

Abstract
A commonly accepted view of gene regulation in bacteria that has emerged over the last decade is that promoters are transcriptionally activated by one of two general mechanisms. The major type involves activator proteins that bind to DNA adjacent to where the RNA polymerase (RNAP) holoenzyme binds, usually assisting in recruitment of the RNAP to the promoter. This holoenzyme uses the housekeeping ς70 or a related factor, which directs the core RNAP to the promoter and assists in melting the DNA near the RNA start site. A second type of mechanism involves the alternative sigma factor (called ς54 or ςN) that directs RNAP to highly conserved promoters. In these cases, an activator protein with an ATPase function oligomerizes at tandem sites far upstream from the promoter. The nitrogen regulatory protein (NtrC) from enteric bacteria has been the model for this family of activators. Activation of the RNAP/ς54 holoenzyme to form the open complex is mediated by the activator, which is tethered upstream. Hence, this class of protein is sometimes called the enhancer binding protein family or the NtrC class. We describe here a third system that has properties of each of these two types. The NtrC enhancer binding protein from the photosynthetic bacterium, Rhodobacter capsulatus, is shown in vitro to activate the housekeeping RNAP/ς70 holoenzyme. Transcriptional activation by this NtrC requires ATP binding but not hydrolysis. Oligomerization at distant tandem binding sites on a supercoiled template is also necessary. Mechanistic and evolutionary questions of these systems are discussed.
Keywords: Gene regulation, enhancer, ATP-dependent, housekeeping holoenzyme
Compilations of data from studies on nearly 150 regulated promoters in enteric bacteria have led to the view that a bacterial promoter is activated by use of one of two systems (for review, see Collado-Vides et al. 1991; Gralla and Collado-Vides 1996). These systems are denoted by where the activator protein binds on the DNA and which sigma factor is used, but a key feature is that the mechanism of activation is quite different for each. The first system involves the RNA polymerase (RNAP) containing the σ70 factor. All bacterial sigma factors are related to this housekeeping sigma (except sigma 54, see below) (Lonetto et al. 1992). In these cases, the activator proteins bind between 30 and 80-bp upstream of the transcription start site, adjacent to the RNAP holoenzyme. These promoters are usually poorly recognized by RNAP without the aided recruitment by the activator protein (Ishihama 1993). Many elegant studies over the last decade have defined the exact residues of specific activators and of the RNAP subunits that make contact with each other. So far, interactions with either the α and/or σ subunits of RNAP have been characterized (for review, see Busby and Ebright 1994). On the basis of these studies, certain rules are beginning to emerge with respect to which RNAP contact is made according to where activator binding is centered (between −30 and −80 ).
A second general type of bacterial activation system involves RNAP containing the rpoN-encoded sigma factor called σ54 (σN) (for review, see Merrick 1993; North et al. 1993; Magasanik 1996). This is the only sigma factor that is unrelated to σ70 and it is responsible for directing RNAP to very highly conserved promoters that have the consensus GG-N10-GC (at −24 and −12 bp). In these cases the activator proteins bind distantly upstream of the promoters and via DNA looping, activate the RNAP/σ54 holoenzyme. A paradigm for this class of activator, for which there are now at least 30 cases in many genera of bacteria, is the nitrogen regulatory protein called NtrC (or NRI). Members of the NtrC class typically bind to DNA at tandem sites centered >100-bp upstream of the start site and these activators are therefore referred to as enhancer-binding proteins (EBPs). In addition to the σ54 and enhancer characteristics, all EBPs contain a nucleotide binding fold for which its ATPase function is required for transcriptional activation (Weiss et al. 1991; Austin and Dixon 1992). For some of the EBP-activated promoters, RNAP/σ54 has been shown to bind the promoter in a stable closed complex, independent of the activator, indicating that recruitment to the promoter is not necessary (Ninfa et al. 1987; Sasse-Dwight and Gralla 1988; Buck and Cannon 1992; Cannon et al. 1993; Syed and Gralla 1997). For NtrC, an unusual multimerization occurs whereby two dimers bind at the tandem sites and then two more dimers bind to these by protein–protein interactions (Wyman et al. 1997). This DNA-mediated oligomerization is necessary for ATPase activity and activation, perhaps prompting an interaction with both the σ54 and β subunits of RNAP (Lee and Hoover 1995).
Here, we describe a third bacterial activation system with properties of both systems. Like the enteric counterpart (Ninfa and Magasanik 1986), the Rhodobacter capsulatus NtrC protein (RcNtrC) is phosphorylated by its cognate kinase, NtrB, in response to nitrogen deprivation (Cullen et al. 1996). This phosphorylation in enterics converts NtrC into the activator with the aforementioned characteristics. The RcNtrC also contains the nucleotide binding fold, which, by genetic analysis, is required for its activation function in vivo (Foster-Hartnett et al. 1994). Two tandem sites that are centered >100-bp upstream of the transcription start site bind purified RcNtrC in each of the five known members of the RcNtrC regulon (nifA1, nifA2, glnB, anfA, and mopA). The upstream tandem sites are necessary for activation in vivo, as demonstrated with the nifA1 and nifA2 promoters (Foster-Hartnett et al. 1994). However, RcNtrC-activated promoters are still expressed in a nitrogen-dependent manner in rpoN mutants and they have no sequence similarity to typical σ54-activated promoters (Foster-Hartnett and Kranz 1992, 1994; Preker et al. 1992; Kutsche et al. 1996). Thus, the RNAP holoenzyme that is activated by the RcNtrC protein has remained an enigma. Here, we show by use of purified components that the R. capsulatus RNAP (RcRNAP)/σ70 holoenzyme is activated by RcNtrC in an NtrB-dependent manner. In vitro transcriptional activation requires a supercoiled template, the upstream tandem binding sites for RcNtrC, and ATP. Our results suggest that this third type of system contains elements of each of the first two and could represent an evolutionary transition between σ54 and σ70 systems that were retained by this α proteobacterial species of photosynthetic bacterium.
Results
RcNtrC activates the RNAP ς70 holoenzyme in an NtrB-dependent manner
To investigate whether the RcRNAP σ70 holoenzyme is activated by the RcNtrC system, we have combined the use of a set of wild-type and mutant nifA1 promoters (Fig. 1A) and hyperactive constitutive RcNtrC mutant proteins. These RcNtrC proteins show increased in vivo activation in response to limiting or sufficient nitrogen. It is known that they are phosphorylated in vitro by NtrB to the same level and bind to the upstream tandem sites with the same affinity as the wild-type RcNtrC (W.C. Bowman, P.J. Cullen, and R.G. Kranz, unpubl.). For in vitro transcription studies, we have recently purified the R. capsulatus housekeeping RNAP containing the major σ70 subunit. This preparation is ∼98% pure by SDS-PAGE and contains only the housekeeping σ70 as determined by Western blot analysis with the 2G10 monoclonal antibody (Cullen et al. 1997) (Fig. 2A,B). We chose the nifA1 promoter because of the in vivo information already known about its upstream DNA including its RcNtrC tandem binding sites (Foster-Hartnett et al. 1994). Moreover, we previously converted the nucleotides in the −35 region of nifA1 toward an ideal −35 hexamer (nifA1Mut3) with the result of high-level basal transcription by the σ70 RNAP (Cullen et al. 1997). Thus, it was known that with only two nucleotide changes, the RcRNAP σ70 could melt this promoter by an RcNtrC-independent process.
Figure 1.

