6), or with buffer (lane 4). As controls, rabbit reticulocyte lysates alone were incubated with hMAD2t (lane 2), hMAD2ΔC (lane 3), or with buffer (lane 1). APC was immunopurified from Xenopus interphase extracts with anti-CDC27 antibody beads, and then incubated with the hMAD2/hCDC20 mixture for 60 min. The APC beads were washed with buffer and assayed for cyclin ubiquitination activity. hCDC20 bound to APC beads was marked on the right. (t) hMAD2 tetramer; (m) hMAD2ΔC monomer. (B) 35S-labeled hCDC20 were incubated with either hMAD2t (lanes 1,2) or hMAD2ΔC (lanes 4,5) for 30 min. Affinity purified anti-hMAD2 antibody (lanes 1,3,4,6) or rabbit total IgG (lanes 2,5) were added to the hMAD2/hCDC20 mixture. After 90-min incubation, Affi-Prep protein A beads (Bio-Rad) were added and incubated for another 90 min. The beads were then washed stringently, and amount of hCDC20 bound to the beads was analyzed by SDS-PAGE. Input lane contains one-tenth amount of hCDC20 added to the binding reactions. For experiments described in A and B, similar results were achieved with purified hCDC20 protein. Briefly, in vitro translated HA-tagged hCDC20 was immunoprecipitated from reticulocyte lysates with anti-HA antibody/protein A beads. hCDC20 protein bound to antibody beads was eluted with the HA peptide and purified to apparent homogeneity as analyzed by silver staining (data not shown).
Given the effects of hMAD2t on the activity of APC, we asked whether hMAD2 can bind directly to hCDC20. To assay direct binding, hMAD2 tetramer and hMAD2ΔC monomer were first incubated with radioactive hCDC20, and then with affinity-purified anti-hMAD2 antibodies or control rabbit IgG. After incubation, hMAD2 complexes were immunopurified, and the amount of bound hCDC20 was analyzed by SDS-PAGE. As shown in Figure 5B, hCDC20 binds to both hMAD2t and hMAD2ΔC. Therefore, binding of hMAD2 to hCDC20 itself cannot be sufficient for inhibition of APC. This experiment also explains why the hMAD2 monomer blocks the ability of hMAD2 tetramer to inhibit APC, presumably by competing for hCDC20 binding. As we expected, an excess of the hMAD2 monomer on a molar basis is required for this dominant effect (data not shown). Our binding experiments are in agreement with a recent report on interactions between yeast Cdc20p and Mad2p found in two-hybrid screens (Hwang et al. 1998; Kim et al. 1998). In addition, our in vitro biochemical analysis suggests that a change in hMAD2 structure, not its association with hCDC20, may be responsible for transducing the checkpoint signal.
hMAD2, hCDC20, and APC form ternary complexes during mitosis
To confirm the importance of the interaction between hMAD2 and hCDC20 in vivo, we examined the cell cycle-dependent association of hMAD2, hCDC20, and APC in vivo. These experiments involve Western blot analysis as well as immunoprecipitations followed by Western blot analysis for MAD2, CDC20, CDH1, cyclin B, and two subunits of APC (APC2 and CDC27) (Fig. 6). Asynchronous cells and S-phase cells are in the first and second lanes. The nocodazole-arrested cells are shown in lane 3. Cells that were released from the prometaphase arrest and harvested at different stages of mitosis and G1 are shown in lanes 4–8. Cyclin B1 protein was, as expected, degraded between 60 and 90 min after release from the nocodazole arrest (Fig. 6A, panel I). At the time of arrest and through the period of cyclin disappearance, APC is present in a phosphorylated form, as assayed by the mobility retardation of CDC27 (panel II). The levels of hCDH1 and hMAD2 proteins do not vary through cell cycle (panels IV,V). The level of hCDC20 drops slowly when cells enter G1 (panel III). We observed a retardation in migration rate of mitotic hCDC20, suggesting that hCDC20 is post-translationally modified during mitosis, consistent with an earlier report on phosphorylation of CDC20 (Weinstein 1997).
