

Abstract
Photolyase uses blue light to restore the major ultraviolet (UV)-induced DNA damage, the cyclobutane pyrimidine dimer (CPD), to two normal bases by splitting the cyclobutane ring. Our earlier studies showed that the overall repair is completed in 700 ps through a cyclic electron-transfer radical mechanism. However, the two fundamental processes, electron-tunneling pathways and cyclobutane ring splitting, were not resolved. Here, we use ultrafast UV absorption spectroscopy to show that the CPD splits in two sequential steps within 90 ps and the electron tunnels between the cofactor and substrate through a remarkable route with an intervening adenine. Site-directed mutagenesis reveals that the active-site residues are critical to achieving high repair efficiency, a unique electrostatic environment to optimize the redox potentials and local flexibility, and thus balance all catalytic reactions to maximize enzyme activity. These key findings reveal the complete spatio-temporal molecular picture of CPD repair by photolyase and elucidate the underlying molecular mechanism of the enzyme’s high repair efficiency.
Keywords: DNA repair photocycle, ultrafast enzyme dynamics, thymine dimer splitting, electron tunneling pathway, active-site mutation
Ultraviolet (UV) component of sunlight irradiation causes DNA damage by inducing the formation of cyclobutane pyrimidine dimer (CPD), which is mutagenic and a leading cause of skin cancer (1–3). CPD can be completely restored by a photoenzyme, photolyase, through absorption of visible blue light (4). In our early work (5–7), we have observed a cyclic electron-transfer (ET) reaction in thymine dimer (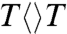 ) repair by photolyase and determined the time scale of 700 ps for the complete repair photocycle (7). However, the central questions of whether the splitting of the cyclobutane ring is synchronously or asynchronously concerted or stepwise and whether the cyclic ET involves specific tunneling pathways were not resolved. Furthermore, the molecular mechanism underlying the high repair efficiency has not been elucidated. Here, using femtosecond spectroscopy and site-directed mutagenesis, we are able to measure the dynamics of all initial reactants, reaction intermediates, and final products with different substrates and with wild-type and active-site mutant enzymes, and thus reveal the complete spatio-temporal molecular picture of thymine dimer repair by photolyase.
) repair by photolyase and determined the time scale of 700 ps for the complete repair photocycle (7). However, the central questions of whether the splitting of the cyclobutane ring is synchronously or asynchronously concerted or stepwise and whether the cyclic ET involves specific tunneling pathways were not resolved. Furthermore, the molecular mechanism underlying the high repair efficiency has not been elucidated. Here, using femtosecond spectroscopy and site-directed mutagenesis, we are able to measure the dynamics of all initial reactants, reaction intermediates, and final products with different substrates and with wild-type and active-site mutant enzymes, and thus reveal the complete spatio-temporal molecular picture of thymine dimer repair by photolyase.
Photolyase contains a fully reduced flavin adenine dinucleotide (FADH-) as the catalytic cofactor and electron donor (4). Based on previous studies (4–10), a sequential repair mechanism of thymine dimer splitting is shown in Fig. 1. Previously, we found that the forward ET from FADH-∗ to 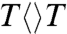 occurs in 250 ps (1/kFET) and the total decay of intermediate FADH• in 700 ps (1/ktotal) (5, 7). These dynamics usually follow a stretched-exponential decay behavior, reflecting heterogeneous ET dynamics controlled by the active-site solvation (5, 6, 11). However, in that study, no thymine-related species could be detected in the visible-light region and no information about the dimer splitting was obtained. To reveal how the thymine dimer splits, we extended our detection wavelengths from visible to deep UV light to catch thymine-related intermediates. To uncover how the electron tunnels in the repair, we used different dimer substrates to follow electron-tunneling pathways. Finally, to elucidate how photolyase achieves such high repair efficiency, we designed a series of active-site mutants to identify the key residues for synergistic catalytic reactions.
occurs in 250 ps (1/kFET) and the total decay of intermediate FADH• in 700 ps (1/ktotal) (5, 7). These dynamics usually follow a stretched-exponential decay behavior, reflecting heterogeneous ET dynamics controlled by the active-site solvation (5, 6, 11). However, in that study, no thymine-related species could be detected in the visible-light region and no information about the dimer splitting was obtained. To reveal how the thymine dimer splits, we extended our detection wavelengths from visible to deep UV light to catch thymine-related intermediates. To uncover how the electron tunnels in the repair, we used different dimer substrates to follow electron-tunneling pathways. Finally, to elucidate how photolyase achieves such high repair efficiency, we designed a series of active-site mutants to identify the key residues for synergistic catalytic reactions.
Fig. 1.