(A) Sequence of the nifA1 promoter region and engineered promoters. (Shaded bars) RcNtrC binding sites, (arrowheads) major DNase I hypersensitive sites. The −35 and −10 hexamers are underlined and the transcriptional start site is marked by a horizontal arrow. The sequences of the nifA1, nifA1Mut1, nifA1Mut2, and nifA1Mut3 promoters are aligned for comparison with the E. coli σ70 consensus. (B) In vitro transcriptional activation of nifA1 wild-type and mutant promoters by RcNtrC. Templates are noted below each set of transcription reactions. (Lanes 1,6,11,16) No activators; (lanes 2,7,12,17) 260 nm RcNtrCC3; (lanes 3,8,13,18) 260 nm RcNtrCC3 and 270 nm MBP–NTRB; (lanes 4,9,14,19) 350 nm RcNtrCC7; (lanes 5,10,15,20) 350 nm RcNtrCC7 and 270 nm MBP–NtrB.
Figure 2.
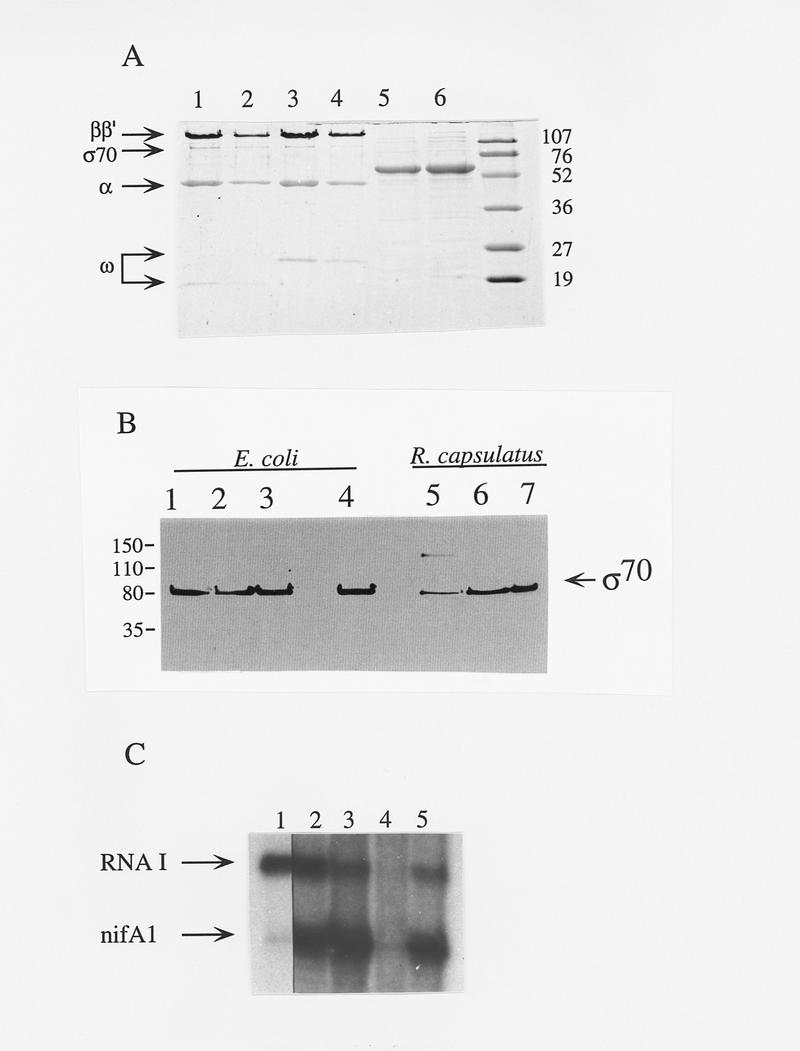
Characterization of the holoenzyme and activator preparations and transcription inhibition by monoclonal antibody 2G10. (A) SDS-PAGE of the E. coli RNAP (lanes 1,2); RcRNAP (lanes 3,4); RcNtrCC3 (lane 5) and RcNtrCC7 (lane 6); standards with sizes indicated in kD. (B) Western analysis of the RcRNAP and EcRNAP purification by use of mAb 2G10, specific for E. coli σ70. Fractions are from E. coli and R. capsulatus as follows: (Lanes 1,5) PEG precipitation fraction; (lanes 2,6) heparin agarose fraction; (lanes 3,7) DEAE–Sepharose fraction; (lane 4) HPLC-purified EcRNAP/σ70 holoenzyme. (C) RcNtrC activated transcription is abolished by σ70 mAb 2G10. The nifA1Mut1 promoter was used. (Lane 1) no activator; (lanes 2–5) 350 nm RcNtrCC7; (lanes 3–5) 270 nm MBP–NtrB; (lane 4) 1 μl of mAb 2G10; (lane 5) 1 μl of control mAb.
For the present study, we converted the wild-type nifA1 promoter toward a consensus −35 hexamer by one (nifA1Mut1, nifA1Mut2) or two nucleotides (nifA1Mut3) and studied transcription in the presence of purified maltose binding protein-NtrB (MBP–NtrB) and two RcNtrC constitutive mutant proteins, designated RcNtrCC3 and RcNtrCC7. These proteins were purified as described previously (Cullen et al. 1996) to >95% homogeneity (Fig. 2A). All transcription reactions shown in Figure 1 were with supercoiled templates in which the indicated nifA1 DNA was cloned upstream of a transcriptional terminator. nifA1 transcription yields an mRNA product of 92 nucleotides that migrates below the 108-nucleotide RNAI transcript. The RNAI transcript is from a well-recognized σ70-type promoter (Scott 1984) that functions with both the Escherichia coli and RcRNAP holoenzymes (Cullen et al. 1997). RNAI transcripts serve as internal controls for each template and all nifA1 transcripts detected in the present study were quantified relative to the RNAI transcript. This corrected for loading differences or degradation and facilitated comparisons with different templates or conditions. In the absence of activator the nifA1Mut3 template exhibited high transcription (Fig. 1B, lane 16). Whereas nifA1Mut1 (Fig. 1B, lane 6) gave a very low basal level of transcription, the wild-type (Fig. 1B, lane 1) and nifA1Mut2 (Fig. 1B, lane 11) exhibited no detectable transcription without activation. Addition of the RcNtrC proteins resulted in at least a 4- to 10-fold increase in transcription at the nifA1 wild-type, nifA1Mut1, and nifA1Mut2 promoters (Fig. 1B). Because no basal level of transcription could be detected with the wild-type or nifA1Mut2 promoters, the fold increase could not be determined. However, at least 10-fold less product from the activated sample could have been detected, indicating at minimum, a 10-fold activation. Because basal levels are so high, activated transcription of the nifA1Mut3 promoter is obscured (Fig. 1B, lanes 16–20). No increase in transcription was observed at the RNAI promoter on addition of the activators, indicating that the activation was specific for nifA1 and the two mutant promoters. The addition of MBP–NtrB to reactions containing each of the three promoters (wild type, nifA1Mut1, and nifA1Mut2) and RcNtrC increased the activation another two- to fourfold, depending on the promoter or RcNtrC allele. However, total activation levels were always higher when the nifA1Mut1 promoter was used, at least fivefold higher than observed with the wild-type and nifA1Mut2 promoters. To define the activation requirements for this system, results with the nifA1Mut1 promoter are presented here, although most activation requirements were also demonstrated with the wild-type nifA1 promoter.