To measure the composition of various complexes of APC with hMAD2, hCDC20, and hCDH1 during the exit from metaphase through G1, we performed immunoprecipitation and Western blotting experiments. When the cell lysates were immunoprecipitated with anti-hMAD2 antibodies, and analyzed with an antibody against APC2, we found that hMAD2 is associated with APC through most of mitosis (Fig. 6B, panel I, lanes 3–6). To determine the temporal association of hCDC20 with hMAD2 and APC during mitosis, we performed immunoprecipitations with hMAD2 or CDC27 and then probed with antibodies to hCDC20. hCDC20 is associated with hMAD2 during the first 60 min after release from nocodazole (Fig. 6B, panel II, lanes 3–5). After that, hCDC20 remains associated with APC for at least 30 min (Fig. 6B, panel III, lanes 3–6). Thus, hMAD2–hCDC20–APC forms a ternary complex for 60 min when cells were first released from prometaphase arrest. Thereafter hMAD2 dissociates from the ternary complex, and a hCDC20–APC binary complex remains. It is during this time, 60–90 min after release from the nocodazole arrest, that APC is active and cyclin is degraded (Fig. 1C and 6A). Association of hCDH1 with APC is very low when cells were first released from prometaphase arrest and in G1 (Fig. 6B, panel IV, lanes 3,4,7,8). However, there is a burst in the level of hCDH1–APC complex late in mitosis (panel IV, lanes 5,6).
We also checked the subcellular localization of hMAD2 and hCDC20 by indirect immuno-fluorescence antibody staining. In addition to diffusive cytoplasmic staining, hMAD2 and hCDC20 colocalize to the kinetochores during prometaphase (Fig. 7). It has been shown that APC also localizes to the kinetochores during mitosis (Jorgensen et al. 1998). Therefore, these data indicated that hMAD2, hCDC20, and APC form a ternary complex at the kinetochores during prometaphase.
Figure 7.
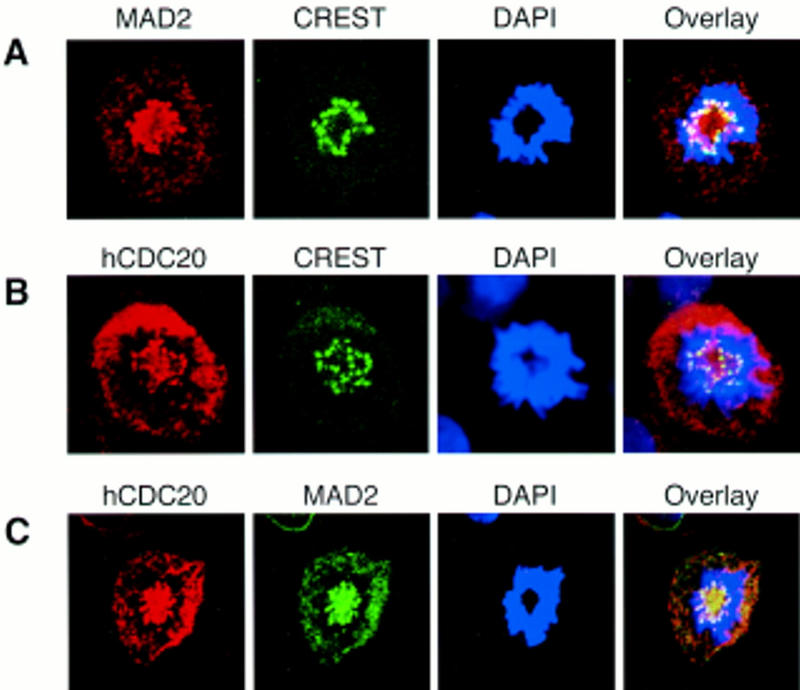
hMAD2 and hCDC20 colocalize to kinetochores at prometaphase. (A) HeLa cells were stained with anti-hMAD2, CREST, and DAPI. (B) Hela cells were stained with anti-hCDC20, CREST, and DAPI. (C) Hela cells were stained with anti-hCDC20, anti-hMAD2, and DAPI.
Discussion
We show here that the checkpoint pathway arrests the cell cycle by inhibiting premature activation of APC before anaphase. The temporal association of hCDC20 and hCDH1 with APC during mitosis suggests a critical role of CDC20 in activation of APC at the metaphase → anaphase transition. The checkpoint protein hMAD2 binds to the hCDC20–APC complex in vivo and prevents activation of APC before anaphase. The recombinant hMAD2 protein exists either as a tetramer or as a monomer in vitro. Significantly, only the tetramer inhibits the CDC20–APC complex. Thus, a change in the conformation or the oligomerization state of hMAD2 may play a role in transducing the checkpoint signal to APC.