Enzyme-substrate complex structure and one sequential repair mechanism with all elementary reactions. X-ray complex structure of A. nidulans photolyase with DNA containing a repaired photoproduct of thymine dimer. E. coli photolyase has a similar structure. Two critical conserved residues in the active site are E283 (E274 in E. coli) near the substrate and N386 (N378 in E. coli) near the cofactor. The thymine dimer is flipped out of DNA and inserted into the active site. A close-up view shows the relative positions of the catalytic cofactor FADH-, the conserved residues E283 and N386, and the repaired substrate with the electron-tunneling pathways in repair. Shown in the sequential repair scheme (bottom box) are forward ET (FET, reaction rate kFET) from FADH-∗ to thymine dimer upon light excitation, followed by back ET (BET, reaction rate kBET) without repair, and the repair channel including splitting of two bonds of C5-C5’ (reaction rate ksp1) and C6-C6’ (reaction rate ksp2) in thymine dimer with subsequent electron return (ER, reaction rate kER) after complete ring splitting. ktotal is the overall decay rate of intermediate state FADH• after the initial charge separation.
Results and Discussion
Sequential Splitting Dynamics of the Cyclobutane Ring.
Fig. 2 shows a striking pattern of the transient absorption signals of the complex of Escherichia coli photolyase with substrate 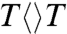 , probed at fifteen wavelengths. At 430 nm, the signal is the summation of all three flavin species (FADH-∗, FADH•, and FADH-) and decays to zero upon completion of repair. From 335 nm in the UV region, we captured the formation and decay of thymine-related intermediates and from 300 nm we clearly observed the long-component formation of final repaired thymines (Fig. 2 B–E insets). By knowing the dynamics of FADH-∗ and the absorption coefficients of FADH• and FADH- (Fig. 2A inset), only with the sequential model shown in Fig. 1 and not any other synchronously or asynchronously concerted schemes of thymine splitting and electron return (8–10), we can systematically fit all the absorption transients from visible to UV (see SI Text), as shown in deconvolution of various species in Fig. 2 B–E insets and the related absorption coefficients in Fig. 2A inset, and thus obtain the entire dynamics of thymine dimer splitting.
, probed at fifteen wavelengths. At 430 nm, the signal is the summation of all three flavin species (FADH-∗, FADH•, and FADH-) and decays to zero upon completion of repair. From 335 nm in the UV region, we captured the formation and decay of thymine-related intermediates and from 300 nm we clearly observed the long-component formation of final repaired thymines (Fig. 2 B–E insets). By knowing the dynamics of FADH-∗ and the absorption coefficients of FADH• and FADH- (Fig. 2A inset), only with the sequential model shown in Fig. 1 and not any other synchronously or asynchronously concerted schemes of thymine splitting and electron return (8–10), we can systematically fit all the absorption transients from visible to UV (see SI Text), as shown in deconvolution of various species in Fig. 2 B–E insets and the related absorption coefficients in Fig. 2A inset, and thus obtain the entire dynamics of thymine dimer splitting.
Fig. 2.

Femtosecond-resolved transient absorption dynamics of reactants, various intermediates, and products involved in repair of thymine dimer. The repair dynamics are probed systematically from 800 to 260 nm and shown are the typical results in UV region with a distinct pattern. Inset (A): The absorption coefficients of all species involved in repair. The absorption spectra of FADH- (red) and FADH• (green) were obtained from steady-state absorption measurements. The absorption spectrum of thymine (dark yellow) shorter than 290 nm was obtained by the steady-state measurements and longer than 290 nm by fitting results. The absorption spectrum of FADH-∗ (dashed blue) and relative absorption spectra of dimer anion intermediates (T-T-, dashed cyan; T-, dashed dark red) are calibrated by the flavin ground-state coefficient, respectively. Insets (B–E): Transient absorption signals probed at 335, 300, 270 and 266 nm. These dynamics are systematically fitted by total flavin-related species (FADH-∗ + FADH• + FADH-, dashed red), thymine dimer intermediate T-T- (dashed cyan), thymine anion (dashed dark red) and thymine products (dashed dark yellow).
Our data indicate that in contrast to the reaction schemes proposed in previous computational studies (9, 10) the thymine dimer splits by a sequential pathway. We note, however, that we were not able to detect the trace signal of 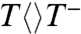 , giving an upper limit of less than 10 ps for the first-bond C5-C5′ breakage (1/ksp1), consistent with the theoretical prediction of a nearly barrierless process (9, 10, 12). This slow formation (kFET) and ultrafast decay (ksp1) result in negligible accumulation of
, giving an upper limit of less than 10 ps for the first-bond C5-C5′ breakage (1/ksp1), consistent with the theoretical prediction of a nearly barrierless process (9, 10, 12). This slow formation (kFET) and ultrafast decay (ksp1) result in negligible accumulation of 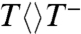 population. However, we did observe the formation and decay of T-T- intermediate after the first-bond C5-C5′ breakage (Fig. 1 and Fig. 2 B, D, and E insets). The decay dynamics in 87 ps mainly represents the second-bond C6-C6′ splitting. Given that the total repair quantum yield is 0.82 and the forward ET yield is 0.85 (4, 5), thus the splitting yield is 0.96, resulting in the second-bond breakage in 90 ps (1/ksp2), much longer than that of theoretical calculations (9, 10, 12), and the back ET without the second-bond splitting in 2.4 ns (1/kBET). The slow formation and fast decay of the T-T- intermediate also cause less accumulation and an apparent reverse kinetics (Fig. 2 B, D, and E insets). After the second-bond cleavage, we observed the signal of T- at around 290 nm that decays in 700 ps (1/kER and Fig. 2C inset), reflecting that the electron return from T- to FADH• is completely decoupled from the second-bond breaking. Thus, the final products of two repaired thymines are formed in two sequential steps in 90 and 700 ps, respectively, upon the initial electron injection (see Fig. 2 D and E insets).