With nifA1Mut1, the RcNtrCC7 protein gives the greatest activation without MBP–NtrB (Fig. 1B, lane 9) and total activated transcription with MBP–NtrB (Figs. 1B, lane 10, and 2C, lanes 2,3). Previously, we have used the σ70 monoclonal antibody 2G10 to inhibit transcription of RcRNAP σ70 promoters as have other groups with E. coli σ70 promoters (for review, see Breyer et al. 1997 and references therein). In vitro transcription of both the RNAI promoter and the activated nifA1Mut1 promoter is inhibited >98% by mAb 2G10 (Fig. 2C, lane 4) but not significantly by a control mAb (lane 5). The 2G10 antibody was also found to inhibit RcNtrC-dependent transcriptional activation of the wild-type nifA1 promoter (data not shown). The only sigma factor in the RcRNAP preparation that reacts with 2G10 is σ70 (Cullen et al. 1997; Fig. 2B, lane 7). The epitope for 2G10 has been mapped to residues 470–486 of the E. coli σ70 factor (Breyer et al. 1997). As expected, the R. capsulatus σ70 is highly conserved in this region (Pasternak et al. 1996), but no σ54 factor, including R. capsulatus σ54 (Jones and Haselkorn 1989), contain this epitope.
To further demonstrate that RcNtrC activates the R. capsulatus σ70 holoenzyme, we constructed two plasmids containing the R. capsulatus rpoD gene that result in overexpression of the R. capsulatus σ70 subunit in E. coli. The σ70 protein expressed from pRGK300 was purified from an SDS gel and renatured by use of the method of Gross et al. (1978). A hexahistidine-tagged σ70 polypeptide was overexpressed from pRGK301 and purified with a nickel affinity column. The purified σ70 proteins were added to in vitro transcription reactions containing R. capsulatus core RNA polymerase that has been shown previously to contain very low levels of σ70 (Cullen et al. 1997). This core preparation gave very low levels of RNAI transcript and no nifA1Mut1 transcript with or without RcNtr proteins (Fig. 3, lanes 1, 2). Addition of either the histidine-tagged (Fig. 3, lanes 3,4) or the renatured σ70 (Fig. 3, lane 5) resulted in a >10-fold increase in the RNAI transcript. When NtrB and NtrC was added to either reaction containing σ70 (Fig. 3, lane 4,5) a significant increase in activation is observed compared with the reaction with core RNAP and σ70 only (Fig. 3, lane 3). These results confirm that it is the R. capsulatus σ70 RNAP that is activated by RcNtrC.
Figure 3.

Purified σ70 added to R. capsulatus core RNAP stimulates transcriptional activation by RcNtrC. In vitro transcription reactions using the supercoiled nifA1Mut1 template with the R. capsulatus core RNAP (lanes 1,2) or core RNAP with the addition of His-tagged σ70 subunit (lanes 3,4) or SDS-gel purified σ70 subunit (lane 5). (Lanes 1,3) no activator; (lanes 2,4,5) 270 nm MBP–NtrB and 260 nm RcNtrCC3.
It is worth noting the presence of a subunit of the RcRNAP preparation, which migrates at ∼23 kD in this SDS-PAGE system (Fig. 2A, lane 3). The E. coli holoenzyme preparation contains a polypeptide that migrates ∼4 kD smaller then this (Fig 2A, lane 1), which is similar in size to the previously noted θ subunit of unknown function (Gentry and Burgess 1986). Amino-terminal sequencing of the R. capsulatus 23-kD polypeptide has demonstrated that it is the θ subunit. The R. capsulatus gene (rpoZ) encoding the θ subunit has been sequenced (R. capsulatus sequencing project at the University of Chicago, URL http://capsulapedia.uchicago.edu). The rpoZ gene sequence indicates a size that is 28 residues larger than the E. coli counterpart, consistent with the size exhibited by SDS-PAGE.
Supercoiled DNA and both upstream tandem sites are required for RcNtrC-mediated activation
To determine if DNA binding by RcNtrC is required for activation, and, if so, whether one dimer site or two are necessary, we constructed plasmids in which either one or both of the RcNtrC binding sites have been removed from the nifA1Mut1 promoter region. The single binding site that was removed was site I, the furthest upstream. This site has been shown to be necessary for in vivo activation (Foster-Hartnett et al. 1994) and deletion of it maintains the sequence/structure of DNA between site II and the promoter. Even on supercoiled templates, removal of one or both RcNtrC binding sites completely abolishes transcriptional activation of the nifA1Mut1 promoter by the RcNtrC proteins (Fig. 4). Basal transcription from the supercoiled templates is unaffected by deletion of the binding sites. No activation was observed with either of the binding site deletion templates when the RcNtrC concentration was increased to 875 nm (data not shown). These data show that the RcNtrC protein must bind DNA to activate transcription and suggest that there is a cooperative interaction between the bound RcNtrC dimers that is also necessary.
Figure 4.

RcNtrC-activated transcription is only observed on supercoiled templates with intact tandem binding sites. In vitro transcription reactions were performed using supercoiled (lanes 1–15) or linear templates (lanes 16–20) that contained 0 (lanes 11–15), 1 (lanes 6–10), or both (lanes 1–5; 16–20) tandem RcNtrC dimer binding sites. (Lanes 1,6,11,16) No activators; (lanes2,7,12,17) 260 nm RcNtrCC3; (lanes 3,8,13,18) 260 nm RcNtrCC3 and 270 nm MBP–NTRB; (lanes 4,9,14,19) 350 nm RcNtrCC7; (lanes 5,10,15,20) 350 nm RcNtrCC7 and 270 nm MBP–NtrB.
Previously, we have shown that phosphorylation of the RcNtrC protein leads to a fourfold increase in binding to the tandem sites located upstream from the glnB promoter (Cullen et al. 1996). We have also shown that unphosphorylated RcNtrC still binds upstream from the nifA1 promoter when one of the two tandem sites has been removed, although approximately fourfold more RcNtrC is required (compared with the binding at tandem sites) (Foster-Hartnett et al. 1994). To determine whether phosphorylation increases the affinity of RcNtrC for a single site, DNase I footprinting was performed with RcNtrCC3 in the presence and absence of MBP–NtrB by use of the EcoRI–HindIII fragment from pALB1, the template that contains a single RcNtrC binding site, as a probe. The concentration of unphosphorylated and phosphorylated RcNtrCC3 that protected from DNase I digestion was found to be 160 nm and 80 nm, respectively (data not shown). These results support the idea that transcriptional activation is dependent on a cooperative interaction that occurs between RcNtrC dimers when they are bound to the two tandem sites (see Discussion).
When the nifA1Mut1 template with intact tandem binding sites was linearized, no transcriptional activation by RcNtrC ± MBP–NtrB was detected (Fig. 4, lanes 16–20). Under these conditions, much less nifA1Mut1 and RNAI basal transcription was observed. Cut and recircularized template also showed no transcriptional activation by RcNtrC, which is consistent with the observation that an increase in nicked template results in poorer transcriptional activation by RcNtrC (data not shown).
ATP binding, but not hydrolysis, is necessary for RcNtrC to activate transcription
To determine whether ATP hydrolysis is necessary for transcriptional activation by RcNtrC, in vitro transcriptional activation reactions were performed by use of the β–γ nonhydrolyzable ATP analog adenylyl imidodiphosphate (AMP–PNP). Because transcription requires hydrolysis of the α–β bond, AMP–PNP can be utilized by the RNA polymerase, but not by RcNtrC for an ATPase activity. This analog has been used previously to investigate the ATPase function for activation by enteric NtrC (e.g., Wang et al. 1995; Syed and Gralla 1997). The RcNtrCC7 protein was used because of its high level of activation that is independent of MBP–NtrB and phosphorylation. With ATP, RcNtrCC7 stimulated transcription at the nifA1Mut1 promoter ∼10-fold over basal levels (Fig. 5A, lanes 1,2). When AMP–PNP was substituted for ATP in the transcription reactions, no activation of the nifA1Mut1 promoter over the basal levels was observed (Fig. 5, lanes 3,4).