Regulation of APC in the cell cycle
It was proposed that APC, like other mitotic activities, is regulated by phosphorylation during mitosis (King et al. 1995; Lahav-Baratz et al. 1995; Peters et al. 1996). However, recent results indicate that regulation of APC involves more than phosphorylation. Mitotic APC is principally controlled by its association with different positive and negative regulatory proteins at different stages of mitosis. Phosphorylation plays an important, but ancillary role (G. Fang, H. Yu, and M.W. Kirschner, in prep.). Binding of these regulatory factors confers a precise temporal regulation of APC activity and a temporal order of substrate degradation. On the basis of work presented here, we can identify at least three different APC complexes during mitosis: an inactive MAD2–CDC20–APC ternary complex before anaphase, a CDC20–APC binary complex active at anaphase, and a CDH1–APC complex active later in mitosis and probably also in G1.
From experiments presented here, we can sketch a pathway of interconversion among these APC states (Fig. 8). Of the components of the APC regulatory network, only the level of the CDC20 protein changes during the cell cycle. CDC20 synthesis begins in G2 and peaks during mitosis. The protein level drops slowly as cells enter G1 (Fig. 6; G. Fang, H. Yu, and M.W. Kirschner, in prep.). The newly synthesized CDC20 may be bound to the checkpoint protein MAD2, and therefore sequestered from activating APC. As cells enter mitosis, APC is phosphorylated under indirect control of the Cdc2 kinase (King et al. 1995; Lahav-Baratz et al. 1995; Peters et al. 1996; data not shown), and phosphorylation increases the affinity for CDC20 to APC (G. Fang, H. Yu, and M.W. Kirschner, in prep.). In addition, phosphorylation of CDC20 during mitosis may also play a role in the control of CDC20 and APC association. Early in mitosis, MAD2, CDC20, and APC form a ternary complex, and this complex is inactive in ubiquitinating substrates. The existence of a MAD2, CDC20, and APC ternary complex in vivo is based on the following observations. First, by immunoprecipitation and Western blot analysis of nocodazole-arrested HeLa cells, we detected associations of MAD2 with CDC20, CDC20 with APC, and MAD2 with APC (Fig. 6). Second, in vitro binding experiments indicate that MAD2 does not interact directly with APC in the absence of CDC20 (data not shown). On the other hand, binding of CDC20 to MAD2 does not affect its ability to associate with APC (Fig. 5A; data not shown). Thus, the observed in vivo association between MAD2 and APC (Fig. 6; Li et al. 1997) is most likely mediated through CDC20.
Figure 8.

A model for a regulatory network of APC in the cell cycle.
Recently, it has been reported that Mad2p associates with Cdc20p throughout the cell cycle in the budding yeast (Hwang et al. 1998). We found that in HeLa cells hMAD2 associates with hCDC20 only during early mitosis, but not in the rest of the cell cycle (Fig. 6). This apparent discrepancy could result either from a difference in the checkpoint mechanism among different organisms, or from different experimental designs. To test the latter possibility, HeLa cells were synchronized at the G1/S boundary by a double thymidine block, and then released to allow progression through the cell cycle. Again, hMAD2 was found to associate with hCDC20 only during early mitosis, but not in G1, S, and G2 (data not shown). In addition, we detected the hMAD2, hCDC20, and APC ternary complex during early mitosis, suggesting that the checkpoint mechanism functions during each cell cycle even in the absence of exogenous spindle-damaging drugs.
When all the chromosomes are aligned at the metaphase plate, an unidentified signal causes the MAD2 protein to dissociate from the ternary complex, leaving CDC20 in a binary complex with APC, which has a high ubiquitination activity. Activated APC in turn ubiquitinates anaphase inhibitors, such as Pds1p in S. cerevisiae, Cut2p in S. pombe, and the putative metazoan equivalent (Cohen-Fix et al. 1996; Funabiki et al. 1996; Yamamoto et al. 1996a; Visintin et al. 1997), and initiates the metaphase → anaphase transition. The CDC20-associated APC has a very limited set of substrates and only recognizes Destruction-box (D-box)-containing proteins (Glotzer et al. 1991; King et al. 1996; G. Fang, H. Yu, and M.W. Kirschner, in prep.).
How is MAD2 released from the MAD2–CDC20–APC complex? We have found that the ternary complex is stable in the presence of 500 mm KCl and 0.5% NP-40 (data not shown), suggesting that MAD2 is unlikely to dissociate from the complex by itself. Instead, we have detected a cell cycle-independent activity in Xenopus egg extracts that reactivates the MAD2-treated APC. Because there is no spindle assembly checkpoint in early embryonic divisions (Minshull et al. 1994; Clute and Masui 1995, 1997), it is not clear whether this activity is responsible for the dissociation of MAD2 from APC at the metaphase → anaphase transition in somatic cells, but it suggests that certain cofactors are required for MAD2 dissociation.