population. However, we did observe the formation and decay of T-T- intermediate after the first-bond C5-C5′ breakage (Fig. 1 and Fig. 2 B, D, and E insets). The decay dynamics in 87 ps mainly represents the second-bond C6-C6′ splitting. Given that the total repair quantum yield is 0.82 and the forward ET yield is 0.85 (4, 5), thus the splitting yield is 0.96, resulting in the second-bond breakage in 90 ps (1/ksp2), much longer than that of theoretical calculations (9, 10, 12), and the back ET without the second-bond splitting in 2.4 ns (1/kBET). The slow formation and fast decay of the T-T- intermediate also cause less accumulation and an apparent reverse kinetics (Fig. 2 B, D, and E insets). After the second-bond cleavage, we observed the signal of T- at around 290 nm that decays in 700 ps (1/kER and Fig. 2C inset), reflecting that the electron return from T- to FADH• is completely decoupled from the second-bond breaking. Thus, the final products of two repaired thymines are formed in two sequential steps in 90 and 700 ps, respectively, upon the initial electron injection (see Fig. 2 D and E insets).
Electron-Tunneling Pathways and Functional Role of Adenine Moiety.
The FADH- cofactor in photolyase has an unusual bent U-shaped conformation with the isoalloxazine and adenine rings in close proximity (Fig. 1). The crystal structure of Aspergillus nidulans photolyase with CPD complex shows that the adenine moiety of FADH- is at van der Waals distances with both base moieties of CPD, 3.1 Å to the 5′ side and 3.2 Å to 3′, and the first carbon atom linked to the isoalloxazine ring at 3.6 Å (dashed red lines in Fig. 1) (13). Intramolecular electron hopping from the isoalloxazine ring to the adenine moiety is unfavorable due to their redox potentials (ΔG ∼ +0.1 eV) (14) and moreover we did not observe any fast quenching of FADH-∗ fluorescence without substrate (5). It is thought that the repair reaction by photolyase involves electron tunneling (15–17). However, the cyclic electron-tunneling pathways, forward (kFET) and backward (kBET) or return (kER), are a matter of some debate. One view is that the electron tunneling is mediated by the intervening adenine with a total distance of about 8 Å (15). An alternative model suggests that tunneling occurs directly from the o-xylene ring of FADH- to the 3′ side of CPD with a shortest distance of 4.3 Å (16, 17). To test the electron-tunneling directionality, we used a series of substrates, 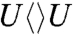 ,
, 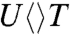 ,
, 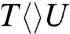 and
and 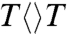 (chemical structures in Fig. 3), as electron acceptors to follow electron-tunneling directions.
(chemical structures in Fig. 3), as electron acceptors to follow electron-tunneling directions.
Fig. 3.

Femtosecond-resolved transient absorption dynamics of DNA repairs with different combination of bases. (A) Transient absorption signals of repair with 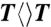 ,
, 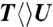 ,
, 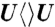 , and
, and 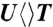 probed at 710 and 620 nm. The dynamics of FADH-∗ (blue line) was probed at 710 nm. The signal at 620 nm is the combination of FADH-∗ and intermediate FADH• (red line) contributions. The chemical structures of various CPD substrates are also shown with highlight at uracil sides. The blue shading of U indicates the forward electron-tunneling to the 5′ side of DNA and the red shading for electron return starting at the 3′ side after the complete two-bond splitting. (B–D) Repair dynamics of
probed at 710 and 620 nm. The dynamics of FADH-∗ (blue line) was probed at 710 nm. The signal at 620 nm is the combination of FADH-∗ and intermediate FADH• (red line) contributions. The chemical structures of various CPD substrates are also shown with highlight at uracil sides. The blue shading of U indicates the forward electron-tunneling to the 5′ side of DNA and the red shading for electron return starting at the 3′ side after the complete two-bond splitting. (B–D) Repair dynamics of 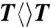 (orange),
(orange), 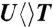 (blue),
(blue), 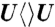 (green), and
(green), and 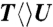 (dark red) probed at 270 nm (B), 300 nm (C) and 335 nm (D). Insets show the deconvolution of total flavin-related species (dashed red), CPD intermediate anions T-U-/U-U- (dashed cyan), and T- (dashed dark red), and the products of T/U (dashed dark yellow) of repair with
(dark red) probed at 270 nm (B), 300 nm (C) and 335 nm (D). Insets show the deconvolution of total flavin-related species (dashed red), CPD intermediate anions T-U-/U-U- (dashed cyan), and T- (dashed dark red), and the products of T/U (dashed dark yellow) of repair with 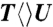 ,
, 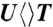 , and
, and 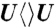 in (B), (C), and (D), respectively.
in (B), (C), and (D), respectively.