Figure 5.



Effects of ADP and nonhydrolyzable ATP analogs on transcriptional activation by RcNtrC. For each set of reactions, lanes 1 and 3 contained no activator and lanes 2 and 4 contained 350 nm RcNtrCC7. (A) In vitro transcription reactions were performed using either ATP (lanes 1,2) or AMP–PNP (lanes 3,4) at 1.5 mm. (B) In vitro transcription reactions were performed in the presence of 1.5 mm ATP (lanes 1,2) or 0.4 mm ATP and 15 mm ADP (lanes 3,4). (C) In vitro transcription reactions were performed in the presence of 1.5 mm ATPγS (lanes 1,2) or 0.4 mm ATPγS and 15 mm ADP (lanes 3,4).
The result with AMP–PNP could be caused by either a requirement for ATP hydrolysis or an inability of RcNtrC to bind AMP–PNP. In fact, transcriptional activation by the enteric NtrC (S. Kustu, pers. comm.) and the EBP, DctD (T.R. Hoover, pers. comm.), is not inhibited by AMP–PNP. This is in contrast to ADP and ATPγS that both inhibit activation by enteric NtrC (Popham et al. 1989; Weiss et al. 1991), suggesting that AMP–PNP may not bind these EBPs. Similarly, we tested whether AMP–PNP, ADP, and ATPγS inhibit the ATP-dependent activation property of RcNtrC. None of the analogs inhibited activation, suggesting that they do not bind RcNtrC, or alternatively, they bind and can function as coactivators (Fig. 5B shows the result with ADP). To resolve this, in place of ATP, we initially tested ATPγS, which can be incorporated into RNA but is considered nonhydrolyzable. Reactions with ATPγS resulted in at least a 10-fold increase in transcription in the presence of RcNtrC (Fig. 5C, lane 2) compared with basal levels (lane 1). Again, ADP did not inhibit this activation (Fig. 5C, cf. lane 4 with lane 3), even at ADP levels 30-fold higher then ATPγS. To confirm that ADP does not act as a coactivator, ADP was added with AMP–PNP and results identical to those shown in Figure 5A were observed (data not shown). Taken together, we conclude that, in addition to phosphorylation by NtrB, RcNtrC requires the specific binding of ATP for transcriptional activation.
The enteric RNAP and RcNtrC activation
We have been unsuccessful in attempts to find conditions in which the E. coli RNAP σ70 is activated by RcNtrC at wild-type or nifA1Mut1 promoters (e.g., see Fig. 6, lanes 4–6). The E. coli RNAP was also not activated when the purified enteric σ54 subunit was added to supercoiled templates containing the wild-type or nifA1Mut1 promoters. This later result with σ54 is not unexpected because none of the RcNtrC-dependent promoters are σ54-type (i.e., GG-N10-GC). The failure of RcNtrC to activate the E. coli σ70 RNAP could be the result of a lack of interaction with RcNtrC or inability to form an open complex at this promoter (or both). To begin to address this, we used the nifA1Mut3 promoter, which yields high basal transcription with the RcRNAP holoenzyme. Surprisingly, the nifA1Mut3 promoter on a supercoiled template was transcribed by E. coli RNAP at a very low basal level, at least 20-fold less than the RNAI promoter and 50-fold less than the R. capsulatus housekeeping enzyme (Fig. 6, lane 7). This result suggests that the E. coli enzyme cannot melt the −10 region of the nifA1 promoter as the R. capsulatus enzyme can. Nevertheless, a basal level of transcription with nifA1Mut3 is observed, and this was not increased on addition of RcNtrC and MBP–NtrB relative to the RNAI transcript (Fig. 6, lanes 7–9) (see Discussion).
Figure 6.

RcNtrC does not activate the E. coli RNAP. In vitro transcription reactions were performed with purified RNAP σ70 from R. capsulatus and E. coli and the MBP–NtrB/RcNtrCC7 transcriptional activation system. Templates: nifA1Mut1 (lanes 1–6) and nifA1Mut3 (lanes 7–9). (Lanes 1,4,7) No activator; (lanes 2,5,8) 350 nm RcNtrCC7; (lanes 3,6,9) 350 nm RcNtrCC7 and 270 nm MBP–NtrB.
Discussion
RcNtrC-activated promoters
During the last 7 years, there has been considerable speculation on the promoters activated by the EBP RcNtrC, and on the holoenzyme(s) that is used (e.g., Kranz and Foster-Hartnett 1990; Morett and Segovia 1993; Foster-Hartnett et al. 1994; Kranz and Cullen 1995; Masepohl and Klipp 1996). In the present study, of the four promoters tested, the −35 hexamer of the nifA1Mut1 promoter is the optimal RcNtrC-activated −35 region. We note that hexamers containing at least four out of six of this optimal sequence are present in each of the five natural RcNtrC-activated promoters (Fig. 7). As in E. coli σ70-activated systems (Busby and Ebright 1994), evolution away from the optimal recognition element results in lower basal levels of expression, as demonstrated here for nifA1. Whereas a consensus −10 region for these promoters remains to be determined, 15- to 18-bp downstream of each of these −35 elements is a potential −10 hexamer with a second position A that is 76% conserved in E. coli σ70 promoters (see Lisser and Margalit 1993). All five −10 regions, including the nifA1 promoter, also contain a GC or a GG dinucleotide. Even if the −10 hexamers shown in Figure 7 have been incorrectly chosen, extended −10 regions show at least 50% GC content. Because the genomic GC content of R. capsulatus is 65%, the R. capsulatus housekeeping holoenzyme may have evolved the ability to melt high GC regions that the E. coli holoenzyme cannot. This ability is probably not limited to RcNtrC-activated promoters, because other natural R. capsulatus promoters with high GC content in the −10 region are poorly transcribed by the E. coli enzyme (Cullen et al. 1997). However, this capability does not reside in the R. capsulatus σ70 regions called 2.4 or 2.5 that have been implicated in melting the −10 hexamer (see Malhotra and Severinova 1996) and extended −10 bp (Barne et al. 1997), respectively; the R. capsulatus sequence of σ70 is identical to that of E. coli in these regions (Pasternak et al. 1996).
Figure 7.

The five natural promoters activated by RcNtrC. (Arrows) In vivo mRNA start sites. Each member of the RcNtrC regulon has tandem sites containing the consensus RcNtrC binding region underlined with dark boxes (Foster-Hartnett and Kranz 1994). The nifA1 and nifA2 tandem sites were shown previously to bind RcNtrC in vitro (Foster-Hartnett et al. 1994) as were the glnB (Foster-Hartnett and Kranz 1994), anfA, and mopA sites (W.C. Bowman and R.G. Kranz, unpubl.). The RcNtrC consensus binding region has been presented previously (Foster-Hartnett and Kranz 1994). The boxed DNA on the left of each promoter region contains at least four of the optimal −35 hexamer (Mut1) sequence. The boxed hexamers (right) are putative −10 regions discussed in the text.
Requirements of upstream tandem binding sites: How does RcNtrC contact RNAP?