CDH1 and APC association occurs after the metaphase → anaphase transition when cells are ready to exit from mitosis (Fig. 6). CDH1-associated APC recognizes substrates with a relaxed D-box specificity (G. Fang, H. Yu, and M.W. Kirschner, in prep.), suggesting that CDH1–APC might have a much broader spectrum of substrates and is likely to be involved in regulation of multiple processes late in mitosis. The identification of these substrates is an important task in understanding late anaphase events.
CDH1 and APC protein levels do not change during the cell cycle, and yet there is a distinct peak of CDH1–APC complex late in mitosis (Fig. 6). Although the mechanism that regulates CDH1 and APC association remains unclear, the absence of association between CDH1 and APC during early mitosis is unlikely because of a competition between CDC20 and CDH1 for APC. We find that APC immunopurified from HeLa cells at different stages of mitosis can be further activated several fold by either CDC20 or CDH1 (data not shown), suggesting that even in mitosis CDC20 and CDH1 are bound to APC at substoichiometric levels. The fact that only a fraction of APC is active at each stage of mitosis raises an interesting question about whether the activated forms of APC are uniformly present in the cell, or whether spatial control is imposed on the observed temporal control.
As cells exit from mitosis, Cdc2 kinase is inactivated because of degradation of mitotic cyclins. Dephosphorylation of APC may cause dissociation of the CDC20–APC complex, and free CDC20 is slowly degraded. A low level of CDH1 is associated with APC in G1 cells, and CDH1 is likely to be responsible for activation of APC in G1 (Fig. 8). As cells enter S-phase, CDH1 dissociates from APC. Some event at the G1/S transition, such as the activation of S-phase kinases, might regulate dissociation of hCDH1 from APC and suppress APC activity in S-phase cells (Amon et al. 1994; Amon 1997; Sigrist and Lehner 1997).
Spindle assembly checkpoint, MAD2, and APC
At the molecular level, little is known about the mechanism of MAD2-mediated inhibition. CDC20 has been shown to be a substrate-specific activator of APC and therefore, might be involved in substrate binding (Visintin et al. 1997). Thus, it is possible that association of MAD2 with CDC20 blocks substrate binding. Another possibility is that MAD2 binding may induce a conformation change in CDC20 so that it is no longer active. Alternatively, as a CDC20-associated kinase activity has been reported (Weinstein et al. 1994), phosphorylation or dephosphorylation of CDC20 might play a role in CDC20 activity and in checkpoint control. Indeed, it has been reported that in S. cerevisiae a regulatory subunit of protein phosphatase 2A, Cdc55p, is required for the spindle assembly checkpoint, and that the loss-of-function mutation in CDC55 suppresses the temperature sensitivity of cdc20-1 (Wang and Burke 1997).
How does MAD2 respond to the checkpoint signal? In vitro, MAD2 protein exists in two forms: a tetramer and a monomer. By limited analytical criteria (1D-NMR, circular dichroism, and protease sensitivity), both forms seem to be folded and have similar amounts of α-helical contents. These two forms only interconvert under denaturing conditions, suggesting that a substantial energy barrier exists between two conformers. Biochemical analysis of APC activity and experiments with Xenopus embryos and extracts indicate that only the oligomeric form of MAD2 is responsible for transducing an inhibitory signal to APC (Figs. 3–5). Although we do not have in vivo evidence for oligomerization of MAD2 upon activation of checkpoint, it is conceivable that either the oligomerization of MAD2 or a conformation associated with the MAD2 tetrameric state may transduce the checkpoint signal. Consistent with this model, we observed an increase in the level of MAD2 protein in high molecular weight complexes in nocodazole-arrested HeLa cell lysates by gel filtration, as compared to lysates from asynchronous cells. However, the majority of MAD2 protein seems to exist as a monomer, even in nocodazole-arrested HeLa cells (data not shown). In S. cerevisiae, Mad1p forms a complex with Mad2p and is hyperphosphorylated upon activation of the checkpoint (Hardwick and Murray 1995). It is possible that Mad1p may transduce the checkpoint signal through inducing a change in Mad2p structure; the tetrameric state of MAD2 in vitro could mimic certain aspects of the checkpoint-induced MAD2 conformation. Recently, it has been reported that human homolog of yeast Mad1p can self-associate (Jin et al. 1998). Thus, we speculate that binding of MAD2 to MAD1 may induce oligomerization of MAD2.