The forward ET dynamics, detected at 710 nm (blue lines in Fig. 3A), occur in 63, 73, 85, and 250 ps for 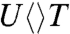 ,
, 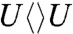 ,
, 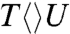 , and
, and 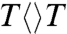 . Generally, uracil has a higher reduction potential by 0.1 V than thymine (14), which provides a larger driving force and results in the faster forward ET dynamics. Significantly, the ET rates increase with U at the 5′ position, indicating that the electron tunneling ends at the 5′ side of CPD (Fig. 1). This observation proves that the adenine moiety mediates forward ET toward the 5′ side of CPD and enhances the ET rate through a superexchange mechanism, ruling out the direct ET from the o-xylene ring of FADH- to the 3′ side of CPD. This result is further supported by the comparison between the repair of CPD and (6-4) photoproduct. The (6-4) photolyase, which specifically repairs the (6-4) photoproduct, exhibits a similar U-shaped cofactor configuration of FADH- and forward ET dynamics (280 ps) as CPD photolyase (18) but the shortest distance from the cofactor to (6-4) photoproduct is 6.3 Å, which is 2 Å longer than that of CPD (19). These observations strongly suggest a general mechanism that the electron from FADH-∗ tunnels through the adenine moiety to substrates.
. Generally, uracil has a higher reduction potential by 0.1 V than thymine (14), which provides a larger driving force and results in the faster forward ET dynamics. Significantly, the ET rates increase with U at the 5′ position, indicating that the electron tunneling ends at the 5′ side of CPD (Fig. 1). This observation proves that the adenine moiety mediates forward ET toward the 5′ side of CPD and enhances the ET rate through a superexchange mechanism, ruling out the direct ET from the o-xylene ring of FADH- to the 3′ side of CPD. This result is further supported by the comparison between the repair of CPD and (6-4) photoproduct. The (6-4) photolyase, which specifically repairs the (6-4) photoproduct, exhibits a similar U-shaped cofactor configuration of FADH- and forward ET dynamics (280 ps) as CPD photolyase (18) but the shortest distance from the cofactor to (6-4) photoproduct is 6.3 Å, which is 2 Å longer than that of CPD (19). These observations strongly suggest a general mechanism that the electron from FADH-∗ tunnels through the adenine moiety to substrates.
Next, we did a series of studies by UV detection for these substrates to gain further information on the splitting of the cyclobutane ring and the subsequent electron return pathway. Fig. 3 B–D show typical three signals probed at 270, 300, and 335 nm. With systematic analyses (SI Text), we obtained the second-bond splitting in 35 ps for both  and
and 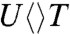 and 75 ps for
and 75 ps for 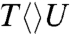 , similar to that of
, similar to that of 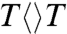 . Thus, after the initial electron tunneling to the 5′ side and the subsequent prompt splitting of the C5-C5′ bond, the resulting radicals are much more stable in
. Thus, after the initial electron tunneling to the 5′ side and the subsequent prompt splitting of the C5-C5′ bond, the resulting radicals are much more stable in 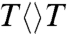 and
and 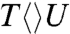 than
than 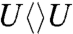 and
and 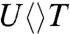 due to the methyl group at the C5 position, on the 5′ side, resulting in slowdown of the second-bond C6-C6′ breakage by a factor of 2. Finally, the electron returns from these repaired substrates take 185 and 210 ps for (T + U)- and (U + U)-, respectively, and 1,220 ps for (U + T)-, leaving 700 ps of (T + T)- in the middle (Fig. 3 A–D). Given that all electron returns in photo-induced ET reactions occur in the Marcus inverted region, thus the electron from U- has a faster tunneling rate than from T- back to the FADH• to restore the active state FADH- and complete the repair photocycle. Clearly, after repair the electron mainly stays on the 3′ side, leading to T + U- and U + U- with the fastest back electron-tunneling rates and U + T- with a longest tunneling time due to the stronger electron affinity of U in proximity. Thus, from the forward ET and second-bond splitting times of these substrates, the forward electron tunneling takes the remarkable pathway from the isoalloxazine ring to the first carbon atom linked to the ring through a covalent bond (1.5 Å) and then to the adenine moiety and finally to the 5′ side of CPD in a total distance of 8.2 Å, rather than taking the shortest distance of 4.3 Å without any bridging molecules, similar to tunneling in vacuum. After the complete breakage of the two C-C bonds, the electron stays at the 3′ side and tunnels back along the original adenine-mediated pathway (Fig. 1). The electron-tunneling, both forward and return, has unique directionality and the adenine moiety has a critical functional role.
due to the methyl group at the C5 position, on the 5′ side, resulting in slowdown of the second-bond C6-C6′ breakage by a factor of 2. Finally, the electron returns from these repaired substrates take 185 and 210 ps for (T + U)- and (U + U)-, respectively, and 1,220 ps for (U + T)-, leaving 700 ps of (T + T)- in the middle (Fig. 3 A–D). Given that all electron returns in photo-induced ET reactions occur in the Marcus inverted region, thus the electron from U- has a faster tunneling rate than from T- back to the FADH• to restore the active state FADH- and complete the repair photocycle. Clearly, after repair the electron mainly stays on the 3′ side, leading to T + U- and U + U- with the fastest back electron-tunneling rates and U + T- with a longest tunneling time due to the stronger electron affinity of U in proximity. Thus, from the forward ET and second-bond splitting times of these substrates, the forward electron tunneling takes the remarkable pathway from the isoalloxazine ring to the first carbon atom linked to the ring through a covalent bond (1.5 Å) and then to the adenine moiety and finally to the 5′ side of CPD in a total distance of 8.2 Å, rather than taking the shortest distance of 4.3 Å without any bridging molecules, similar to tunneling in vacuum. After the complete breakage of the two C-C bonds, the electron stays at the 3′ side and tunnels back along the original adenine-mediated pathway (Fig. 1). The electron-tunneling, both forward and return, has unique directionality and the adenine moiety has a critical functional role.