We have demonstrated that two RcNtrC dimers bound to the upstream tandem sites are necessary for activation. Phosphorylated RcNtrC at 875 nm concentration was still unable to activate transcription from supercoiled templates with only one dimer binding site. This result is similar to that initially demonstrated for the E. coli NtrC EBP (Ninfa et al. 1987). In the R. capsulatus system, as low as 160 nm unphosphorylated RcNtrC (Foster-Hartnett et al. 1994) and 80 nm of phosphorylated RcNtrC was able to protect the single site from DNAse on linear DNA. Moreover, as low as 80 nm of phosphorylated RcNtrC could activate transcription of nifA1 on the template containing tandem sites. These results suggest that oligomerization of at least two dimers is required for transcriptional activation. We also note conserved phasing of the tandem sites for all five RcNtrC-activated promoters with either 5, 6, or 15 bp separating each RcNtrC binding site (see Fig. 7). Such phasing was shown to be necessary for the EBP XylR (Perez-Martin and de Lorenzo 1996).
All five promoters contain tandem sites that bind RcNtrC located distant from the promoter. We consider three possible mechanisms by which RcNtrC might contact RNAP. (1) It is possible that the DNA between RNAP and RcNtrC binding sites loops out naturally as is the case for the glnA promoter and enteric NtrC (Su et al. 1990; Wedel et al. 1990). Magasanik and colleagues have reported elegant studies recently on promoters activated by the enteric NtrC, demonstrating that some contain natural curvature of the DNA that is necessary to optimize contact with RNAP (Carmona and Magasanik 1996; Carmona et al. 1997). (2) Some of the RNAP/σ54 systems require that IHF bind and loop the DNA between the promoter and EBP binding sites, thereby increasing the frequency of contact between EBP and RNAP (e.g., Santero et al. 1992). We have shown previously that IHF does not bind to the nifA1 DNA (Foster-Hartnett et al. 1994) and no such potential IHF binding sites are found upstream of any of these promoters (Fig. 7). The RcNtrC and RNAP preparations used here are >95% pure, although we cannot rule out a minor contaminating factor that plays such a role. (3) It is possible that RcNtrC multimerizes on the DNA from the upstream tandem binding sites toward the RNAP. This could involve less specific binding to DNA, particularly because no RcNtrC-consensus binding sites are present in the intervening DNA between the tandem sites and promoter. We have not observed such binding on linear templates. We have engineered nine different DNA inserts of sizes from 4 to 130 bp into positions −84 and −47, relative to the transcript start site of nifA1 (Foster-Hartnett et al. 1994). All nine inserts resulted in loss of activation in vivo by RcNtrC. This result indicates that the structure and/or sequence of the intervening DNA is important, as might be expected for mechanisms (1) or (2).
The mechanism by which RcNtrC activates open complex formation by the R. capsulatus holoenzyme and the protein–protein contacts that are made, remain important questions. It is intriguing that the E. coli enzyme is not activated at this promoter. Even with a −35 hexamer of the nifA1 promoter (i.e., Mut3), which yields a low level of basal transcription with the E. coli holoenzyme, it cannot be further activated by RcNtrC. It is therefore possible that RcNtrC does not interact with the E. coli holoenzyme. In a similar line of experiments, the Salmonella NtrCS160F protein did not activate RcRNAP (not shown) even at concentrations in which DNA binding of NtrCS160F is not necessary for activation of the enteric σ54 RNAP (Weiss et al. 1991). These results suggest that the RcNtrC and RcRNAP may have coevolved specific sites for interaction, although more studies will be necessary to confirm this.
Requirement for ATP binding but not ATP hydrolysis
We used the in vitro activation system to determine whether ATP binding or ATP hydrolysis is required by RcNtrC. Inhibition with various ATP analogs of the ATP-dependent activation by RcNtrC was analyzed. The ability of analogs to act as coactivators was investigated, as has been carried out with the enteric EBPs (Popham et al. 1991; Weiss et al. 1991). The results indicate that AMP–PNP and ADP do not bind RcNtrC. Importantly, the nonhydrolyzable analog ATPγS supports activation by RcNtrC (Fig. 5C). Because the nonhydrolyzable AMP–PNP does not support activation when it replaces ATP in the transcription reaction, it can also be concluded that only ATP and not other nucleotide triphosphates are coactivators. We conclude that the RcNtrC protein specifically requires ATP binding for activation, but unlike the EBP activators of σ54 RNAP, ATP hydrolysis is not essential. This is consistent with our observation that none of the purified RcNtrC proteins, wild-type or constitutive, exhibit a detectable ATPase activity, with or without DNA containing tandem binding sites (P.J. Cullen and R.G. Kranz, unpubl.).
Beside the EBP family of σ54 RNAP activators, two other bacterial regulators contain nucleotide binding folds and activate the σ70 RNAP. TyrR binds to ATP and has an ATPase activity (Cui et al. 1993). However, mutational analysis of Walker motif A indicates that ATP binding is not necessary for its activation function (Pittard and Davidson 1991; Yang et al. 1993). The TyrR protein appears to bind between −35- and −80-bp upstream of the transcription start site when it is an activator (Wilson et al. 1994; Gralla and Collado-Vides 1996). MalT binds to both maltotriose and ATP as coactivators (Richet and Raibaud 1989). MalT always binds to the promoters that it activates near −38-bp upstream of the start site (Danot and Raibaud 1994) and this binding only occurs when the coactivators are present (Richet and Raibaud 1989). It is quite clear that the RcNtrC protein binds to its upstream tandem sites in the absence of ATP, indicating that the ATP-mediated mechanism for activation will be different from that of MalT. RcNtrC wild-type, RcNtrC with mutations in the nucleotide binding fold, and RcNtrCC3 and RcNtrCC7 all bind to the upstream tandem sites with similar affinities in the presence or absence of ATP (W.C. Bowman and R.G. Kranz, unpubl.). We suggest that the RcNtrC protein simultaneously senses two states of the cell. One is the nitrogen status that is mediated by the classic two-component NtrB kinase pathway (via GlnB). A second is the concentration of ATP or energy status. This may not be too surprising because it is crucial that a photosynthetic bacterium knows the levels of ATP that are available for nitrogen fixation, for example, before it induces the >36 genes necessary for this energy-intense process (Kranz and Cullen 1995).
Evolutionary aspects
Two important properties that are essential for σ54/EBP systems are retained by the RcNtrC system: (1) cooperative binding to tandem sites upstream to induce oligomerization and (2) nucleotide binding fold in the central domain that requires ATP for activation. It is shown that the natural holoenzyme used for activation by RcNtrC is the housekeeping RNAP and a −35 consensus region is now better defined for this unique system. Therefore, we conclude that R. capsulatus has three general types of activation systems: (1) the traditional σ70-type in which activators bind the DNA adjacent to the RNAP (Bauer 1995; Cullen et al. 1997), (2) a bonafide σ54 RNAP and EBP activators (e.g., The NifA and AnfA proteins) (Cullen et al. 1994; Kutsche et al. 1996), and (3) the RcNtrC system defined here with properties of 1 and 2. Because R. capsulatus is considered to be one of the most metabolically versatile microorganisms, it is possible that it has evolved these systems to add to its control repertoire (i.e., for regulatory versatility). It is worth considering that a system like the RcNtrC activator pathway evolved into the classic σ54/EBP and was retained by this α proteobacterium, along with the classical system.