In summary, we have shown that APC is regulated by positively and negatively acting regulatory factors in the cell cycle and in checkpoint control. Different complexes of APC control different events in mitosis and in G1, presumably by ubiquitinating and degrading specific substrates. These findings have greatly enhanced our understanding of the control of mitosis, and also raise many new questions about the regulation of APC-dependent proteolysis. For example, what is the exact role of phosphorylation of CDC20, CDH1, and several APC subunits in the regulation of cell cycle progression and in checkpoint control? What determines the temporal order of MAD2, CDC20, CDH1, and APC association and dissociation during mitosis? What controls the release of MAD2 from the inactive MAD2–CDC20–APC ternary complex at the metaphase to anaphase transition? Does MAD2 in vivo exist in two functional states and if so, what factors drive this allosteric transition? What is the function of APC in G1? For example, does it prevent premature DNA replication? What represses the APC activity in S-phase cells? Does APC play a role in cellular differentiation? At the molecular level, we still do not understand how CDC20 and CDH1 activate APC and how MAD2 inhibits this activation. The identification of the APC regulatory network and its connection through MAD2 to mitotic checkpoint controls will allow us to address these important questions. Further studies of these regulatory interventions should lead to a better understanding of the role of proteolysis.
Materials and methods
Antibodies
Affinity-purified antibodies against human cyclin B1 and hCDC20 were purchased from Santa Cruz and used at a final concentration of 1 μg/ml in immunoblotting experiments. The anti-hMAD2 polyclonal rabbit serum was raised against the recombinant hMAD2 protein (a mixture of tetramer and monomer). Anti-hMAD2 antibodies were affinity-purified with the hMAD2 monomer covalently coupled to Affi-Gel 15 (Bio-Rad) (Harlow and Lane 1988). The anti-hCDH1 polyclonal rabbit serum was raised against two peptides derived from the hCDH1 protein: residues 65–79 and residues 108–123. The antibodies were affinity-purified with each peptide antigen coupled separately to Sulfolink Gel (Pierce), and in Western blot analysis both antibodies recognize a single band with molecular mass expected for hCDH1, demonstrating the specificity of the antibodies. The rabbit anti-APC2 antibodies were raised against a carboxy-terminal fragment (residues 701–823) of the human APC2, which was subcloned into pGEX–2TK (Pharmacia) and expressed as a GST fusion protein. The fusion protein was purified and cleaved with thrombin, and the APC2 fragment was separated from GST by gel filtration chromatography. The rabbit anti-CDC27 antibodies were made against a carboxy-terminal fragment containing six tetratrico peptide repeats of human CDC27, which was subcloned into pET-28a (Novagen), expressed with an amino-terminal His-tag, purified with nickel–agarose beads (Qiagen).
Expression and purification of the recombinant hMAD2 protein
The hMAD2 protein was tagged with His6 at its amino terminus by subcloning the gene into the expression vector pET-28a (Novagen). The recombinant protein was expressed in BL21(DE3)pLysS and purified with a nickel–agarose column (Qiagen). The hMAD2 protein was then loaded onto a gel filtration column, either S100 or Superdex 200 (Pharmacia), and eluted with a buffer containing 10 mm HEPES (pH 7.7), 100 mm KCl, 1 mm MgCl2, and 1 mm DTT. The peak fractions for the monomer and tetramer were pooled separately and concentrated. hMAD2ΔN and hMAD2ΔC mutants were constructed by PCR mutagenesis, expressed, and purified as described above.
Microinjection of hMAD2 into Xenopus embryos and histone H1 kinase assay
Xenopus eggs were fertilized with sperms by standard procedures, and embryos were incubated in 0.1×MMR [5 mm HEPES (pH 7.8), 100 mm NaCl, 2 mm KCl, 0.1 mM EDTA, 2 mm CaCl2, and 1 mm MgCl2]. Immediately after the first cell division, 25 nl of 10 mg/ml hMAD2t or hMAD2m proteins were microinjected into one of the blastomeres, and the injected embryos were transferred to 0.1×MMR with 1% Ficoll. For histone H1 kinase assay, both blastomeres in two-cell stage embryos were injected. Four injected embryos were pooled in 15 μl of EB buffer [80 mm β-glycerophosphate (pH 7.4), 15 mm MgCl2, 10 mm EGTA, and 0.1% NP-40] in the presence of 10 μg/ml each of leupeptin, pepstatin, and chymostatin. Embryos were frozen and thawed once, and then spun at 20,000g for 3 min. Supernatants 10 μl were used for histone H1 kinase assay. Xenopus egg extracts were prepared as previously described (King et al. 1995).