Active-Site Mutation and Repair Efficiency Modulation.
The repair efficiency (0.82) of thymine dimer by photolyase is higher than those (0.004–0.41) of all chemical model systems synthesized so far (20–22), indicating that the amino acids in the active site must significantly contribute to the repair efficiency by modulating the redox properties of the flavin/CPD pair or by steric effects. To examine how the protein active site controls the higher repair efficiency, we mutated a series of residues (E274A, R226A, R342A, N378C, and M345A) at the active site and here showed two typical mutants, N378C near the cofactor side and E274A near the substrate side, which make critical contributions to the repair efficiency (Fig. 4A). We systematically studied these two mutants by probing from visible to UV wavelengths. Four typical results are shown in Fig. 4 B–D at 800, 620, 270, and 266 nm. The final results of forward ET, back ET, second-bond splitting, and electron return are shown in Fig. 5B with the measured total repair quantum yields in Fig. 5A. Both mutants exhibit the lower quantum yields of 0.69 for N378C and 0.40 for E274A, resulting from a combination of two-step quantum yields, forward ET relative to lifetime emission (kLT) and second-bond splitting relative to back ET (two pairs of dashed lines in Fig. 5B). Both mutants modulate the ET redox potentials, N378C for FADH- at the cofactor side and E274A for 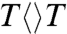 at the 5′ side, leading to longer forward ET times and thus resulting in the lower first-step quantum yields. Furthermore, the backward ET processes significantly become faster, reducing the chance for the second-bond splitting and again causing a decrease in the second-step splitting quantum yields. For N378C, we obtained the same second-bond splitting time in 90 ps as the wild type, consistent with the fact that the mutation affects only the cofactor. For E274A we observed a faster second-bond splitting time of 30 ps, probably due to the destabilization of the splitting transition state by the mutant of E274A that abolishes two hydrogen-bonds with
at the 5′ side, leading to longer forward ET times and thus resulting in the lower first-step quantum yields. Furthermore, the backward ET processes significantly become faster, reducing the chance for the second-bond splitting and again causing a decrease in the second-step splitting quantum yields. For N378C, we obtained the same second-bond splitting time in 90 ps as the wild type, consistent with the fact that the mutation affects only the cofactor. For E274A we observed a faster second-bond splitting time of 30 ps, probably due to the destabilization of the splitting transition state by the mutant of E274A that abolishes two hydrogen-bonds with 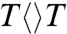 at the 5′ side (Fig. 4A). The observation of thymine dimer repair by mutant E274A also excludes any possibility of proposed proton transfer(s) between E274 with
at the 5′ side (Fig. 4A). The observation of thymine dimer repair by mutant E274A also excludes any possibility of proposed proton transfer(s) between E274 with 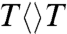 during repair that has been suggested based on theoretical consideration (10, 23).
during repair that has been suggested based on theoretical consideration (10, 23).
Fig. 4.

Effect of active-site mutations on repair dynamics. (A) X-ray structure of the active site of A. nidulans photolyase with two critical residues of N386 (N378 in E. coli) and E283 (E274 in E. coli). The hydrogen-bonding distances of the two residues with FADH- and CPD are also shown, respectively. (B–D) Femtosecond-resolved absorption signals of the repair of damaged CPD by the wild type and two mutants (N378C and E274A) probed at 800 and 620 nm (B), 270 nm (C), and 266 nm (D). Insets in (B) and (C) show the deconvolution of various species’ contributions of N378C mutant probed at 620 and 270 nm, respectively, while the inset in (D) for E274A mutant probed at 260 nm.
Fig. 5.

Quantum yields and various reactions times of four elementary steps. (A) The overall repair quantum yields (QY) of the two mutants N378C and E274A were measured, relative to the known wild-type one (0.82), by monitoring the formation of thymine bases at 266-nm absorption with certain visible-light irradiation of the enzyme-substrate solution. (B) The reaction times of each elementary step in CPD repair by the wild type and two mutants with our measured total QY from (A). The vertical dashed lines represent the two-step repair efficiency of the FET to lifetime emission (LT) and the second-bond splitting (SP2) to BET. The ER is decoupled from the CPD splitting.