Materials and methods
Plasmids
pUct–nifA1, pA1M1, pA1M2, and pA1M3 have been described previously (Cullen et al. 1997). pALB1 was made by PCR of the nifA1Mut1 promoter region in plasmid pA1M1 with the upstream oligonucleotide 5′-CCCGGTACCGGTTCGCCGCATAATTG-3′ and the downstream oligonucleotide 5′-TGACCGGCAGCAAAATG-3′. The 0.3-kb product was digested with KpnI and HindIII and cloned into pUC118. pALB2 was made by PCR of the nifA1Mut1 promoter region in plasmid pA1M1 by the upstream oligonucleotide 5′-CCCGGTACCCTTGCAAAAATGAACC-3′ and the same downstream oligonucleotide as pALB1. The 0.3-kb product was digested with KpnI and HindIII and cloned into pUC118. pALB1 and pALB2 were sequenced to confirm the removal of 1 or both RcNtrC binding sites, respectively. Plasmid pRGK300, which allows overexpression of the R. capsulatus σ70 protein, was made by PCR of the rpoD gene (Pasternak et al. 1996) from the R. capsulatus chromosome by the upstream oligonucleotide 5′-TGCGCAGCCCCGATGCAGCCCGACGAGGAG-3′ and the downstream oligonucleotide 5′-GCATCTTCAGATCTTCGGGGCCTTACTGGT-3′. These oligonucleotides contain NcoI and BglII sites that facilitated cloning the 2-kb PCR product downstream of the T7 promoter in pET15 (Novagen). Plasmid pRGK301, which allows overexpression of the R. capsulatus σ70 protein that contains a hexahistidine tag, was made by PCR of the rpoD gene from the R. capsulatus chromosome by use of the upstream oligonucleo tide 5′-CGAGGAGCGCATATGGCCGCCAAGGACATC-3′ and the same downstream oligonucleotide that was used for pRGK300. The 2.0-kb PCR product was digested with NdeI and BglII and ligated in frame to the amino-terminal hexahistidine tag encoded by pET15.
RNAP purification
Purification of RNAP from E. coli and R. capsulatus has been described previously (Cullen et al. 1997). Both heparin–agarose pure and DEAE–Sepharose pure RcRNAP holoenzyme were used in in vitro transcription reactions, as well as DEAE pure R. capsulatus core RNA polymerase. For in vitro transcription reactions that utilized the E. coli RNA polymerase EcRNAP, only the DEAE-Sepharose pure fractions were used.
σ70 purification
The σ70 subunit from R. capsulatus without a hexahistadine tag was overexpressed in E. coli strain BL21–δDE3 containing pRGK300 by the addition of 1 mm IPTG for 3 hr at 37°C. Cells were sonicated in 20 mls of buffer (20 mm Tris-HCl, 2 mm EDTA, at pH 8) and cell debris was removed by centrifugation at 12,000g for 15 min at 4°C in a Sorvall centrifuge. The supernatant contains a major polypeptide that is not present in BL21–δDE3 containing no plasmid; this polypeptide migrates at the same size as the σ70 from the R. capsulatus holoenzyme preparation (not shown). The supernatant (0.5 ml) was run on an 8% polyacrylamide gel and the σ70 protein was purified from the gel and renatured by use of the method of Gross et al. (1978).
Hexahistidine-tagged σ70 subunit from R. capsulatus was overexpressed in E. coli strain BL21–δDE3 containing pRGK301 by the addition of 1 mm IPTG for 3 hr at 37°C. Cells were sonicated in 20 mls of buffer (20 mm Tris-HCl, 2 mm EDTA at pH 8) and cell debris was removed by centrifugation at 12,000g for 15 min at 4°C in a Sorvall centrifuge. The supernatant contains a major polypeptide that is not present in BL21–δDE3 containing no plasmid; this polypeptide was similar in size by SDS-PAGE to the R. capsulatus σ70 subunit in the holoenzyme preparation (not shown). The supernatant was loaded onto a His-Bind (Novagen) column and the column was washed with 10 volumes of binding buffer (5 mm imidazole, 0.5 m NaCl, 20 mm Tris-HCl at pH 7.9), followed by 10 volumes of wash buffer (60 mm imidazole, 0.5 m NaCl, 20 mm Tris-HCl at pH 7.9). The histidine-tagged σ70 was eluted in 6 volumes of elution buffer (250 mm imidazole, 0.5 m NaCl, 20 mm Tris-HCl at pH 7.9). The protein was concentrated to 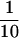 of the original volume in a Centricon 30, diluted to 1 ml with storage buffer (50 mm Tris-HCl, 0.5 mm EDTA at pH 8, 1 mm DTT, 50% glycerol), and stored at −80°C.
of the original volume in a Centricon 30, diluted to 1 ml with storage buffer (50 mm Tris-HCl, 0.5 mm EDTA at pH 8, 1 mm DTT, 50% glycerol), and stored at −80°C.
Purification of MBP–NtrB and RcNtrCC proteins
Purification of the MBP–NtrB has been described previously (Cullen et al. 1996). RcNtrC constitutive mutant proteins (RcNtrCC) were purified through the DEAE–Sepharose step by use of the method of Cullen et al. (1996). The isolation of genes encoding R. capsulatus NtrCC alleles will be described elsewhere.
In vitro transcription
In vitro transcription reactions were performed in transcription buffer [50 mm Tris-HCl at pH 8, 100 mm potassium acetate, 10 mm magnesium acetate, 1 mm ATP, 10 mm DTT and 0.5 μl RNAsin (Promega)] by the method of Cullen et al. (1997). The concentration of linear or supercoiled templates was ∼40 nm for all reactions reported here. For in vitro transcriptional activation reactions, MBP–NtrB (270 nm) was incubated in transcription buffer for 10 min at 37°C prior to the addition of RcNtrCC (80–875 nm as noted) and RNAP (40 nm). RcNtrCC and RNAP were added simultaneously and the reactions were incubated at 24°C for 30 min. Purified σ70 proteins were added to the core RNAP prior to the start of the reactions.
DNase I footprinting
DNase I footprinting analysis of phosphorylated and unphosphorylated RcNtrCC at the nifA1 promoter region was performed by the method of Cullen et al. (1996). Probes were prepared by digesting plasmids (pA1M1 of pALB1) with EcoRI and dephosphorylating the ends with calf intestinal phosphatase. The 5′ ends were labeled with [γ-32P]ATP and T4 polynucleotide kinase, then the labeled DNA was digested with HindIII. After separation on a 4% native acrylamide gel (50 mm Tris–borate EDTA), gel sliced that contained the appropriate fragments were excised and the probes were eluted overnight at 37°C in Tris–EDTA buffer. Approximately 0.2 nm of probe (30,000–60,000 cpm) was used in each footprinting reaction.
Other methods
Western analysis was performed with peroxidase detection reagents from Pierce. Use of monoclonal antibody 2G10, a gift from Nancy Thompsen and Richard Burgess (University of Wisconsin, Madison) and monoclonal antibodies to β-galactosidase have been described previously (Cullen et al. 1997). Transcripts were quantitated by scanning the autoradiograms with an HP scanjet 4C and analyzing the bands by use of IP Labgel software from Data Analysis Corp (Malek et al. 1997). The scanning and software could easily distinguish differences in transcript levels twofold or above. Protein sequencing was carried out with an Applied Biosystems 470A protein sequencer.
Acknowledgments
We thank Paul Cullen and Charles Kaufman for the housekeeping RNAP preparation and Western blotting; Barry Goldman for the construction of pRGK300 and pRGK301 and the purification of the histidine-tagged σ70 protein; Anne Bradburn for the construction of pALB1 and pALB2; Sydney Kustu and Andrea Shauger for providing σ54 and NtrCS160F proteins; Nancy Thompsen and Richard Burgess for mAb 2G10; Steve Slater and Richard Thomas at Monsanto for protein sequencing; Sydney Kustu for comments on the manuscript and advice on the ATP-dependence function of RcNtrC; Ray Dixon, Tim Hoover, Mike Merrick, Alex Ninfa for comments on the manuscript. This work was supported by U.S. Department of Agriculture National Research Initiative grant 9703754 (to R.G.K.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Kranz@biodec.wustl.edu; FAX (314) 935-4432.