Preparation of synchronized HeLa cell extracts
HeLa S3 cells (ATCC) were grown in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 100 μg/ml penicillin, and streptomycin. For synchronization with a double thymidine block, cells were grown in the presence of 2 mm thymidine (Sigma) for 18 hr, washed with PBS, and grown in fresh medium without thymidine for 8 hr. Cells were then incubated with 2 mm thymidine for 18 hr to block cells at the G1/S boundary. To arrest cells in mitosis, cells were first treated with 2 mm thymidine for 18 hr, released into fresh medium for 3–4 hrs, and then blocked with medium containing 100 ng/ml nocodazole (Sigma) for 12 hr. Cells were washed with PBS twice, either harvested immediately or transferred into fresh medium for 0.5, 1, 1.5, 4, or 10 hr, and then harvested. The cell cycle status of the samples was determined by FACS analysis of the DNA content.
For preparation of extracts, cells were lysed with seven volumes of the NP-40 lysis buffer [50 mm Tris-HCl (pH 7.7), 150 mm NaCl, 0.5% NP-40, 1 mm DTT, 10% glycerol, 0.5 μm okadaic acid, and 10 μg/ml each of leupeptin, pepstatin, and chymostatin]. The lysates were then centrifuged for 30 min at 200,000g to make the high speed supernatant.
Immunoprecipitation and immunofluorescence
Anti-CDC27 antibodies and anti-hMAD2 antibodies were coupled covalently to Affi-Prep protein A beads (Bio-Rad) as described (Harlow and Lane 1988). The beads were washed twice with 10 volumes of 100 mm glycine (pH 2.5), and neutralized with 10 mm Tris-HCl (pH 7.5). Three microliters of anti-CDC27 or anti-hMAD2 antibody beads were incubated with 250 μl of HeLa cell S100 supernatants at 4°C for 2 hr. The beads were washed five times with 20 volumes of XB buffer [10 mm HEPES (pH 7.7), 100 mm KCl, 0.1 mm CaCl2, 1 mm MgCl2, 50 mm sucrose] containing 500 mm KCl and 0.5% NP-40, and three times with XB alone. Proteins bound to beads were eluted with SDS sample buffer, separated by SDS-PAGE (5%–15%), and analyzed by Western blotting.
Asynchronous HeLa cells growing on chambered slides (Lab Tech) were fixed with 4% paraformaldehyde in PBS for 5 min, permeabilized with PBS containing 0.1% Triton X-100, and blocked with PBS containing 0.1% Triton X-100 and 3% BSA. Cells were then incubated with 5 μg/ml anti-hMAD2, 1 μg/ml anti-hCDC20, or 1:500 dilution of CREST serum for overnight at 4°C in the presence of the blocking buffer. After washing with PBS containing 0.1% Triton X-100, the slides were blocked with PBS containing 0.1% Triton X-100 and 5% donkey serum and stained with 1:200 dilution of FITC- or Cy3-coupled secondary antibodies (Jackson Laboratories) for 1 hr at room temperature. Slides were washed twice with PBS, once with PBS containing 1 μg/ml DAPI (Sigma), twice with PBS, and mounted. The images were viewed with a X100 objective lens on a Zeiss LSM confocal microscope.
Cyclin degradation and ubiquitination assays
To assay cyclin degradation in crude mitotic extracts, equal volumes of 2 mg/ml hMAD2 and Xenopus mitotic extracts were mixed and incubated at room temperature for 20 min. The substrate used in the assay was an amino-terminal fragment of Xenopus cyclin B1 (residues 1–102), and was labeled with 125I to a specific activity of 100 μCi/μg using the chloramine T procedure (Parker 1990). To initiate the cyclin degradation reaction, bovine ubiquitin (Sigma) and the labeled substrate were added to the hMAD2-preincubated extracts at final concentrations of 1.25 mg/ml and 12.5 ng/ml, respectively. Reactions were stopped by SDS sample buffer at different time after substrate addition and analyzed by SDS-PAGE (5%–15%). To assay dominant negative effect of hMAD2m, hMAD2t was incubated with hMAD2m or hMAD2ΔC for 5 min at room temperature. The protein mixture was then added to mitotic extracts and incubated for another 20 min before addition of the substrate to initiate degradation reactions. The final concentrations of hMAD2t, hMAD2ΔC, and hMAD2m protein in extracts were 0.25, 0.125, and 0.8 mg/ml, respectively.