Conclusion
We reported our direct observation of ultrafast sequential splitting dynamics of the cyclobutane ring in a few and 90 picoseconds and identification of unique electron-tunneling pathways in dimer repair. Such identification reveals the critical functional role of the adenine moiety as an efficient electron-tunneling mediator in the unique bent U-shape conformation of flavin cofactor. During the repair, the back ET without the second-bond splitting tremendously slows down to 2.4 ns to enhance the repair channel and the final electron return after the repair is in 700 ps, completely decoupled from the ring splitting. Thus, to maximize the repair quantum yield and balance the four elementary processes between the forward ET and lifetime emission, and between the second-bond splitting and back ET, the active-site electrostatics of photolyase must contain the appropriate functional groups to optimize redox potentials and active-site mobility for electron tunneling (5, 6, 24). Clearly, the active-site environment in photolyase seems ideal for CPD repair and is well optimized over the course of evolution. Any mutation would break the delicate balance of the four processes and is unlikely to speed up the forward ET and slow down the back ET (25, 26). The best combination of the four processes is shown in Fig. 6, a complete photocycle for the maximum repair of thymine dimer by photolyase on the ultrafast time scale (27).
Fig. 6.

Complete photocycle of CPD repair by photolyase. All resolved elementary steps of CPD (thymine dimer) repair with reaction times, showing the complete repair photocycle on the ultrafast time scales and the elucidated molecular mechanism.
Materials and Methods
CPD Photolyase and Mutants.
The purification of E. coli CPD photolyase with depletion of the antenna cofactor has been reported previously (28, 29). For mutant studies, we mutated a series of critical residues (E274A, R226A, R342A, N378C, and M345A) at the active site, including two typical mutants of N378C near the flavin cofactor side and E274A near the substrate side, to examine enzyme activities. Mutant plasmids were constructed using QuikChange II XL kit (Stratagene) based on the plasmid of wild-type enzyme. All mutated DNA plasmids were sequenced to ensure correct results. In femtosecond UV absorption studies, 100 μM of enzyme (or 50 μM in experiments with probe wavelengths of shorter than 300 nm) was used in a reaction buffer containing 100 mM NaCl, 50 mM Tris-HCl at pH 7.5, 20 mM dithiothreitol, 1 mM EDTA, and 50% (v/v) glycerol. For some visible-light measurements, a higher enzyme concentration of 300 μM was used.
For absorption spectra of FADH• and FADH- at longer than 300 nm (see inset Fig. 2A), we directly obtained them after purification from holoenzyme with FADH• and then the active form FADH- by photoactivation. The absorption spectra of FADH• and FADH- at shorter than 300 nm were acquired as described elsewhere with some modifications (30). In short, we dissolved 10 μM of photolyase in 50 mM KPi buffer with pH 3.5 for half hour. The flavin cofactor was released from the binding pocket and was completely removed by concentrating the sample solution in Amicon centrifugal filter devices [30,000 molecular weight cut-off (MWCO)] to 10% of the volume and then restoring back to the original volume, and these processes were repeated by three times. The absorption spectrum of apoenzyme was then obtained. The absorption spectra of FADH• and FADH- below 300 nm were obtained by subtracting the absorption spectrum of apoenzyme from holoenzyme absorption spectra at the respective redox states.
CPD Substrates.
We prepared various CPD substrates (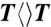 ,
, 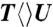 ,
, 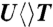 ,
, 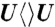 ) as described elsewhere with some modifications (31). The dinucleotide dTpdT, dTpdU, dUpdT, dUpdU (Sigma-Aldrich) were dissolved in 15% aqueous acetone (v/v) with a concentration of 100 optical density (OD) units per mL. The argon-purged DNA solutions were irradiated on ice with a UVB lamp (302 nm, General Electric) at a distance of 2 cm for 1.5–2 h to eliminate the distinctive 260 nm absorption peak. After irradiation, we purified the cyclobutane pyrimidine dimers by HPLC, using a C18 reversed-phase column (Grace, 250 mm × 10 mm) with a 75 mM potassium phosphate buffer (pH 6.8). The potassium salt was removed later by washing and eluting with water twice through the same C-18 column. The final concentration of all CPD substrates used in femtosecond and steady-state repair experiments is 18 mM. For all experiments, the ratio of the substrate to photolyase is at least 60∶1 and such mixing can be used for several hours for femtosecond-resolved measurements without any notice of change of the repair dynamics.
) as described elsewhere with some modifications (31). The dinucleotide dTpdT, dTpdU, dUpdT, dUpdU (Sigma-Aldrich) were dissolved in 15% aqueous acetone (v/v) with a concentration of 100 optical density (OD) units per mL. The argon-purged DNA solutions were irradiated on ice with a UVB lamp (302 nm, General Electric) at a distance of 2 cm for 1.5–2 h to eliminate the distinctive 260 nm absorption peak. After irradiation, we purified the cyclobutane pyrimidine dimers by HPLC, using a C18 reversed-phase column (Grace, 250 mm × 10 mm) with a 75 mM potassium phosphate buffer (pH 6.8). The potassium salt was removed later by washing and eluting with water twice through the same C-18 column. The final concentration of all CPD substrates used in femtosecond and steady-state repair experiments is 18 mM. For all experiments, the ratio of the substrate to photolyase is at least 60∶1 and such mixing can be used for several hours for femtosecond-resolved measurements without any notice of change of the repair dynamics.
Enzyme Activities.