References
- Austin S, Dixon R. The prokaryotic enhancer binding protein NtrC has an ATPase activity which is phosphorylation and DNA dependent. EMBO J. 1992;11:2219–2228. doi: 10.1002/j.1460-2075.1992.tb05281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barne KA, Bown JA, Busby JW, Minchin SD. Region 2.5 of the Escherichia coli RNA polymerase sigma 70 subunit is responsible for the recognition of the ’extended -10’ motif at promoters. EMBO J. 1997;16:4034–4040. doi: 10.1093/emboj/16.13.4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CE. Regulation of photosynthesis gene expression. In: Blankenship RE, Madigan MT, Bauer CE, editors. Anoxygenic photosynthetic bacteria. Dordrecht, The Netherlands: Kluwer; 1995. pp. 1221–1234. [Google Scholar]
- Breyer MJ, Thompson NE, Burgess RR. Identification of the epitope for a highly cross-reactive monoclonal antibody on the major sigma factor of bacterial RNA polymerase. J Bacteriol. 1997;179:1404–1408. doi: 10.1128/jb.179.4.1404-1408.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M, Cannon W. Specific binding of the transcription factor sigma-54 to promoter DNA. Nature. 1992;358:422–424. doi: 10.1038/358422a0. [DOI] [PubMed] [Google Scholar]
- Busby S, Ebright RH. Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell. 1994;79:743–746. doi: 10.1016/0092-8674(94)90063-9. [DOI] [PubMed] [Google Scholar]
- Cannon W, Claverie-Martin F, Austin S, Buck M. Core RNA polymerase assists binding of the transcription factor σ54 to promoter DNA. Mol Microbiol. 1993;8:287–298. doi: 10.1111/j.1365-2958.1993.tb01573.x. [DOI] [PubMed] [Google Scholar]
- Carmona M, Magasanik B. Activation of transcription at sigma 54-dependent promoters on linear templates requires intrinsic or induced bending of the DNA. J Mol Biol. 1996;261:348–356. doi: 10.1006/jmbi.1996.0468. [DOI] [PubMed] [Google Scholar]
- Carmona M, Claverie-Martin F, Magasanik B. DNA bending and the initiation of transcription at sigma 54-dependent bacterial promoters. Proc Natl Acad Sci. 1997;94:9568–9572. doi: 10.1073/pnas.94.18.9568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado-Vides J, Magasanik B, Gralla J. Control site location and transcriptional regulation in Escherichia coli. Microbiol Rev. 1991;55:371–394. doi: 10.1128/mr.55.3.371-394.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Ni L, Somerville RL. ATPase activity of TyrR, a transcriptional regulatory protein for sigma 70 RNA polymerase. J Biol Chem. 1993;268:13023–13025. [PubMed] [Google Scholar]
- Cullen PJ, Foster-Hartnett D, Gabbert K, Kranz RG. Structure and expression of the alternative sigma factor, RpoN, in Rhodobacter capsulatus; physiological relavance of an autoactivated nifU2-rpoN superoperon. Mol Microbiol. 1994;11:51–65. doi: 10.1111/j.1365-2958.1994.tb00289.x. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, Bowman WC, Kranz RG. In vitro reconstitution and characterization of the Rhodobacter capsulatus NtrB and NtrC two-componet system. J Biol Chem. 1996;271:6530–6536. doi: 10.1074/jbc.271.11.6530. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, Kaufman CK, Bowman WC, Kranz RG. Characterization of the Rhodobacter capsulatus housekeeping RNA polymerase. In vitro transcription of photosynthesis and other genes. J Biol Chem. 1997;272:27266–27273. doi: 10.1074/jbc.272.43.27266. [DOI] [PubMed] [Google Scholar]
- Danot O, Raibaud O. Multiple protein-DNA and protein-protein interactions are involved in transcriptional activation by MalT. Mol Microbiol. 1994;14:335–346. doi: 10.1111/j.1365-2958.1994.tb01294.x. [DOI] [PubMed] [Google Scholar]
- Foster-Hartnett D, Kranz RG. Analysis of the promoters and upstream sequences of nifA1 and nifA2 in Rhodobacter capsulatus; activation requires NtrC but not RpoN. Mol Microbiol. 1992;6:1049–1060. doi: 10.1111/j.1365-2958.1992.tb02170.x. [DOI] [PubMed] [Google Scholar]
- ————— The Rhodobacter capsulatus glnB gene is regulated by NtrC at tandem RpoN-independent promoters. J Bacteriol. 1994;176:5171–5176. doi: 10.1128/jb.176.16.5171-5176.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster-Hartnett D, Cullen PJ, Monika EM, Kranz RG. A new type of NtrC transcriptional activator. J Bacteriol. 1994;176:6175–6187. doi: 10.1128/jb.176.20.6175-6187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry DR, Burgess RR. The cloning and sequencing of the gene encoding the omega subunit of Escherichia coli RNA polymerase. Gene. 1986;48:33–40. doi: 10.1016/0378-1119(86)90349-5. [DOI] [PubMed] [Google Scholar]
- Gralla JD, Collado-Vides J. Organization and function of transcription regulatory elements. In: Neidhardt FC, editor. Escherichia coli and Salmonella. Washington D.C.: ASM Press; 1996. pp. 1232–1245. [Google Scholar]
- Gross C, Hoffman J, Ward C, Hager D, Burdick G, Berger H, Burgess R. Mutation affecting thermostability of sigma subunit of Escherichia coli RNA polymerase lies near the dnaG locus at about 66 min on the E. coli genetic map. Proc Natl Acad Sci. 1978;75:427–431. doi: 10.1073/pnas.75.1.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama A. Protein-protein communication with the transcription apparatus. J Bacteriol. 1993;175:2483–2489. doi: 10.1128/jb.175.9.2483-2489.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R, Haselkorn R. The DNA sequence of the Rhodobacter capsulatus ntrA, ntrB and ntrC gene analogues required for nitrogen fixation. Mol & Gen Genet. 1989;215:507–516. doi: 10.1007/BF00427050. [DOI] [PubMed] [Google Scholar]
- Kranz RG, Foster-Hartnett D. Transcriptional regulatory cascade of nitrogen-fixation genes in anoxygenic photosynthetic bacteria: Oxygen- and nitrogen-responsive factors. Mol Microbiol. 1990;4:1793–1800. doi: 10.1111/j.1365-2958.1990.tb02027.x. [DOI] [PubMed] [Google Scholar]
- Kranz RG, Cullen PJ. Regulation of nitrogen fixation genes. In: Blankenship RE, Madigan M, Bauer CE, editors. Anoxygenic photosynthetic bacteria. Dordrecht, The Netherlands: Kluwer Academic Publishing; 1995. pp. 1191–1208. [Google Scholar]
- Kutsche M, Leimkuhler S, Angermuller S, Klipp W. Promoters controlling expression of the alternative nitrogenase and the molybdenum uptake system in Rhodobacter capsulatus are activated by NtrC, independent of sigma 54, and repressed by molybdenum. J Bacteriol. 1996;178:2010–2017. doi: 10.1128/jb.178.7.2010-2017.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Hoover TR. Protein crosslinking studies suggest that Rhizobium meliloti C4-dicarboxylic acid transport protein D, a σ54-dependent transcriptional activator, interacts with σ54 and the β subunit of RNA polymerase. Proc Natl Acad Sci. 1995;92:9702–9706. doi: 10.1073/pnas.92.21.9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisser S, Margalit H. Compliation of E. coli mRNA promoter sequences. Nucleic Acids Res. 1993;21:1507–1516. doi: 10.1093/nar/21.7.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonetto M, Gribskov MC, Gross C. The σ70 family: Sequence conservation and evolutionary relationships. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magasanik B. Regulation of nitrogen utilization. In: Neidhardt FC, editor. Escherichia coli and Salmonella. Washington D.C.: ASM Press; 1996. pp. 1344–1356. [Google Scholar]