To assay the APC activity in HeLa cells, 3 μl of anti-CDC27 antibody beads were incubated with 500 μl of HeLa cell S100 supernatants at 4°C for 2 hr. The APC beads were washed five times with 20 volumes of XB buffer containing 500 mm KCl and 0.5% NP-40, and twice with XB. The APC beads were then assayed for cyclin ubiquitination activity. Ubiquitination assays were performed in a total volume of 5 μl. The reaction mixture contains an energy regenerating system, 1.25 mg/ml of bovine ubiquitin, 12.5 ng/ml of labeled substrate, 200 μg/ml wheat E1, 50 μg/ml Xenopus UBCx, and 2 μl of APC beads. The reactions were incubated at room temperature for 1 hr, quenched with SDS sample buffer, and analyzed by SDS-PAGE (5%–15% gradient gels). Gels were scanned with a PhosphorImager (Molecular Dynamics).
To purify interphase APC, the anti-CDC27 beads were incubated with 10 volumes of interphase Xenopus egg extracts for 2 hr at 4°C and washed five times with 20 volumes of XB containing 500 mm KCl and 0.5% NP-40 and twice with XB. The interphase APC beads were then incubated for 1 hr at room temperature with in vitro translated hCDC20 that had been preincubated with the hMAD2 protein. After incubation, APC beads were washed twice with XB, and assayed for cyclin ubiquitination activities.
Acknowledgments
We thank X. Luo for help with the CD spectroscopy analysis; J. Roberts, L. Ma, and R. Davis for assistance with immunofluorescence; and the Harvard Microchemistry Laboratory for the mass spectroscopic analysis of hMAD2 protein. We are grateful to R. Li for discussion and F. McKeon and S. Taylor for comments on the manuscript. G.F. is a recipient of a Helen Hay Whitney Postdoctoral Fellowship. H.Y. is supported by the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation Fellowship, DRG-1340. This work is supported by grants GM39023-08 and GM26875-17 from the National Institutes of Health to M.W.K.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL marc@hms.harvard.edu; FAX (617) 432-0420.
References
- Amon A. Regulation of B-type cyclin proteolysis by Cdc28-associated kinases in budding yeast. EMBO J. 1997;16:2693–2702. doi: 10.1093/emboj/16.10.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon A, Irniger S, Nasmyth K. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 1994;77:1037–1050. doi: 10.1016/0092-8674(94)90443-x. [DOI] [PubMed] [Google Scholar]
- Brandeis M, Hunt T. The proteolysis of mitotic cyclins in mammalian cells persists from the end of mitosis until the onset of S phase. EMBO J. 1996;15:5280–5289. [PMC free article] [PubMed] [Google Scholar]
- Chen RH, Waters JC, Salmon ED, Murray AW. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science. 1996;274:242–246. doi: 10.1126/science.274.5285.242. [DOI] [PubMed] [Google Scholar]
- Clute P, Masui Y. Regulation of the appearance of division asynchrony and microtubule-dependent chromosome cycles in Xenopus laevis embryos. Dev Biol. 1995;171:273–285. doi: 10.1006/dbio.1995.1280. [DOI] [PubMed] [Google Scholar]
- ————— Microtubule dependence of chromosome cycles in Xenopus laevis blastomeres under the influence of a DNA synthesis inhibitor, aphidicolin. Dev Biol. 1997;185:1–13. doi: 10.1006/dbio.1997.8540. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Peters J-M, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes & Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- Dawson IA, Roth S, Artavanis TS. The Drosophila cell cycle gene fizzy is required for normal degradation of cyclins A and B during mitosis and has homology to the CDC20 gene of Saccharomyces cerevisiae. J Cell Biol. 1995;129:725–737. doi: 10.1083/jcb.129.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints—preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Funabiki H, Yamano H, Kumada K, Nagao K, Hunt T, Yanagida M. Cut2 proteolysis required for sister-chromatid separation in fission yeast. Nature. 1996;381:438–441. doi: 10.1038/381438a0. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Hardwick KG, Murray AW. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J Cell Biol. 1995;131:709–720. doi: 10.1083/jcb.131.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science. 1996;273:953–956. doi: 10.1126/science.273.5277.953. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- He X, Patterson TE, Sazer S. The Schizosaccharomyces pombe spindle checkpoint protein mad2p blocks anaphase and genetically interacts with the anaphase-promoting complex. Proc Natl Acad Sci. 1997;94:7965–7970. doi: 10.1073/pnas.94.15.7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway SL, Glotzer M, King RW, Murray AW. Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell. 1993;73:1393–1402. doi: 10.1016/0092-8674(93)90364-v. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–517. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. Budding yeast Cdc20: A target of the spindle checkpoint. Science. 1998;279:1041–1044. doi: 10.1126/science.279.5353.1041. [DOI] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;81:269–277. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- Jin DY, Spencer F, Jeang KT. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell. 1998;93:81–91. doi: 10.1016/s0092-8674(00)81148-4. [DOI] [PubMed] [Google Scholar]