We measured the enzyme activities of thymine dimer (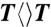 ) repair by photolyase mutants as follows. Two mixtures of samples in two cuvettes were prepared with 1 μM concentrations of the wild-type and mutant photolyase (N378C or E274A) with 18 mM
) repair by photolyase mutants as follows. Two mixtures of samples in two cuvettes were prepared with 1 μM concentrations of the wild-type and mutant photolyase (N378C or E274A) with 18 mM 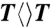 substrate, respectively. Then, the two cuvettes were purged with argon and irradiated at room temperature using a white-light lamp (General Electric) with the same distance of 6 cm. We measured the change of absorption spectra of the two mixtures and recorded the absorption change at 266 nm with the illumination time. Such steady-state repair experiments were repeated for multiple times (one typical result is shown in Fig. S1). The change of absorption at 266 nm was averaged by these measurements and the slope of the average-absorption change against time is proportional to the repair quantum yield of the enzyme. By knowing the repair quantum yield of
substrate, respectively. Then, the two cuvettes were purged with argon and irradiated at room temperature using a white-light lamp (General Electric) with the same distance of 6 cm. We measured the change of absorption spectra of the two mixtures and recorded the absorption change at 266 nm with the illumination time. Such steady-state repair experiments were repeated for multiple times (one typical result is shown in Fig. S1). The change of absorption at 266 nm was averaged by these measurements and the slope of the average-absorption change against time is proportional to the repair quantum yield of the enzyme. By knowing the repair quantum yield of 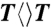 by the wild type (0.82), we then obtained the mutant repair quantum yield. In addition, we did not observe any absorption change at 266 nm in the control experiments with only
by the wild type (0.82), we then obtained the mutant repair quantum yield. In addition, we did not observe any absorption change at 266 nm in the control experiments with only 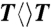 at the same concentration under all the exactly same conditions.
at the same concentration under all the exactly same conditions.
Femtosecond Absorption Spectroscopy.
All the femtosecond-resolved measurements were carried out using the transient absorption methods. The experimental layout has been detailed previously (32, 33). Briefly, for all measurements, the pump pulse at 400 nm in 1 kHz was generated by the doubling of 800 nm in a 0.2 mm thick β-barium borate crystal (BBO, type I). The pump pulse energy was typically attenuated to 140–200 nJ/pulse before being focused into the sample cell. All desired probe wavelengths, from visible to ultraviolet, were generated from optical parametric amplifiers (OPA-800C and TOPAS, Spectra-Physics). The instrument response time is about 250 fs and all experiments were done at the magic angle (54.7°). Samples were kept fast stirring during irradiation to maintain the fresh complex concentration as well as to avoid heating and photobleaching. All enzyme reactions in the femtosecond-resolved measurements were carried out under anaerobic conditions.
Supplementary Material
Acknowledgments.
We thank Dr. Chaitanya Sexena for the initial help with experiment in Fig. 3A. This work is supported in part by the National Institute of Health (Grant GM074813), the Packard fellowship (to D.Z.), the American Heart Association fellowship (to Z.L.) and the Ohio State University Pelotonia fellowship (to C.T. and J.L.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110927108/-/DCSupplemental.
References
- 1.Taylor JS. Unraveling the molecular pathway from sunlight to skin cancer. Acc Chem Res. 1994;27:76–82. [Google Scholar]
- 2.Daya-Grosjean L, Dumaz N, Sarasin A. The specificity of p53 mutation spectra in sunlight-induced human cancers. J Photochem Photobiol B. 1995;28:115–124. doi: 10.1016/1011-1344(95)07130-t. [DOI] [PubMed] [Google Scholar]
- 3.Lima-Bessa KM, Menck CFM. Skin cancer: lights on genome lesions. Curr Biol. 2005;15:R58–R61. doi: 10.1016/j.cub.2004.12.056. [DOI] [PubMed] [Google Scholar]
- 4.Sancar A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem Rev. 2003;103:2203–2237. doi: 10.1021/cr0204348. [DOI] [PubMed] [Google Scholar]
- 5.Kao Y-T, Saxena C, Wang L, Sancar A, Zhong D. Direct observation of thymine dimer repair in DNA by photolyase. Proc Natl Acad Sci USA. 2005;102:16128–16132. doi: 10.1073/pnas.0506586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang C-W, et al. Ultrafast solvation dynamics at binding and active sites of photolyases. Proc Natl Acad Sci USA. 2010;107:2914–2919. doi: 10.1073/pnas.1000001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kao Y-T, Saxena C, Wang L, Sancar A, Zhong D. Femtochemistry in enzyme catalysis: DNA photolyase. Cell Biochem Biophys. 2007;48:32–44. doi: 10.1007/s12013-007-0034-5. [DOI] [PubMed] [Google Scholar]