- Malek AM, Izumo S, Alper SL. Quantitative densitometric analysis using a commercially available handheld cd digital camera. BioTechniques. 1997;22:1150–1153. [PubMed] [Google Scholar]
- Malhotra A, Severinova E. Crystal structure of a sigma 70 subunit fragment from E. coli RNA polymerase. Cell. 1996;87:127–136. doi: 10.1016/s0092-8674(00)81329-x. [DOI] [PubMed] [Google Scholar]
- Masepohl B, Klipp W. Organization and regulation of genes encoding the molybdenum nitrogenase and the alternative nitrogenase in Rhodobacter capsulatus.Arch. Microbiol. 1996;165:80–90. [Google Scholar]
- Merrick MJ. In a class of its own—the RNA polymerase sigma factor sigma 54 (sigma N) Mol Microbiol. 1993;10:903–909. doi: 10.1111/j.1365-2958.1993.tb00961.x. [DOI] [PubMed] [Google Scholar]
- Morett E, Segovia L. The σ54 bacterial enhancer-binding protein family: Mechanism of action and phylogenetic relationship of their functional domains. J Bacteriol. 1993;175:6067–6074. doi: 10.1128/jb.175.19.6067-6074.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninfa AJ, Magasanik B. Covalent modification of the glnG product, NRI, by the glnL product, NRII, regulates the transcription of the glnALG operon in Escherichia coli. Proc Natl Acad Sci. 1986;83:5909–5913. doi: 10.1073/pnas.83.16.5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninfa AJ, Reitzer LJ, Magasanik B. Initiation of transcription at the bacterial glnAp2 promoter by purified E. coli components is facilitated by enhancers. Cell. 1987;50:1039–1046. doi: 10.1016/0092-8674(87)90170-x. [DOI] [PubMed] [Google Scholar]
- North AK, Klose KE, Stedman KM, Kustu S. Prokaryotic enhancer-binding proteins reflect eukaryote-like modularity: The puzzle of nitrogen regulatory protein C. J Bacteriol. 1993;175:4267–4273. doi: 10.1128/jb.175.14.4267-4273.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak C, Chen W, Heck C, Klug G. Cloning, nucleotide sequence and characterization of the rpoD gene encoding the primary sigma factor of Rhodobacter capsulauts. Gene. 1996;176:177–184. doi: 10.1016/0378-1119(96)00243-0. [DOI] [PubMed] [Google Scholar]
- Perez-Martin J, de Lorenzo V. ATP binding to the sigma 54-dependent Activator XylR triggers a protein multimerization cycle catalyzed by UAS DNA. Cell. 1996;86:331–339. doi: 10.1016/s0092-8674(00)80104-x. [DOI] [PubMed] [Google Scholar]
- Pittard AJ, Davidson BE. TyrR protein of Escherichia coli and its role as repressor and activator. Mol Microbiol. 1991;5:1585–1592. doi: 10.1111/j.1365-2958.1991.tb01904.x. [DOI] [PubMed] [Google Scholar]
- Popham DL, Szeto D, Keener J, Kustu S. Function of a bacterial activator protein that binds to transcriptional enhancers. Science. 1989;243:629–635. doi: 10.1126/science.2563595. [DOI] [PubMed] [Google Scholar]
- Popham D, Keener J, Kustu S. Purification of the alternative σ factor, σ54, from Salmonella typhimurium and characterization of the σ54-holoenzyme. J Bio Chem. 1991;256:19510–19518. [PubMed] [Google Scholar]
- Preker P, Hübner P, Schmehl M, Klipp W, Bickle TA. Mapping and characterization of the promoter elements of the regulatory nif genes rpoN, nifA1 and nifA2 in Rhodobacter capsulatus. Mol Microbiol. 1992;6:1035–1048. doi: 10.1111/j.1365-2958.1992.tb02169.x. [DOI] [PubMed] [Google Scholar]
- Richet E, Raibaud O. MalT, the regulatory protein of the Escherichia coli maltose system, is an ATP-dependant transcriptional activator. EMBO J. 1989;8:981–987. doi: 10.1002/j.1460-2075.1989.tb03461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santero E, Hoover T, North A, Berger D, Porter S, Kustu S. Role of integration host factor in stimulating transcription from the σ54-dependent nifH promoter. J Mol Biol. 1992;227:602–620. doi: 10.1016/0022-2836(92)90211-2. [DOI] [PubMed] [Google Scholar]
- Sasse-Dwight S, Gralla JD. Probing the Escherichia coli glnALG upstream activation mechanism in vivo. Proc Natl Acad Sci. 1988;85:8934–8938. doi: 10.1073/pnas.85.23.8934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JR. Regulation of plasmid replication. Microbiol Rev. 1984;48:1–23. doi: 10.1016/b978-0-12-048850-6.50006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Porter S, Kustu S, Echols H. DNA-looping and enhancer activity: Association between DNA-bound NtrC activator and RNA polymerase at the bacterial glnA promoter. Proc Natl Acad Sci. 1990;87:5504–5508. doi: 10.1073/pnas.87.14.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed A, Gralla JD. Isolation and properties of enahncer-bypass mutants of sigma 54. Mol Microbiol. 1997;23:987–995. doi: 10.1046/j.1365-2958.1997.2851651.x. [DOI] [PubMed] [Google Scholar]
- Wang JT, Syed A, Hsieh M, Gralla JD. Converting Escherichia coli RNA polymerase into an enhancer-responsive enzyme: Role of an amino-terminal leucine patch in sigma 54. Science. 1995;270:992–994. doi: 10.1126/science.270.5238.992. [DOI] [PubMed] [Google Scholar]
- Wedel A, Weiss DS, Popham D, Droge P, Kustu S. A bacterial enhancer functions to tether a transcriptional activator near a promoter. Science. 1990;248:486–490. doi: 10.1126/science.1970441. [DOI] [PubMed] [Google Scholar]
- Weiss DS, Batut J, Klose KE, Keener J, Kustu S. The phosphorylated form of the enhancer-binding protein NTRC has an ATPase activity that is essential for activation of transcription. Cell. 1991;67:155–167. doi: 10.1016/0092-8674(91)90579-n. [DOI] [PubMed] [Google Scholar]
- Wilson TJ, Maroudas P, Howlett GJ, Davidson BE. Ligand-induced self-association of the Escherichia coli regulatory protein TyrR. J Mol Biol. 1994;238:309–318. doi: 10.1006/jmbi.1994.1294. [DOI] [PubMed] [Google Scholar]
- Wyman C, Rombel I, North AK, Bustamante C, Kustu S. Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science. 1997;275:1658–1661. doi: 10.1126/science.275.5306.1658. [DOI] [PubMed] [Google Scholar]
- Yang J, Ganesan S, Sarsero J, Pittard AJ. A genetic analysis of various functions of the TyrR protein of Escherichia coli. J Bacteriol. 1993;175:1767–1776. doi: 10.1128/jb.175.6.1767-1776.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]