- Jorgensen PM, Brundell E, Starborg M, Höög C. A subunit of the anaphase-promoting complex is a centromere-associated protein in mammalian cells. Mol Cell Biol. 1998;18:468–476. doi: 10.1128/mcb.18.1.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Lin DP, Matsumoto S, Kitazono A, Matsumoto T. Fission yeast Slp1: An effector of the Mad2-dependent spindle checkpoint. Science. 1998;279:1045–1047. doi: 10.1126/science.279.5353.1045. [DOI] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- King RW, Glotzer M, Kirschner MW. Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol Biol Cell. 1996;7:1343–1357. doi: 10.1091/mbc.7.9.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav-Baratz S, Sudakin V, Ruderman JV, Hershko A. Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase. Proc Natl Acad Sci. 1995;92:9303–9307. doi: 10.1073/pnas.92.20.9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- Li Y, Gorbea C, Mahaffey D, Rechsteiner M, Benezra R. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc Natl Acad Sci. 1997;94:12431–12436. doi: 10.1073/pnas.94.23.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T. A fission yeast homolog of CDC20/p55CDC/Fizzy is required for recovery from DNA damage and genetically interacts with p34cdc2. Mol Cell Biol. 1997;17:742–750. doi: 10.1128/mcb.17.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshull J, Sun H, Tonks NK, Murray AW. A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell. 1994;79:475–486. doi: 10.1016/0092-8674(94)90256-9. [DOI] [PubMed] [Google Scholar]
- Murray AW. Cell cycle checkpoints. Curr Opin Cell Biol. 1994;6:872–876. doi: 10.1016/0955-0674(94)90059-0. [DOI] [PubMed] [Google Scholar]
- ————— The genetics of cell cycle checkpoints. Curr Opin Genet Dev. 1995;5:5–11. doi: 10.1016/s0959-437x(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Nicklas RB. How cells get the right chromosomes. Science. 1997;275:632–637. doi: 10.1126/science.275.5300.632. [DOI] [PubMed] [Google Scholar]
- Parker CW. Radiolabeling of proteins. Methods Enzymol. 1990;182:721–737. doi: 10.1016/0076-6879(90)82056-8. [DOI] [PubMed] [Google Scholar]
- Peters J-M, King RW, Höög C, Kirschner MW. Identification of BIME as a subunit of the anaphase-promoting complex. Science. 1996;274:1199–1201. doi: 10.1126/science.274.5290.1199. [DOI] [PubMed] [Google Scholar]
- Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–780. doi: 10.1016/s0955-0674(96)80077-9. [DOI] [PubMed] [Google Scholar]
- Schwab M, Lutum AS, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- Sigrist SJ, Lehner CF. Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell. 1997;90:671–681. doi: 10.1016/s0092-8674(00)80528-0. [DOI] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–198. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Tugendreich S, Tomkiel J, Earnshaw W, Hieter P. The CDC27HS protein co-localizes with the CDC16HS protein to the centrosome and mitotic spindle and is essential for the metaphase to anaphase transtion. Cell. 1995;81:261–268. doi: 10.1016/0092-8674(95)90336-4. [DOI] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1—a family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Wang Y, Burke DJ. Cdc55p, the B-type regulatory subunit of protein phosphatase 2A, has multiple functions in mitosis and is required for the kinetochore/spindle checkpoint in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:620–626. doi: 10.1128/mcb.17.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J. Cell cycle-regulated expression, phosphorylation, and degradation of p55CDC. A mammalian homolog of CDC20/Fizzy/slp1. J Biol Chem. 1997;272:28501–28511. doi: 10.1074/jbc.272.45.28501. [DOI] [PubMed] [Google Scholar]
- Weinstein J, Jacobsen FW, Hsu CJ, Wu T, Baum LG. A novel mammalian protein, p55CDC, present in dividing cells is associated with protein kinase activity and has homology to the Saccharomyces cerevisiae cell division cycle proteins Cdc20 and Cdc4. Mol Cell Biol. 1994;14:3350–3363. doi: 10.1128/mcb.14.5.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Murakami H, Okayama H. A WD repeat protein controls the cell cycle and differentiation by negatively regulating Cdc2/B-type cyclin complexes. Mol Biol Cell. 1997;8:2475–2486. doi: 10.1091/mbc.8.12.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Guacci V, Koshland D. Pds1p is required for faithful execution of anaphase in the yeast, Saccharomyces cerevisiae. J Cell Biol. 1996a;133:85–97. doi: 10.1083/jcb.133.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s) J Cell Biol. 1996b;133:99–110. doi: 10.1083/jcb.133.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]