- 8.MacFarlane AW, Stanley RJ. Cis-syn thymidine dimer repair by DNA photolyase in real time. Biochemistry. 2003;42:8558–8568. doi: 10.1021/bi034015w. [DOI] [PubMed] [Google Scholar]
- 9.Hassanali AA, Zhong D, Singer SJ. An AIMD study of the CPD repair mechanism in water: reaction free energy surface and mechanistic implications. J Phys Chem B. 2011;115:3848–3859. doi: 10.1021/jp107722z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masson F, Laino T, Rothlisberger U, Hutter J. A QM/MM investigation of thymine dimer radical anion splitting catalyzed by DNA photolyase. ChemPhysChem. 2009;10:400–410. doi: 10.1002/cphc.200800624. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, et al. Protein dynamics control the kinetics of initial electron transfer in photosynthesis. Science. 2007;316:747–750. doi: 10.1126/science.1140030. [DOI] [PubMed] [Google Scholar]
- 12.Harrison CB, O’Neil LL, Wiest O. Computational studies of DNA photolyase. J Phys Chem A. 2005;109:7001–7012. doi: 10.1021/jp051075y. [DOI] [PubMed] [Google Scholar]
- 13.Mees A, et al. Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science. 2004;306:1789–1793. doi: 10.1126/science.1101598. [DOI] [PubMed] [Google Scholar]
- 14.Seidel CAM, Schulz A, Sauer MHM. Nucleobase-specific quenching of fluorescent dyes. 1. Nucleobase one-electron redox potentials and their correlation with static and dynamic quenching efficiencies. J Phys Chem. 1996;100:5541–5553. [Google Scholar]
- 15.Antony J, Medvedev DM, Stuchebrukhov AA. Theoretical study of electron transfer between the photolyase catalytic cofactor FADH- and DNA thymine dimer. J Am Chem Soc. 2000;122:1057–1065. [Google Scholar]
- 16.Prytkova TR, Beratan DN, Skourtis SS. Photoselected electron transfer pathways in DNA photolyase. Proc Natl Acad Sci USA. 2007;104:802–807. doi: 10.1073/pnas.0605319104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Acocella A, Jones GA, Zerbetto F. What is adenine doing in photolyase? J Phys Chem B. 2010;114:4101–4106. doi: 10.1021/jp101093z. [DOI] [PubMed] [Google Scholar]
- 18.Li J, et al. Dynamics and mechanism of repair of ultraviolet-induced (6-4) photoproduct by photolyase. Nature. 2010;466:887–891. doi: 10.1038/nature09192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maul MJ, et al. Crystal structure and mechanism of a DNA (6-4) photolyase. Angew Chemie Int Ed Engl. 2008;47:10076–10080. doi: 10.1002/anie.200804268. [DOI] [PubMed] [Google Scholar]
- 20.Kim ST, Hartman RF, Rose SD. Solvent dependence of pyrimidine dimer splitting in a covalently linked dimer-indole system. Photochem Photobiol. 1990;52:789–794. doi: 10.1111/j.1751-1097.1990.tb08683.x. [DOI] [PubMed] [Google Scholar]
- 21.Carell T, Epple R. Repair of UV light induced DNA lesions: a comparative study with model compounds. Eur J Org Chem. 1998;1998:1245–1258. [Google Scholar]
- 22.Song QH, et al. Efficient photosensitized splitting of thymine dimer by a covalently linked tryptophan in solvents of high polarity. Eur J Org Chem. 2005;2005:1097–1106. [Google Scholar]
- 23.Essen LO, Klar T. Light-driven DNA repair by photolyases. Cell Mol Life Sci. 2006;63:1266–1277. doi: 10.1007/s00018-005-5447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hassanali AA, Zhong D, Singer SJ. An AIMD study of CPD repair mechanism in water: role of solvent in ring splitting. J Phys Chem B. 2011;115:3860–3871. doi: 10.1021/jp107723w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 26.Gray HB, Winkler JR. Electron transfer in proteins. Annu Rev Biochem. 1996;65:537–561. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 27.Zhong D. Ultrafast catalytic processes in enzymes. Curr Opin Chem Biol. 2007;11:174–181. doi: 10.1016/j.cbpa.2007.02.034. [DOI] [PubMed] [Google Scholar]
- 28.Sancar A, Smith FW, Sancar GB. Purification of Escherichia coli DNA photolyase. J Biol Chem. 1984;259:6028–6032. [PubMed] [Google Scholar]
- 29.Heelis PF, Payne G, Sancar A. Photochemical properties of Escherichia coli DNA photolyase: selective photodecomposition of the second chromophore. Biochemistry. 1987;26:4634–4640. doi: 10.1021/bi00389a007. [DOI] [PubMed] [Google Scholar]
- 30.Jorns MS, Wang BY, Jordan SP, Chanderkar LP. Chromophore function and interaction in Escherichia coli DNA photolyase: reconstitution of the apoenzyme with pterin and/or flavin derivatives. Biochemistry. 1990;29:552–561. doi: 10.1021/bi00454a032. [DOI] [PubMed] [Google Scholar]
- 31.Langenbacher T, et al. Substrate and temperature dependence of DNA photolyase repair activity examined with ultrafast spectroscopy. J Am Chem Soc. 1997;119:10532–10536. [Google Scholar]
- 32.Kao Y-T, et al. Ultrafast dynamics and anionic active states of the flavin cofactor in cryptochrome and photolyase. J Am Chem Soc. 2008;130:7695–7701. doi: 10.1021/ja801152h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saxena C, Sancar A, Zhong D. Femtosecond dynamics of DNA photolyase: Energy transfer of antenna initiation and electron transfer of cofactor reduction. J Phys Chem B. 2004;108:18026–18033. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.